")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 17
Diversities of Mechanism in Patients with VHL Syndrome and diabetes: A Report of Two Cases and Literature Review
Authors Wang Y, Liu Z , Zhao W , Cao C, Xiao L , Xiao J
Received 7 October 2023
Accepted for publication 23 March 2024
Published 9 April 2024 Volume 2024:17 Pages 1611—1619
DOI https://doi.org/10.2147/DMSO.S443495
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Yanlei Wang,* Zhaoxiang Liu,* Wenhui Zhao, Chenxiang Cao, Luqi Xiao, Jianzhong Xiao
Department of Endocrinology, Beijing Tsinghua Changgung Hospital, School of Clinical Medicine, Tsinghua University, Beijing, 102218, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianzhong Xiao, Department of Endocrinology, Beijing Tsinghua Changgung Hospital, School of Clinical Medicine, Tsinghua University, No. 168 Litang Road, Changping District, Beijing, 102218, People’s Republic of China, Tel/Fax +8610-5611-9057, Email [email protected]
Background: Von Hippel-Lindau (VHL) syndrome is characterized by tumorous lesions affecting multiple organs. Pancreatic involvement in VHL syndrome can present as endocrine tumors and pancreatic cysts, which can interfere with both exocrine and endocrine functions of the pancreas. Diabetes is an uncommon complication of VHL syndrome.
Purpose: This study aims to summarize the various mechanisms of diabetes in VHL syndrome by reporting two cases and conducting a literature review.
Methods: We analyzed the clinical and imaging data of two patients with VHL syndrome and diabetes. Additionally, we reviewed the existing literature to explore the clinical diversities and management strategies for VHL syndrome complicated with diabetes.
Results: The first patient presented with liver metastasis of pancreatic neuroendocrine tumor and multiple pheochromocytoma. After surgery, the patient’s diabetic control improved, as evidenced by a significant reduction in insulin dosage. This indicates a potential insulin resistance due to elevated metanephrine levels prior to surgery and partial insulin deficiency caused by distal pancreatectomy. The second patient had multiple hemangioblastomas, as well as multiple pancreatic cysts and positive pancreatic islet autoantibodies. Diabetes in this case may be attributed to pancreatic lesions and the coexistence of autoimmune insulitis. A literature review of other patients with VHL combined with diabetes revealed multiple mechanisms, including increased catecholamine levels, pancreatic lesions, surgical removal of pancreatic tissue, endocrine treatment, and possibly the coexistence of autoimmune insulitis.
Conclusion: VHL syndrome complicated with diabetes involves diverse mechanisms.
Keywords: von Hippel-Lindau syndrome, diabetes, pancreatic neuroendocrine tumor
Introduction
Von Hippel-Lindau (VHL) syndrome is an autosomal dominant inherited disease with a prevalence of 1 in 36,000 live births and an average onset age of 26 years. It is caused by mutations in the VHL gene on the short arm of chromosome 3, leading to the inactivation of VHL protein. The normal VHL protein works to degrade hypoxia-inducible factors (HIFs). When VHL protein is inactivated, the inhibition of HIFs is weakened, causing hypoxic responses in the body. This affects glucose uptake, metabolism, angiogenesis, extracellular matrix generation, cell proliferation and directly promotes tumor formation.1,2 VHL syndrome is characterized by tumorous lesions affecting multiple organs, such as central nervous system and retinal hemangioblastomas (60–84%), pheochromocytoma (10–20%), renal cysts and renal cancer (25–60%), pancreatic neuroendocrine tumors (12–17%), etc.3 Depending on the severity of HIF accumulation, VHL syndrome can manifest as type 1 (low risk for pheochromocytoma), type 2A (high risk for pheochromocytoma and low risk for renal cell carcinoma), type 2B (high risk for pheochromocytoma and renal cell carcinoma), and type 2C (high risk for pheochromocytoma only).4
Pancreatic involvement in VHL syndrome can present as endocrine tumors and pancreatic cysts, along with other abnormalities in the pancreas. These lesions can interfere with both exocrine and endocrine functions of the pancreas. This article focuses on the features of VHL syndrome in combination with diabetes through the analysis of two case studies. We will examine existing data regarding VHL syndrome and its lesser-known correlation with significant glycemic abnormalities in the preoperative, perioperative, and postoperative stages. Additionally, we will discuss the clinical management of these cases.
Case 1
On August 5, 2020, a 28-year-old female patient was admitted to our hospital with liver metastasis of pancreatic neuroendocrine tumor and combined with bladder and adrenal pheochromocytoma. In 2017, at the age of 25, the patient underwent transurethral resection of a bladder tumor after a routine physical examination revealing a mass in the bladder. At that time, her blood pressure and glucose levels were within normal range, and the pathology confirmed pheochromocytoma. During a follow-up examination in 2018, a solid cystic mass measuring approximately 4.8 × 3.8 × 5.6 cm was found in the right adrenal gland, along with a fasting blood glucose level of 7.44mmol/L, which remained untreated. In March 2020, an MRI revealed multiple occupying lesions in the right adrenal gland, liver, and pancreas. Her fasting blood glucose level was 13.0 mmol/L, glycosylated hemoglobin (HbA1c) was 9.5%, fasting C-peptide was 0.68ng/mL, and 2-hour C-peptide was 4.65ng/mL. Additionally, levels of free methoxy norepinephrine (NMN) were 2020.0pg/mL (<145), and free methoxyepinephrine (MN) were 37.4pg/mL (<62). She was diagnosed with diabetes and received 11-9-8 units of Regular Insulin before meals and 24 units of NPH Insulin at night, with fasting blood glucose ranging from 9–10mmol/L and postprandial blood glucose ranging from 8–13mmol/L. She had also experienced a weight loss of 7 kg over the past three years.
The patient had a family history of VHL syndrome. Her father had undergone surgery for brainstem and spinal cord hemangioblastoma and had a pheochromocytoma in the left adrenal gland removed. Her brother had bilateral adrenal pheochromocytomas and had also undergone surgery. He was currently receiving monthly infusion of Sandostatin and daily oral treatment with Lenvatinib, and his blood glucose levels were normal.
On physical examination, the patient presented with a pulse rate of 101 beats/min, blood pressure of 113/70mmHg, height of 160m, weight of 45kg, and BMI of 17.6kg/m2. The patient’s skin was moist, and no abnormalities were detected during the cardiac and pulmonary exam. The abdomen was soft, and an approximately 8cm diameter hard mass was palpable in the upper abdomen with unclear boundaries, poor mobility, and no obvious tenderness. The blood test showed an elevated metanephrine level (Table 1). An enhanced CT scan revealed a possible right adrenal pheochromocytoma with multiple lesions in the pancreas and liver (Figure 1).
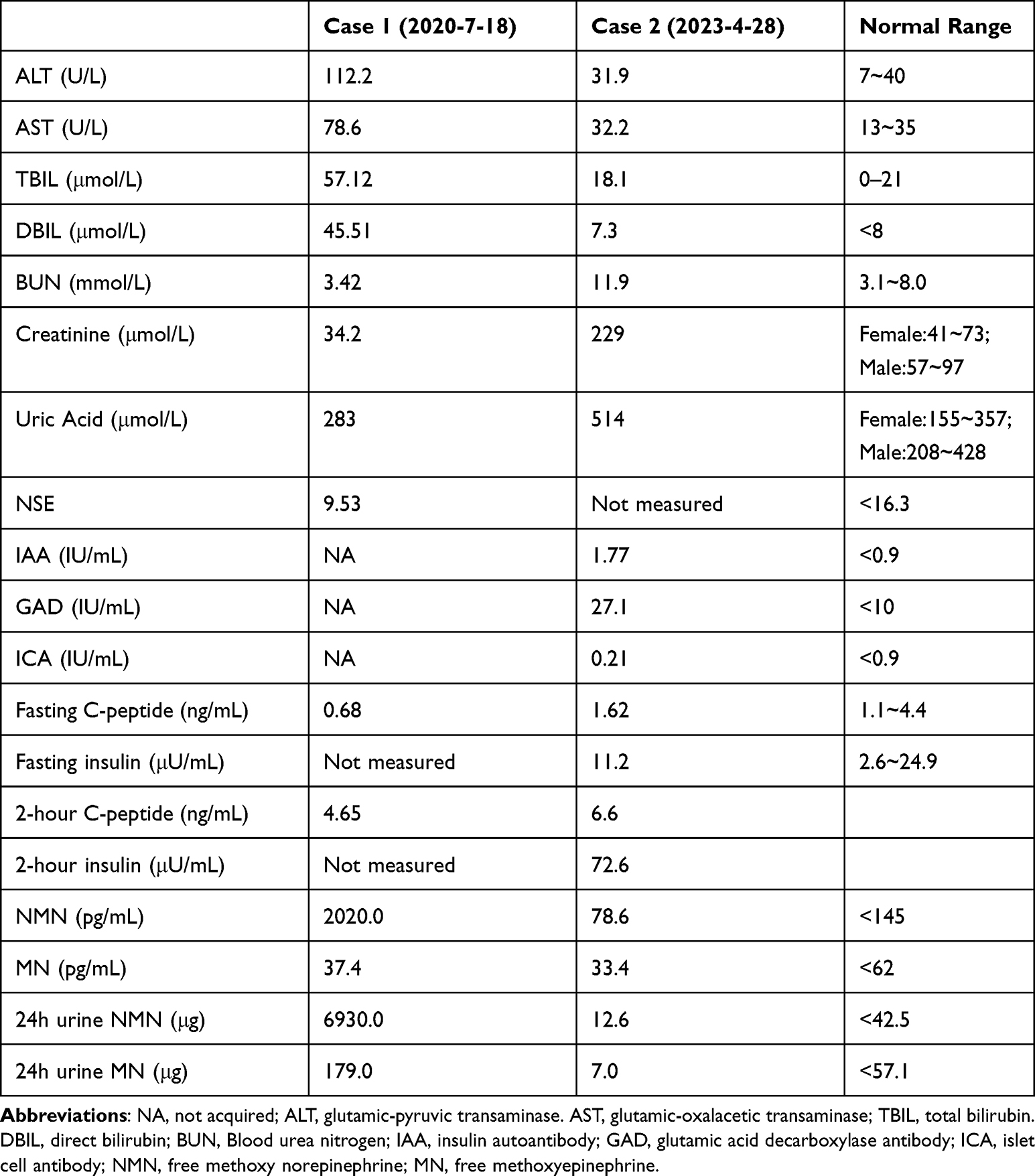 |
Table 1 Laboratory Measurements of the Two Cases at Presentation |
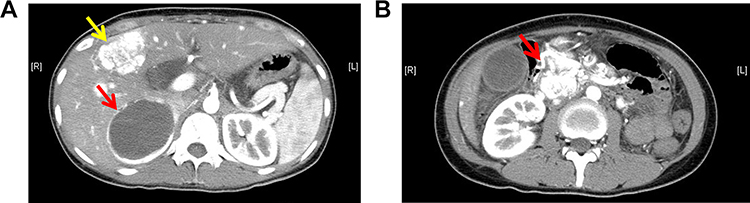 |
Figure 1 Enhanced CT scan of the abdomen (patient one) revealed a mass in the right adrenal gland measuring 7.9cm×5.6cm×8.8cm, which showed obvious enhancement on the arterial phase and a non-enhancing necrotic center, consistent with a pheochromocytoma (A) (the red arrow). The scan also showed multiple metastases in the pancreas (the largest measuring approximately 6.0m×6.0cm×5.7cm, with irregular enhancement on the arterial phase) (B) (the red arrow), and possible multiple liver metastases (A) (the yellow arrow). |
On August 12, 2020, the patient underwent surgery for complete resection of the right adrenal tumor, left liver S4 segment tumor, and distal pancreatectomy with resection of the pancreatic tail, while preserving approximately 4 cm of pancreatic tissue and the spleen. The postoperative pathology report revealed a pheochromocytoma in the adrenal gland and pancreatic neuroendocrine tumor (pNET) with liver metastasis. The patient and family members (father and brother) all tested positive for a heterozygous mutation of the VHL gene in intron 1 of the short arm of chromosome 3, specifically c.340+3_340+10delinsCG (a deletion-insertion variant).
MN and NMN were found to be normal following the latest surgery. The patient was treated with Lenvatinib and Sandostatin and required a reduced insulin dosage from 52 IU in whole day to IDegAsp insulin 8 units before the main meal at noon, with fasting blood glucose of 5.1 mmol/L and postprandial blood glucose of 8.3 mmol/L. Glycosylated hemoglobin was monitored at 5.8–6.3%. Follow-up PET/CT one year after surgery showed no new tumors.
Case 2
On April 27, 2023, a 41-year-old male patient was admitted to our hospital with complaints of fatigue, decreased vision, and polydipsia. The patient’s medical history dated back to 1999, when he underwent brain surgery because of dizziness, and the pathology confirmed cerebellar hemangioblastoma. In 2008, he was diagnosed with retinal hemangioblastoma and received laser therapy. In 2010, he was diagnosed with VHL syndrome after a mutation (c.642+70C>A in 3’-untranslated region of exon 3) was detected in the VHL gene. Subsequently, renal clear cell carcinoma was discovered and treated with percutaneous radiofrequency ablation (RFA) under full ultrasound monitoring. In 2014, the patient underwent surgical removal of cervical hemangioblastoma due to difficulty swallowing. Regular annual medical examinations were conducted, and recurrent tumors necessitated multiple surgeries. During a review in 2022, a spinal cord mass was discovered, and an enhanced magnetic resonance imaging (MRI) revealed the presence of multiple hemangioblastomas (Figure 2) in the spinal cord and cerebellar hemispheres, which were subsequently treated surgically.
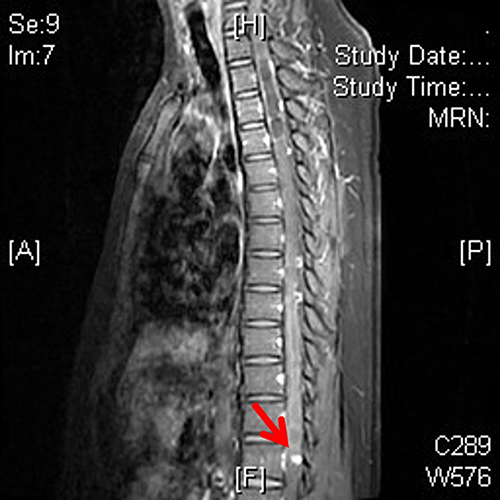 |
Figure 2 Multiple hemangioblastomas (the red arrow) seen on thoracic spine MRI (patient two). |
One month before admission, the patient experienced fatigue and thirst, and his self-measured blood glucose level was 34 mmol/L, leading to a diagnosis of diabetes. He was initiated on insulin therapy, including 8–12 IU of aspartate insulin before meals and 18 IU of glargine insulin before bed. His fasting blood glucose ranged between 7 and 8 mmol/L, while his postprandial blood glucose 10–11 mmol/L. The patient did not experience any spontaneous tendency for ketosis or hypoglycemic episodes.
The patient denied any family history of diabetes or hemangioblastoma, and genetic testing of his parents and brother did not reveal any similar mutation in the VHL gene. On examination, the patient had a blood pressure of 132/94 mmHg, weighed 70 kg, had a height of 170 cm, BMI of 24 kg/m2 and a waist circumference of 80 cm. No obvious abnormalities were detected during cardiac, pulmonary, or abdominal examination. However, an abdominal MRI revealed multiple cysts in the adrenal gland, liver, gallbladder, pancreas, and kidneys (Figure 3). Blood test showed positive pancreatic islet autoantibodies (Table 1). The patient was prescribed 14 units of IDegAsp insulin before the main meal at noon, along with 5mg of Sitagliptin once daily. Subsequent blood glucose monitoring indicated fasting blood glucose of 6–7mmol/l and postprandial blood glucose of 8–10mmol/l.
 |
Figure 3 Abdominal MRI of patient two revealed multiple cysts in the pancreas (A) (the red arrow), as well as multiple cysts and solid nodules in both kidneys (B) (the red arrows). |
Discussion
The Von Hippel-Lindau (VHL) syndrome encompasses various subtypes characterized by specific tumor types involved. Our first patient was classified as VHL type 2A although exhibiting clinical features similar to those associated with type 2B described in existing literature.5 The second case was designated as VHL type 1. The co-occurrence of VHL syndrome and diabetes is rare. Even in cases involving partial pancreatic surgery or complete pancreatic replacement due to multiple cysts, diabetes may not be present.6 However, diabetes can also be the primary manifestation of VHL syndrome.7 In VHL syndrome patients with pheochromocytoma, the prevalence of diabetes was found to be 16% (3/16),8 while in patients with VHL undergoing surgery for hemangioblastoma, the prevalence was 10.3% (6/58).9
In the case of our first patient, diabetes initially resulted from the secretion of norepinephrine by pheochromocytomas, leading to secondary diabetes. After surgery, the patient required much less insulin dosage, indicating a pronounced insulin-resistant state prior to the surgery. Literature reports suggest that catecholamines produced by pheochromocytomas can stimulate gluconeogenesis through β-receptor action, reduce insulin release through α-2 receptor interaction, and potentially induce insulin resistance by desensitizing β-adrenergic receptors.10 Blood glucose levels can be normalized after the removal of pheochromocytomas in VHL patients,11 confirming their leading role in precipitating diabetes in certain cases of VHL.
In our first patient, diabetes persisted after pheochromocytoma surgery. Given the normal levels of catecholamines, it is likely attributed to pancreatic-related diabetes resulting from the partial removal of the pancreas. Similarly, our second patient developed pancreatic-related diabetes due to extensive cyst replacement of normal pancreatic tissue. VHL syndrome can cause various pancreatic lesions, including 47% pancreatic cysts, 11% serous cystadenomas, and 15% neuroendocrine tumors known as VHL-pNET.4 These VHL-pNET are typically non-functional and often present as multiple lesions.11 While cases of diabetes associated with these pancreatic lesions are sporadically documented in the literature (Table 2), it is intriguing to note that these cases generally lack the manifestations of exocrine pancreatic dysfunction, such as steatorrhea, which differentiates them from conventional type 3C diabetes. Within VHL-related pancreatic lesions, endocrine dysfunction appears to be more prominent than exocrine dysfunction. Furthermore, our second patient also exhibited positive pancreatic islet autoantibodies, an observation previously unreported in the literature. This suggests the presence of potential autoimmune pancreatic insulitis in VHL patients.
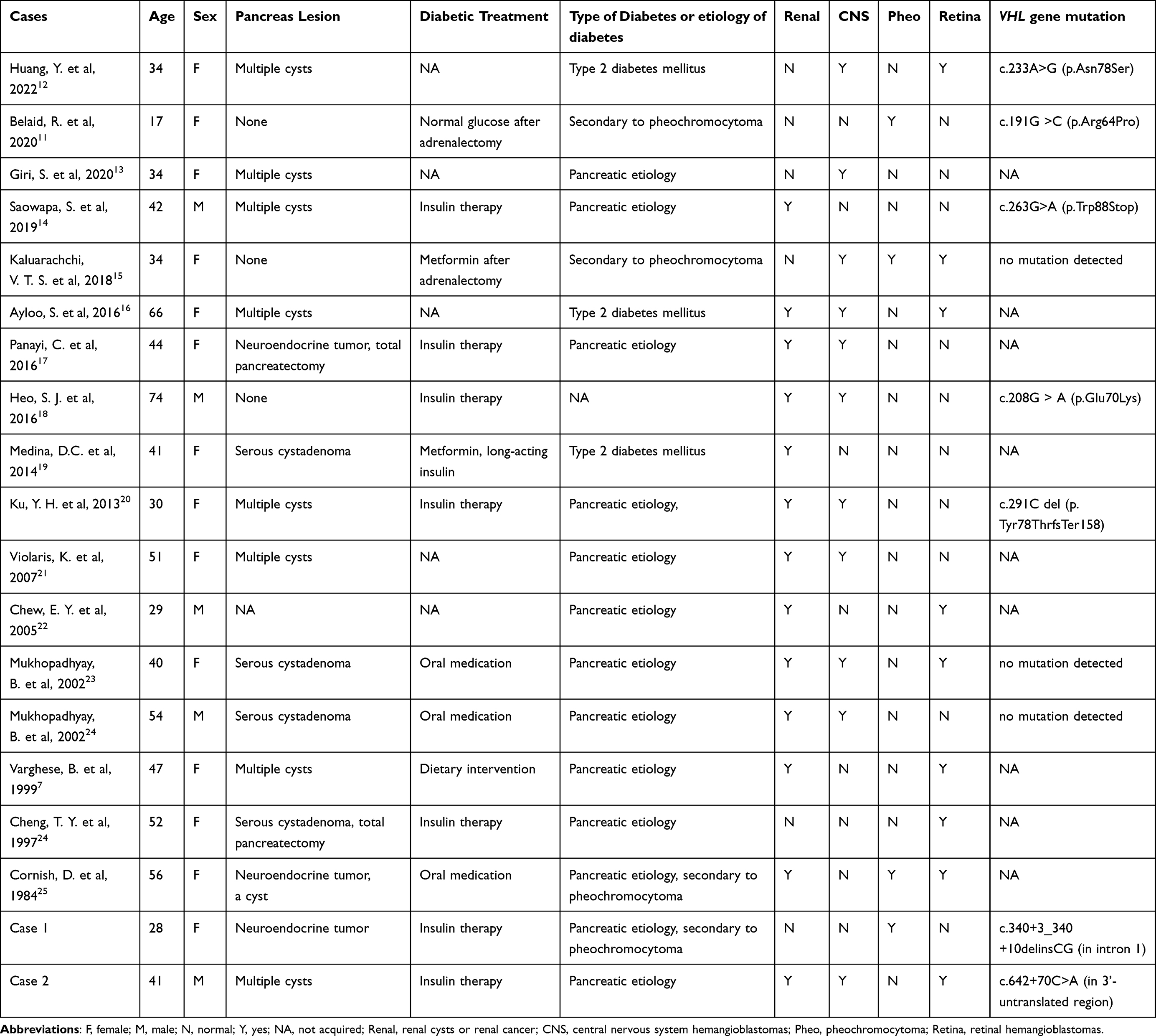 |
Table 2 Literature Review of Patients with VHL and Diabetes |
A comprehensive search was conducted on PubMed. Two hundred articles were found by searching for “Von Hippel-Lindau syndrome” along with “diabetes”, “glucose”, or “insulin”. Among them, 188 articles were irrelevant, 2 articles were not found, and 10 articles were included in the statistics. Additionally, 6 relevant articles were found from reviews, making a total of 16 articles included (Table 2). Based on the findings from these reports, the relationship between VHL syndrome and diabetes can be summarized as follows: (1) pheochromocytoma manifestation in VHL syndrome can lead to secondary diabetes; (2) pancreatic diabetes may occur when VHL syndrome presents as pancreatic neuroendocrine tumors or multiple cysts; (3) insulin-dependent diabetes mellitus may occur in combination with pancreatic islet autoantibodies; (4) non-insulin dependent diabetes mellitus may occur in individuals with a family history of diabetes or insulin resistance, which can precede the onset of VHL symptoms; (5) there is a possibility of developing diabetes after the use of somatostatin analogs, with an incidence rate of approximately 5% (5/101).26 Despite severe pancreatic lesions were observed in the reported cases, some residual pancreatic function remained, indicating the compensatory ability of the remaining pancreatic tissue. Blood sugar levels can be effectively controlled using a simple regimen of insulin combined with oral medications.
Conclusions
In conclusion, we presented two cases of VHL syndrome with distinct disease subtypes, pancreatic lesions, and diabetic mechanisms. Despite these differences, both cases demonstrated abnormal glucose metabolism prior to the onset of exocrine pancreatic dysfunction. Additionally, after undergoing treatment, individuals with VHL syndrome can effectively manage their blood glucose using relatively straightforward methodologies for glycemic reduction. This phenomenon may be attributed to the improvement in insulin resistance following the removal of pheochromocytoma, as well as the preservation of residual pancreatic function. This observation holds significance for the future management of blood glucose control in individuals with VHL syndrome.
Data Sharing Statement
All the data are fully available without restriction. All data generated or analyzed during this study are included in this published article.
Ethics Approval and Informed Consent
All procedures performed in this study involving human participants were in accordance with the ethical standards of the ethics committee of Tsinghua Changgung Hospital, and with the 1964 Helsinki declaration and its later amendments. Written informed consent was obtained from the patient and her parents included in the study. Publication of case report does not need ethical review in our institution.
Consent for Publication
Written informed consent for publication was obtained from the two patients. A copy of the written consent is available by request.
Acknowledgments
We thank the patient for granting permission to publish this information.
Author Contributions
All authors were involved in the care of the patient, made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Jianzhong Xiao is the guarantor of this work and had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This study was financially supported by the Tsinghua Precision Medicine Foundation (Grant No. 100010104). The funding organization had no role in design or conduct of the study, collection, management, analysis, or interpretation of the data, preparation, review, or approval of the manuscript, or decision to submit the manuscript for publication.
Disclosure
The authors declare that there are no conflicts of interest regarding the publication of this article.
References
1. Rechsteiner MP, von Teichman A, Nowicka A, Sulser T, Schraml P, Moch H. VHL gene mutations and their effects on hypoxia inducible factor HIFalpha: identification of potential driver and passenger mutations. Cancer Res. 2011;71(16):5500–5511. doi:10.1158/0008-5472.CAN-11-0757
2. Anthony BD, Amit T, Kristin H, et al. Guidelines for surveillance of patients with von Hippel‐Lindau disease: consensus statement of the international VHL surveillance guidelines consortium and VHL Alliance. Cancer. 2023;129(19):2927–29403. doi:10.1002/cncr.34896
3. Penitenti F, Landoni L, Scardoni M, et al. Clinical presentation, genotype-phenotype correlations, and outcome of pancreatic neuroendocrine tumors in Von Hippel-Lindau syndrome. Endocrine. 2021;74(1):180–187. doi:10.1007/s12020-021-02752-8
4. Ganeshan D, Menias CO, Pickhardt PJ, et al. Tumors in von Hippel-Lindau syndrome: from head to toe-comprehensive state-of-the-art review. Radiographics. 2018;38(3):849–866. doi:10.1148/rg.2018170156
5. Wang X, Zhang N, Ning X, et al. Higher prevalence of novel mutations in VHL gene in Chinese von Hippel-Lindau disease patients. Urology. 2014;83(3):675 e671–675. doi:10.1016/j.urology.2013.09.069
6. Maeda S, Motoi F, Oana S, et al. Pancreatic neuroendocrine tumor with complete replacement of the pancreas by serous cystic neoplasms in a patient with von Hippel-Lindau disease: a case report. Surg Case Rep. 2017;3(1):105. doi:10.1186/s40792-017-0381-4
7. Varghese B, Stephens WP. A new diabetic patient with an abdominal mass. Postgrad Med J. 1999;75(882):249–250. doi:10.1136/pgmj.75.882.249
8. Butz JJ, Yan Q, McKenzie TJ, et al. Perioperative outcomes of syndromic paraganglioma and pheochromocytoma resection in patients with von Hippel-Lindau disease, multiple endocrine neoplasia type 2, or neurofibromatosis type 1. Surgery. 2017;162(6):1259–1269. doi:10.1016/j.surg.2017.08.002
9. Hidaka T, Ikawa F, Michihata N, et al. Perioperative surgical risks in patients with hemangioblastomas: a retrospective nationwide review in Japan. World Neurosurg. 2023;170:e21–e27. doi:10.1016/j.wneu.2022.10.042
10. Ronen JA, Gavin M, Ruppert MD, Peiris AN. Glycemic Disturbances in Pheochromocytoma and Paraganglioma. Cureus. 2019;11(4):e4551. doi:10.7759/cureus.4551
11. Belaid R, Oueslati I, Chihaoui M, Yazidi M, Grira W, Chaker F. A case of von Hippel-Lindau disease with bilateral pheochromocytoma and ectopic hypersecretion of intact parathyroid hormone in an adolescent girl. Case Rep Endocrinol. 2020;2020:8824640. doi:10.1155/2020/8824640
12. Huang Y, Hu W, Huang X. Retinal hemangioblastoma in a patient with Von Hippel-Lindau disease: a case report and literature review. Front Oncol. 2022;12:963469. doi:10.3389/fonc.2022.963469
13. Giri S, Sundaram S, Darak H, Kumar S, Bhatia S. Von Hippel-Lindau disease presenting as obstructive jaundice. ACG Case Rep J. 2020;7(2):e00324. doi:10.14309/crj.0000000000000324
14. Saowapa S, Ativitavas T, Sriphrapradang C. Bilateral scrotal masses as a clue to diagnosis of genetic disease. Am J Med Sci. 2020;359(4):250–251. doi:10.1016/j.amjms.2019.11.014
15. Kaluarachchi VTS, Bulugahapitiya U, Arambewela M, Gunathilake S. Successful management of pheochromocytoma detected in pregnancy by interval adrenalectomy in a VHL patient. Case Rep Endocrinol. 2018;2018:9014585. doi:10.1155/2018/9014585
16. Ayloo S, Molinari M. Pancreatic manifestations in von Hippel-Lindau disease: a case report. Int J Surg Case Rep. 2016;21:70–72. doi:10.1016/j.ijscr.2016.02.031
17. Panayi C, Antoun N, Sandford R. Bulbar dysfunction and aspiration pneumonia due to a brainstem haemangioblastoma: an unusual complication of von Hippel-Lindau disease. BMJ Case Rep. 2016;2016. doi:10.1136/bcr-2016-217076
18. Heo SJ, Lee CK, Hahn KY, et al. A case of von Hippel-Lindau disease with colorectal adenocarcinoma, renal cell carcinoma and hemangioblastomas. Cancer Res Treat. 2016;48(1):409–414. doi:10.4143/crt.2014.299
19. Medina DC, Osorno R, Boutros CN. Biliary and gastric drainage in advanced pancreatic serous cystadenoma and portal hypertension in Von Hippel-Lindau syndrome. J Gastrointest Oncol. 2014;5(2):E50–3. doi:10.3978/j.issn.2078-6891.2014.017
20. Ku YH, Ahn CH, Jung CH, et al. A novel mutation in the von Hippel-Lindau tumor suppressor gene identified in a patient presenting with gestational diabetes mellitus. Endocrinol Metab. 2013;28(4):320–325. doi:10.3803/EnM.2013.28.4.320
21. Violaris K, Siozos T, Skoulios N, Sakellariou P. A case report of a family with 7 patients of the Von Hippel-Lindau disease. Surg Neurol. 2007;68(6):650–654. doi:10.1016/j.surneu.2006.11.040
22. Chew EY. Ocular manifestations of von Hippel-Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc. 2005;103:495–511.
23. Mukhopadhyay B, Sahdev A, Monson JP, Besser GM, Reznek RH, Chew SL. Pancreatic lesions in von Hippel-Lindau disease. Clin Endocrinol. 2002;57(5):603–608. doi:10.1046/j.1365-2265.2002.01637.x
24. Cheng TY, Su CH, Shyr YM, Lui WY. Management of pancreatic lesions in von Hippel-Lindau disease. World J Surg. 1997;21(3):307–312. doi:10.1007/s002689900234
25. Cornish D, Pont A, Minor D, Coombs JL, Bennington J. Metastatic islet cell tumor in von Hippel-Lindau disease. Am J Med. 1984;77(1):147–150. doi:10.1016/0002-9343(84)90450-9
26. Halperin R, Tirosh A. Non-interventional management of advanced pancreatic neuroendocrine neoplasms in patients with von Hippel-Lindau disease. Cancers. 2023;15(6):1739. doi:10.3390/cancers15061739
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.