")
Back to Journals » OncoTargets and Therapy » Volume 10
DNA hypermethylated status and gene expression of PAX1/SOX1 in patients with colorectal carcinoma
Authors Huang J, Tan ZR , Yu J, Li H, Lv Q, Shao Y, Zhou H
Received 6 June 2017
Accepted for publication 14 July 2017
Published 26 September 2017 Volume 2017:10 Pages 4739—4751
DOI https://doi.org/10.2147/OTT.S143389
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Jin Huang,1,2 Zhi-Rong Tan,1,2 Jing Yu,1,2 He Li,1,2 Qiao-Li Lv,1,2 Ying-Ying Shao,3 Hong-Hao Zhou1,2
1Department of Clinical Pharmacology, Xiangya Hospital, Central South University, Changsha, Hunan, 2Institute of Clinical Pharmacology, Central South University, Hunan Key Laboratory of Pharmacogenetics, Changsha, Hunan, 3Institute of Life Science, Chongqing Medical University, Yuzhong District, Chongqing, China
Background: Colorectal cancer (CRC) is a widespread and aggressive carcinoma with poor prognosis. Hypermethylation of specific gene promoters is an important mechanism of CRC. In this study, we investigated the hypermethylation of paired boxed gene 1 (PAX1) and sex-determining region Y-related high-mobility group box 1 (SOX1) genes in CRC tissues.
Methods: DNA methylation at cg2,09,07,471 PAX1 and cg0,66,75,478 SOX1 from 166 cancer tissues and 37 normal tissues from CRC patients were compared using datasets downloaded from The Cancer Genome Atlas. Quantitative methylation-specific polymerase chain reaction and assay of PAX1 and SOX1 were performed in dissected tumor and paracancerous tissues by surgery from 41 CRC patients. Quantitative reverse transcription polymerase chain reaction and immunohistochemistry assay were performed in both CRC and paired normal tissues to detect mRNA and protein expression, respectively.
Results: Methylation levels of PAX1/SOX1 genes were significantly higher in cancer tissues than in paired normal tissues. PAX1 and SOX1 genes were methylated in 28 (68.3%) of the 41 CRC samples but in 5 (12.2%) and 0 (0%) of the paired normal control samples (both P<0.001), respectively. Sensitivities and specificities of PAX1 methylation for the detection of cancer were 68.3% and 87.8%, respectively, whereas the corresponding values for SOX1 were 68.3% and 100%. However, the Kaplan–Meier analysis illustrated no significant difference in the overall survivals between patients with high and low methylation levels of SOX1 or PAX1 (P>0.5). In addition, the methylation level of PAX1/SOX1 was significantly higher in CRC patients with high TNM stage (TNM stage III/IV, 3.11±2.43) than those with low TNM stage (TNM stage I/II, 1.26±2.94, P<0.05). Relative RNA and protein expression levels of PAX1/SOX1 were both significantly lower in CRC tissues than in their paired normal tissue.
Conclusions: This study is the first analysis of the methylation of PAX1/SOX1, which may be new biomarkers for CRC screening.
Keywords: colorectal cancer, PAX1, SOX1, molecular biomarker, DNA methylation, epigenetic
Introduction
Colorectal cancer (CRC) is a widespread carcinoma and has more than 600,000 associated mortalities annually worldwide.1,2 CRC is one of the most common cancers of the gastrointestinal tract. After lung and breast cancers, CRC ranks third among the most common cancers worldwide.3,4 Colon cancer mortality ranks fourth among cancer-related mortalities. CRC incidence and mortality increase with age. In China, the incidence of CRC has been rising because of improved living standards and the shift to high-protein, high-calorie, and low-fiber diet. According to the cancer statistics in 2016, the incidence of CRC in China was 33.1 million, which ranked fourth in all malignant tumors. The mortality of CRC in China was 15.9 million, which ranked fifth in cancer deaths.5 Colon cancer mortality increased by 15.22% in China, among which female colon cancer mortality ranked third among malignant carcinomas, whereas that of males ranked fourth.6 Paired boxed gene 1 (PAX1) and sex-determining region Y-related high-mobility group box 1 (SOX1) are tumor suppressor genes. Previous studies demonstrated that PAX1 was silenced because of hypermethylated status in many carcinomas, especially in cervical, ovarian, and oral cancers.7–10 PAX1 methylation is a potential biomarker to screen cervical cancer, as well as oral squamous cell carcinoma, with high sensitivity and specificity.8,11,12 SOX1 is a tumor suppressor that was suppressed by the hypermethylation of its promoter region in various carcinomas, including cervical, hepatocellular, and nasopharyngeal cancers.10,12,13 However, the methylation of PAX1 or SOX1 in CRC has not been analyzed yet.
CRC and other carcinomas are often caused by an accumulation of genetic and epigenetic alterations, among which DNA methylation is the most frequent epigenetic alteration. DNA methylation usually occurs at carbon 5 of the cytosine ring within the CpG dinucleotides, and this process is a major epigenetic modification in the human genome.14–16 DNA hypermethylation is an important mechanism to inactivate tumor suppressor genes that could be involved in DNA repair, metabolism of cancer, cell cycle control, apoptosis, and angiogenesis.15,17,18 However, a few hypermethylated genes have been associated with CRC, and a limited number of studies have focused on prognostic methylation biomarker in CRC. To the best of our knowledge, APC, RASSF1A, FAS, ZNF582, GFRA1, and GSTM2 promoter hypermethylation has been reported to be involved in the development with CRC.19–22 However, whether the promoter methylation status of the PAX1 and SOX1 genes is associated with CRC remains to be elucidated.
In this study, the methylation status of PAX1 and SOX1 in CRC tissues and paired paracarcinoma tissues was investigated using quantitative methylation-specific polymerase chain reaction (qMSP). The results were verified using datasets from The Cancer Genome Atlas (TCGA) database.
Materials and methods
Patients and samples
Samples of CRC tissues and adjacent normal tissues were collected from 41 patients with CRC. Written informed consents were signed by all patients before collection of specimens. This study was approved by the Institutional Review Board of Department of Clinical Pharmacology, Xiangya Hospital, Central South University (registration number: CTXY-1,50,001–2) and the Chinese Clinical Trial Registry (registration number: ChiCTR-DCD-1,50,06,289). Tumor tissues were defined as the sections next to the tumor diagnosed by histopathology. Paracancerous tissues were taken from surgically dissected tissues ~2 cm away from the defined tumor area without tumor invasion. The dissected tumor and paracancerous tissues were further evaluated histologically.
Genomic DNA was extracted from the tissues with the iSat Nucleic Acid Extraction kit (iStat Biomedical Co., Ltd., New Taipei City, Taiwan) according to the manufacturer’s manual. The concentration of DNA was determined using BioSpec-Nano spectrophotometer (Shimadzu Company, Kyoto, Japan). Samples with a DNA yield of more than 500 ng were considered for further testing. Total RNA was extracted using TRIZOL reagent (Invitrogen, San Diego, CA, USA) according to the manufacturer’s protocol and dissolved in 30 μL of diethylpyrocarbonate-treated water.
Determination of DNA methylation
Sodium bisulfite conversion of 500 ng of genomic DNA was performed using the iStat Bisulfite Conversion Kit (iStat Biomedical Co., Ltd) according to the manufacturer’s manual. The bisulfited DNA was stored at −80°C until used. The DNA methylation of PAX1 and SOX1 genes was analyzed using PAX1 and SOX1 DNA Detection Kits (iSat Biomedical Co., Ltd), which use simplified technology based on TaqMan technologies for qMSP. The qMSP reaction was conducted in a total volume of 20 μL containing 2 μL of bisulfite-converted template gDNA (50 ng), 1 μL of 2× custom detection mix, and 10 μL of 2× custom universal polymerase chain reaction (PCR) master mix. The reaction was performed using the LightCycler 480 Real-Time PCR System (Roche Applied Science, Penzberg, Germany) under the following conditions. The reaction systems were first subjected to an initial incubation at 95°C for 10 min, followed by 50 cycles at 95°C for 10 s, and annealing and extension at 60°C for 40 s. DNA methylation levels were assessed as the methylation index (meth-index) was calculated as follows: 10,000×2(Cp value of gene-Cp value of COL2A). Testing results with Cp values of COL2A >36 were defined as detection failures.
Quantitative reverse transcription-PCR
Single-stranded cDNA was prepared using a PrimeScript RT Reagent Kit (Takara, Otsu, Japan) according to the manufacturer’s protocol. Quantitative reverse transcription-PCR (qRT-PCR) was performed using LightCycler 480 Real-Time PCR System (Roche Applied Science) by SYBR Premix DimerErase™ kit (Takara) to detect the expression of SOX1 and PAX1, with PPIA and B2M as normalizing controls. The sequences of the primers used were as follows: SOX1: forward, 5′-CAGTACAGCCCCATCTCCAAC-3′ and reverse, 5′-GCGGGCAAGTACATGCTGA-3′; PAX1: forward, 5′-TCGCTATGGAGCAGACG TATG-3′ and reverse, 5′-GCTGCCGACTGATGTCACA-3′; PPIA: forward, 5′-CC CACCGTGTTCTTCGACATT-3′ and reverse, 5′-GGACCCGTATGCTTTAGGATGA-3′; and B2M: forward, 5′-GAGGCTATCCAGCGTACTCCA-3′ and reverse, 5′-CG GCAGGCATACTCATCTTTT-3′. The qRT-PCR reaction systems were performed with incubation at 95°C for 10 min, followed by 45 cycles at 95°C for 10 s, and annealing and extension at 60°C for 60 s. Relative quantification results were determined by 2−ΔΔCt relative to the normalizing control genes.
Validation using TCGA data
The dataset of DNA methylation was obtained from the TCGA database, which included clinical information from 166 CRC tissues and 37 normal tissues. Level 3 datasets were obtained through Infinium HumanMethylation 27 BeadChip and analyzed using R software (http://www.R-project.org/).
Statistical analysis
Paired-sampled t-test was performed to compare the methylation levels between the CRC and normal tissue samples. Correlations between the frequency of DNA methylation occurrence and clinicopathological characteristics of CRC patients were analyzed by χ2 test. Associations between DNA methylation status and clinicopathological characteristics of CRC patients were analyzed by Mann–Whitney U-test. Wilcoxon signed-rank test was performed to compare the methylation levels between CRC and paired normal tissues. P-values <0.05 were considered statistically significant. Kaplan–Meier method was performed to evaluate the overall survival rates. Log-rank test was used to calculate the difference in survival. The association between methylation levels and relative mRNA expression was analyzed by calculating Pearson’s correlation coefficient and P-values. All statistical analyses were performed using SPSS Statistics 19.0 software (IBM Corporation, Armonk, NY, USA).
Results
Analysis of DNA methylation of PAX1/SOX1 in CRC and paracancer tissues
DNA methylation status of PAX1/SOX1 in 166 CRC patients was analyzed using TCGA datasets. Figure 1A shows that the methylation levels of PAX1 and SOX1 genes were significantly greater in the 166 CRC samples than in the 37 normal samples (both P<0.05). In addition, the methylation levels of the two genes were higher in the majority of the CRC samples than in their paired normal samples (Figure 1B). The areas under the curve (AUCs) of the receiver-operating characteristic (ROC) plots for the methylation of SOX1 and PAX1 in 166 CRC and 37 normal tissues were 0.515 and 0.805, respectively (Figure 1C). SOX1 methylation testing achieved 78.3% sensitivity and 83.8% specificity, and PAX1 methylation testing achieved 31.9% sensitivity and 100% specificity.
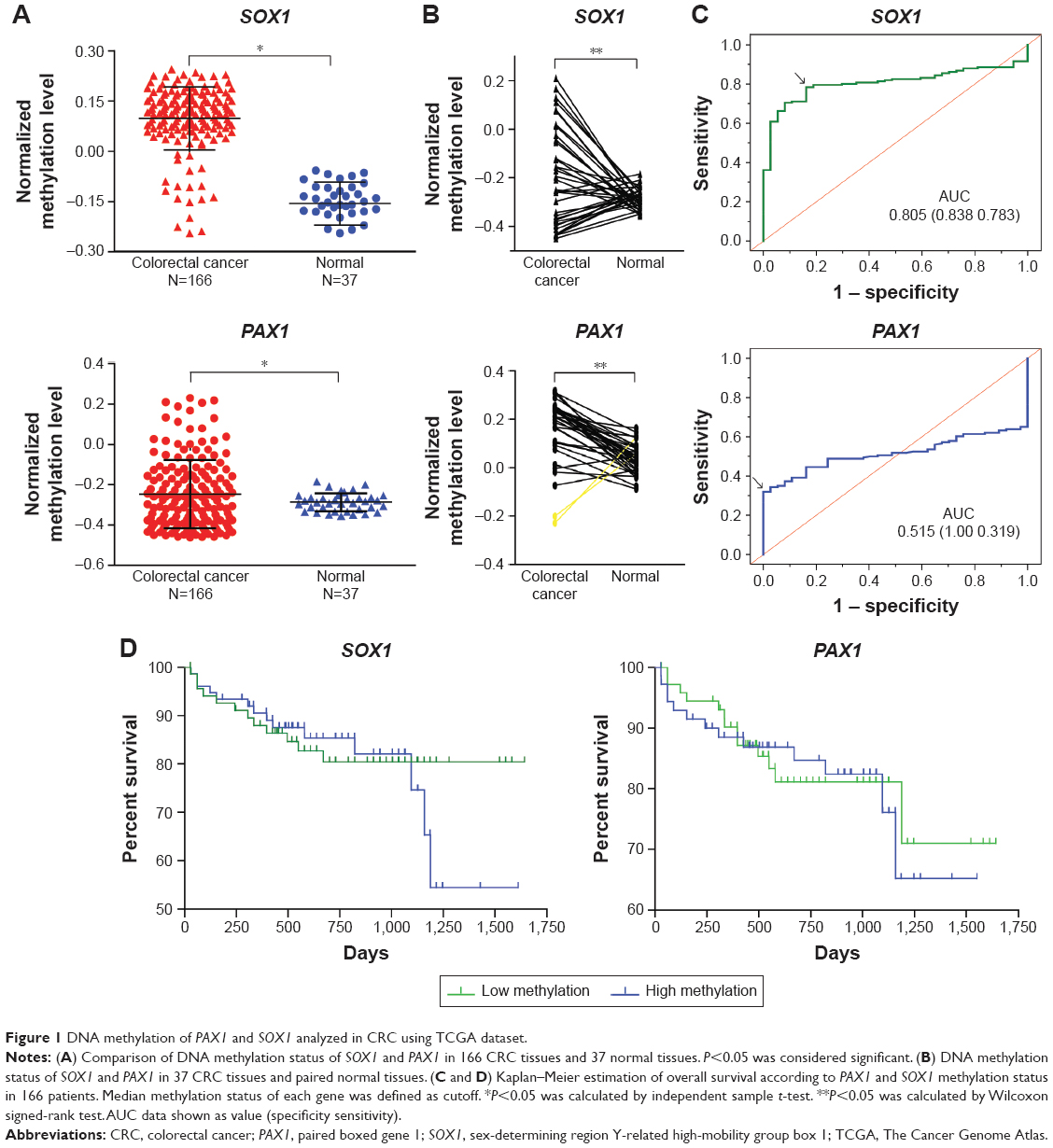 | Figure 1 DNA methylation of PAX1 and SOX1 analyzed in CRC using TCGA dataset. |
The results from the TCGA datasets were verified by detecting the methylation levels of PAX1 and SOX1 in the tissues of patients with CRC. A total of 41 patients with CRC were recruited from Xiangya Hospital in Changsha, China, between May 2015 and June 2016. Patient ages ranged from 24 to 84 years (median age, 57 years). The clinicopathological characteristics of these patients are presented in Tables 1 and 4. The CRC tissues showed a significantly greater level of hypermethylated SOX1/PAX1 compared with their paired normal tissues as detected by qMSP (both P<0.05, Figure 2A and B). The methylation status of samples and frequencies were determined based on the best meth-index cut-off values calculated using the ROC curves (Figure 2C and D). Log (meth-index) ≥3.6 for SOX1 or log (meth-index) ≥3.7 for PAX1 were considered to be methylation positivity. PAX1 and SOX1 genes were both methylated in 28 (68.3%) of the 41 CRCs samples but in 5 (12.2%) and 0 (0%) of paired normal control samples (both P<0.001), respectively. The AUCs of the ROC plots in Figure 2C and D for the methylation of PAX1 and SOX1 to detect CRC from normal tissues were 0.743 (95% CI 0.627–0.858) and 0.798 (95% CI 0.689–0.907), respectively (Table 2). At meth-index of 1.56, PAX1 methylation testing achieved 68.3% sensitivity and 87.8% specificity. At meth-index of 3.01, SOX1 methylation testing achieved 68.3% sensitivity and 100% specificity.
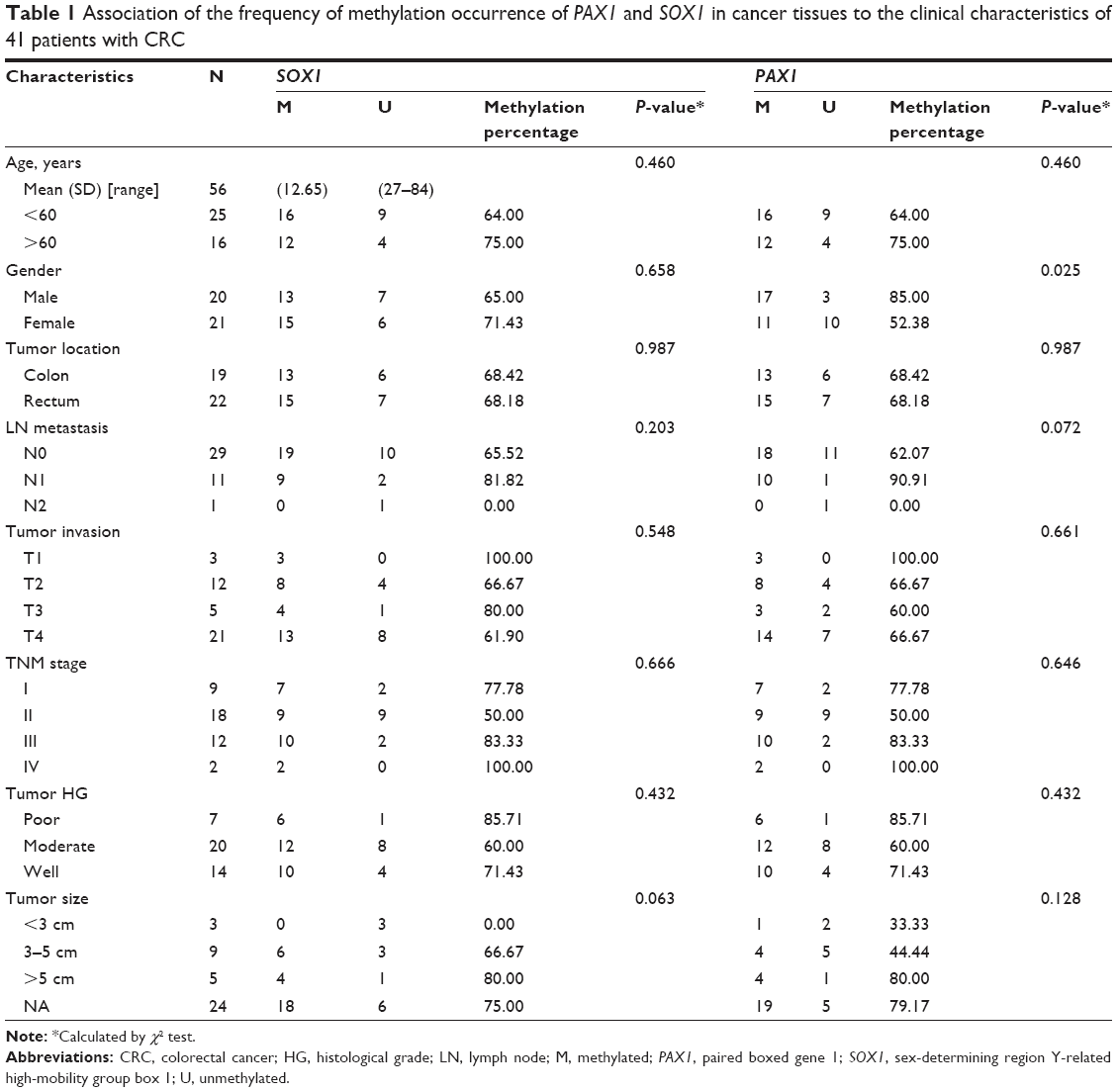 | Table 1 Association of the frequency of methylation occurrence of PAX1 and SOX1 in cancer tissues to the clinical characteristics of 41 patients with CRC |
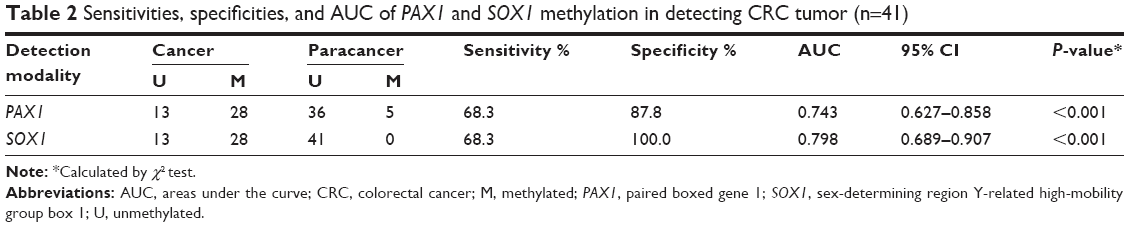 | Table 2 Sensitivities, specificities, and AUC of PAX1 and SOX1 methylation in detecting CRC tumor (n=41) |
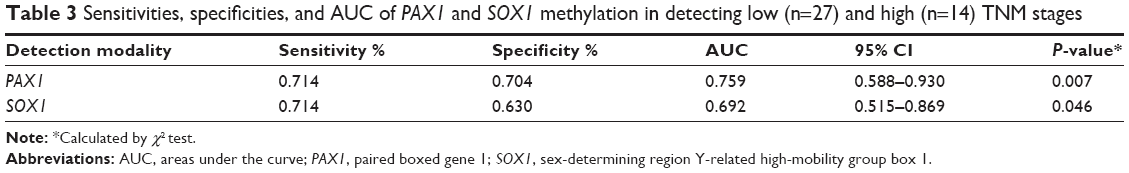 | Table 3 Sensitivities, specificities, and AUC of PAX1 and SOX1 methylation in detecting low (n=27) and high (n=14) TNM stages |
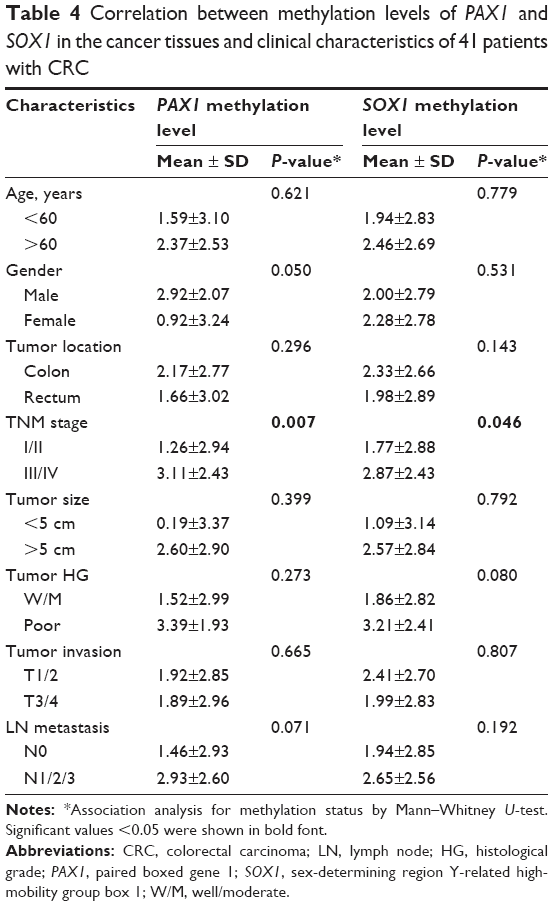 | Table 4 Correlation between methylation levels of PAX1 and SOX1 in the cancer tissues and clinical characteristics of 41 patients with CRC |
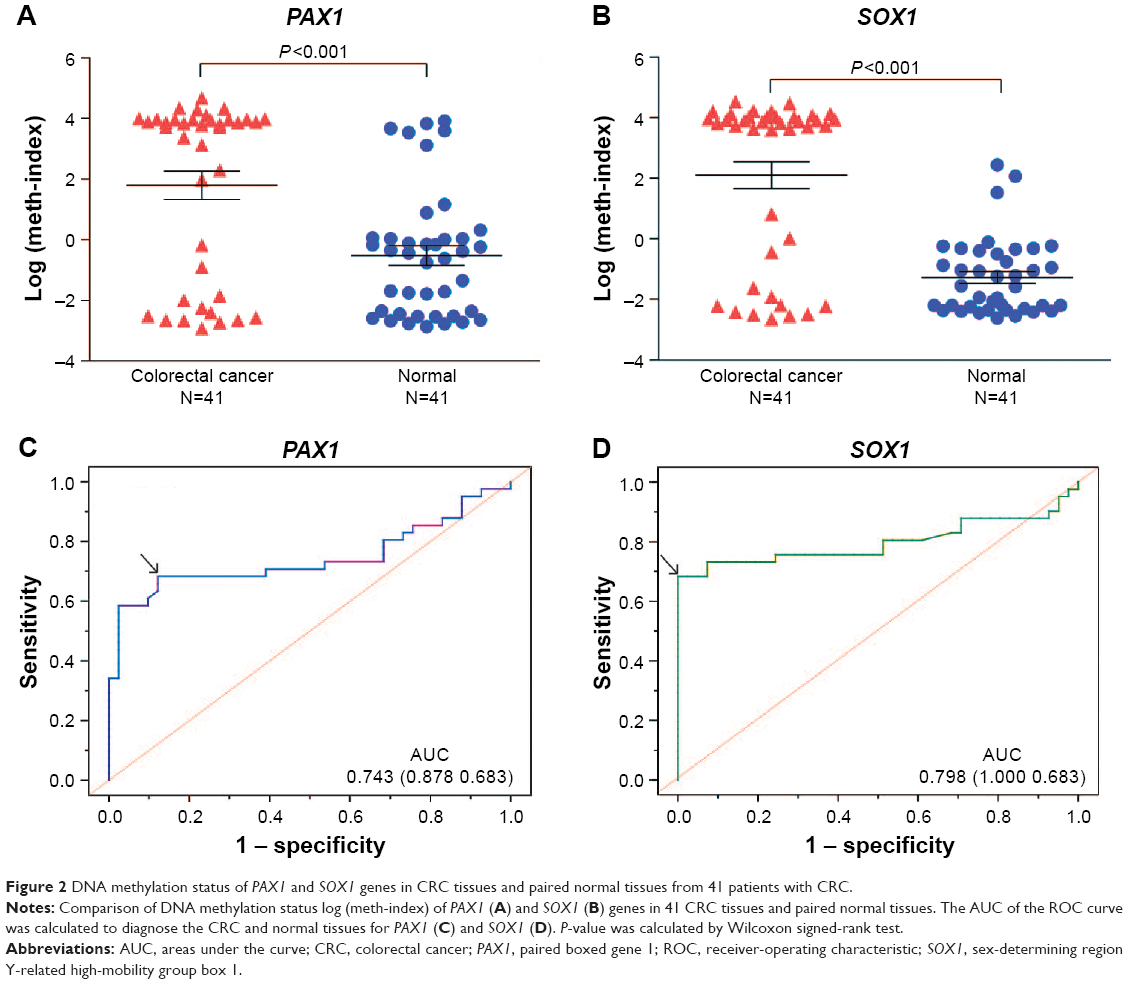 | Figure 2 DNA methylation status of PAX1 and SOX1 genes in CRC tissues and paired normal tissues from 41 patients with CRC. |
Furthermore, the correlation between the methylation status of SOX1 and PAX1 in the same CRC samples is analyzed in Figure S1. Pearson’s correlation analysis showed that there is no linear relationship between the methylation status of PAX1 and SOX1 in the same CRC samples.
PAX1/SOX1 methylation status and survival in CRC patients using TCGA datasets
We used the TCGA dataset to investigate whether the methylation status of SOX1 or PAX1 influenced prognosis. The results from the Kaplan–Meier analysis illustrated no significant difference in the overall survival between patients with high and low methylation levels of SOX1 or PAX1 (P>0.5, Figure 1D). The mean survival time ± standard error (SE) of patients with CRC and low methylation level of SOX1 was 619.0±42.2 days, while that of patients with CRC and high methylation level of SOX1 was 602.2±49.8 days. The mean survival time ± SE of patients with CRC and low methylation level of PAX1 was 603.4±44.9 days, while that of patients with CRC and high methylation level of PAX1 was 617.8±47.5 days.
Correlation between methylation status of PAX1/SOX1 and clinicopathological characteristics of CRC patients
The association of patients’ demographic and clinicopathological characteristics with PAX1/SOX1 methylation status was analyzed using the TCGA dataset and clinical samples. The relationship between methylation and clinicopathological characteristics in CRC is summarized in Table S1 for TCGA datasets and Table 3 for clinical samples. No significant association was found between the PAX1 or SOX1 methylation frequency and the clinicopathological characteristics of patients with CRC in TCGA datasets and clinical samples.
The correlation between methylation status of PAX1/SOX1 in cancer tissues and clinical characteristics of CRC patients is summarized in Table 4. The methylated status of SOX1 and PAX1 was significantly associated with TNM stage. The methylation distributions in the CRC tissues from the 41 patients with CRC at various TNM stages are illustrated in Figure 3. The methylation level log (meth-index) of PAX1 in patients with CRC and high TNM stage (TNM stage III/IV) was 3.11±2.43, which was significantly higher than that of patients with low TNM stage (TNM stage I/II, 1.26±2.94, P<0.05). The AUCs under ROC plots for the methylation of SOX1 and PAX1 to detect high TNM stage of CRC were 0.759 (95% CI 0.515–0.869) and 0.692 (95% CI 0.588–0.930), respectively. At log (meth-index) of 3.84, PAX1 methylation testing achieved 71.4% sensitivity and 70.4% specificity. At log (meth-index) of 3.82, PAX1 methylation testing achieved 63.0% sensitivity and 69.2% specificity.
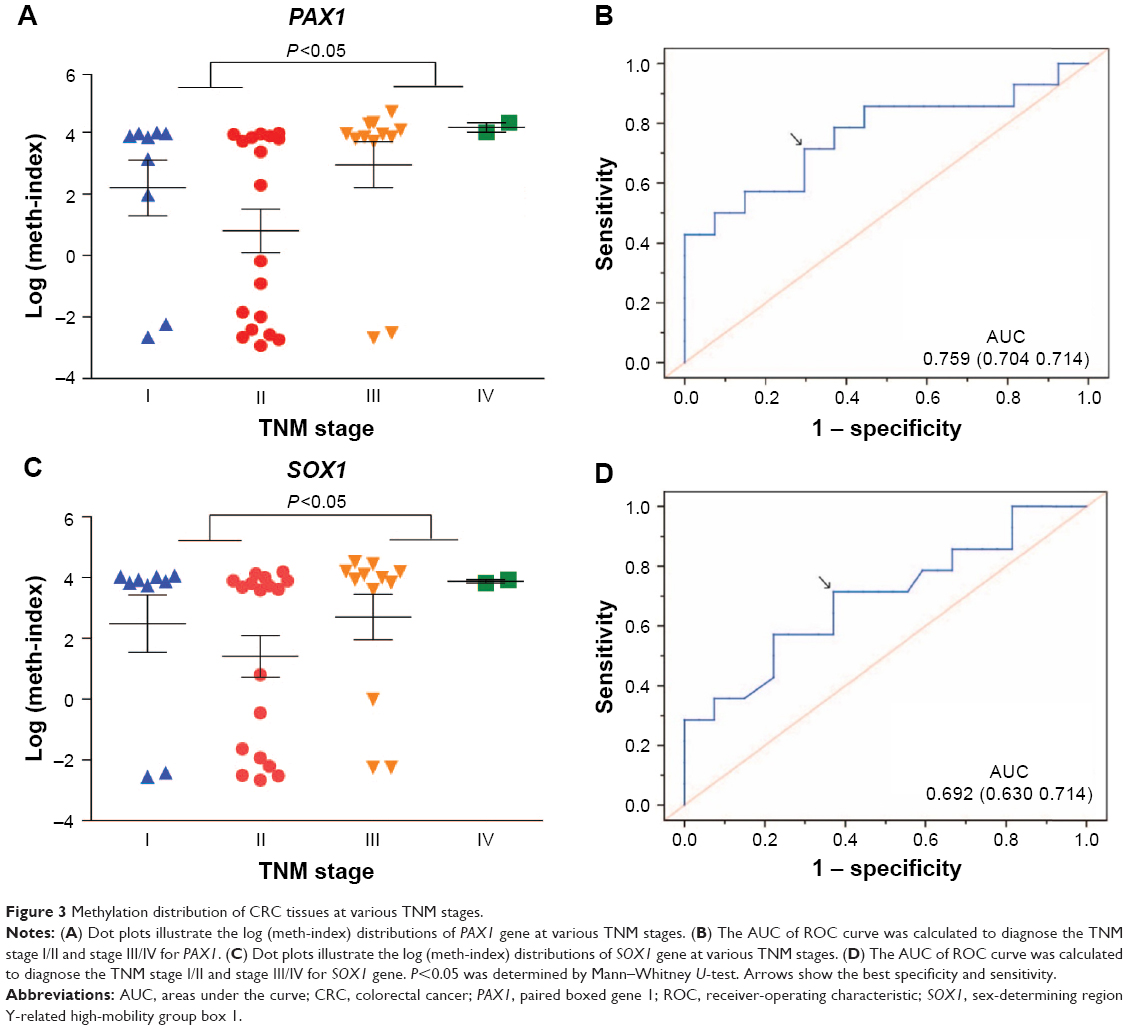 | Figure 3 Methylation distribution of CRC tissues at various TNM stages. |
Correlations between mRNA expression and DNA methylation of PAX1/SOX1 in CRC samples
The mRNA expression level of PAX1/SOX1 in the cancer and paired normal tissues of the 41 patients was quantified using quantitative RT-PCR to determine whether methylation changes in these two genes affected gene expression. The results indicated significant difference in the mRNA levels of PAX1/SOX1 between CRC and paired normal tissues (P<0.05, Figure 4A and B). The mRNA levels of both genes were lower in the CRC samples than in their paired normal samples.
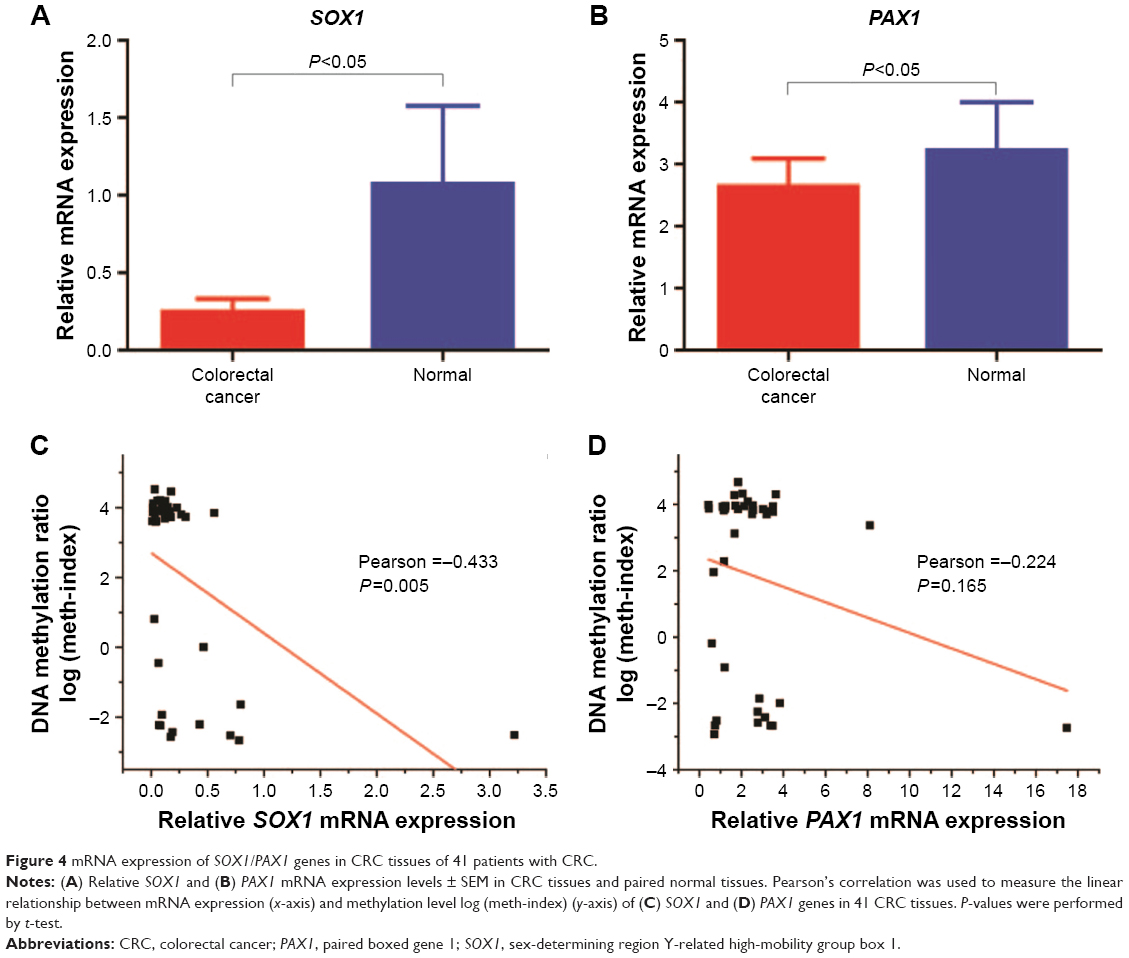 | Figure 4 mRNA expression of SOX1/PAX1 genes in CRC tissues of 41 patients with CRC. |
Pearson’s correlation analysis was also performed to analyze the relationship between mRNA expression levels and DNA methylation (Figure 4C and D). The results showed no linear relationship between methylation levels and mRNA expression of PAX1 or SOX1 in the same CRC sample.
Immunohistochemical expression of PAX1/SOX1
Immunohistochemical examination of the 41 pairs of cancer and normal tissues of patients with CRC was performed to evaluate whether methylation changes in the two genes affected protein expression. High SOX1 expression was detected in 9 (22.0%) of 41 CRC tissues and in 28 (68.2%) of 41 paracancer tissues. High SOX1 expression was detected in 9 (22.0%) of 41 CRC tissues and in 22 (53.6%) of 41 paracancer tissues. The results indicated significantly lower PAX1 and SOX1 protein expression in CRC compared with their paired normal tissues (P>0.05). Typical immunohistochemical (IHC) staining images for SOX1 and PAX1 in CRC tissues and paracancer tissues are shown in Figure 5.
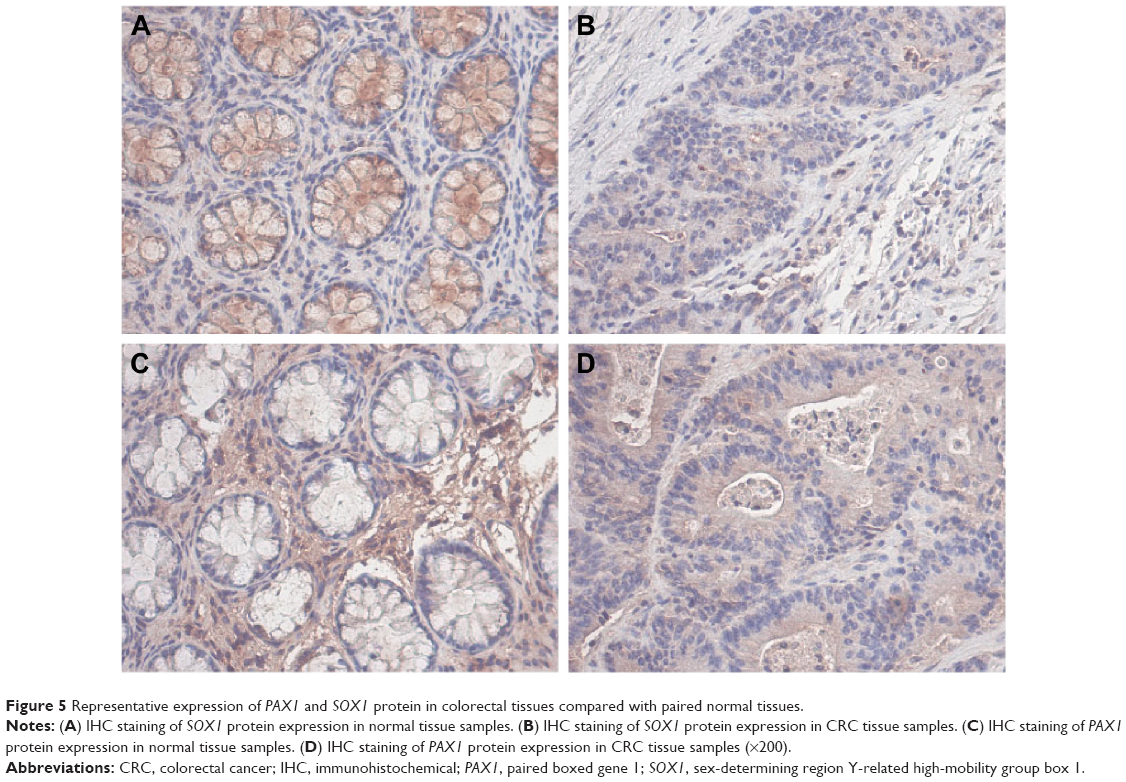 | Figure 5 Representative expression of PAX1 and SOX1 protein in colorectal tissues compared with paired normal tissues. |
Discussion
DNA methylation is the most frequent epigenetic alteration. Hypermethylation of specific gene promoters is an important mechanism of carcinogenesis. High frequency of promoter methylation of PAX1 and SOX1 genes has been detected in cervical and oral cancers. In the present study, we demonstrated that the methylation levels of PAX1 and SOX1 are higher in colorectal tumor tissues compared with the paired normal tissues from the patients with CRC. DNA methylation at cg2,09,07,471 (PAX1) and cg0,66,75,478 (SOX1) from TCGA datasets supports our results.
PAX1 is a member of the PAX family, which plays a key role during fetal development. The PAX gene family is classified into four groups based on the gene structure.7 PAX1 is categorized into group 1, which is the most frequently methylated gene in the PAX family. PAX1 may be involved in carcinogenesis and tumor progression in invasive or aggressive carcinomas. PAX1, as a tumor suppressed gene, was reported to inhibit cervical, oral, and ovarian carcinomas silenced by promoter hypermethylation. A study on head and neck squamous cell carcinoma illustrated that PAX1 participated in the Notch pathway.23 PAX1 activation was blocked by the silence of HOX family of transcription factors, which are downstream targets of Notch signaling.
SOX1 is a member of the SOX transcription factor family, which plays crucial roles in embryonic and postnatal development and stem cell regulation.24 Several members of the SOX family have been implicated in cancer development. SOX10 is a novel oncogene in hepatocellular carcinoma, which could activate the Wnt/β-catenin signaling pathway.25 SOX9 is a transcription factor expressed in human fetal prostate epithelium and enhances prostate cancer invasion.26 SOX1 has been reported as a tumor suppressor to promoters in various carcinoma tissues depending on tumor status and type. SOX1 was reported to inhibit cervical, liver, and nasopharyngeal carcinomas via the Wnt/β-catenin signaling pathway.13 Moreover, SOX1 gene could regulate prostate cancer stem cell invasion by interfering with STAT3 signaling pathway.27,28
The association of PAX1/SOX1 methylation to the clinicopathological characteristics of patients with CRC was analyzed to further evaluate the clinical significance of PAX1 and SOX1 methylation in CRC development and progression. The results showed significant association between PAX1/SOX1 and TNM stage of these patients. Besides, the sensitivity and specificity of SOX1 and PAX1 may play important roles in the poor prognosis and tumor development in CRCs. These results were consistent with a previous study on other carcinomas. A study on hepatocellular cancer has also indicated that SOX1 is correlated with TNM stage of hepatocellular cancer because low SOX1 expression in cancer tissues was significantly associated with advanced TNM stage.29 The methylation of PAX1 was significantly associated with the cervical intraepithelial neoplasia (CIN) grade, and the methylation level of PAX1 increased as the grade of CIN increased.7,30
Hypermethylation was believed to play key roles in gene expression. The methylation status of SOX1 and PAX1 was observed to be significantly higher in cancer tissues compared with paired paracancer tissues, whereas the mRNA expression of these genes was lower in the cancer tissues. However, no evident linear relationship was found with methylation status and mRNA expression in the same sample of CRC. This phenomenon may be due to the fact that the methylation of specific CpG site in a promoter could not affect the expression of the whole gene. Nevertheless, mRNA expression of SOX1 and PAX1 genes was inhibited by promoter hypermethylation. This observation is in agreement with previous hepatocellular cancer studies.29 In these studies, the mRNA expression of SOX1 gene was significantly lower in hepatocellular cancer tissues than in the paired normal tissues based on the semiquantitative RT-PCR detection in 15 paired cancer tissues and paracancer tissues in hepatocellular cancer patients. In addition, mRNA expression of SOX1 was downregulated in nasopharyngeal cancer tissues compared with their paired paracancer tissues because of the hypermethylation of SOX1 in nasopharyngeal cancer tissues.13
We further detected the expression of PAX1 and SOX1 proteins in 41 paired CRC and paracancer tissues by IHC. The expression level of PAX1/SOX1 protein was significantly lower in CRC tissues than in their paired normal tissues. It can be inferred that the hypermethylation may induce the low protein expression in CRC tissues. These results were in agreement with previous studies on hepatocellular cancer.29 Previous results showed that the expression level of SOX1 protein was significantly lower in the tissues of hepatocellular cancer than in their paired normal tissues as shown by IHC and Western blot assays.
Conclusions
For the first time, the hypermethylation levels of PAX1/SOX1 genes in CRC are reported in this study. The methylation level of PAX1/SOX1 was significantly higher in patients with CRC at high TNM stage than those at low TNM stage. Relative mRNA and protein expressions of PAX1/SOX1 were downregulated by the aberrant methylation in CRC compared with their paired normal tissue. Thus, aberrant DNA methylation of PAX1/SOX1 could be a potential molecular tumor biomarker for CRC screening.
Acknowledgments
The authors are indebted to all the donors whose names were not included in the author list, but who participated in our study. This study was supported by the National Natural Science Foundation of China (No 8,15,03,563). Literature collection was performed by using electronic database PubMed. All statistical analyses were performed using SPSS Statistics 19.0 software (IBM Corporation, Armonk, NY, USA). Raw and processed data are stored with the corresponding author of this paper and are available upon request.
Author contributions
HHZ designed the study. JH carried out the molecular experiments and data analysis and wrote the manuscript. ZRT and YYS contributed in data collection. QLL, JY, and HL provided technical assistance. All authors read and approved the final manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang ZM, Guan X, Li YJ, Zhu MC, Yang XJ, Zou X. HLA Class I Expressions on Peripheral Blood Mononuclear Cells in Colorectal Cancer Patients. Chin J Cancer Res. 2012;24(1):77–82. | ||
Marley AR, Nan H. Epidemiology of colorectal cancer. Int J Mol Epidemiol Genet. 2016;7(3):105–114. | ||
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. | ||
Hwang C, Lee S, Kim A, Kim YG, Ahn SJ, Park DY. Venous invasion in colorectal cancer: impact of morphologic findings on detection rate. Cancer Res Treat. 2016;48(4):1222–1228. | ||
Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2012;62(1):10–29. | ||
Song D, Huang R, Ma T, Chen Y, Li J, Wang X. [Analysis of screening for colorectal cancer high-risk groups in Harbin, China 2012–2015]. Zhonghua wei chang wai ke za zhi. 2016;19(10):1139–1143. Chinese. | ||
Chang CC, Huang RL, Wang HC, Liao YP, Yu MH, Lai HC. High methylation rate of LMX1A, NKX6–1, PAX1, PTPRR, SOX1, and ZNF582 genes in cervical adenocarcinoma. Int J Gynecol Cancer. 2014;24(2):201–209. | ||
Huang YK, Peng BY, Wu CY, Su CT, Wang HC, Lai HC. DNA methylation of PAX1 as a biomarker for oral squamous cell carcinoma. Clin Oral Investig. 2014;18(3):801–808. | ||
Chao TK, Ke FY, Liao YP, Wang HC, Yu CP, Lai HC. Triage of cervical cytological diagnoses of atypical squamous cells by DNA methylation of paired boxed gene 1 (PAX1). Diagn Cytopathol. 2013;41(1):41–46. | ||
Su HY, Lai HC, Lin YW, Chou YC, Liu CY, Yu MH. An epigenetic marker panel for screening and prognostic prediction of ovarian cancer. Int J Cancer. 2009;124(2):387–393. | ||
Kan YY, Liou YL, Wang HJ, et al. PAX1 methylation as a potential biomarker for cervical cancer screening. Int J Gynecol Cancer. 2014;24(5):928–934. | ||
Lai HC, Ou YC, Chen TC, et al. PAX1/SOX1 DNA methylation and cervical neoplasia detection: a Taiwanese Gynecologic Oncology Group (TGOG) study. Cancer Med. 2014;3(4):1062–1074. | ||
Guan Z, Zhang J, Wang J, et al. SOX1 down-regulates beta-catenin and reverses malignant phenotype in nasopharyngeal carcinoma. Mol Cancer. 2014;13:257. | ||
Chela-Flores J, Migoni RL. CG methylation in DNA transcription. Int J Theor Phys. 1990;29(8):853–862. | ||
Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. | ||
Teodoridis JM, Strathdee G, Brown R. Epigenetic silencing mediated by CpG island methylation: potential as a therapeutic target and as a biomarker. Drug Resist Updat. 2004;7(4–5):267–278. | ||
Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. | ||
Dayeh T, Tuomi T, Almgren P, et al. DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics. 2016;11(7):482–488. | ||
Matthaios D, Balgkouranidou I, Karayiannakis A, et al. Methylation status of the APC and RASSF1A promoter in cell-free circulating DNA and its prognostic role in patients with colorectal cancer. Oncol Lett. 2016;12(1):748–756. | ||
Manoochehri M, Borhani N, Karbasi A, Koochaki A, Kazemi B. Promoter hypermethylation and downregulation of the FAS gene may be involved in colorectal carcinogenesis. Oncol Lett. 2016;12(1):285–290. | ||
Harada T, Yamamoto E, Yamano HO, et al. Analysis of DNA methylation in bowel lavage fluid for detection of colorectal cancer. Cancer Prev Res (Phila). 2014;7(10):1002–1010. | ||
Wei J, Li G, Zhang J, et al. Integrated analysis of genome-wide DNA methylation and gene expression profiles identifies potential novel biomarkers of rectal cancer. Oncotarget. 2016;7(38):62547–62558. | ||
Guerrero-Preston R, Michailidi C, Marchionni L, et al. Key tumor suppressor genes inactivated by “greater promoter” methylation and somatic mutations in head and neck cancer. Epigenetics. 2014;9(7):1031–1046. | ||
Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12(1):15–30. | ||
Zhou D, Bai F, Zhang X, et al. SOX10 is a novel oncogene in hepatocellular carcinoma through Wnt/beta-catenin/TCF4 cascade. Tumour Biol. 2014;35(10):9935–9940. | ||
Wang H, Leav I, Ibaragi S, et al. SOX9 is expressed in human fetal prostate epithelium and enhances prostate cancer invasion. Cancer Res. 2008;68(6):1625–1630. | ||
Tsao CM, Yan MD, Shih YL, et al. SOX1 functions as a tumor suppressor by antagonizing the WNT/beta-catenin signaling pathway in hepatocellular carcinoma. Hepatology. 2012;56(6):2277–2287. | ||
Mathews LA, Hurt EM, Zhang X, Farrar WL. Epigenetic regulation of CpG promoter methylation in invasive prostate cancer cells. Mol Cancer. 2010;9:267. | ||
Lou J, Zhang K, Chen J, Gao Y, Wang R, Chen LB. Prognostic significance of SOX-1 expression in human hepatocelluar cancer. Int J Clin Exp Pathol. 2015;8(5):5411–5418. | ||
Lai HC, Lin YW, Huang RL, et al. Quantitative DNA methylation analysis detects cervical intraepithelial neoplasms type 3 and worse. Cancer. 2010;116(18):4266–4274. |
Supplementary materials
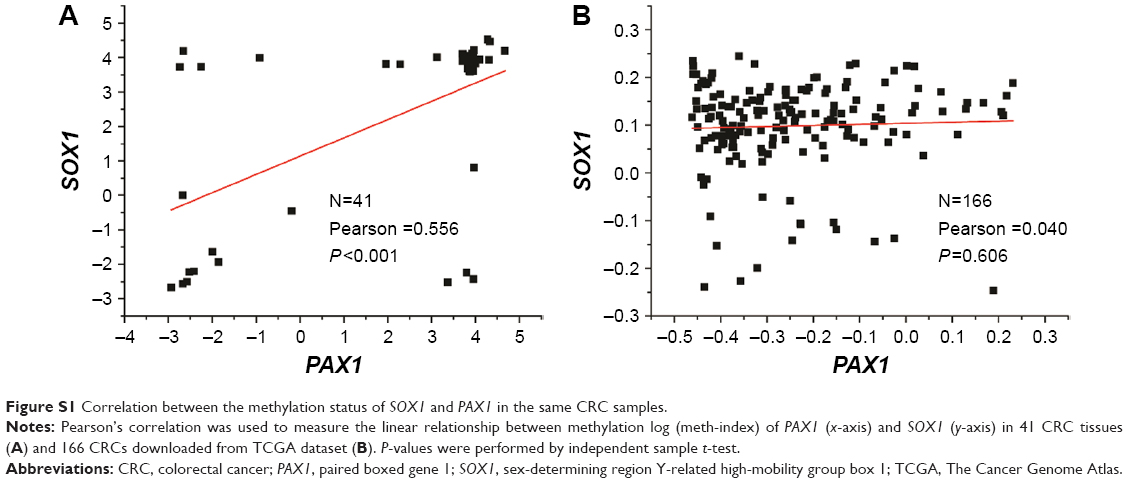 | Figure S1 Correlation between the methylation status of SOX1 and PAX1 in the same CRC samples. |
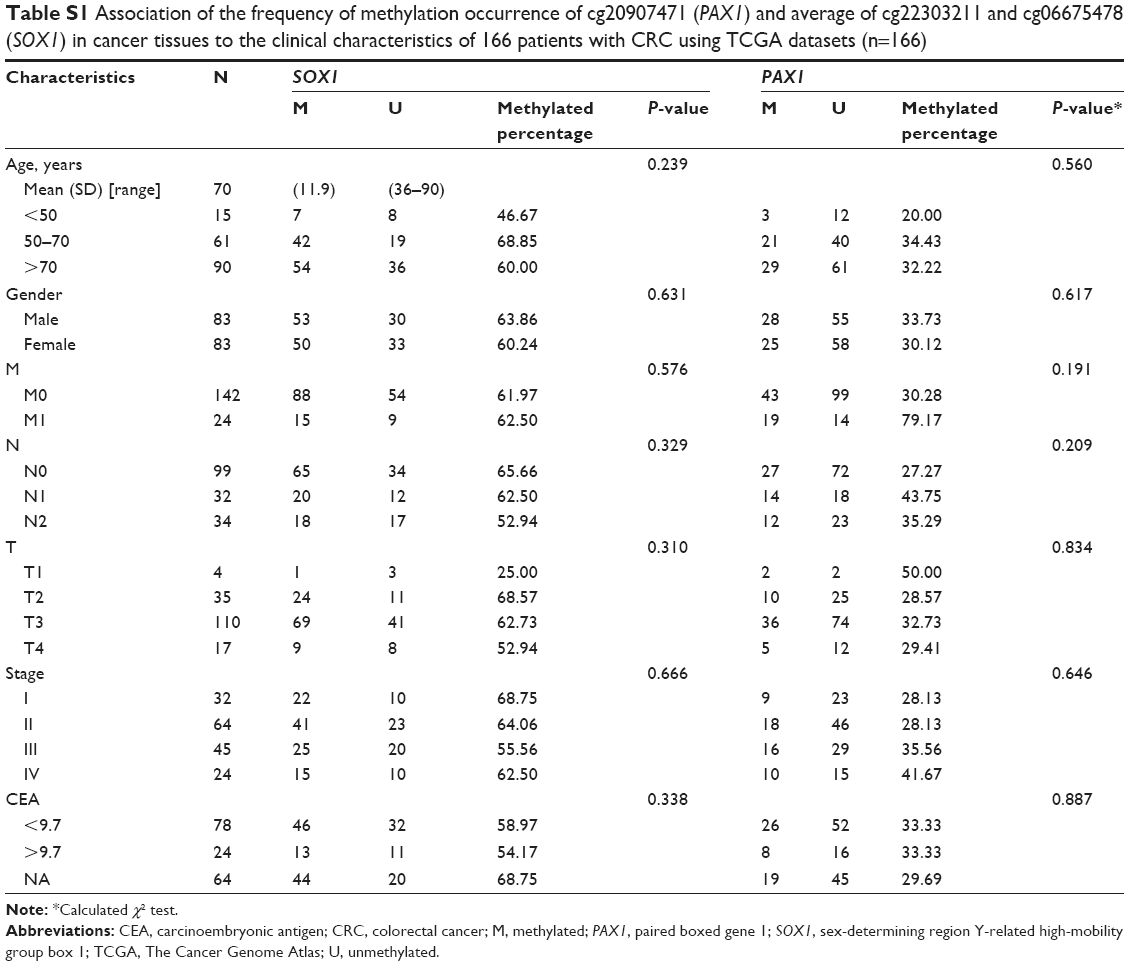 | Table S1 Association of the frequency of methylation occurrence of cg20907471 (PAX1) and average of cg22303211 and cg06675478 (SOX1) in cancer tissues to the clinical characteristics of 166 patients with CRC using TCGA datasets (n=166) |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.