Back to Journals » Journal of Pain Research » Volume 13
DNA Microarray Analysis of Differential Gene Expression in the Dorsal Root Ganglia of Four Different Neuropathic Pain Mouse Models
Authors Yokoyama H, Hirai T ![]() , Nagata T, Enomoto M, Kaburagi H
, Nagata T, Enomoto M, Kaburagi H ![]() , Leiyo L, Motoyoshi T, Yoshii T, Okawa A, Yokota T
, Leiyo L, Motoyoshi T, Yoshii T, Okawa A, Yokota T
Received 2 August 2020
Accepted for publication 22 October 2020
Published 20 November 2020 Volume 2020:13 Pages 3031—3043
DOI https://doi.org/10.2147/JPR.S272952
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Robert B. Raffa
Hiroyuki Yokoyama,1 Takashi Hirai,1 Tetsuya Nagata,2 Mitsuhiro Enomoto,1 Hidetoshi Kaburagi,1 Li Leiyo,1 Takayuki Motoyoshi,1 Toshitaka Yoshii,1 Atsushi Okawa,1 Takanori Yokota2
1Department of Orthopedic Surgery, Tokyo Medical and Dental University, Tokyo 113-8519, Japan; 2Department of Neurology and Neurological Science, Tokyo Medical and Dental University, Tokyo 113-8519, Japan
Correspondence: Takashi Hirai; Takanori Yokota Email [email protected]; [email protected]
Purpose: Pathological stimuli or injury to the peripheral nervous system can trigger neuropathic pain with common clinical features such as allodynia and hypersensitivity. Although various studies have identified molecules or genes related to neuropathic pain, the essential components are still unclear. Therefore, in this study, we investigated the molecular and genetic factors related to neuropathic pain.
Methods: We extracted candidate genes in the dorsal root ganglion (DRG) from three nerve injury mouse models and a sham-operated model (sciatic nerve ligation and resection, sural nerve resection, spared nerve injury [SNI], and sham) using DNA microarray to elucidate the genes responsible for the neuropathic pain mechanism in the SNI model, which exhibits hypersensitivity in the hindpaw of the preserved sural nerve area. We eliminated as many biases as possible. We then focused on an upregulated endogenous vasopressin receptor and clarified whether it is closely associated with traumatic neuropathic pain using a knockout mouse and drug-mediated suppression of the gene.
Results: Algorithm analysis of DNA microarray results identified 50 genes significantly upregulated in the DRG of the SNI model. Two independent genes—cyclin-dependent kinase-1 (CDK-1) and arginine vasopressin receptor 1A (V1a)—were subsequently identified as candidate SNI-specific genes in the DRG by quantitative PCR analysis. Administration of V1a agonist to wild-type SNI mice significantly alleviated neuropathic pain. However, V1a knockout mice did not exhibit higher hypersensitivity to mechanical stimulation than wild-type mice. In addition, V1a knockout mice showed similar pain behaviors after SNI to wild-type mice.
Conclusion: Through the DNA microarray analysis of several neuropathic models, we detected specific genes related to chronic pain. In particular, our results suggest that V1a in the DRG may partially contribute to the mechanism of neuropathic pain.
Keywords: neuropathic pain, arginine vasopressin 1a, peripheral nerve injury, dorsal root ganglion, microarray, molecular target
Introduction
Neuropathic pain is a form of chronic pain caused by conditions affecting the somatosensory nervous system.1 It is clinically characterized by allodynia (ie, hypersensitivity to nociceptive stimulation). Various drugs, including opioids, gabapentinoid, duloxetine, and acetaminophen, are widely used for the treatment of chronic neuropathic pain.2–5 However, neuropathic pain remains a chronic refractory disease with no known curative treatment. Indeed, although numerous studies have identified various putative receptors, neurotrophic factors, and neuropeptides that are considered to be important molecules underlying the mechanisms of chronic pain, debate continues regarding the essential therapeutic target in neuropathic pain. Accordingly, numerous research groups around the world are attempting to unravel the molecular mechanisms underlying the onset and maintenance of painful neuropathy.
Several animal models of neuropathic pain involving sciatic nerve injury are widely used worldwide. In the spared nerve injury (SNI) model,6 the common and tibial nerves are cut and the sural nerve is preserved. The SNI model is a unique model of nerve injury because it exhibits strong mechanical hypersensitivity in the sural nerve dominant area. Interestingly, the sciatic nerve transection model,7 which involves total cross section of the sciatic nerve, shows lost perception in the hindpaw plantar. From this evidence, we hypothesized that some transcriptional changes that mediate mechanical hypersensitivities might occur in the dorsal root ganglion (DRG) of the preserved sural nerve.8 Therefore, we wanted to determine the mechanism underlying the neuropathic pain. New strategies based on global analysis of nucleic acids have been developed and applied to identify crucial factors in specific pathologies, and numerous studies have documented alterations in the expression of candidate genes for diseases in the peripheral nervous system. To date, most investigations have simply compared pathological and sham-operated or normal tissues. However, there are likely to be some biases in such head-to-head comparisons.
Arginine vasopressin (AVP) is a seven-transmembrane domain G-coupled polypeptide that has been demonstrated to play roles in many neurological functions, such as autism,9 sex behavior,10 schizophrenia,11 aggression, and pain. Intracerebroventricular12 and intrathecal13 administration of AVP produces behavioral analgesia in rodent models. A canonical animal study14 demonstrated that AVP-deficient Brattleboro rats have hyperalgesia and diminished stress-induced analgesia. Thus, systemic AVP contributes to animal behaviors that attempt to avoid nociceptive pain in normal and pathological states of various animal models. In particular, arginine vasopressin receptor 1A (V1a) is involved in nociceptive pain modulation. V1a has been characterized as a receptor that can bind not only vasopressin, but also oxytocin with high affinity.15 Some work16 has revealed that V1a activation by either oxytocin or vasopressin can alleviate painful behaviors in rodent models and humans. In a human study, patients with back pain reported amelioration of self-reported pain after intrathecal administration of oxytocin.17 A rodent model study demonstrated that upregulation of peripheral V1a caused by endogenous oxytocin decreases depolarization-induced calcium transients in DRG neurons, suggesting that oxytocin may suppress peripheral nerve activity via V1a receptor in inflammatory pain states.16,18 However, no study has investigated the association between V1a and neuropathic pain.
Accordingly, in this preclinical study, we compared three different nerve injury models and a sham-operated model in the mouse DRG using DNA microarray analysis. We then generated a knockout (KO) mouse for the identified pain-associated neuropathy-specific gene and evaluated whether it is closely associated with traumatic neuropathic pain.
Materials and Methods
Ethics Statement
All animal experiments were approved by the Animal Experiment Committee of Tokyo Medical and Dental University and performed in accordance with its Ethical and Safety Guidelines.
Model Establishment
A total of 64 male and female C57BL/6J mice aged 8 to 14 weeks and weighing approximately 20 g were used in this study. In total, 24 mice were used for behavioral tests, 12 for microarray analysis, 16 for PCR assay, and 12 for immunohistochemical experiments. In addition, 8 male and female B6.129Sv-Avpr1atm1 (V1a KO mice) aged 8 to 14 weeks and weighing approximately 20 g were also used. V1a KO mice were obtained in the form of frozen embryos from the Institute of Resource Development and Analysis at Kumamoto University.19 All mice were kept in standard cages at a constant temperature and light/dark cycle of 12 h each, with the light/dark hours changing automatically, and were given water and food ad libitum throughout the experimental period. From 1 week before and throughout the experimental period, the mice were housed in a sterile room with an ambient temperature of 25°C to prevent infection and minimize external stimulation. The mice were randomly divided into three different peripheral nerve injury models and a sham-operated model: the sciatic nerve ligation and resection model (group 1, n = 3), the sural nerve resection model (group 2, n = 3), the SNI model (group 3, n = 3), and the sham model (group 4, n = 3) (Figure 1). Hindpaw hypersensitivity was observed only in the SNI model (Supplementary Figure 1). In addition to these models, a partial sciatic nerve ligation (PSNL) model was used to compare neuropathic pain models in terms of V1a expression in the DRG.20
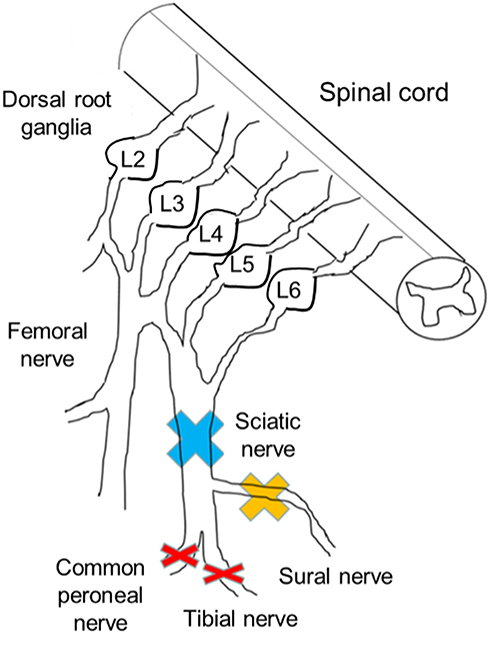 |
Figure 1 Injury sites of each peripheral nerve injury model. The sciatic nerve branches into the tibial nerve, peroneal nerve, and sural nerve. The sural nerve includes only sensory fibers. In model 1, the sciatic nerve was ligated and resected (blue cross). In model 2, the sural nerve was resected (yellow cross). In model 3 (the spared nerve injury [SNI] model), both motor nerves were resected and the sensory nerve was preserved (red cross). In model 4, the animals underwent a sham operation. Only the SNI models showed mechanical hypersensitivity. Three weeks after the surgery, we isolated lumbar dorsal root ganglia (L3, 4, and 5) and compared DNA expression patterns among the groups. |
Surgical Procedure
The mice were anesthetized under a 1.5% isoflurane (Forane; Baxter, Deerfield, IL) gas mixture with 1.5 L/min room airflow. An incision was made on the left lateral mid-thigh, and the underlying muscles were separated to expose the sciatic nerve. In the sciatic nerve ligation and resection model, the sciatic nerve was tight-ligated with 8–0 silk and resected. In the sural nerve resection model, only the sural nerve was resected. In the SNI group, the common peroneal and tibial nerves were tight-ligated with 8.0 silk and sectioned distally to the ligation to remove 2–4 mm of the distal nerve stump. Care was taken to avoid any contact with or stretching of the intact sural nerve. In the sham group, only the nerves were exposed. Then, the muscles and skin were closed in two layers using 4–0 monofilament nylon sutures. After the operation, the animals were allowed to recover in their own cages. The observation period was 6 weeks.
DNA Microarray for the Lumbar DRG
Three different nerve injury models and a sham-operated model were created for wild-type (WT) C57BL/6J mice (n = 3 each) and DNA microarrays were performed. The relationships between the increased or decreased genes in each model are shown in a Venn diagram (Figure 2A and B). Briefly, we first compared the SNI and sciatic nerve transection models to detect signals associated with hypersensitivities caused by sural nerve preservation. We then compared the SNI and sham models to clarify signals related to hypersensitivities mediated with ligation of the tibial and peroneal nerves. Of these genes, we excluded those in which there were significant differences between the sham and sural nerve transection models to eliminate death signals in the sural nerve. The area of the right oval minus the center circle was extracted as SNI-specific genes (Figure 2A and B).
 |
Figure 2 Venn diagram of nerve injury models. The right oval area subtracted from the center circle was extracted as spared nerve injury-specific genes ((A), upregulated; (B), downregulated). SNI, spared nerve injury. (C) Of the 50 SNI-specific genes showing an increase, 2 were validated using real-time PCR. Cdk1 and Avpr1a were significantly increased compared with the sham model. |
The total RNA of different groups was individually hybridized with gene chips. Briefly, each chip was hybridized with 1.65 μg of Cy3-labeled cRNA by using a Gene Expression Hybridization Kit (Cat# 5188–5242, Agilent Technologies, Santa Clara, CA, USA) in a hybridization oven (Cat# G2545A, Agilent Technologies) according to the manufacturer’s instructions. After 17 h of hybridization, the chips were washed in staining dishes (Cat# 121, Thermo Shandon, Waltham, MA, USA) with a Gene Expression Wash Buffer Kit (Cat# 5188–5327, Agilent Technologies) following the manufacturer’s instructions. The chips were scanned with an Agilent Microarray Scanner (Cat# G2565CA, Agilent Technologies). Array data were processed using SurePrint G3 Mouse (Agilent Technologies) and imported into GeneSpring (Agilent Technologies) for analysis. Data were filtered to remove probes that did not reach a threshold. Each gene group was compared, and genes with 1.5-fold or higher and 0.5-fold or lower changes and showing a significant difference were extracted while controlling for the false discovery rate (p<0.05, t-test with Benjamini–Hochberg multiple testing correction).
Tactile Threshold
Mechanical sensitivity was measured by applying a series of calibrated von Frey filaments (0.16–6 g) to the plantar aspect of the hindpaw.21 Each filament was applied once to each mouse. Beginning with the 0.16 g filament, each filament was applied perpendicularly to the hindpaw for 4–6 s. A brisk withdrawal of the hindpaw indicated a positive response, and a lack of withdrawal indicated a negative response. The filament testing was performed three times, and at least two responses to the filament out of the three trials indicated an overall positive response. If no overall positive response was observed (0 or 1 of 3 responses), the filament with the next highest force was applied as described above.
Measurement of mRNA Expression by Quantitative RT-PCR
Total RNA was extracted from the harvested tissues, including the lumbar DRG, using Isogen (Nippon Gene, Tokyo, Japan). The DRG RNA samples were obtained from the L3, L4, and L5 DRG. From the 50 upregulated genes in the DRG, we performed quantitative RT-PCR analysis to identify the candidate genes for generating knockout mice. SNI model cDNA was amplified using the quantitative SYBR Green system on a LightCycler 480 Real-Time PCR Instrument according to the manufacturer’s protocol (Roche Diagnostics, Basel, Switzerland). As an internal control, GAPDH cDNA was also quantified.
Pharmacology and Compounds
For pharmacological experiments with V1a agonist and antagonist, baseline measures were taken before intraperitoneal injection of the drugs. The agonist was [Phe2, Ile3, Orn8]-vasopressin (AVP)/[Phe2, Orn8]-vasotocin. The antagonist was SR49059 ((2S) 1-[(2R 3S)-5-chloro-3-(2-chlorophenyl)-1-(3,4-dimethoxybenzene-sulfonyl)-3-hydroxy-2,3-dihydro-1H-indole-2-carbonyl]-pyrrolidine-2-carboxamide).22,23 Measurements were obtained at 30-min intervals up to 120 min. Agonist experiments were conducted at three doses (0.1 mg/kg, 0.5 mg/kg, and 1.0 mg/kg), whereas antagonist experiments were conducted at two doses (1.0 mg/kg and 5.0 mg/kg) on different days.11
Immunohistochemistry of the Lumbar DRG
Mice were sacrificed and subjected to transcardial perfusion of phosphate-buffered saline (PBS) for 5 min at room temperature, followed by 4% paraformaldehyde in PBS for 15 min at 4°C. After perfusion, the lumbar DRGs were immediately removed and postfixed in 4% paraformaldehyde in PBS at 4°C overnight.
After postfixation, the samples were transferred to PBS containing 30% sucrose and dehydrated for 3 days. The tissues were then embedded in optimal cutting temperature compound and cut into 20-μm longitudinal sections using a cryostat. After treatment with 0.2% Triton X-100 for 5 min, the DRG sections were blocked with 5% normal goat serum (NGS; Vector Laboratories, Burlingame, CA) and then incubated with a mouse monoclonal anti-V1a receptor (1:300; LSBio, Seattle, US) and rabbit polyclonal anti-calcitonin gene-related peptide (CGRP, 1:300; BMA Biomedicals, Augst, Switzerland) or rabbit polyclonal anti-S100 (1:300, Leica Biosystems, Wetzlar, Germany) for 24 h at 4°C. Following incubation, the antibody was visualized with goat anti-mouse IgM Alexa 594 (1:300; Invitrogen, Carlsbad, CA) for 1 h at room temperature. Images were obtained on an AX70 Olympus microscope (Olympus Optical Co., Ltd., Tokyo, Japan). ImageJ (https://imagej.nih.gov/ij/, Wayne Rasband, NIH) was used to quantify the V1a-positive area in each model.
Statistical Analysis
The Student’s t-test for between-group comparisons and one- or two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons for multiple-group comparisons were used to analyze RT-PCR data, with p<0.05 accepted as statistically significant. Repeated-measures ANOVA was used to compare the behavioral test results of the four animal models.
Results
DNA Microarray for the Lumbar DRG and Its Validation by Real-Time PCR
Three different nerve injury models and a sham-operated model were created using WT C57BL/6J mice (n = 3 each) and DNA microarrays were performed. The results of the DNA microarray were available (Supplementary file) and analyzed according to the indicated flowchart (Figure 2A). Compared with the sciatic nerve transection model, 220 genes were significantly upregulated in the SNI model. Of these genes, 54 also showed a significant increase in the SNI model compared with the sham model. To eliminate death signaling pathways from the affected DRG, the gap between the sham and sural nerve transection model was removed from these genes. Finally, 50 upregulated genes were extracted as differentially expressed genes involved in SNI neuropathic pain (Table 1). In addition, 20 downregulated genes were identified as differentially expressed genes after their analysis under the same strategy (Figure 2B and Table 2).
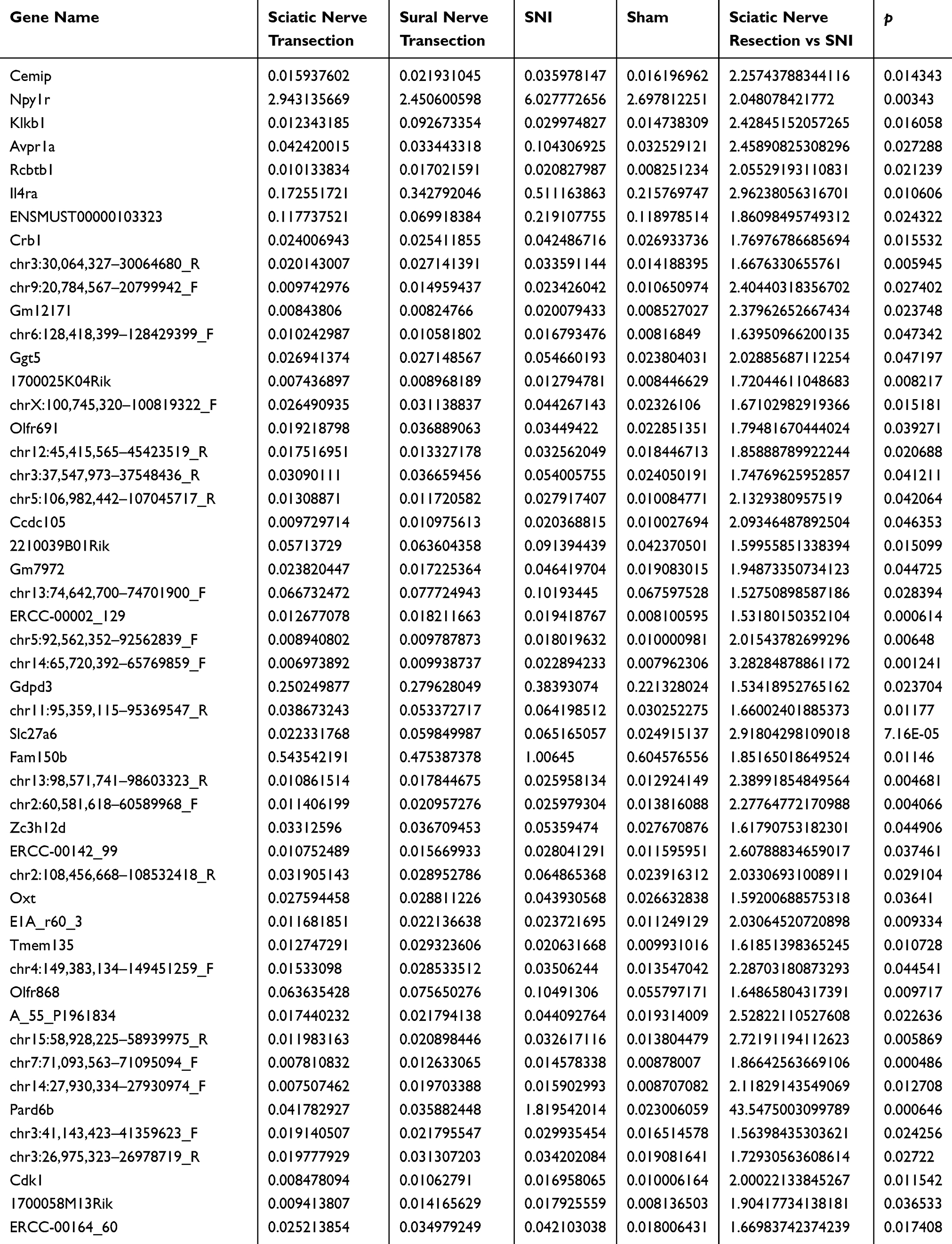 |
Table 1 SNI-Specific Upregulated Genes (n = 50) Extracted by Analysis of DNA Microarray Results |
 |
Table 2 SNI-Specific Downregulated Genes (n = 20) Extracted by Analysis of DNA Microarray Results |
To validate the genes upregulated in neuropathic pain models via a canonical strategy to generate knockout mice of the candidate upregulated genes, 2 of the 50 genes, which encode common proteins, specifically CDK1 and V1a, were quantified by real-time PCR (Figure 2C). We then compared the results with those of the sham model, and CDK1 and V1a showed a significant increase in SNI animals.
Altered Expression of Avpr1a in the DRG
As a result of the microarray results, we focused on V1a as a putative gene related to the neuropathic pain mechanism. We, therefore, quantified chronological changes in V1a between SNI and sham models in the affected L3–5 DRG by real-time PCR. V1a was increased by about 2.2-fold at 3 weeks and by about 1.5-fold at 6 weeks after surgery in the SNI model (Figure 3). To clarify whether the differential expression was also evident in a different neuropathic pain model, V1a expression was investigated in the DRG of the PSNL model. Quantitative RT-PCR demonstrated that the PSNL model also exhibited an increase in V1a (Supplementary Figure 2).
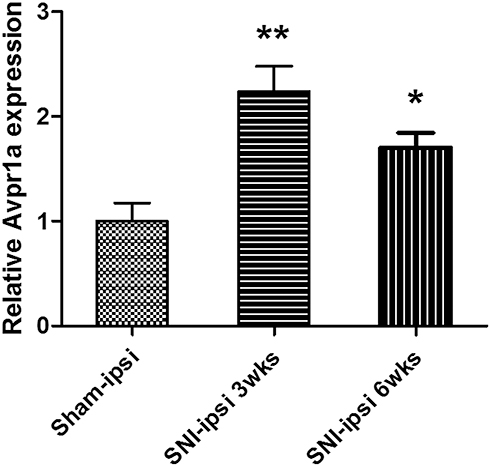 |
Figure 3 V1a expression in the dorsal root ganglia (L3, 4, and 5) 3 weeks and 6 weeks after SNI surgery. The SNI model showed a significant 2.23±0.8-fold increase in expression at 3 weeks. The SNI model also showed a significant 1.53±0.34-fold increase at 6 weeks. |
Behavioral Analysis of Mechanical Stimulation in WT and V1a KO Mice
To investigate the relationship between V1a and mechanical hypersensitivity, we performed von Frey testing in three groups: the WT sham model, the WT SNI model, and the SNI model of V1a KO mice (Figure 4). At 1 week after surgery, the response threshold to mechanical stimulation was slightly but not significantly decreased in the WT mouse sham model. Responses to mechanical stimuli were unchanged in the sham model during the observation period. In the SNI model of WT mice, the response threshold to mechanical stimulation was significantly reduced 1 week after surgery. The reaction threshold continued to decrease until 3 weeks after surgery and increased mechanical irritation was observed. The threshold decrease reached a plateau 3 weeks after surgery and persisted until the last observation 6 weeks after surgery.
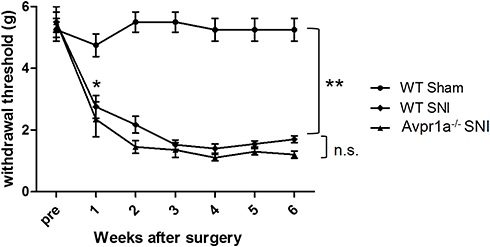 |
Figure 4 Dorsal skin sensitivity changes. Changes in the mechanical flexor reflex withdrawal response to stimulation of the dorsal surface of the hindpaw (sural nerve territory) after spared nerve injury (SNI) (n = 8) and sham (n = 8) procedures. The withdrawal threshold of the dorsal skin had a higher threshold in the control period and a smaller reduction after the SNI. This change was similar in vasopressin receptor 1A knockout SNI mice. |
Preoperatively, V1a KO mice did not exhibit significant hypersensitivity to mechanical stimulation compared with WT mice (Figure 4). After SNI surgery, the response threshold was decreased significantly from 1 week after surgery, similar to the SNI model in WT mice. The lower threshold for mechanical stimulation was maintained until the final observation 6 weeks after surgery.
Changes in Mechanical Stimulation Hypersensitivity Induced by Avpr1a Agonist
To evaluate the function of V1a, we administered a selective V1a agonist and investigated the response to mechanical stimulation. We analyzed the chronological changes in behavioral test results before and after intraperitoneal administration with three different doses of V1a agonist using the SNI model of WT mice. We also evaluated the differences according to sex. In the SNI model of WT male mice 3 weeks after surgery, the group intraperitoneally administered 0.1 mg/kg V1a selective agonist showed no significant change in response to mechanical stimulation compared with the PBS group (Figure 5A). However, the threshold of the mechanical stimulation response was significantly increased 30 min after administration in the groups administered 0.5 mg/kg and 1.0 mg/kg of the V1a selective agonist compared with the PBS group. There were no significant changes at 60 min, 90 min, and 120 min after administration. The dose-dependent analgesic effect was confirmed 30 min after administration. The same experiments were performed in an SNI model of WT female mice 3 weeks after surgery. The behavioral changes in the PBS group and each group administered agonist were similar to those in the male mice, indicating no clear sex difference (Figure 5B).
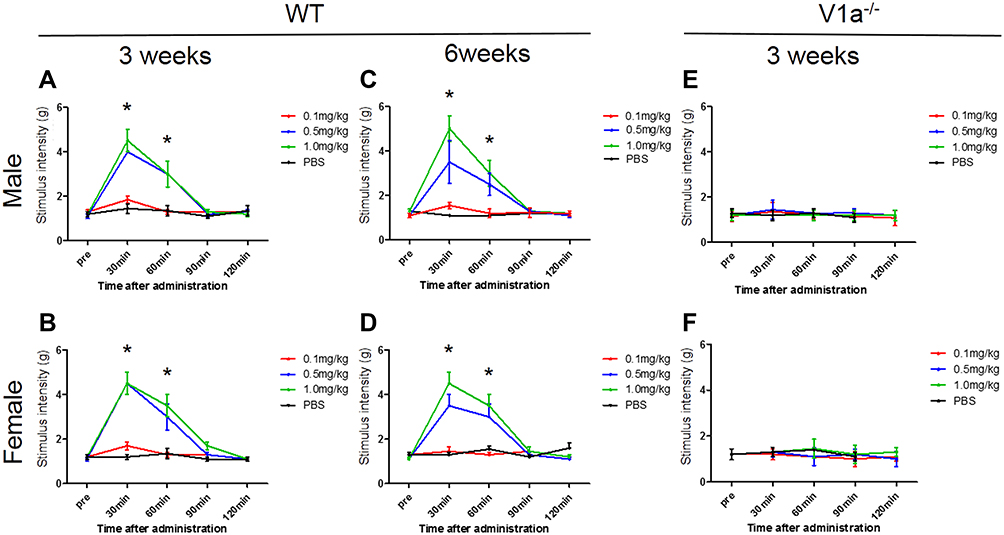 |
Figure 5 Changes in mechanical stimulation hypersensitivity with vasopressin receptor 1A (V1a) agonist. The groups intraperitoneally administered 0.5 mg/kg and 1.0 mg/kg V1a selective agonist showed a significant increase in the threshold of the mechanical stimulation response 30 min after administration compared with the phosphate-buffered saline group for both males (A) and females (B). The change in the threshold 30 and 60 min after administration tended to be volume-dependent. Similar results were seen in spared nerve injury models 6 weeks after injury in both males (C) and females (D). There were no significant changes in the response to mechanical stimulation in male (E) or female (F) V1a knockout mice at any dose. |
Next, the same evaluation was performed in an SNI model of WT male and female mice 6 weeks after surgery. Similar to the results in male and female mice 3 weeks after SNI, V1a agonist dose-dependently ameliorated mechanical hypersensitivity in both male and female mice (Figure 5C and D). From these results, V1a agonist can provide a dose-dependent analgesic effect for neuropathic pain mice in relatively acute and chronic phases. In addition, our experiments with both sexes found that there were no significant differences in its analgesic effect according to sex.
To clarify whether there is a sex difference in the function of V1a, male and female V1a KO mice were used as experimental animals, and changes in the thresholds for agonist administration and mechanical stimulation were evaluated 3 weeks after surgery (Figure 5E and F). Responses to mechanical stimulation hypersensitivity were compared between a PBS-administered group and a V1a selective agonist-administered group in SNI models of male and female V1a KO mice. The results showed no significant changes in the response to mechanical stimulation in males and females at all doses.
Changes in the Response to Mechanical Stimulation by Avpr1a Antagonist Administration to WT Mice
To evaluate whether the response threshold to mechanical stimuli fell after V1a blockage, we intraperitoneally administered a V1a selective antagonist to the WT sham and SNI mouse groups 3 weeks after surgery. Neither the WT SNI nor sham mice of either sex showed any apparent mechanical hypersensitivity 30, 60, 90, or 120 min after administration (Figure 6A and B).
 |
Figure 6 Changes in mechanical stimulation hypersensitivity induced by V1a antagonist. Wild-type (WT) sham (A) and spared nerve injury (SNI) mouse (B) groups 3 weeks after surgery. Neither the WT SNI nor sham mice of either sex showed any apparent mechanical hypersensitivity 30, 60, 90, or 120 min after administration. |
Immunohistochemical Analysis of Avpr1a in the DRG 3 Weeks After Surgery
The L4 DRG was collected from each model mouse and subjected to immunostaining to verify protein levels. V1a was mainly expressed in the rim of DRG cells in both the SNI and sham-operated animals (Figure 7A). It was also confirmed that the KO mice did not express V1a. There were significant differences in terms of the cross-sectional area of V1a expression between SNI mice (2445±46.2 μm2) and the sham-operated model (1582±50.7 μm2, p<0.001; Figure 7B). V1a was mostly localized to Schwann cells expressing S-100, but not to DRG cells expressing CGRP (Figure 7C).
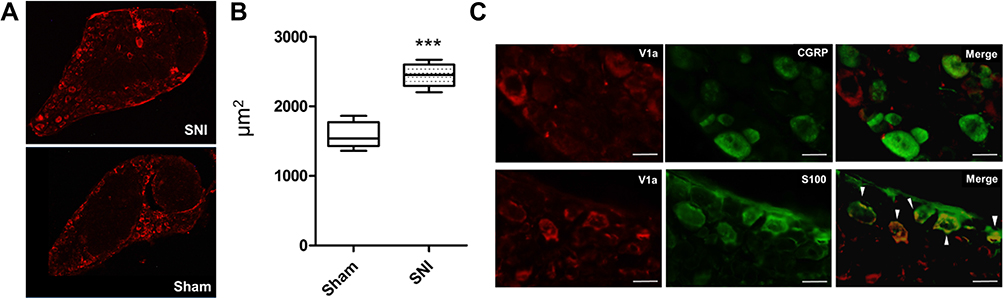 |
Figure 7 Immunohistochemistry of the lumbar dorsal root ganglia with vasopressin receptor 1A (V1a) antibody. (A) V1a was mainly expressed in the rim of DRG cells in both SNI and sham-operated animals. (B) A significant difference was found in the cross-sectional area of V1a expression between SNI mice (2445±46.2 μm2) and the sham-operated model (1582 ± 50.7 μm2, p<0.001). (C) Double-immunostaining assay showed that V1a colocalized with S-100 (arrowheads), but not with CGRP. |
Discussion
Comparison and Examination of Multiple Nerve Injury Models Using DNA Microarray
We created three different nerve injury models and a sham-operated model and performed microarray analysis of the lumbar DRG in the models to identify genetic changes in the DRG linked to SNI-specific hypersensitivity. Multiple nerve injury models were considered necessary to remove bias and noise and extract pure mechanical hypersensitivity signals. First, the sciatic nerve transection model and the SNI model were analyzed. The SNI model exhibits stronger mechanical hypersensitivity early after injury, whereas the sciatic nerve transection model generally loses perception.7 The difference between these two nerve injury models lies in whether the sural nerve is preserved. Interestingly, in the present study, the sural nerve transaction model showed normal sensation and no painful neuropathic behaviors. This finding indicates that preservation of the tibial and peroneal nerves with ligation of the sural nerve does not lead to hypersensitivities in the hindpaw. Taking the findings of the sciatic transection and sural transection models together, we believe that the mechanical hypersensitivity seen in the SNI is caused by hyperexcitation of the DRG related to sural nerve preservation.8
By extracting the genes whose expression was specifically increased in the SNI animals after comparing the two models, we obtained 222 candidate genes. Next, we compared the SNI and sham models to remove the bias associated with invasive surgery and inflammatory reactions. Accordingly, 166 genes were eliminated. To eliminate neural death signaling from the remaining 56 genes, we compared the sural nerve transection and sham models. Finally, we identified 50 genes as SNI-specific genes.
DNA microarray has been used to identify differential gene expression in specific disease models. Vega-Avelaira et al24 used microarray analysis to compare adult and young nerve injury rats in terms of gene expression in the L4/L5 DRG and demonstrated that several genes involved in the immune response to injury were upregulated in adult rats compared with infant rats, which do not experience painful neuropathy. They concluded that this regulated expression may be associated with the onset and maintenance of neuropathic pain. Cheng et al25 investigated specific modules and hub genes associated with neuropathic pain caused by tibial nerve transection based on weighted gene co-expression network analysis and protein–protein networks. They ultimately identified that the module involved in the pathogenesis of neuropathic pain regulates the defense response and calcium binding. Recently, numerous molecular scientists interested in next-generation sequencing have focused not only on identifying differential gene expression, but also on specifying the molecular variants involved of the genes of interest. Mao et al26 investigated long non-coding RNA (lncRNA) expression in the DRG of neuropathic pain rats using high-throughput RNA sequencing and found that as many as 112 lncRNAs were differentially expressed in their spared sciatic nerve injury model. Another study compared whole transcriptomes in the lumbar DRG between spinal nerve ligation mice and sham mice using RNA sequencing and demonstrated that the regulated expression mostly involved protein-coding genes.27
Thus, various investigations associated with neuropathic pain-related molecules have been performed. Although we were able to extract differential genes of neuropathic pain using DNA microarray, further experiments using next-generation sequencing are needed to identify cell-specific alterations in both the central and peripheral nervous systems for unraveling the molecular mechanisms underlying neuropathic pain.
Relationship Between Neuropathic Pain and Avpr1a
This study shows that V1a and CDK1 expression were increased in the DRG. CDK1 is essential for mitotic events.28 Although this master gene is likely associated with neuropathic pain mechanisms, CDK1 general knockout mice exhibit viviparous lethality. On the other hand, because V1a has been associated with inflammatory pain, we speculated that V1a was also likely to be associated with neuropathic pain and selected this gene for analysis from among the two. Upregulation of V1a alleviates hypersensitivities in a neuropathic pain model of WT mice.29 Various studies have demonstrated that vasopressin can regulate the pain process in the brain through central cholinergic and opioidergic systems.30 Notably, V1a plays an important role in the mechanism of inflammatory pain. In peripheral sensory neurons, ASICs (acid-sensing ion channels) have been found on cell bodies and sensory terminals, where they have been suggested to be important for nociception.31 The present study demonstrated that V1a is mainly expressed in S-100-positive cells. This finding suggests that ASICs may be localized not only to neurons, but also to the Schwann cells surrounding neurons and contribute to the mechanisms of chronic pain.
Inflammation and tissue injury are reported to increase the expression levels of ASIC mRNA in DRG neurons and thereby contribute to hyperalgesia. In addition, oxytocin inhibits ASIC activity through vasopressin, particularly V1a receptor, in primary sensory neurons.32,33 The results of our DNA microarray suggest that V1a is also involved as a molecule related to SNI-specific neuropathic pain. The previous study demonstrated that oxytocin effectively suppresses painful behaviors in infraorbital nerve injury mice, likely by modulation of voltage-gated K channels through V1a.34 Therefore, we investigated the relationship between neuropathic pain and V1a by focusing on the peripheral nervous system, particularly the DRG (rather than the spinal cord), which is often associated with spinal cord and spinal cord diseases, among pain transmission pathways. We validated the V1a expression change in the SNI DRG using RT-PCR after the DNA microarray. The SNI model showed a significant 2.23-fold increase in expression 3 weeks postoperatively and a 1.53-fold increase 6 weeks postoperatively compared with sham. These results suggest that V1a in DRG is associated with neuropathic pain.
Changes in Neuropathic Pain Model Mice Caused by Avpr1a Agonist
We confirmed that the SNI model mice exhibited stronger mechanical stimulation hypersensitivity in the early postoperative period than the sham mice. Various studies have suggested that intraventricular injection of V1a produces antinociception and that V1a antagonists weaken antinociception.12,35,36 Other studies also observed that intrathecal administration of V1a causes antinociception in rats.37–39 These previous studies suggest that V1a protects against various pain stimuli in both the central and peripheral nervous systems. Our results showed that the expression of V1a was significantly increased in the DRG of the nerve injury model. We then intraperitoneally administered V1a agonist to the SNI model mouse; the threshold for the response to the mechanical stimulus rapidly increased in a dose-dependent manner, and the neuropathic pain was alleviated. Therefore, our findings suggest that V1a may alleviate the mechanical hypersensitivity caused by peripheral nerve injury in the DRG. V1a is widely expressed throughout the body. Mogil et al30 quantified V1a mRNA in the nerve tissues of two different strains of mice and demonstrated that V1a mRNA expression is higher in the tissues of the pain-resistant A/J mouse, particularly in the DRG and spinal cord, than in the brain. This evidence suggests that pain resistance may be mediated by V1a upregulation mainly in the peripheral nervous system. We, therefore, speculated that administration of V1a agonist was likely to activate V1a in the peripheral nervous system, which was related to neuropathic pain in the SNI model. Differential down- or upregulation of V1a in peripheral nerves, including the DRG and sciatic nerve, will be required to clarify whether peripheral V1a plays an important role in the onset and maintenance of neuropathic pain.
Avpr1a Antagonists Do Not Exacerbate Mechanical Hypersensitivity
To investigate the inhibition of the antinociceptive effect of V1a in neuropathic pain, we administered V1a antagonist to sham and SNI models of WT male mice. The V1a antagonist did not impair mechanical hypersensitivity in sham mice. Similarly, V1a KO mice demonstrated a normal response to mechanical stimulation before peripheral nerve injury (Figure 4). We also confirmed that there was little expression of V1a in the DRG of not only KO mouse, but also in the sham and contralateral DRG to the SNI based on immunohistochemistry and PCR analysis of the physiological DRG (Figure 7). Because of this low expression of V1a in the intact DRG (and even irrespective of whether its expression is increased in a pathological state), it is possible that the endogenous ligand bound to V1a is also low, so we could not confirm the inhibitory effect of the antagonist. These results indicate that V1a does not directly influence the onset of neuropathic pain under normal conditions. A previous study reported that painful behaviors worsened after inflammatory nociception in V1a KO mouse.
This evidence and our results indicate that the absence or low levels of V1a exacerbate inflammatory pain but not neuropathic pain and suggest that V1a may have a protective effect on neuropathic pain alone. These results raise two points. First, because V1a receptor expression may also be low in the DRG, the effects of antagonists may be relatively modest in the physiological DRG. It has long been known that V1a receptors are associated with inflammatory pain and are densely distributed in skin and peripheral nerve endings.40 In contrast, V1a is reported to be expressed in about 20% of all cells in the trigeminal ganglion. Even if its expression was to increase due to nerve injury, this would still affect less than 40% of all cells.34 Together, these findings suggest that V1a may be strongly involved in the mechanism of inflammatory pain but only partially contribute to the mechanism of neuropathic pain. Second, the threshold change in hypersensitivity after administration of the antagonist may have been too small to be detected in the SNI mice used in this study.
Conclusion
In this preclinical study, we applied microarray analysis to three different nerve injury models and a sham-operated model to extract specific genes related to neuropathic pain in the mouse lumbar DRG. Microarray and PCR analyses revealed that the V1a gene was linked to neuropathic pain. Although V1a KO mouse did not show any behavioral changes in response to the SNI compared with WT mice, administration of V1a agonist to SNI WT mice revealed that V1a plays a protective role in neuropathic pain model mice. These results suggest that V1a in the DRG may partially contribute to the mechanism of neuropathic pain.
Acknowledgments
We thank Dr Akito Tanoue of the Department of Pharmacology, National Research Institute for Child Health and Development, for kindly providing the Avpr1a knockout mice. This work was supported by the following grants: grants from the Japan Agency for Medical Research and Development (#18ek0610013h0002 and #19ek0610013h0003), a grant from the Takeda Science Foundation, and a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (#18K16609).
Author Contributions
All authors made substantial contributions to the conception and design of the study; acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52:77–92. doi:10.1016/j.neuron.2006.09.021
2. Kupfer M, Formal CS. Non-opioid pharmacologic treatment of chronic spinal cord injury-related pain. J Spinal Cord Med. 2020;1–10. doi:10.1080/10790268.2020.1730109
3. Kushnarev M, Pirvulescu IP, Candido KD, et al. Neuropathic pain: preclinical and early clinical progress with voltage-gated sodium channel blockers. Expert Opin Investig Drugs. 2020;29:259–271. doi:10.1080/13543784.2020.1728254
4. Lara CO, Burgos CF, Moraga-Cid G, et al. Pentameric ligand-gated ion channels as pharmacological targets against chronic pain. Front Pharmacol. 2020;11:167. doi:10.3389/fphar.2020.00167
5. Urru M, Muzzi M, Coppi E, et al. Dexpramipexole blocks Nav1.8 sodium channels and provides analgesia in multiple nociceptive and neuropathic pain models. Pain. 2020;161:831–841. doi:10.1097/j.pain.0000000000001774
6. Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi:10.1016/s0304-3959(00)00276-1
7. Wall PD, Devor M, Inbal R, et al. Autotomy following peripheral nerve lesions: experimental anaesthesia dolorosa. Pain. 1979;7:103–111. doi:10.1016/0304-3959(79)90002-2
8. Hirai T, Enomoto M, Kaburagi H, et al. Intrathecal AAV serotype 9-mediated delivery of shRNA against TRPV1 attenuates thermal hyperalgesia in a mouse model of peripheral nerve injury. Mol Ther. 2014;22:409–419. doi:10.1038/mt.2013.247
9. Voinsky I, Bennuri SC, Svigals J, et al. Peripheral blood mononuclear cell oxytocin and vasopressin receptor expression positively correlates with social and behavioral function in children with autism. Sci Rep. 2019;9:13443. doi:10.1038/s41598-019-49617-9
10. Rigney N, Beaumont R, Petrulis A. Sex differences in vasopressin 1a receptor regulation of social communication within the lateral habenula and dorsal raphe of mice. Horm Behav. 2020;121:104715. doi:10.1016/j.yhbeh.2020.104715
11. Golimbet V, Alfimova M, Abramova L, et al. Arginine vasopressin 1a receptor RS3 promoter microsatellites in schizophrenia: a study of the effect of the “risk” allele on clinical symptoms and facial affect recognition. Psychiatry Res. 2015;225:739–740. doi:10.1016/j.psychres.2014.11.043
12. Kordower JH, Bodnar RJ. Vasopressin analgesia: specificity of action and non-opioid effects. Peptides. 1984;5:747–756. doi:10.1016/0196-9781(84)90017-2
13. Peng F, Qu ZW, Qiu CY, et al. Spinal vasopressin alleviates formalin-induced nociception by enhancing GABAA receptor function in mice. Neurosci Lett. 2015;593:61–65. doi:10.1016/j.neulet.2015.03.023
14. Bodnar RJ, Wallace MM, Kordower JH, et al. Modulation of nociceptive thresholds by vasopressin in the Brattleboro and normal rat. Ann N Y Acad Sci. 1982;394:735–739. doi:10.1111/j.1749-6632.1982.tb37491.x
15. Schorscher-Petcu A, Sotocinal S, Ciura S, et al. Oxytocin-induced analgesia and scratching are mediated by the vasopressin-1A receptor in the mouse. J Neurosci. 2010;30:8274–8284. doi:10.1523/JNEUROSCI.1594-10.2010
16. Han RT, Kim HB, Kim YB, et al. Oxytocin produces thermal analgesia via vasopressin-1a receptor by modulating TRPV1 and potassium conductance in the dorsal root ganglion neurons. Korean J Physiol Pharmacol. 2018;22:173–182. doi:10.4196/kjpp.2018.22.2.173
17. Yang J. Intrathecal administration of oxytocin induces analgesia in low back pain involving the endogenous opiate peptide system. Spine (Phila Pa 1976). 1994;19:867–871. doi:10.1097/00007632-199404150-00001
18. Hobo S, Hayashida K, Eisenach JC. Oxytocin inhibits the membrane depolarization-induced increase in intracellular calcium in capsaicin sensitive sensory neurons: a peripheral mechanism of analgesic action. Anesth Analg. 2012;114:442–449. doi:10.1213/ANE.0b013e31823b1bc8
19. Sanbe A, Takagi N, Fujiwara Y, et al. Alcohol preference in mice lacking the Avpr1a vasopressin receptor. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1482–1490. doi:10.1152/ajpregu.00708.2007
20. Shir Y, Seltzer Z. A-fibers mediate mechanical hyperesthesia and allodynia and C-fibers mediate thermal hyperalgesia in a new model of causalgiform pain disorders in rats. Neurosci Lett. 1990;115:62–67. doi:10.1016/0304-3940(90)90518-e
21. Cichon J, Sun L, Yang G. Spared nerve injury model of neuropathic pain in mice. Bio Protoc. 2018;8:e2777. doi:10.21769/bioprotoc.2777
22. Rao T, Shao Y, Hamada N, et al. Pharmacokinetic study based on a matrix-assisted laser desorption/ionization quadrupole ion trap time-of-flight imaging mass microscope combined with a novel relative exposure approach: a case of octreotide in mouse target tissues. Anal Chim Acta. 2017;952:71–80. doi:10.1016/j.aca.2016.11.056
23. Ponzoni L, Braida D, Bondiolotti G, et al. The non-peptide arginine-vasopressin v1a selective receptor antagonist, SR49059, blocks the rewarding, prosocial, and anxiolytic effects of 3,4-methylenedioxymethamphetamine and its derivatives in zebra fish. Front Psychiatry. 2017;8:146. doi:10.3389/fpsyt.2017.00146
24. Vega-Avelaira D, Géranton SM, Fitzgerald M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol Pain. 2009;5:70. doi:10.1186/1744-8069-5-70
25. Cheng N, Zhang Z, Guo Y, et al. Weighted gene co-expression network analysis reveals specific modules and hub genes related to neuropathic pain in dorsal root ganglions. Biosci Rep. 2019;39. doi10.1042/BSR20191511.
26. Mao P, Li CR, Zhang SZ, Zhang Y, Liu BT, Fan BF. Transcriptomic differential lncRNA expression is involved in neuropathic pain in rat dorsal root ganglion after spared sciatic nerve injury. Braz J Med Biol Res. 2018;51:e7113. doi:10.1590/1414-431X20187113
27. Wu S, Marie Lutz B, Miao X, et al. Dorsal root ganglion transcriptome analysis following peripheral nerve injury in mice. Mol Pain. 2016;12. doi:10.1177/1744806916629048
28. Diril MK, Ratnacaram CK, Padmakumar VC. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. PNAS. 2012;109(10):3826–3831. doi:10.1037/pnas.1115201109
29. Bagdas D, Yucel-Ozboluk H, Orhan F, et al. Role of central arginine vasopressin receptors in the analgesic effect of CDP-choline on acute and neuropathic pain. Neuroreport. 2013;24:941–946. doi:10.1097/WNR.0000000000000009
30. Mogil JS, Sorge RE, LaCroix-Fralish ML, et al. Pain sensitivity and vasopressin analgesia are mediated by a gene-sex-environment interaction. Nat Neurosci. 2011;14:1569–1573. doi:10.1038/nn.2941
31. Alvarez de la Rosa D, Zhang P, Shao D, et al. Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci U S A. 2002;99:2326–2331. doi:10.1073/pnas.042688199
32. Qiu F, Hu WP, Yang ZF. Enhancement of GABA-activated currents by arginine vasopressin in rat dorsal root ganglion neurons. Sheng Li Xue Bao. 2014;66:647–657. doi:10.13294/j.aps.2014.0077
33. Qiu F, Qiu CY, Cai H, et al. Oxytocin inhibits the activity of acid-sensing ion channels through the vasopressin, V1A receptor in primary sensory neurons. Br J Pharmacol. 2014;171:3065–3076. doi:10.1111/bph.12635
34. Kubo A, Shinoda M, Katagiri A, et al. Oxytocin alleviates orofacial mechanical hypersensitivity associated with infraorbital nerve injury through vasopressin-1A receptors of the rat trigeminal ganglia. Pain. 2017;158:649–659. doi:10.1097/j.pain.0000000000000808
35. Bodnar RJ, Nilaver G, Wallace MM, et al. Pain threshold changes in rats following central injection of beta-endorphin, met-enkephalin, vasopressin or oxytocin antisera. Int J Neurosci. 1984;24:149–160. doi:10.3109/00207458409089803
36. Madrazo I, Franco-Bourland RE, León-Meza VM, et al. Intraventricular somatostatin-14, arginine vasopressin, and oxytocin: analgesic effect in a patient with intractable cancer pain. Appl Neurophysiol. 1987;50:427–431. doi:10.1159/000100753
37. Watkins LR, Suberg SN, Thurston CL, et al. Role of spinal cord neuropeptides in pain sensitivity and analgesia: thyrotropin releasing hormone and vasopressin. Brain Res. 1986;362:308–317. doi:10.1016/0006-8993(86)90455-5
38. Thurston CL, Culhane ES, Suberg SN, et al. Antinociception vs motor effects of intrathecal vasopressin as measured by four pain tests. Brain Res. 1988;463:1–11. doi:10.1016/0006-8993(88)90520-3
39. Thurston CL, Campbell IG, Culhane ES, et al. Characterization of intrathecal vasopressin-induced antinociception, scratching behavior, and motor suppression. Peptides. 1992;13:17–25. doi:10.1016/0196-9781(92)90135-p
40. Manzano-García A, González-Hernández A, Tello-García IA, et al. The role of peripheral vasopressin 1A and oxytocin receptors on the subcutaneous vasopressin antinociceptive effects. Eur J Pain. 2018;22:511–526. doi:10.1002/ejp.1134
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.