")
Back to Journals » Cancer Management and Research » Volume 12
DNAM1 and 2B4 Costimulatory Domains Enhance the Cytotoxicity of Anti-GPC3 Chimeric Antigen Receptor-Modified Natural Killer Cells Against Hepatocellular Cancer Cells in vitro
Authors Huang Y , Zeng J, Liu T, Xu Q, Song X , Zeng J
Received 12 March 2020
Accepted for publication 4 April 2020
Published 8 May 2020 Volume 2020:12 Pages 3247—3255
DOI https://doi.org/10.2147/CMAR.S253565
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eileen O'Reilly
Yao Huang,1,2 Jianxing Zeng,2 Teng Liu,2 Qingyi Xu,2 Xianglin Song,2 Jinhua Zeng1,2
1Department of Hepatobiliary Surgery, The First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian, People’s Republic of China; 2Department of Hepatic Surgery, Mengchao Hepatobiliary Hospital of Fujian Medical University, Fuzhou, Fujian, People’s Republic of China
Correspondence: Jinhua Zeng
Tel +86 13905918632
Email [email protected]
Purpose: Hepatocellular cancer (HCC) is the sixth most prevalent cancer and the third leading cause of cancer-related death worldwide. Cellular immunotherapy against glypican 3 (GPC3) has recently been used in the treatment of HCC, following the success of chimeric antigen receptor (CAR)-T therapy in treatment of B cell malignancy. However, CAR-T cells are not “off-the-shelf” and always cause cytokine release syndrome, which can be eliminated by using natural killer (NK) cells as effector cells. Since a costimulatory signal is necessary for the activation, persistence, or cytotoxicity of CAR-T cells, we speculated that the costimulatory signal is also required for CAR-NK cells in HCC treatment.
Methods: Five anti-GPC3 CAR plasmids containing different costimulatory domains were constructed. They included Z (only the CD3ζ domain, no costimulatory domain), CD28.Z (T-cell costimulatory domain CD28), DNAM1/2B4.Z (NK-cell-associated costimulatory domain DNAM1 or 2B4), and DNAM1.2B4.Z (both NK-cell-associated costimulatory domains). Respective CAR-NK-92 cells were generated. The MTT viability assay was performed to evaluate the effect of the different costimulatory domains on CAR-NK-cell proliferation. The effect on persistence was analyzed using an apoptosis assay and flow cytometry. Special cytotoxicity against normal hepatocellular cells and GPC3+ malignant cells was investigated in vitro. The concentration of cytokines (TNF-α and IFN-γ) released by CAR-NK-92 cells was also measured by ELISA.
Results: NK-cell-associated costimulatory signal was necessary for CAR-NK-92 cells. CAR-NK-92 cells with DNAM1 and/or 2B4 expanded more quickly and persisted with a lower apoptotic ratio, compared to the presence of CD28 or no costimulatory signal. All CAR-NK-92 cells showed special cellular cytotoxicity in vitro. CAR-NK-92 cells with NK-cell-associated costimulatory domains exhibited higher cytotoxic ability compared with those without any costimulatory domain or with T-cell costimulatory domain. CAR-NK-92 cells with both DNAM1 and 2B4 displayed the highest cytotoxicity. The cytokine release assay results were consistent with those of the cytotoxicity assay.
Conclusion: We provided the first evidence supporting a strategy using DNAM1 and 2B4 costimulatory domains to generate anti-GPC3 CAR-NK-92 cells, which exhibits enhanced cytotoxicity against hepatocellular cancer cells in vitro.
Keywords: HCC, CAR-NK, DNAM1, 2B4, proliferation, apoptosis, cytotoxicity
Introduction
Liver cancer is the sixth most prevalent cancer, with 850,000 new cases diagnosed annually worldwide.1 Most primary liver cancers are hepatocellular carcinoma (HCC), which constitutes 70% to 90% of liver cancers.2 Risk factors for HCC include hepatitis B or hepatitis C infection, cirrhosis, heavy drinking, obesity, diabetes, iron storage disease, and aflatoxin.3 The prognosis of HCC is usually poor with a mean survival time of 6 to 20 months and the ratio of mortality/incidence is close to 1.4 HCC is the cause of the third highest number of cancer-related deaths globally.5
In recent years, chimeric antigen receptor-T-cell (CAR-T) therapy has been rapidly developed for the treatment of cancer.6–10 Because of its success in the treatment of CD19+ hematological malignancies,11 CAR-T therapy has also been used to treat solid tumors, including HCC.12 However, clinical trials of CAR-T therapy for solid tumors have been disappointing. This is partially due to immunosuppressive tumor microenvironment, which is not present in blood cancers and which is an obstacle to treatment.13 Moreover, autologous T cells are expensive and delivery of CAR-T cells from the factory to patients is logistically challenging.14 Thus, for HCC immunotherapy, it is necessary to exploit allogeneic immune cells as an “off-the-shelf” product, which can be manufactured and then readily distributed to hospitals.
Natural killer (NK) cells may be candidate allogeneic immune cells, as they do not require human leukocyte antigen match or induce graft-versus-host disease. Furthermore, usage of CAR-NK cells does not induce cytokine release syndrome, which is an inevitable safety concern in all clinical trials of CAR-T therapy.15 However, isolation, purification, and expansion of primary NK cells are difficult,16 and the transfection efficiency of primary NK cells is poor.17 Thus, the NK-92, one of the NK cell lines, has been used as the effector cell in CAR-NK therapy, as it can expand more easily in vitro and is more effectively transfected.18
Glypican 3 (GPC3) is a member of the glypican family and an oncofetal glycoprotein that is attached to the cell surface by a glycophosphatidylinositol anchor.19 GPC3 is overexpressed in several types of tumors, particularly HCC.18 GPC3 has been used as a therapeutic target in immunotherapy for HCC.20
CAR commonly contains three domains. They are an extracellular antigen-binding domain composed of fragments of monoclonal antibodies that specially recognize tumor antigen (eg, GPC3 for HCC), a transmembrane domain, and an intracellular signaling transduction domain that is necessary to activate the effector function of the CAR-modified immune cells.21 In the first-generation CAR-T, the signal transduction domain contained only a CD3ζ, capable of transmitting “signal 1” for T-cell activation. Second- and third-generation CAR included one or more costimulatory molecules, such as 4-1BB or CD28, respectively.21 The costimulatory signal is important for the activation of CAR-T cells. These cells present different characteristics depending on the different costimulatory signals. The 4-1BB-costimulatory signal is necessary for CAR-T cells to persist for a longer time, whereas the CD28-costimulatory signal increases the cytotoxic ability of CAR-T cells.22
DNAM1, also named as CD226, was first discovered as a costimulatory receptor in cytotoxic T cells. It is also expressed by NK cells and other immune cells such as monocytes.23 The extracellular portion of DNAM1 contains two Ig-like domains and its cytoplasmic tail has three tyrosine residues. NK cell cytotoxicity is triggered by DNAM1 cross-linking which results in tyrosine-phosphorylation.24 It is reported that DNAM1 is strongly linked to the effector functions of NK cells. DNAM1+ NK cells produce significantly more IFN-γ IL-6, CCL5, and GM-CSF than DNAM1- NK cells.25 2B4, also known as CD244, is expressed by all NK cells, γδT cells, basophils, monocytes, and a subset of CD8+ αβT cells.26 CD244 is an Ig Superfamily Signaling Lymphocyte Activation Molecule (SLAM) family receptor. Its cytoplasmic domain contains four Immunoreceptor Tyrosine-based Switch Motifs (ITSMs) that interact with a variety of signaling adaptor molecules and transmit activating signals.27 It has been reported that 2B4 functions as a coreceptor in the activation of NK cells.28 Activation of 2B4 signaling induces the production of cytokines and invasiveness and modulates the cytotoxicity.29 In a word, DNAM1 and 2B4 are NK-cell-associated costimulatory molecules.
To present, the costimulatory domains of CAR structure used in the clinical study of CAR-NK therapy (NCT00995137, NCT01974479, NCT02892695, NCT02944162, NCT02742727, NCT02839954, NCT03383978, NCT03056339, NCT03579927) are T-cell costimulatory molecules. In our study, we used DNAM1 and 2B4 as costimulatory molecules. We hypothesized that DNAM1 and 2B4 are more suitable for CAR-NK (especially CAR-NK-92) and they play essential roles in the expansion and cytotoxic effect of CAR-NK-92 cells. To explore this, we constructed anti-GPC3 CARs with different costimulatory domains in NK-92 cells. As expected, 2B4 and DNAM1 were both suitable for CAR-NK-92 cells. These molecules promoted the proliferation and inhibited the apoptosis of NK-92 cells. Moreover, CAR-NK-92 cells with NK-cell-associated costimulatory molecules exhibited higher cytotoxicity, compared to cells without costimulatory molecules or with T-cell costimulatory molecule (CD28). The data will provide insight into the strategy of CAR construction in CAR-NK therapy for HCC.
Materials and Methods
Construction of CAR and Lentivirus Production
Figure 1A shows the schematic diagrams of the CARs. All the CARs carry a single-chain variable fragment (scFv) that recognizes human GPC3. The sequence of scFv was derived from the monoclonal antibody GC33.30 The scFv was fused to the hinge and transmembrane domains of human CD8 (nucleotides 412-609, GenBank NM 001768.6), which were directly linked to the intracellular signal domain of CD3ζ (nucleotides 154-492, GenBank NM 198253.2) in the Z CAR plasmid. In the CD28.Z CAR plasmid, the transmembrane and intracellular domains of CD28 (nucleotides 398-658, NM 006139.4) were fused to the intracellular signaling domain of CD3ζ. The sequences of DNAM1 (nucleotides 835-1080, GenBank NM 006566) and 2B4 (nucleotides 914-1335, GenBank NM 001166663) were cloned into the vector plasmid pCDH-EF1α-MCS to construct 2B4.Z CAR and DNAM1.Z CAR, respectively. The DNAM1.2B4.Z CAR plasmid contained the transmembrane and intracellular domains of DNAM1 and intracellular domain of 2B4 (nucleotides 976-1335, GenBank NM 001166663). Lentivirus was produced in 293T cells by a calcium phosphate transfection system31 and was concentrated by ultracentrifugation.32
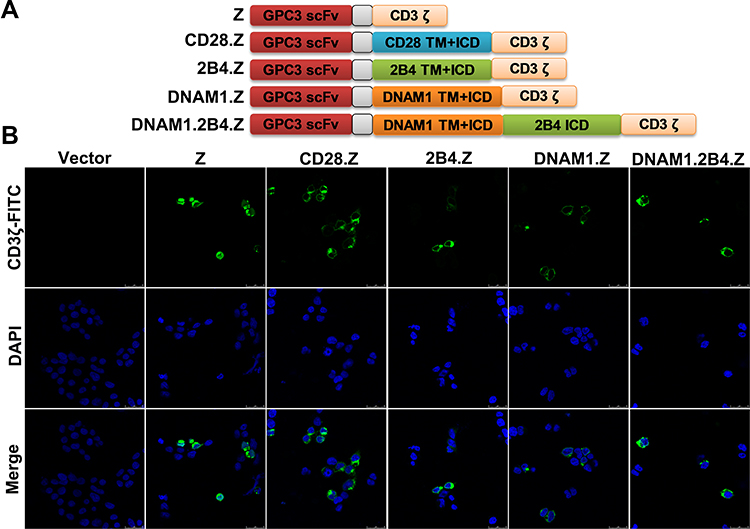 |
Figure 1 Construction of GPC3. (A) Schematic diagram of CAR structures based on lentiviral plasmid. (B) Expression of CAR structures. 293T cells were collected and stained 48 h after transfection with CAR plasmids. The CAR structures were predominantly expressed on the surface of 293T cells. |
Cell Lines and Cell Culture
HepG2, Hep3B, NK-92, and 293T cells were purchased from American Type Culture Collection (Manassas, VA, USA). L-02 and Huh7 cells were obtained from Antihela (Xiamen, Fujian, China). HepG2, Hep3B, 293T, L-02, and Huh7 cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco, Detroit, MI, USA) containing 10% fetal bovine serum (FBS, Gibco), 100 U/mL penicillin, 100 U/mL streptomycin, and 2 mM L-glutamine. NK-92 cells were cultured in Minimum Essential Medium-alpha (Gibco) without nucleosides but supplemented with 0.1 mM 2-mercaptoethanol, 0.2 mM inositol, 0.02 mM folic acid, 200 U/mL recombinant human interleukin-2 (IL-2) (Novoprotein, Shanghai, China), 12.5% horse serum (Gibco), and 12.5% FBS.
Immunofluorescent Staining
293T cells were fixed using 4% formaldehyde 24 h after transfection with CAR plasmids. To reduce non-specific binding, 5% bovine serum albumin (BSA, Gibco) in phosphate-buffered saline (PBS) was used to block the cells for 1 h after a 10-min treatment with 0.1% Triton X-100 at 28°C. The cells were stained with fluorescein isothiocyanate (FITC) coagulated anti-CD3ζ antibody (Biolegend, San Diego, CA, USA) for 30 min at 28°C, followed by staining of the nucleus using 5 ug/mL 4′,6-diamidino-2-phenylindol (Yeasen, Shanghai, China) for 10 min after three washes with PBS containing 0.1% Tween-20. After three washes, photographs were taken using a model TCS SP8 DLS camera (Leica, Wetzlar, Germany).
Establishment of Stable Cell Lines
NK-92 cells were infected with lentivirus carrying the different CAR plasmids at a multiplicity of infection of 20 by centrifugation (800×g, 2 h). The medium was supplemented with 10 ug/mL polybrene (Solarbio Life Science & Technology Co., Ltd., Beijing, China).
Staining of Membrane-Associated Proteins and Flow Cytometry Examination
L-02, HepG2, Huh7, and Hep3B cells were harvested and washed once with PBS containing 2% BSA. FITC conjugated anti-GPC3 antibody (R&D Systems, Shanghai, China) was used to stain cells at 25°C for 10 min. The stained cells were washed three times and analyzed by flow cytometry.
To stain NK-92 cells that stably expressed CAR, the cells were exposed to biotinylated human GPC3 antigen (AcroBiosystems, Beijing, China) to recognize the GPC3-scFv, followed by staining with FITC conjugated avidin (Thermo Fisher Scientific, Waltham, MA, USA).
Cell Proliferation Assay
NK-92 cells were seeded into 96-well plates (104 cells/well). After 0, 24, 48, and 72 h of culture, cell viability was detected using an CCK8 assay kit (A311-01; Vazyme, Nanjing, China) according to the manufacturer’s instructions. Absorbance was measured at 450 nm using a SpectraMax Absorbance Reader (Molecular Devices, Sunnyvale, CA, USA).
Apoptosis Assay
Harvested NK-92 cells were (1.0 × 106) were stained in the dark for 10 min at 28°C using 200 µg/mL Annexin V-FITC and 30 µg/mL propidium iodide (PI, Cat: C1062S; Beyotime, Shanghai, China). The cells were then subjected to flow cytometry analysis using a NovoCyte flow cytometer (Cat 1300; ACEA, San Diego, CA, USA). The results are presented as the percentage of apoptotic cells relative to all the analyzed cells.
Analysis of Direct Cytotoxicity
L-02, Huh7, and Hep3B cells were stained with 5 μM carboxyfluorosuccinimide ester (CFSE; Biolegend, San Diego, CA, USA). NK-92/Z, NK-92/CD28.Z, NK-92/2B4.Z, NK92/DNAM1.Z, or NK92/DNAM1.2B4.Z cells were incubated with these target cells at an effector-to target (E:T) ratio of 0.5:1, 1:1, 2:1, and 4:1. After a 12-h coculture, the cells were collected and stained with 30 µg/mL PI. The cell mixture was analyzed using the aforementioned NovoCyte flow cytometer. The proportion of CFSE+PI− cells was the residual of the target cells.
Detection of Released Cytokines by Enzyme-Linked Immunosorbent Assay (ELISA)
NK-92/Z, NK-92/CD28.Z, NK-92/2B4.Z, NK92/DNAM1.Z, or NK92/DNAM1.2B4.Z cells were cocultured with target cells at an E:T ratio of 1:1 for 24 h. The supernatant was harvested and the concentrations of interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) were measured using ELISA kits (R&D Systems).
Statistical Analysis
Analysis was performed using SPSS software 25.0 (IBM SPSS, Armonk, NY, USA). Unpaired Student’s t-test was used to compare the difference between pairwise samples. A p-value <0.05 was considered statistically significant.
Results
Construction of GPC3 CAR
Different generations of CARs were constructed in NK-92 cells against CPC3+ HCC cells (Figure 1A). The first-generation CAR contained only the CD3ζ domain, which was designated Z. Second-generation CARs also contained costimulatory domains. CAR with the T-cell costimulatory domain CD28 was designated CD28.Z, and CAR with NK-cell-associated costimulatory domain DNAM1 or 2B4 was designated DNAM1.Z and 2B4.Z, respectively. The third-generation CAR consisted of two costimulatory domains, DNAM1 and 2B4, and was designated DNAM1.2B4.Z. After construction, these CAR plasmids were transfected into 293T cells and their expression was assessed using immunofluorescence staining. The CAR structures were predominantly expressed on the surfaces of 293T cells (Figure 1B). These CAR plasmids could be packaged in CAR-structure lentivirus.
Production of Stably CAR-Expressing NK-92 Cells
NK-92 Cells Were Cultured and Infected with Lentivirus Carrying CAR Genes to Generated Stably CAR-Expressing NK Cells
As shown in Figure 2A, staining and flow cytometry demonstrated that each group had a similar proportion of CAR+ NK-92 cells. There was no significant difference in the median fluorescence intensity (MFI) of GPC3-scFv between each group, which excluded the effect of expression intensity of CAR structures (Figure 2B).
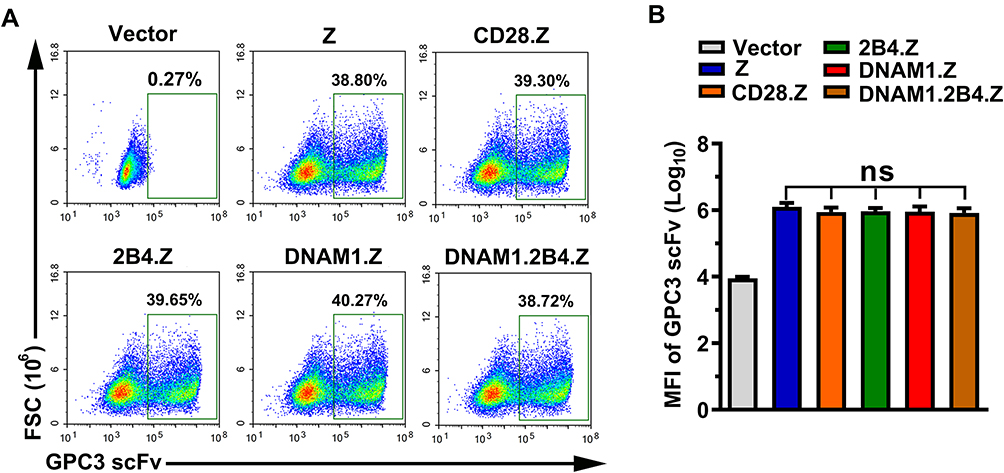 |
Figure 2 Generation of CAR+ NK cells. (A) Representative images obtained using flow cytometry. After a 72-h infection with lentivirus, NK-92 cells were stained and analyzed by flow cytometry. The positive population represented CAR+ NK-92 cells. (B) Median fluorescence intensity (MFI) of GPC3-scFv on CAR+ NK-92 cells in each group (n=3; ns, no significance). |
NK-Cell-Associated Costimulatory Signal Facilitates Proliferation of NK-92 Cells
To study the effect of different costimulatory signals on cellular proliferation in NK-92 cells, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed. CAR+ NK-92 cells with NK-cell-associated costimulatory signal significantly grew more rapidly, compared to cells with or without the T-cell costimulatory signal (Figure 3A). The CD28 costimulatory signal also promoted the growth of NK-92 cells (Figure 3A).
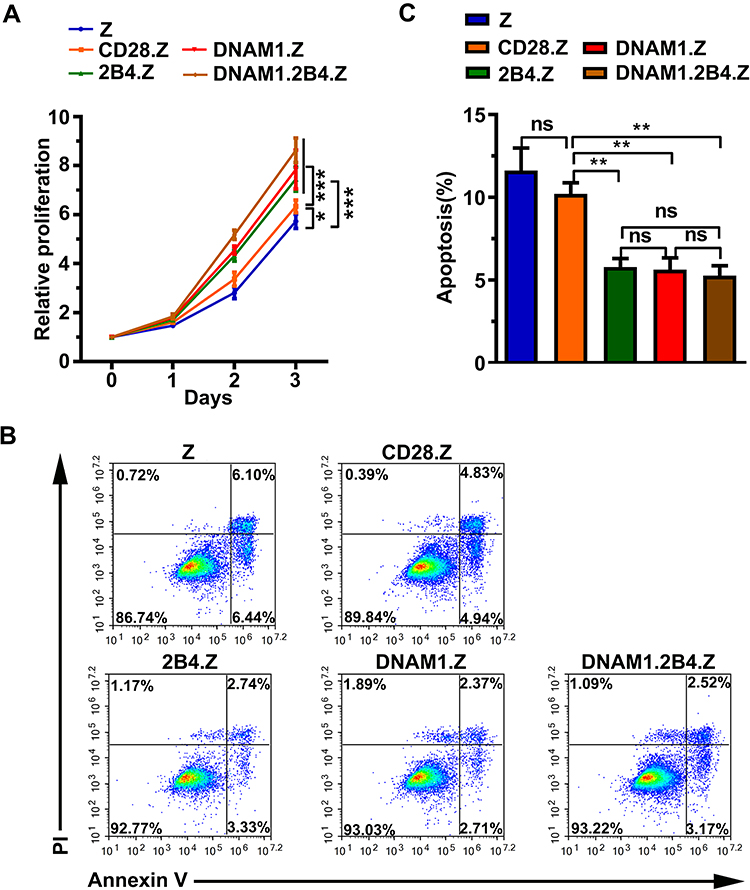 |
Figure 3 NK-cell costimulatory signal promotes cell proliferation and inhibits cell apoptosis in NK-92 cells. (A) CCK8 assay of cell proliferation. The data are presented as the mean ± standard deviation (s.d.) of quadruple wells. Unpaired two-tailed t-test: *p < 0.05, ***p < 0.001. (B) Representative images of apoptosis analysis by flow cytometry showing basal apoptosis of NK-92 cells. (C) Histogram of basal apoptosis in each group. The data are presented as the mean ± s.d. of triple wells. Unpaired two-tailed t-test: **p < 0.01. |
NK-Cell-Associated Costimulatory Signal Inhibits Apoptosis of NK-92 Cells
The flow cytometry-based apoptosis assay was performed to determine whether the NK-cell-associated costimulatory signal could inhibit cell apoptosis. The proportion of apoptotic cells in the DNAM1.Z, 2B4.Z, and DNAM1.2B4.Z groups was obviously smaller than that in the Z group (Figure 3B–C), indicating that the NK-cell-associated costimulatory signal suppressed the apoptosis of NK-92 cells.
Expression Level of GPC3 in HCC Cell Lines
Target cells must be chosen according to the expression level of GPC3. Therefore, we evaluated the expression of GPC3 in the L-02 normal hepatic cell line and HepG2, Huh7, and Hep3B HCC cell lines. As shown in Figure 4A and B, the expression of GPC3 was nearly undetectable in L-02 cells, while all the HCC cells expressed GPC3 on their membranes. The expression of GPC3 was highest for HepG2 cells, followed by Hep3B and Huh7 cells (Figure 4C). Based on these data, L-02 cells were chosen as the negative control and HepG2 and Huh7 as positive target cells.
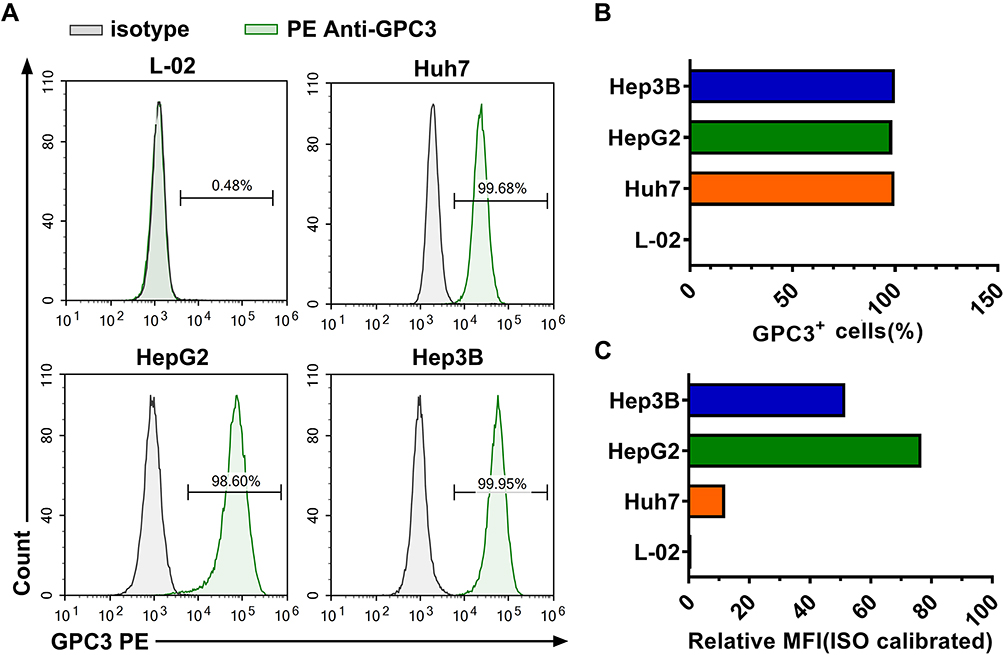 |
Figure 4 Expression level of GPC3 in different cell lines. (A) Representative images of flow cytometry analysis of GPC3 expression. (B) Proportion of GPC3+ cells in different cell lines. (C) Histogram of GPC3 MFI in different cell lines. |
Enhanced Cytotoxicity of CAR+NK-92 Cells with the NK-Cell-Associated Costimulatory Domain
Flow cytometry was used to determine whether CAR+NK-92 cells specifically recognized and killed GPC3-positive HCC cells (Supplement Figures 1–3). CAR+NK-92 cells displayed no significant cytotoxicity against L-02 cells, even at the high E:T ratio of 4:1, whereas obvious cytotoxicity was evident against Huh7 and HepG2 cells (Figure 5A). HepG2 cells were more sensitive to CAR+NK-92 cells. Moreover, CAR+NK-92 cells with the NK-cell-associated costimulatory domain possessed higher cytotoxicity than those with T-cell costimulatory domain (Figure 5A). NK-92/DNAM1.2B4.Z exhibited the highest cytotoxic activity (Figure 5A). The collective data suggested that the anti-tumor activity of CAR+NK-92 cells depended on the expression of special antigens on target cells, and that the NK-cell-associated costimulatory signal was more efficacious for the cytotoxicity of NK-92 cells. TNF-α and IFN-γ are important cytokines secreted by NK cells that are associated with the cytotoxic activity of NK cells.33,34 Thus, the levels of both cytokines were detected. As shown in Figure 5B, CAR+NK-92 cells secreted these cytokines only when cocultured with Huh7 and HepG2 cells, and the levels of cytokine released by CAR+NK-92 cells with NK-cell-associated costimulatory domain were higher, compared to with T-cell costimulatory domain. NK-92/DNAM1.2B4 secreted the highest levels of IFN-γ and TNF-α (Figure 5B). The results were consistent with the flow cytometry cytotoxic analysis.
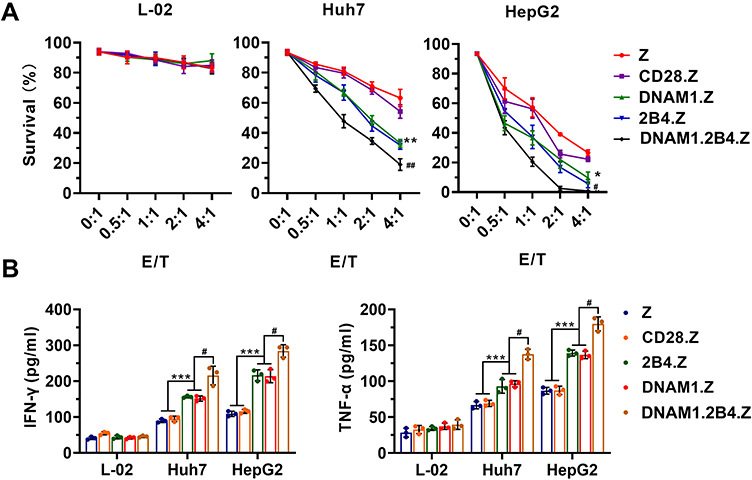 |
Figure 5 Enhanced cytotoxicity of CAR+NK-92 cells with NK-cell costimulatory domain. (A) Semi-quantification of residual target cells at indicated E/T ratio. The data are presented as the mean ± s.d. of triplicate wells. *p < 0.05, **p < 0.01, Z or CD28.Z group vs DNAM1.Z or 2B4.Z group; #p < 0.05, ##p < 0.05, DNAM1.Z or 2B4.Z group vs DNAM1.2B4.Z group. (B) Detection of released IFN-γ and TNF-α by ELISA after a 24-h coculture at an E/T of 1/1. The data are presented as the mean ± s.d. of triplicate wells. Unpaired two-tailed t-test: ***p < 0.001, Z or CD28.Z group vs DNAM1.Z or 2B4.Z group; #p < 0.05, DNAM1.Z or 2B4.Z group vs DNAM1.2B4.Z group. |
Discussion
HCC constitutes the majority of primary liver cancers. It is the sixth most prevalent cancer worldwide and the third leading cause of cancer-related death.35 Treatment of HCC is limited and the prognosis is usually poor. The development of immunotherapy, especially CAR-T therapy, has appreciably improved the success of the therapy of CD19+ hematological malignancies, and has created new choices for HCC therapy. However, clinical trials have revealed inevitable drawbacks in CAR-T therapy, which has increased the focus on the CAR-NK therapy. The data from many studies have proven that a costimulatory signal is necessary for the activation and anti-malignant ability. Whether this is the case with CAR-NK therapy has been unclear. The present findings confirm that the costimulatory signal is essential for the expansion, persistence, and cytotoxicity of CAR-NK-92 cells. DNAM1 and 2B4 appear to be efficient costimulatory molecules. We are constructing animal models with orthotopic hepatocellular carcinoma, and our next work is to investigate the effect of anti-GPC3 CAR-NK-92 in vivo.
Both NK cells derived from peripheral blood and the NK-92 cell line have been successfully engineered to express CARs. Both types have disadvantages and advantages. NK-92 cells seem more economical and convenient. However, CAR-NK-92 cells must be irradiated before injection to prevent potential carcinogenicity, which shortens their survival in patients.16,17 Multiple injections may help prolong the survival time.18
Rational selection of tumor-specific antigens is the initial and key step. GPC3 is highly expressed in 70% of HCCs and is not detected in normal liver tissues.19 We confirmed this in the present study (Figure 4). The level of GPC3 is usually linked to the progression of HCC, and it expression can become high again after relapse.19 Antibodies against GPC3 and GPC3-peptide vaccines have been demonstrated to be safe and effective.36,37 Presently, CAR-NK-92 against GPC3 especially killed GPC3+ HCC cells and did not harm GPC3− normal hepatocellular cells, suggesting that GPC3 is a suitable target in CAR-NK therapy.
Solid tumors have a complex immune microenvironment that differs from hematological malignancies. This environment can impede filtration and immune suppression, limiting the efficacy of CAR-NK therapy.38 Combined treatments with radiotherapy, chemotherapy, or immune checkpoint inhibitors will be a promising strategy to improve treatment efficacy.39
Conclusion
We provided the first evidence supporting a strategy using DNAM1 and 2B4 costimulatory domains to generate anti-GPC3 CAR-NK-92 cells, which exhibits enhanced cytotoxicity against hepatocellular cancer cells in vitro.
Funding
This work is supported by the Backbone Talents Training Project of Fujian Health Department (Grant No. 2019-ZQN-65).
Disclosure
All authors report no conflicts of interest in this work.
References
1. Zhu RX, Seto WK, Lai CL, Yuen MF. Epidemiology of hepatocellular carcinoma in the Asia-Pacific Region. Gut Liver. 2016;10(3):332–339. doi:10.5009/gnl15257
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
3. Tang A, Hallouch O, Chernyak V, Kamaya A, Sirlin CB. Epidemiology of hepatocellular carcinoma: target population for surveillance and diagnosis. Abdom Radiol (NY). 2018;43(1):13–25. doi:10.1007/s00261-017-1209-1
4. Bialecki ES, Di Bisceglie AM. Diagnosis of hepatocellular carcinoma. HPB (Oxford). 2005;7(1):26–34. doi:10.1080/13651820410024049
5. Zhang J-N, Qin X, Liu X-G, et al. Identification of abnormally expressed genes and their roles in invasion and migration of hepatocellular carcinoma. Aging (Albany NY). 2020;12. doi:10.18632/aging.102727
6. Brudno JN, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for lymphoma. Nat Rev Clin Oncol. 2018;15(1):31–46. doi:10.1038/nrclinonc.2017.128
7. Ghobadi A. Chimeric antigen receptor T cell therapy for non-Hodgkin lymphoma. Curr Res Transl Med. 2018;66(2):43–49. doi:10.1016/j.retram.2018.03.005
8. Hosen N. Chimeric antigen receptor T-cell therapy for multiple myeloma. Rinsho Ketsueki. 2018;59(10):2189–2194. doi:10.11406/rinketsu.59.2189
9. Chen Y, Chang-Yong E, Gong Z-W, et al. Chimeric antigen receptor-engineered T-cell therapy for liver cancer. Hepatobiliary Pancreat Dis Int. 2018;17(4):301–309. doi:10.1016/j.hbpd.2018.05.005
10. Gorchakov AA, Kulemzin SV, Kochneva GV, Taranin AV. Challenges and prospects of chimeric antigen receptor T-cell therapy for metastatic prostate cancer. Eur Urol. 2019. S0302-2838(0319)30658-X
11. Jakobczyk H, Sciortino F, Chevance S, Gauffre F, Troadec M-B. Promises and limitations of nanoparticles in the era of cell therapy: example with CD19-targeting chimeric antigen receptor (CAR)-modified T cells. Int J Pharm. 2017;532(2):813–824. doi:10.1016/j.ijpharm.2017.07.075
12. Ma W, Wu L, Zhou F, Hong Z, Yuan Y, Liu Z. T cell-associated immunotherapy for hepatocellular carcinoma. Cell Physiol Biochem. 2017;41(2):609–622. doi:10.1159/000457883
13. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423–431. doi:10.1038/nature22395
14. Siegler EL, Zhu Y, Wang P, Yang L. Off-the-shelf CAR-NK cells for cancer immunotherapy. Cell Stem Cell. 2018;23(2):160–161. doi:10.1016/j.stem.2018.07.007
15. Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25(8):1769–1781. doi:10.1016/j.ymthe.2017.06.012
16. Zeng J, Tang SY, Toh LL, Wang S. Generation of “Off-the-Shelf” natural killer cells from peripheral blood cell-derived induced pluripotent stem cells. Stem Cell Rep. 2017;9(6):1796–1812. doi:10.1016/j.stemcr.2017.10.020
17. Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy - advantages of the NK-92 cell line over blood NK cells. Front Immunol. 2016;7:91. doi:10.3389/fimmu.2016.00091
18. Xu Y, Liu Q, Zhong M, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12(1):49. doi:10.1186/s13045-019-0732-7
19. Filmus J, Capurro M. Glypican-3: a marker and a therapeutic target in hepatocellular carcinoma. FEBS J. 2013;280(10):2471–2476. doi:10.1111/febs.12126
20. Nishida T, Kataoka H. Glypican 3-targeted therapy in hepatocellular carcinoma. Cancers (Basel). 2019;11(9):1339. doi:10.3390/cancers11091339
21. Stoiber S, Cadilha BL, Benmebarek M-R, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. 2019;8(5):472. doi:10.3390/cells8050472
22. Guedan S, Posey AD
23. Sanchez-Correa B, Valhondo I, Hassouneh F, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers (Basel). 2019;11:6. doi:10.3390/cancers11060877
24. Shibuya A, Campbell D, Hannum C, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity. 1996;4(6):573–581. doi:10.1016/S1074-7613(00)70060-4
25. Huntington ND, Martinet L, Smyth MJ. DNAM-1: would the real natural killer cell please stand up! Oncotarget. 2015;6(30):28537–28538. doi:10.18632/oncotarget.5952
26. Assarsson E, Kambayashi T, Persson CM, Ljunggren HG, Chambers BJ. 2B4 co-stimulation: NK cells and their control of adaptive immune responses. Mol Immunol. 2005;42(4):419–423. doi:10.1016/j.molimm.2004.07.021
27. Agresta L, Hoebe KHN, Janssen EM. The emerging role of CD244 signaling in immune cells of the tumor microenvironment. Front Immunol. 2018;9:2809. doi:10.3389/fimmu.2018.02809
28. Sivori S, Parolini S, Falco M, et al. 2B4 functions as a co-receptor in human NK cell activation. Eur J Immunol. 2000;30(3):787–793. doi:10.1002/1521-4141(200003)30:3<787::AID-IMMU787>3.0.CO;2-I
29. Chuang SS, Kumaresan PR, Mathew PA. 2B4 (CD244)-mediated activation of cytotoxicity and IFN-gamma release in human NK cells involves distinct pathways. J Immunol. 2001;167(11):6210–6216. doi:10.4049/jimmunol.167.11.6210
30. Nakano K, Ishiguro T, Konishi H, et al. Generation of a humanized anti-glypican 3 antibody by CDR grafting and stability optimization. Anticancer Drugs. 2010;21(10):907–916. doi:10.1097/CAD.0b013e32833f5d68
31. Wang H, Zhou M, Shi B, et al. Identification of an exon 4-deletion variant of epidermal growth factor receptor with increased metastasis-promoting capacity. Neoplasia (New York, NY). 2011;13(5):461–471. doi:10.1593/neo.101744
32. Ichim CV, Wells RA. Generation of high-titer viral preparations by concentration using successive rounds of ultracentrifugation. J Transl Med. 2011;9:137. doi:10.1186/1479-5876-9-137
33. Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65(10):3980–3985. (). doi:10.1158/0008-5472.CAN-04-3995
34. Wang R, Jaw JJ, Stutzman NC, Zou Z, Sun PD. Natural killer cell-produced IFN-gamma and TNF-alpha induce target cell cytolysis through up-regulation of ICAM-1. J Leukoc Biol. 2012;91(2):299–309. doi:10.1189/jlb.0611308
35. Genco C, Cabibbo G, Maida M, et al. Treatment of hepatocellular carcinoma: present and future. Expert Rev Anticancer Ther. 2013;13(4):469–479. doi:10.1586/era.13.21
36. Ikeda M, Ohkawa S, Okusaka T, et al. Japanese Phase I study of GC33, a humanized antibody against glypican-3 for advanced hepatocellular carcinoma. Cancer Sci. 2014;105(4):455–462. doi:10.1111/cas.12368
37. Wu Q, Pi L, Le Trinh T, et al. A novel vaccine targeting glypican-3 as a treatment for hepatocellular carcinoma. Mol Ther. 2017;25(10):2299–2308. doi:10.1016/j.ymthe.2017.08.005
38. Zhang Q, Zhang H, Ding J, et al. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J Immunol Res. 2018;2018:4263520. doi:10.1155/2018/4263520
39. Xu J, Tian K, Zhang H, et al. Chimeric antigen receptor-T cell therapy for solid tumors require new clinical regimens. Expert Rev Anticancer Ther. 2017;17(12):1099–1106. doi:10.1080/14737140.2017.1395285
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.