Back to Journals » Journal of Inflammation Research » Volume 19
Disulfiram Attenuates Bleomycin-Induced Lung Inflammation and Fibrosis with Reduced Endothelial Pyroptosis and Neutrophil Adhesion
Authors Sun CY ![]() , Yang WL, Shen HC
, Yang WL, Shen HC ![]() , Yu WK, Chen WC, Yang KY
, Yu WK, Chen WC, Yang KY ![]()
Received 19 October 2025
Accepted for publication 18 April 2026
Published 28 May 2026 Volume 2026:19 569219
DOI https://doi.org/10.2147/JIR.S569219
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Yuhan Xing
Chuan-Yen Sun,1,2 Wan-Ling Yang,3 Hsiao-Chin Shen,1,2,4,5 Wen-Kuang Yu,1,2 Wei-Chih Chen,1,2,* Kuang-Yao Yang1,2,*
1Department of Chest Medicine, Taipei Veterans General Hospital, Taipei, Taiwan; 2School of Medicine, College of Medicine, National Yang Ming Chiao Tung University, Taipei, Taiwan; 3Taipei Hospital, Ministry of Health and Welfare, Taipei, Taiwan; 4Division of Evidence-Based Medicine, Department of Medical Education, Taipei Veterans General Hospital, Taipei, Taiwan; 5Institute of Microbiology and Immunology, School of Life Sciences, National Yang Ming Chiao Tung University, Taipei, Taiwan
*These authors contributed equally to this work
Correspondence: Wei-Chih Chen, Email [email protected]
Objective: Pulmonary fibrosis is a progressive disease characterized by chronic inflammation and ultimately leads to irreversible scarring of lung tissue. Current therapies primarily target fibrotic pathways, such as the antifibrotic agents nintedanib and pirfenidone. In bleomycin-induced lung fibrosis models, neutrophils are critical contributors, yet anti-neutrophilic strategies remain underexplored. To address this unmet need, this study characterizes the associations between neutrophilic inflammation and neutrophil adhesion, lung inflammation, and fibrosis in bleomycin models. It further evaluates the effects of disulfiram on neutrophil-driven responses, focusing on pyroptosis-related endothelial dysfunction.
Methods: Disulfiram was administered to 8– 12-week-old male C57BL/6 mice by intraperitoneal injection at a dose of 25 or 50 mg/kg daily for five days per week, starting 24 hours after intratracheal injection of bleomycin at 1.5 U/kg. Lung histopathology, chemokine receptor expression, and regulatory signaling pathways were analyzed at 7 or 14 days following bleomycin administration with and without disulfiram treatment.
Results: Histological analysis demonstrated that disulfiram significantly reduced lung inflammation and fibrosis, as evidenced by hematoxylin and eosin staining and Ashcroft scoring of lung sections. Disulfiram treatment also decreased N-terminal gasdermin D (N-GSDMD) levels, a main player in the mechanism of pyroptosis. Furthermore, significant downregulation of key markers, including lymphocyte antigen 6G, myeloperoxidase, phospho-nuclear factor kappa-light-chain-enhancer of activated B cells, vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1, was observed in disulfiram-treated bleomycin-induced pulmonary fibrosis mice. Additionally, disulfiram suppressed pyroptosis activation, which in turn reduced endothelial damage and neutrophil adhesion in human pulmonary microvascular endothelial cells, as evidenced by decreased expression of N-GSDMD, and VCAM-1, together with restoration of CD31 expression.
Conclusion: In conclusion, disulfiram reduced bleomycin-induced lung inflammation and fibrosis by modulating endothelial pyroptosis-related neutrophilic inflammation and adhesion.
Keywords: disulfiram, pulmonary fibrosis, neutrophil adhesion, endothelial pyroptosis
Introduction
Pulmonary fibrosis is a progressive disease that follows pulmonary inflammation and is characterized by the abnormal deposition of collagen and extracellular matrix.1,2 The pathophysiology of pulmonary inflammation and fibrosis is complex involving various cellular and molecular pathways and still not fully understood. Current treatments for pulmonary fibrosis primarily focus on antifibrotic agents, including nintedanib and pirfenidone, which are currently approved for clinical use.3,4 However, the early-stage intervention of targeting neutrophilic inflammation remains underexplored and warrants further investigation. Recent research has demonstrated that the abnormal activation of neutrophils and macrophages, along with the cytokines they release, plays a crucial role in lung inflammation and subsequent fibrosis.5,6 Among the various mechanisms and hypotheses, pyroptosis, an inflammatory form of programmed cell death driven by pore formation, has garnered attention as a potential link to lung inflammation and fibrosis formation.7,8 Gasdermin D (GSDMD) is a critical mediator of pyroptosis, consisting of an N-terminal pore-forming domain and a C-terminal autoinhibitory domain.9,10 Previous studies have shown that cleavage of GSDMD releases the active pore-forming fragment, N-GSDMD, which inserts into the plasma membrane and forms pores, leading to pyroptosis.11,12 This process further induces the release of various cytokines, contributing to sustained inflammation.
Bleomycin (BLM)-induced pulmonary fibrosis has long been used as a model for fibrotic lung diseases in animal studies.13,14 This model involves acute inflammation in the alveolar epithelium, followed by fibrosis, a process also observed in idiopathic pulmonary fibrosis (IPF) and acute respiratory distress syndrome (ARDS). Previous studies have shown that BLM may also induce endothelial inflammation through the upregulation of fibrogenic mediators, including transforming growth factor (TGF)-β, connective tissue growth factor, and platelet-derived growth factor-C.15,16 These mediators are elevated in cells from BLM-treated lungs compared to untreated lungs, contributing to the progression of lung fibrosis.17 However, current investigations examining the relationship between GSDMD-mediated endothelial damage and lung fibrosis remain limited. Disulfiram (DSF), initially developed for the treatment of alcohol use disorder,18,19 has attracted attention for its potential protective and therapeutic effects in various diseases, particularly its impact on lung inflammation and fibrosis.20,21 A previous study demonstrated that DSF could suppress the NOD-like receptor protein-3 (NLRP3)-mediated pyroptosis pathway induced by BLM, thereby alleviating lung inflammation.22 Additionally, Meiyue Song et al reported that DSF inhibits pyroptosis in alveolar macrophages, reducing pathological damage and inflammatory diseases.22 However, these studies primarily focused on macrophages. While DSF is classically known as an aldehyde dehydrogenase inhibitor and has been shown to possess antioxidant properties,23,24 the specific role of DSF in regulating endothelial pyroptosis-mediated neutrophil adhesion remains a critical knowledge gap. To address this knowledge gap, we aim to evaluate the relationship between DSF and neutrophilic inflammation and adhesion in bleomycin-induced pulmonary fibrosis.
Materials and Methods
Experimental Animals and Animal Models
Male C57BL/6 mice (8–12 weeks old, weighing 20–25 g) were purchased from the National Experimental Animal Center (Taipei, Taiwan) and housed in standard plastic cages with husk bedding at 25±2°C, under a 12-hour light/dark cycle. The mice were maintained under specific pathogen-free conditions and provided with food and water ad libitum. All experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee (TVGH IACUC No. 2021–160).
To induce pulmonary fibrosis, the mice were anesthetized and given an intratracheal injection of bleomycin sulfate (Merck, Darmstadt, Germany) at a dose of 1.5 U/kg in 50 µL phosphate-buffered saline (PBS), following a protocol adapted from our previous experiments.25,26 Control mice received an intratracheal injection of 50 µL PBS and did not receive any subsequent treatment. In dose-response experiments, DSF was prepared in 200 µL of vehicle (160 µL dimethyl sulfoxide [DMSO] and 40 µL PBS) and administered at a dose of 25 or 50 mg/kg by intraperitoneal injection (i.p) for five days each week, beginning 24 hours after BLM administration, as modified from a previous study.27 Mice in the BLM group did not receive DSF treatment. For the DSF-alone group, mice received intratracheal PBS followed by i.p. DSF at 50 mg/kg. The animals were sacrificed on days 7 or 14 after BLM administration, corresponding to the phases of maximal inflammation and fibrosis, respectively.
Histology and Immunohistochemistry
Lungs from both the DSF-treated and control groups were excised at 7 and 14 days following BLM-induced pulmonary fibrosis. The lung tissue was then fixed in 4% paraformaldehyde for 10 minutes, embedded in paraffin, and cut into 3 µm thick sections. Hematoxylin and eosin (HE) and immunohistochemical (IHC) staining were performed on 3 µm paraffin sections of formalin-fixed lung samples. Staining for GSDMD (1:100, ab219800, abcam, Cambridge, UK), N-GSDMD (1:100, #10137, Cell Signaling, Danvers, MA, USA), lymphocyte antigen 6G (Ly6G) (1:100, LS-C112469, LSBio, Washington, USA), myeloperoxidase (MPO) (1:100, sc-52707, Santa Cruz, Dallas, Texas, USA), phospho-nuclear factor kappa-light-chain-enhancer of activated B cells (p-NF-κB) (1:100, #3033, Cell Signaling, Danvers, MA, USA), and vascular cell adhesion molecule 1 (VCAM-1) (1:100, ARG42059, arigo, Zhubei City, Hsinchu County, R.O.C), and intercellular adhesion molecule 1 (ICAM-1) (1:100, 16,174-1-AP, Proteintech, Rosemont, IL, USA) were carried out using Envision® + Dual Link System-HRP (DAB+) kits (K4065, DAKO, Carpinteria, CA, USA). Briefly, the sections were deparaffinized with xylene, dehydrated with ethanol, and heated in 0.01 M citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked using 3% hydrogen peroxide for 10 minutes at room temperature, followed by blocking with a blocking buffer (K4065 kit). The sections were then incubated with primary antibodies at room temperature for 1 hour. Secondary anti-mouse antibodies and polymer peroxidase complexes (K4065 kit) were applied for 30 minutes at room temperature, followed by substrate/chromogen treatment (K4065 kit) and a 5- to 15-second incubation at room temperature. The slides were counterstained with hematoxylin (109249, Merck, Darmstadt, Germany) for 10 seconds and washed in running water for 10 minutes. The sections were observed and photographed using a Leica microscope (Leica Camera AG, Wetzlar, Germany) and a SPOT RT camera (Diagnostic Instruments, Inc., Sterling Heights, MI, USA). To quantitatively analyze the IHC intensity in the stained area, the percentage of IHC signal per photographed field was calculated using image processing software (Image-Pro Plus, Media Cybernetics, Inc., Silver Spring, MD, USA).
Lung Injury Score
To quantify the severity of lung fibrosis histologically, a lung injury score was assessed. Two investigators independently evaluated each HE-stained slide in a blinded manner. The lung injury score was generated by counting 300 alveoli on each slide at 400× magnification. Points were assigned within each field based on predetermined criteria from a previous study: lung injury score = ([alveolar hemorrhage points/number of fields] + 2 × [alveolar infiltrate points/number of fields] + 3 × [fibrin points/number of fields] + [alveolar septal congestion/number of fields]) / total number of alveoli counted.28 Three to five mice were used in each group.
Masson’s Trichrome Staining and Ashcroft Scale
Lung sections were fixed in 4% paraformaldehyde for 10 minutes, embedded in paraffin, and cut into 3 µm thick sections. These sections were then stained using the Trichrome Stain Kit (ab150686, Abcam, Cambridge, UK) according to the manufacturer’s instructions. Two investigators independently evaluated each Masson’s trichrome-stained slide in a blinded manner. Points were assigned within each field based on predetermined criteria from a previous study,29 graded on a scale from 0 to 8, where Grade 0 represents normal lung, Grades 1–3 indicate minimal fibrous thickening, Grades 4–6 denote moderate fibrosis, and Grades 7–8 represent severe distortion of structure and total fibrous obliteration of the field.
Western Blotting
Homogenized lung tissue in lysis buffer (475 µL RIPA, 5 µL reagent cocktail, and 20 µL 0.1 M Na3VO4) was centrifuged at 12,000 rpm for 20 minutes at 4°C and the supernatants were collected and stored at −80°C until use. Equal amounts of protein homogenate were resolved on 7.5–10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred onto polyvinylidene fluoride (PVDF) membranes. The blots were blocked in Tris-buffered saline and polysorbate 20 (TBST, with 1% Tween 20) containing 5% milk and probed with primary antibodies against NF-κB (1:1000, #8242, Cell Signaling, Danvers, MA, USA), p-NF-κB (1:1000, #3033, Cell Signaling, Danvers, MA, USA), GSDMD (1:1000, ab219800, abcam, Cambridge, UK), N-GSDMD (1:1000, #10137, Cell Signaling, Danvers, MA, USA), VLA-4 (1:1000, #8440, Cell Signaling Technology, Danvers, MA, USA), lymphocyte function-associated antigen-1 (LFA-1) (1:1000, 10,554-1-AP, Proteintech, Rosemont, IL, USA), VCAM-1 (1:1000, ARG42059, arigo, Zhubei City, Hsinchu County, R.O.C), ICAM-1 (1:1000, 16,174-1-AP, Proteintech, Rosemont, IL, USA), and β-actin (1:5000, 20,536-1-A, Proteintech, Rosemont, IL, USA). The blots were then washed in TBST, incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10000, goat anti-rabbit IgG H+L, ab205718, Abcam, Cambridge, UK), and detected using enhanced chemiluminescence (Pierce Biochemicals, 32106, Thermo Fisher Scientific, Waltham, MA, USA). Each blot was exposed to film, and densitometry of immunoreactive bands was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence (IF) Staining
The sections were deparaffinized with xylene, dehydrated with ethanol, and then heated in 0.01 M citrate buffer (pH 6.0). After blocking with 3% fetal bovine serum (FBS) in PBS for 60 minutes at room temperature, primary antibodies against Ly6G (1:100, LS-C112469, LSBio, Washington, USA), very late antigen 4 (VLA-4) (1:100, 8440S, Cell Signaling Technology, Danvers, MA, USA), chemokine (C-X-C motif) receptor 2 (CXCR2) (1:100, ab217314, Abcam, Cambridge, UK), and G protein-coupled receptor kinase 2 (GRK2) (1:100, orb669177, Biorbyt, Cambridge, UK) were applied overnight at 4°C. The following day, secondary antibodies, goat anti-rabbit immunoglobulin G (IgG) H+L (Alexa Fluor® 488) (1:400, ab150077, Abcam, Cambridge, UK) and goat anti-rabbit IgG H+L (Cy5®) (1:400, ab6564, Abcam, Cambridge, UK), were incubated at 37°C for 2 hours. The slides were then mounted with mounting medium containing 4’,6-diamidino-2-phenylindole (DAPI) (H-1200, Vector Laboratories, Burlingame, CA, USA) for nuclear staining. Images of the cells were captured using a FLUOVIEW confocal microscope (FV10i, Olympus America, Melville, NY, USA).
Culture of Human Pulmonary Microvascular Endothelial Cells (HPMECs)
HPMECs at a density of 5×104 were seeded and cultured in each well of an 8-well chamber slide coated with fibronectin until they reached confluency. The cells were then treated according to the experimental design. After treatment, the cells from different groups were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 (in PBS), and blocked with 3% FBS (in PBS) for 60 minutes at room temperature prior to IF staining.
Neutrophil Adhesion Assay
Neutrophil adhesion methods were modified from a previous approach.30 Peripheral blood was collected from a healthy individual, and neutrophils (purity > 98%) were isolated using plasma-Percoll gradients following dextran sedimentation of erythrocytes. The neutrophils were resuspended in RPMI 1640 containing 5% fetal calf serum at a final concentration of 5 × 106 cells/mL. HPMECs were seeded in a 3 cm dish to cover 95% of the bottom surface. Prior to the adhesion experiment, HPMECs were treated with or without BLM and DSF for 6 hours. Neutrophils were stained with 4 μg/mL Calcein-AM (BD Pharmingen, #564061) for 1 hour. After washing twice with 1x PBS, neutrophils were co-cultured with HPMECs for 2 hours at 37°C. The liquid was then removed, and the cells were washed twice with 1x PBS. Fluorescence cell counts were measured after fixation with 4% paraformaldehyde. Specifically, the adhered neutrophils were manually counted in three to five randomly selected, non-overlapping fields per well by independent investigators blinded to the treatment groups, and the average number of cells per field was calculated.
Lactate Dehydrogenase (LDH) Release Assay
Cytotoxicity was evaluated by measuring LDH release using the CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega, Madison, WI, USA). HPMECs (3 × 104 cells/well) were cultured in 96-well plates and subjected to the indicated treatments for 24 hours. To determine the maximum LDH release, control cells were completely lysed with the provided 10X Lysis Solution for 45 minutes prior to supernatant collection. Subsequently, 50 μL of cell culture supernatant from each well was transferred to a fresh plate and mixed with 50 μL of CytoTox 96® Reagent. After a 30-minute incubation at room temperature in the dark, the reaction was terminated by adding 50 μL of Stop Solution. Absorbance was measured at 490 nm using a microplate reader. The percentage of LDH release was calculated as [(Experimental OD - Background OD) / (Maximum OD - Background OD)] × 100.
Statistical Analysis
The mice were prepared and studied concurrently. Separate mice were used for IHC, lung injury score, Ashcroft scale, immunofluorescence, and Western blotting analyses. Data are presented as the mean ± standard deviation for each experimental group. One-way analysis of variance (ANOVA) followed by the Tukey–Kramer multiple comparisons test or a pairwise Student’s t-test was used for statistical analysis. A p-value of less than 0.05 was considered statistically significant.
Results
Effects of DSF on the Histopathological Changes in Mice with BLM-Induced Pulmonary Fibrosis
Lung inflammation and fibrosis induced by pyroptosis were triggered by intratracheal injection of BLM (Supplementary Figure 1). Figure 1A shows that IF of N-GSDMD was increased following BLM administration. Additionally, progressive lung fibrosis was observed, particularly on day 14, when compared to mice that did not receive BLM.
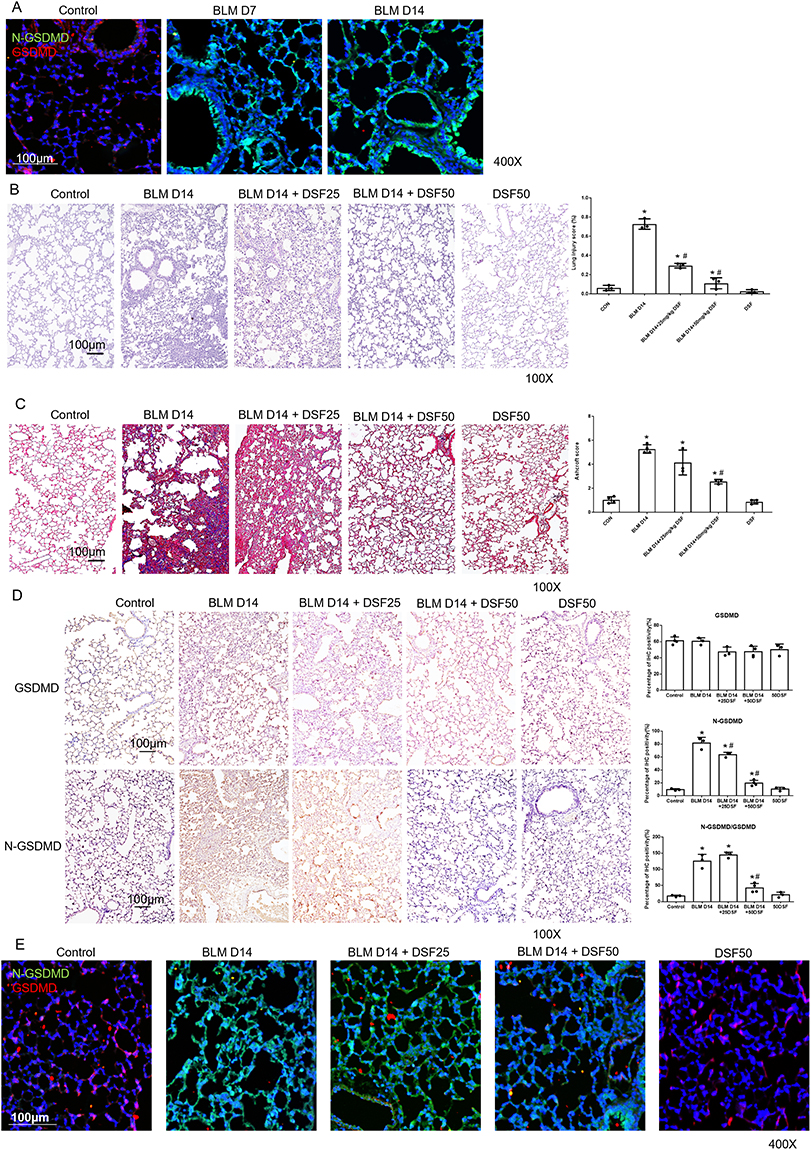 |
Figure 1 Histological assessment and GSDMD/N-GSDMD expression in bleomycin (BLM)-induced pulmonary fibrosis with disulfiram (DSF) treatment. (A) Representative immunofluorescence (IF) images of lung sections stained for N-GSDMD (green) and GSDMD (red) in control lungs and in BLM-treated lungs on days 7 and 14. Nuclei are stained with DAPI (blue). Scale bar = 100 μm. (B) Representative Hematoxylin and Eosin (HE) stained lung sections (left panels) and quantitative analysis of lung injury scores (right panel) in Control, BLM, BLM+DSF25, BLM+DSF50 and DSF50 groups on day 14. Scale bar = 100 μm. (C) Representative Masson’s trichrome stained lung sections (left panels) for collagen deposition (blue) and quantitative analysis using the Ashcroft score (right panel) in Control, BLM, BLM+DSF25, BLM+DSF50 and DSF50 groups on day 14. Scale bar = 100 μm. (D) Representative immunohistochemical (IHC) images and corresponding quantitative analysis of GSDMD expression, N-GSDMD expression, and the ratio of N-GSDMD to GSDMD in lung tissues from the Control, BLM, BLM+DSF25, BLM+DSF50 and DSF50 groups on day 14. Upper and lower image panels show GSDMD and N-GSDMD staining, respectively; the right panels show the corresponding quantification and the N-GSDMD/GSDMD ratio. Scale bar = 100 μm. (E) Representative IF images (left panels) of N-GSDMD (Alexa Fluor 488, green) and GSDMD (Cy5, red) expression in lung tissues from Control, BLM, BLM+DSF25, BLM+DSF50 and DSF50 groups on day 14. Nuclei are stained with DAPI (blue). Scale bar = 100 μm. Data are presented as mean ± SD. *p < 0.05 vs. Control group; #p < 0.05 vs. BLM group (n = 3–4 per group). DSF25: 25 mg/kg DSF; DSF50: 50 mg/kg DSF. Symbols represent individual mice. |
To investigate the effect of DSF on pyroptosis in lung fibrosis, lung sections were obtained from mice with BLM-induced lung fibrosis on day 14. The mice received varying doses of DSF via i.p., including 25 mg/kg and 50 mg/kg, along with a control group that did not receive treatment. Histological evaluation by HE staining showed a significant reduction in lung inflammation and fibrosis after DSF treatment, with a more pronounced effect observed at the 50 mg/kg dose compared to the 25 mg/kg dose. The lung injury score was also lower in the DSF-treated groups compared to the untreated group (Figure 1B). Masson’s trichrome staining and the Ashcroft score both showed the progression of pulmonary fibrosis 14 days after BLM injection. Treatment with DSF significantly attenuated BLM-induced pulmonary fibrosis, as demonstrated by reduced collagen deposition in Masson’s trichrome staining and lower fibrosis grades in the Ashcroft score (Figure 1C).
We further investigated the relationship between DSF and the GSDMD pathway using IHC staining. Compared to the control group, lung sections from BLM-injected mice demonstrated significantly higher levels of N-GSDMD and increased neutrophil infiltration. Following DSF treatment, a marked reduction in N-GSDMD levels was observed, along with a significantly lower ratio of N-GSDMD to GSDMD (Figure 1D). Additionally, IF staining revealed that BLM-induced pulmonary fibrosis mice treated with DSF also showed a significant decrease in N-GSDMD compared to untreated mice (Figure 1E). These findings suggest that DSF effectively inhibits BLM-induced lung inflammation and fibrosis by reducing the cleavage of N-GSDMD from GSDMD.
Effects of DSF on BLM-Induced Neutrophilic Inflammation and Adhesion
IHC staining of lung sections was performed to examine neutrophilic inflammation in mice with pulmonary fibrosis. The expression of Ly6G, a specific marker for neutrophil infiltration, and MPO, an indicator of neutrophil activation, were significantly increased 14 days after BLM injection, but both markers were suppressed following DSF treatment at a dose of 50 mg/kg (Figure 2A and B). Additionally, p-NF-κB showed a similar expression pattern, suggesting that DSF may reduce BLM-induced neutrophilic inflammation and accumulation in the lungs (Figure 2C).
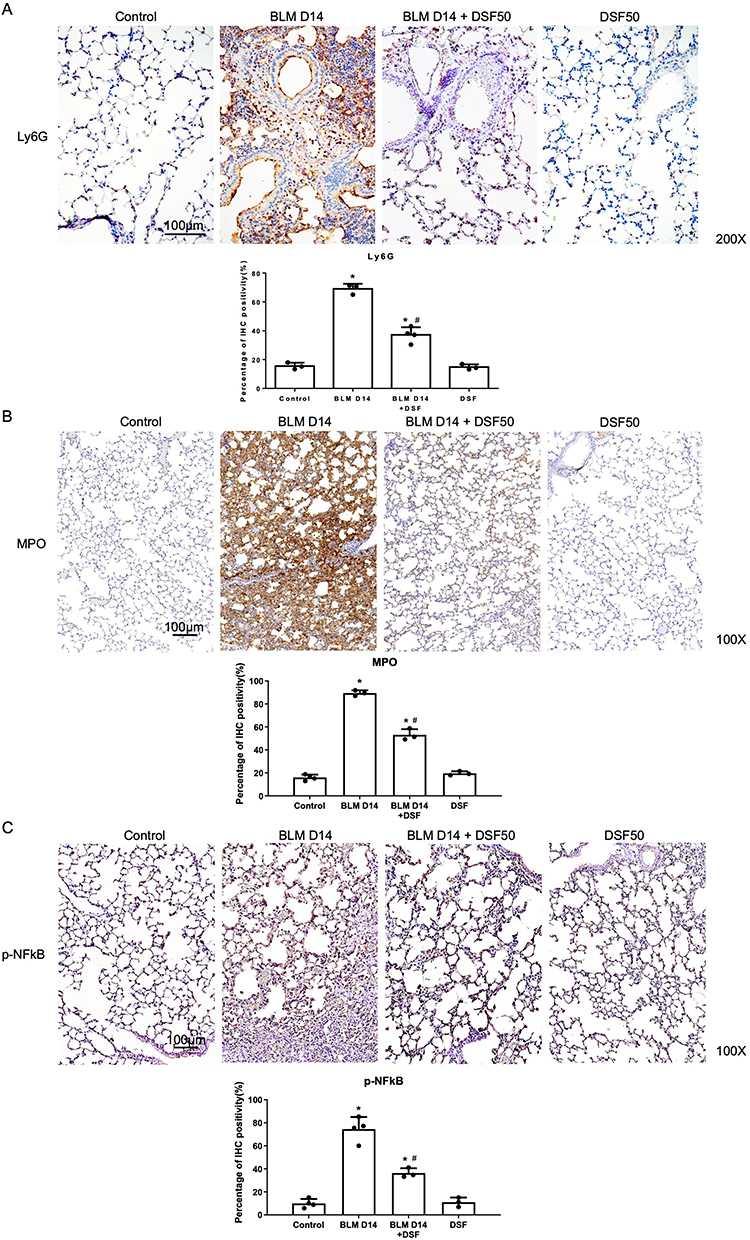 |
Figure 2 Assessment of neutrophilic inflammation and accumulation in lung tissues of mice with bleomycin (BLM)-induced pulmonary fibrosis. (A) Representative immunohistochemical (IHC) images (upper panels) and the corresponding quantitative analysis (lower panel) of lymphocyte antigen 6G (Ly6G) expression in lung tissues from the Control, BLM, BLM+DSF50 and DSF50 groups on day 14. Scale bar = 100 μm. (B) Representative IHC images (upper panels) and quantitative analysis (lower panel) of myeloperoxidase (MPO) expression in lung tissues from the Control, BLM, BLM+DSF50 and DSF50 groups on day 14. Scale bar = 100 μm. (C) Representative IHC images (upper panels) and quantitative analysis (lower panel) of phospho-nuclear factor kappa-light-chain-enhancer of activated B cells (p-NF-κB) expression in lung tissues from the Control, BLM, BLM+DSF50 and DSF50 groups on day 14. Scale bar = 100 μm. Data are presented as mean ± SD. *p < 0.05 compared to the control group; #p < 0.05 compared to the BLM group (n = 3–4 per group). DSF50: DSF at a dose of 50 mg/kg. Symbols represent individual mice. |
In addition to neutrophilic inflammation, we investigated the effect of DSF on neutrophil adhesion by evaluating the expression of VCAM-1 and ICAM-1 as these are key endothelial adhesion molecules essential for the firm adhesion and recruitment of neutrophils to inflamed tissues. Figure 3A shows that 14 days after intratracheal BLM administration, both VCAM-1 and ICAM-1 were significantly expressed compared to the control group. DSF treatment notably reduced the expression levels of both adhesion markers, as observed in the IHC staining. Moreover, bronchoalveolar lavage fluid (BALF) analysis revealed a marked accumulation and adhesion of neutrophils in the lungs of mice with BLM-induced lung fibrosis, evidenced by increased expression of Ly6G and VLA-4, a neutrophil integrin and marker of neutrophil adhesion. DSF effectively decreased polymorphonuclear neutrophil (PMN) aggregation and adhesion compared to the untreated group (Figure 3B). Western blot analysis of lung tissue further confirmed these findings, showing that DSF inhibits the active form of NF-κB (p-NF-κB) and prevents the cleavage of GSDMD to N-GSDMD (Figure 4A). Additionally, DSF reduced neutrophil accumulation and adhesion by down-regulating VLA-4, LFA-1, VCAM-1, and ICAM-1 (Figure 4B).
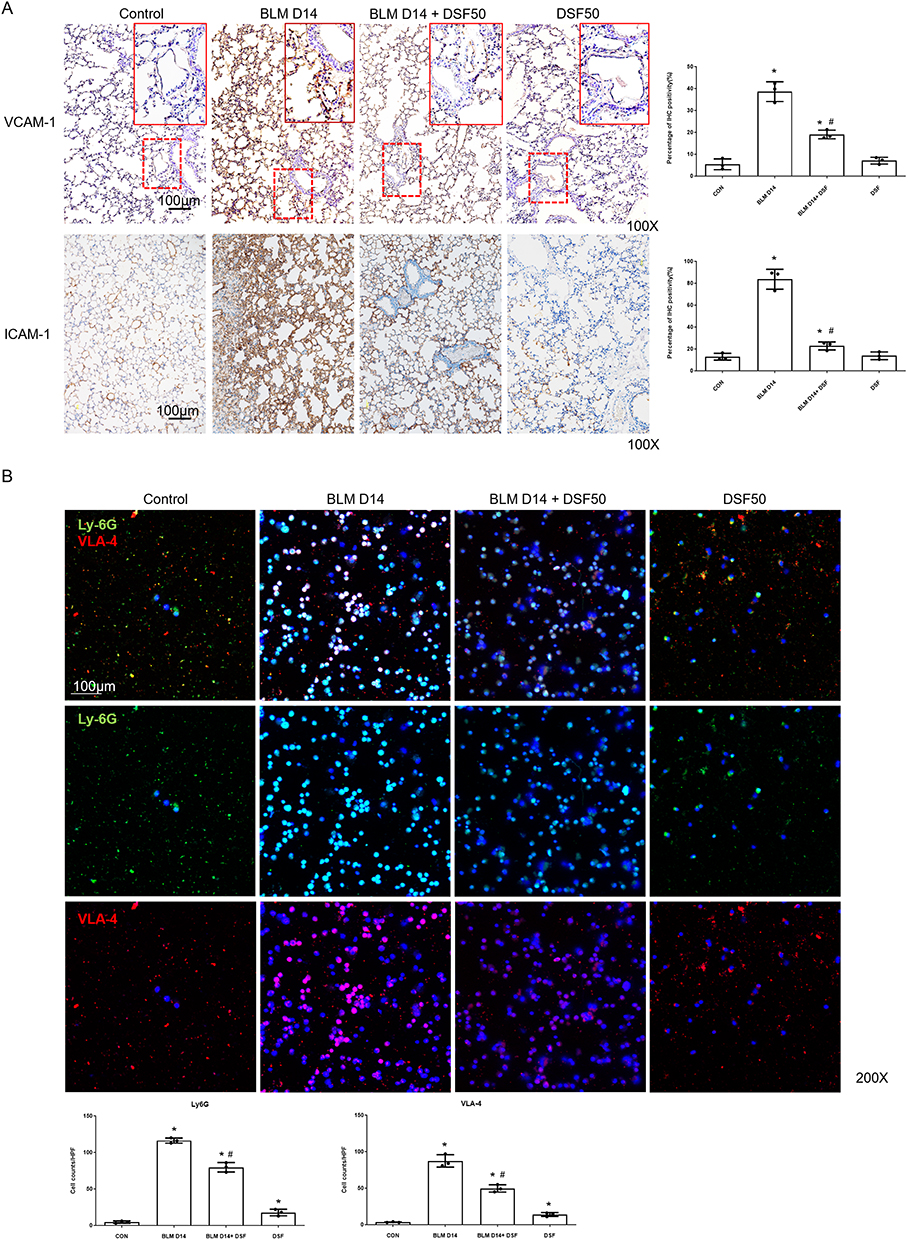 |
Figure 3 Assessment of endothelial adhesion molecules in lung tissues and neutrophil accumulation in bronchoalveolar lavage fluid (BALF). (A) Representative immunohistochemical (IHC) images (left panels) and the corresponding quantitative analysis (right panels) of vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) expression in lung tissues from the Control, BLM, BLM+DSF50 and DSF50 groups on day 14. Scale bar = 100 μm. DSF50: DSF dose of 50 mg/kg. (B) Representative immunofluorescence (IF) double staining images of cells isolated from BALF on day 14. Cells were stained for lymphocyte antigen 6G (Ly6G, green) and very late antigen 4 (VLA-4, red). Nuclei were counterstained with DAPI (blue) to indicate the total cell count. Scale bar = 100 μm. The lower panels display the quantitative analysis, representing the number of Ly6G+ cells and VLA-4+ cells evaluated against the total cell count (DAPI). Data are presented as mean ± SD. *p < 0.05 compared to the control group; #p < 0.05 compared to the BLM group (n = 3–4 per group). DSF50: DSF at a dose of 50 mg/kg. Symbols represent individual mice. |
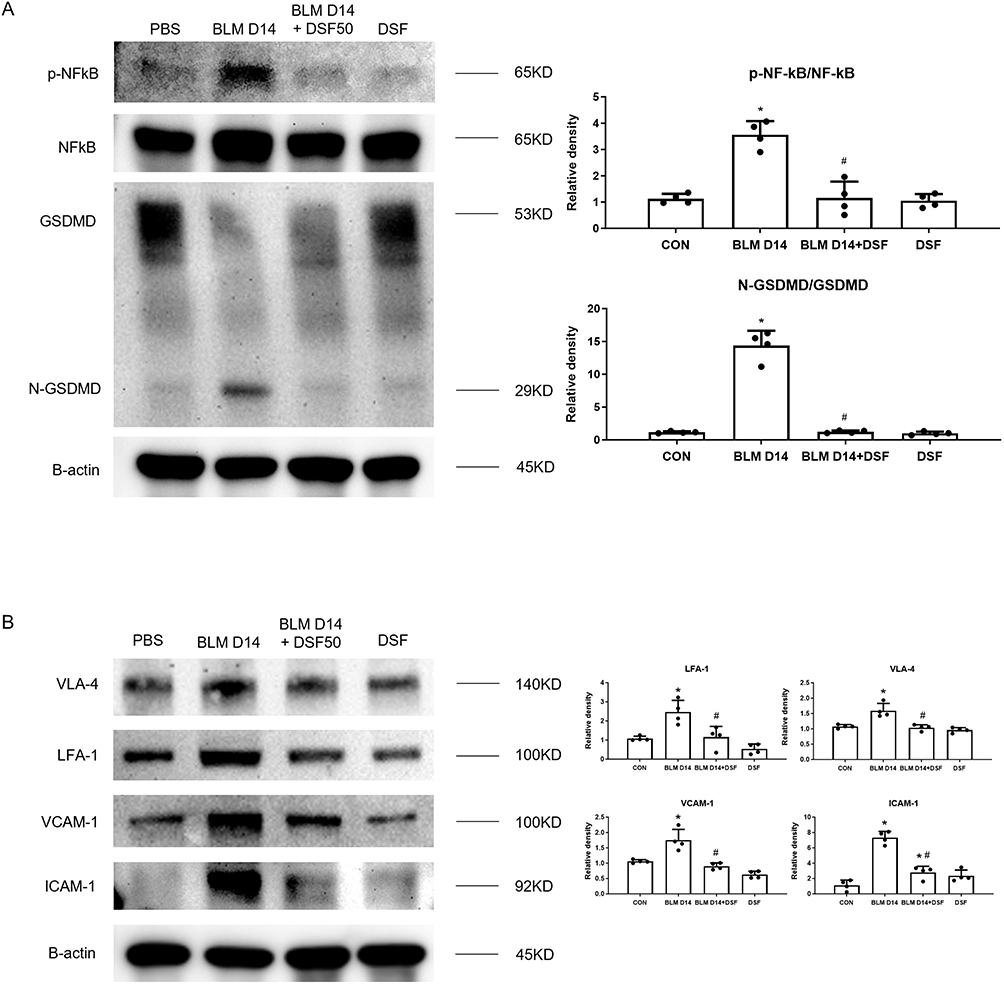 |
Figure 4 Disulfiram (DSF) modulates pyroptosis-induced neutrophilic inflammation and adhesion in mice with bleomycin (BLM)-induced pulmonary fibrosis. (A) Representative Western blot images (left panels) and corresponding densitometric analysis (right panels) of the p-NF-κB/NF-κB ratio and the N-GSDMD/GSDMD ratio in lung tissues from the Control, BLM, BLM+DSF50, and DSF50 groups on day 14. β-actin was used as a loading control. (B) Representative Western blot images (left panels) and the corresponding quantitative analysis (right panels) of very late antigen 4 (VLA-4), lymphocyte function-associated antigen-1 (LFA-1), vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1 (ICAM-1) expression in lung tissues from the Control, BLM, BLM+DSF50 and DSF50 groups on day 14. β-actin was used as an internal loading control. Data are presented as mean ± SD. *p < 0.05 compared to the control group; #p < 0.05 compared to the BLM group (n = 3–4 per group). DSF50: DSF at a dose of 50 mg/kg. Symbols represent individual mice. |
We also observed that BLM enhanced PMN chemotaxis by increasing CXCR2 expression and down-regulating GRK2 in peripheral blood. IF staining showed that DSF suppressed the expression of CXCR2 and upregulated GRK2 in peripheral blood in mice with BLM-induced lung fibrosis, compared to the untreated group. These findings suggest that DSF may inhibit chemotaxis (Supplementary Figure 2).
Effects of DSF on Inflammation and Adhesion in HPMECs
To explore the optimal dose and duration of DSF treatment on HPMECs following BLM administration, we divided the doses into 2.5 µM, 5 µM, and 10 µM, and treated the cells at various time points after BLM administration (3 hours, 6 hours, and 12 hours). We observed that HPMECs treated with DSF at 5 µM and 10 µM, 6 hours after BLM stimulation, exhibited the most optimal response (Figure 5). The expression levels of N-GSDMD in HPMECs increased following BLM instillation but were suppressed after treatment with DSF at 5 µM and 10 µM. These findings were consistent with the results observed in the whole lung tissue (Figure 6A). Furthermore, BLM reduced endothelial integrity and enhanced PMN adhesion, as evidenced by decreased CD31 levels and increased VCAM-1 expression. DSF treatment effectively mitigated these effects induced by BLM on HPMECs (Figure 6B). Figure 6C demonstrates that neutrophil adhesion was suppressed by DSF.
 |
Figure 5 N-GSDMD immunofluorescence (IF) in HPMECs after bleomycin (BLM) stimulation with disulfiram (DSF) treatment. Representative IF images of N-GSDMD in HPMECs. Cells were stimulated with BLM and treated with DSF (2.5, 5, or 10 μM) for the indicated durations (3, 6, or 12 h). N-GSDMD is shown in red and nuclei are counterstained with DAPI (blue). Scale bar = 100 μm. |
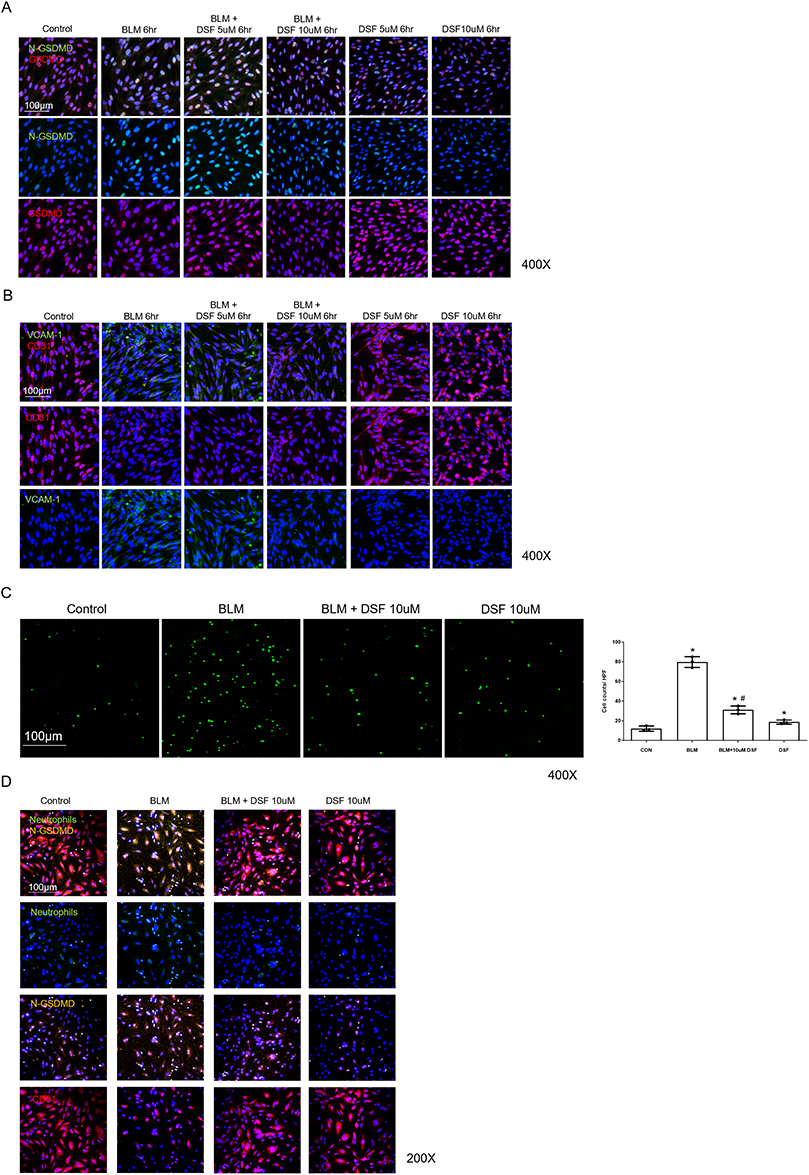 |
Figure 6 Markers of endothelial injury and neutrophil adhesion in human pulmonary microvascular endothelial cells (HPMECs) after bleomycin (BLM) stimulation with disulfiram (DSF) treatment. (A) Representative immunofluorescence (IF) images of N-terminal gasdermin D (N-GSDMD, green) and gasdermin D (GSDMD, red) expression in HPMECs. The cells were stimulated with BLM and treated with the indicated concentrations of DSF for 6 hours. Nuclei were counterstained with DAPI (blue). Scale bar = 100 μm. (B) Representative IF images of vascular cell adhesion molecule 1 (VCAM-1, green) and CD31 (red) expression in HPMECs. The cells were stimulated with BLM and treated with DSF for 6 hours. Top row: merged images; middle row: CD31; bottom row: VCAM-1. Nuclei were counterstained with DAPI (blue). Scale bar = 100 μm. (C) Representative images (left panels) and corresponding quantitative analysis (right panel) of Calcein-AM–positive neutrophils adhered to HPMECs following 6 h of BLM stimulation and DSF treatment. Scale bar = 100 μm. (D) Representative triple IF images of HPMECs co-cultured with neutrophils. Cells were stained for CD31 (red, endothelial marker), N-GSDMD (yellow, pyroptosis marker), and adhered neutrophils (green, labeled with Calcein-AM). Nuclei were counterstained with DAPI (blue). Top, second, third, and bottom rows show merged images, neutrophils, N-GSDMD, and CD31, respectively. Scale bar = 100 μm. Data are presented as mean ± SD. *p < 0.05 compared to the control group; #p < 0.05 compared to the BLM group. Symbols represent independent experiments. |
To further investigate the relationship between endothelial pyroptosis and neutrophil adhesion, triple IF staining was performed to morphologically evaluate the cells. As shown in Figure 6D, the control group exhibited continuous CD31 expression (red) with minimal N-GSDMD signals (yellow) and few adhered neutrophils (green). Following BLM stimulation, the continuity of CD31 staining was disrupted, and N-GSDMD signals were markedly increased. In this group, an increased number of adhered neutrophils was observed. In the DSF-treated BLM group, both the disruption of CD31 and the elevation of N-GSDMD signals were visibly attenuated compared to the BLM group. Concurrently, fewer adhered neutrophils were observed. Furthermore, to evaluate the biochemical impact of these treatments on overall endothelial cell membrane integrity, we measured LDH release in HPMECs at 24 hours post-treatment. Consistent with the functional and morphological observations, BLM stimulation significantly increased LDH release compared to the control group. Treatment with DSF resulted in a trend of reduced LDH release (Supplementary Figure 3).
Discussion
To the best of our knowledge, this is the first study to explore the role of DSF in regulating neutrophilic inflammation and adhesion, which are associated with BLM-induced endothelial damage and pulmonary fibrosis. We found that DSF reduced lung fibrosis driven by neutrophilic inflammation through the inhibition of GSDMD cleavage to N-GSDMD. Additionally, we observed that DSF decreased the expression of adhesion markers, including VCAM-1 and ICAM-1, thereby reducing PMN adhesion.
GSDMD, a key mediator of inflammasome-dependent pyroptotic cell death, is activated by proteolytic cleavage, releasing N-GSDMD to form membrane pores, which can induce the release of further cytokines and activate inflammatory pathways.31 Peng L et al demonstrated that Scutellarin inhibits the NF-κB/NLRP3 pathway, thereby suppressing GSDMD activation and reducing BLM-induced lung fibrosis in mice.32 Additionally, another study observed GSDMD-induced pyroptosis in macrophages of silicosis lung tissue, providing preliminary evidence that caspase-1 and caspase-4/5/11 mediate GSDMD activation. Mice lacking Gsdmd were found to be resistant to the development and progression of silicosis.22 DSF, originally developed for the treatment of alcohol use disorder,33 has recently gained attention for its potential to modulate immune responses.34 It targets the Cys191/192 residues in GSDMD,35 which are essential for pore formation in the cell membrane.36 Previous studies have shown that DSF effectively inhibits pyroptosis by blocking IL-1β release and prolonging survival during sepsis.37 Yugo Okabe et al also noted that DSF inhibits monocyte/macrophage migration and reduces the accumulation of monocyte-derived macrophages in lung tissue during inflammation in BLM-induced lung fibrosis.38 Our study further demonstrates that DSF blocks GSDMD activation not only in mouse lungs but also in HPMECs, suggesting that DSF reduces endothelial damage and alleviates lung inflammation and fibrosis. These findings indicate that DSF may exert effects through pathways beyond those identified in previous studies.
Neutrophil accumulation driven by cytokines plays a crucial role in lung inflammation associated with the development of fibrosis.39 Mice with BLM-induced fibrosis exhibited increased levels of N-GSDMD and neutrophil markers, including Ly6G and MPO, suggesting that GSDMD-mediated pyroptosis may contribute to neutrophilic inflammation. Our study shows that DSF effectively reduces neutrophil accumulation by inhibiting GSDMD cleavage, as evidenced by the significant decrease in MPO and Ly6G expression in lung tissues. Moreover, in BALF, we observed that BLM administration was associated with increased Ly6G and VLA-4 levels, which were lowered by DSF treatment. These findings suggest that DSF effectively reduces neutrophil numbers, thereby suppressing the progression of lung inflammation. Recent evidence indicates that DSF can inhibit the formation of neutrophil extracellular traps (NETs) in anti-neutrophil cytoplasmic antibody-associated vasculitis.40 Jose M. Adrover et al reported that DSF also reduced NETs caused by neutrophil infiltration in the lungs during acute lung injury.21 Our finding that DSF reduces neutrophil aggregation may partially explain the reduction in NETs observed in their study. While previous research indicates the direct inhibition of GSDMD-dependent NETosis in neutrophils or GSDMD-mediated pyroptosis in macrophages, our study provides an additional perspective. Our data demonstrate that DSF inhibits GSDMD cleavage in endothelial cells, leading to the downregulation of adhesion molecules including VCAM-1 and ICAM-1. We suggest that by preserving endothelial integrity and attenuating the recruitment and adhesion of neutrophils, DSF may potentially reduce neutrophils available for NETosis in the pulmonary microvasculature. Further investigations are warranted to elucidate the precise mechanistic cross-talk between endothelial pyroptosis and NET formation, and to determine how early-stage interventions like DSF modulate this axis during the pathogenesis of pulmonary fibrosis.
In addition to reducing neutrophil accumulation, we also observed a decrease in neutrophil adhesion following DSF treatment, as evidenced by the reduced expression of adhesion markers, including VCAM-1 and ICAM-1, which are involved in neutrophil adhesion to the endothelial cells in the lungs. Furthermore, we found that DSF-treated HPMECs showed restored endothelial integrity, as indicated by the increased expression of CD31 after treatment. Previous studies have demonstrated that serum from preeclamptic women induces endothelial dysfunction, as evidenced by increased VCAM-1 mRNA expression and enhanced adhesion of peripheral blood mononuclear cells (PBMCs) to human umbilical vein endothelial cells (HUVECs). The research found that the addition of DSF reduced VCAM-1 mRNA expression, suggesting that DSF may have a beneficial effect on improving endothelial dysfunction, which is consistent with our findings.41 By reducing neutrophilic adhesion and accumulation, DSF helps alleviate BLM-induced lung inflammation and subsequent fibrosis.
A recent review highlighted a notable shift since 2008 in the BLM model, with an increasing number of studies evaluating drugs in a therapeutic rather than a preventive setting.42 Currently, the primary treatment for pulmonary fibrosis includes monotherapy with antifibrotic agents such as nintedanib and pirfenidone, emphasizing the central role of antifibrotic strategies. However, unlike the BLM-induced pulmonary fibrosis murine model, which exposes a single intratracheal instillation of BLM, human pulmonary fibrosis develops through repetitive cycles of inflammation and scarring. This discrepancy suggests that adjunctive therapy combining antifibrotic and anti-inflammatory agents may have greater potential to mitigate disease progression. In this study, we explored the preventive benefit of DSF in alleviating the neutrophilic inflammatory phase of pulmonary fibrosis by inhibiting endothelial pyroptosis. Further translational research is warranted to investigate the combination strategies involving antifibrotic and anti-inflammatory therapies in treating pulmonary fibrosis.
This study still has several limitations. The pathophysiology of lung fibrosis is complex, involving various signaling pathways and mechanisms. While we have explored the effects of DSF on neutrophil adhesion, these results do not encompass all of DSF’s effects on inflammation in the context of BLM-induced lung fibrosis. Further studies focusing on the broader pathophysiology of lung fibrosis are warranted. Second, we used the BLM-induced murine model to evaluate the effects of DSF on lung inflammation and fibrosis. Although widely used, this model primarily reflects an acute inflammatory injury followed by fibrotic remodeling and does not fully recapitulate the chronic, repetitive epithelial injury and progressive scarring typical of human fibrotic lung disease. Therefore, clinical extrapolation should be made cautiously, and future work should test DSF in progressive fibrotic models while defining stage-specific treatment timing and dosing. Third, although this study focused on the inhibition of GSDMD cleavage and neutrophil adhesion by DSF, it is possible that DSF has other biological effects, such as its impact on immune responses, antioxidation, or other cell death pathways (eg, autophagy or apoptosis). These effects may contribute additional benefits to the improvement of pulmonary fibrosis, but their mechanisms are not fully understood at present and warrant further investigation. Fourth, while our triple IF co-staining provides evidence linking N-GSDMD-mediated endothelial pyroptosis to neutrophil adhesion, we did not perform rescue experiments such as GSDMD siRNA knockdown. Therefore, we cannot definitively conclude that endothelial pyroptosis is the absolute trigger, as other concomitant inflammatory pathways may also contribute. Future studies utilizing genetic approaches will be essential to establish strict causality. Finally, from a clinical translational perspective, DSF requires further evaluation as a potential adjunct to established antifibrotic agents such as nintedanib and pirfenidone, which slow disease progression but do not fully halt it. Future studies should assess combination strategies and define an appropriate therapeutic window and dosing, and prospective clinical studies are ultimately required to determine long-term safety and clinical utility in pulmonary fibrosis.
Conclusions
In this study, we demonstrated that DSF inhibits GSDMD cleavage to N-GSDMD, thereby reducing neutrophil adhesion in BLM-induced lung fibrosis. By suppressing pyroptosis and modulating inflammatory pathways, DSF alleviates BLM-induced lung inflammation and fibrosis, presenting a potential early-stage intervention for diseases driven by excessive neutrophilic inflammation. Future research is necessary to explore its full potential and clinical applicability.
Abbreviations
GSDMD, Gasdermin D; BLM, Bleomycin; IPF, Idiopathic pulmonary fibrosis; ARDS, Acute respiratory distress syndrome; TGF-β, Transforming growth factor-β; DSF, Disulfiram; NLRP3, NOD-like receptor protein-3; PBS, Phosphate-buffered saline; HE, Hematoxylin and eosin; IHC, Immunohistochemistry; Ly6G, Lymphocyte antigen 6G; MPO, Myeloperoxidase; p-NF-κB, Phospho-nuclear factor kappa-light-chain-enhancer of activated B cells; VCAM-1, Vascular cell adhesion molecule 1; ICAM-1, Intercellular adhesion molecule 1; SDS-PAGE, Sodium dodecyl sulfate–polyacrylamide gel electrophoresis; PVDF, Polyvinylidene fluoride; LFA-1, Lymphocyte function-associated antigen-1; FBS, Fetal bovine serum; VLA-4, Very late antigen 4; CXCR2, Chemokine (C-X-C motif) receptor 2; GRK2, G protein-coupled receptor kinase 2; HPMECs, Human pulmonary microvascular endothelial cells; IgG, Immunoglobulin G; DAPI, Diamidino-2-phenylindole; ANOVA, One-way analysis of variance; BALF, Bronchoalveolar lavage fluid; PMN, Polymorphonuclear neutrophil; PBMCs, Peripheral blood mononuclear cells; HUVECs, Human umbilical vein endothelial cells.
Generative Artificial Intelligence
GPT-5 was used solely for English grammar and style polishing. No new content, data analysis, or interpretation was generated.
Data Sharing Statement
The datasets used or analyzed in the current study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
All experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee (TVGH IACUC No. 2021-160).
Author Contributions
Sun CY - Conceptualization, Methodology, Investigation, Formal analysis, Data curation, Writing – original draft
Yang KY - Conceptualization, Methodology, Writing – review & editing, Supervision, Funding acquisition
Chen WC - Methodology, Formal analysis, Writing – review & editing, Supervision
Yang WL - Investigation, Data curation, Writing – review & editing
Shen HC - Investigation, Writing – review & editing
Yu WK - Investigation, Writing – review & editing
Chen WC and Yang KY contributed equally to this work.
All authors agreed on the journal to which the paper was submitted and agreed to be accountable for all aspects of the work. All authors have read and approved the final manuscript.
Funding
This research was funded by grants from Taiwan National Science and Technology Council (NSTC 112-2314-B-075-050, NSTC 113-2314-B-075-045, NSTC 114-2314-B-075-084, NSTC 113-2314-B-A49-074-MY2), and Taipei Veterans General Hospital (V111B-024, V112B-031, V113B-015, V114B-009, V113C-007, V115C-180 and V114C-014). Additionally, this work was supported by grants for Cancer and Immunology Research Center (113W031101, and 114W031101, KYY) of National Yang Ming Chiao Tung University from the Feature Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Disclosure
The authors declare that they have no competing interests.
References
1. Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogen Tissue Repair. 2012;5(1):11. doi:10.1186/1755-1536-5-11
2. Wuyts WA, Agostini C, Antoniou KM, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. 2013;41(5):1207–16. doi:10.1183/09031936.00073012
3. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi:10.1056/NEJMoa1402584
4. King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A Phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. doi:10.1056/NEJMoa1402582
5. Laskin DL, Malaviya R, Laskin JD. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol Sci. 2019;168(2):287–301. doi:10.1093/toxsci/kfy309
6. Lv J, Xiong Y, Li W, Yang W, Zhao L, He R. BLT1 mediates bleomycin-induced lung fibrosis independently of neutrophils and CD4+ T cells. J Immunol. 2017;198(4):1673–1684. doi:10.4049/jimmunol.1600465
7. Gao J, Peng S, Shan X, et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis. 2019;10(12):957. doi:10.1038/s41419-019-2195-8
8. Ouyang B, Deng L, Yang F, et al. Albumin-based formononetin nanomedicines for lung injury and fibrosis therapy via blocking macrophage pyroptosis. Mater Today Bio. 2023;20:100643. doi:10.1016/j.mtbio.2023.100643
9. Chu X, Xiao X, Wang G, et al. Gasdermin D-mediated pyroptosis is regulated by AMPK-mediated phosphorylation in tumor cells. Cell Death Dis. 2023;14(7):469. doi:10.1038/s41419-023-06013-6
10. Zuo Y, Chen L, Gu H, et al. GSDMD-mediated pyroptosis: a critical mechanism of diabetic nephropathy. Expert Rev Mol Med. 2021;23:e23. doi:10.1017/erm.2021.27
11. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48(1):35–44.e6. doi:10.1016/j.immuni.2017.11.013
12. Devant P, Boršić E, Ngwa EM, et al. Gasdermin D pore-forming activity is redox-sensitive. Cell Rep. 2023;42(1):112008. doi:10.1016/j.celrep.2023.112008
13. Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L152–60. doi:10.1152/ajplung.00313.2007
14. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L379–99. doi:10.1152/ajplung.00010.2008
15. Hashimoto N, Phan SH, Imaizumi K, et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;43(2):161–172. doi:10.1165/rcmb.2009-0031OC
16. Ishida Y, Kuninaka Y, Mukaida N, Kondo T. Immune mechanisms of pulmonary fibrosis with bleomycin. Int J Mol Sci. 2023;24(4):3149.
17. Kato S, Inui N, Hakamata A, et al. Changes in pulmonary endothelial cell properties during bleomycin-induced pulmonary fibrosis. Respir Res. 2018;19(1):127. doi:10.1186/s12931-018-0831-y
18. Kranzler HR. Overview of Alcohol Use Disorder. Am J Psychiatry. 2023;180(8):565–572. doi:10.1176/appi.ajp.20230488
19. Jørgensen CH, Pedersen B, Tønnesen H. The efficacy of disulfiram for the treatment of alcohol use disorder. Alcohol Clin Exp Res. 2011;35(10):1749–1758. doi:10.1111/j.1530-0277.2011.01523.x
20. Hamidi N, Feizi F, Azadmehr A, et al. Disulfiram ameliorates bleomycin induced pulmonary inflammation and fibrosis in rats. Biotech Histochem. 2023;98(8):584–592. doi:10.1080/10520295.2023.2261367
21. Adrover JM, Carrau L, Daßler-Plenker J, et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight. 2022;7(5). doi:10.1172/jci.insight.157342.
22. Song M, Wang J, Sun Y, et al. Inhibition of gasdermin D-dependent pyroptosis attenuates the progression of silica-induced pulmonary inflammation and fibrosis. Acta Pharm Sin B. 2022;12(3):1213–1224. doi:10.1016/j.apsb.2021.10.006
23. Veverka KA, Johnson KL, Mays DC, Lipsky JJ, Naylor S. Inhibition of aldehyde dehydrogenase by disulfiram and its metabolite methyl diethylthiocarbamoyl-sulfoxide. Biochem Pharmacol. 1997;53(4):511–518. doi:10.1016/S0006-2952(96)00767-8
24. Wei S, Xiao Z, Huang J, Peng Z, Zhang B, Li W. Disulfiram inhibits oxidative stress and NLRP3 inflammasome activation to prevent LPS-induced cardiac injury. Int Immunopharmacol. 2022;105:108545. doi:10.1016/j.intimp.2022.108545
25. How CK, Chien Y, Yang KY, et al. Induced pluripotent stem cells mediate the release of interferon gamma-induced protein 10 and alleviate bleomycin-induced lung inflammation and fibrosis. Shock. 2013;39(3):261–270. doi:10.1097/SHK.0b013e318285f2e2
26. Yu WK, Yang YP, Chen WC, et al. Induced pluripotent stem cell-derived conditioned medium, as well as nintedanib, ameliorates bleomycin-induced pulmonary fibrosis via suppressing endothelial-mesenchymal transition. Respir Investig. 2025;63(5):904–914. doi:10.1016/j.resinv.2025.07.005
27. Chen WC, Chen NJ, Chen HP, et al. Nintedanib reduces neutrophil chemotaxis via activating GRK2 in bleomycin-induced pulmonary fibrosis. Int J Mol Sci. 2020;21(13):4735.
28. Yang KY, Shih HC, How CK, et al. IV delivery of induced pluripotent stem cells attenuates endotoxin-induced acute lung injury in mice. Chest. 2011;140(5):1243–1253. doi:10.1378/chest.11-0539
29. Hübner RH, Gitter W, El Mokhtari NE, et al. Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques. 2008;44(4):507–11, 14–7. doi:10.2144/000112729
30. Su VY, Chiou SH, Chen WC, et al. Induced pluripotent stem cell-derived conditioned medium promotes endogenous leukemia inhibitory factor to attenuate endotoxin-induced acute lung injury. Int J Mol Sci. 2021;22(11):5554. doi:10.3390/ijms22115554
31. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B. 2021;11(9):2768–2782. doi:10.1016/j.apsb.2021.02.006
32. Peng L, Wen L, Shi QF, et al. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis. 2020;11(11):978. doi:10.1038/s41419-020-03178-2
33. Skinner MD, Lahmek P, Pham H, Aubin HJ. Disulfiram efficacy in the treatment of alcohol dependence: a meta-analysis. PLoS One. 2014;9(2):e87366. doi:10.1371/journal.pone.0087366
34. Zhang S, Zong Y, Chen L, Li Q, Li Z, Meng R. The immunomodulatory function and antitumor effect of disulfiram: paving the way for novel cancer therapeutics. Discov Oncol. 2023;14(1):103. doi:10.1007/s12672-023-00729-9
35. Sun Z, Hornung V. A critical role for palmitoylation in pyroptosis. Mol Cell. 2024;84(12):2218–2220. doi:10.1016/j.molcel.2024.05.023
36. Kappelhoff S, Margheritis EG, Cosentino K. New insights into Gasdermin D pore formation. Biochem Soc Trans. 2024;52(2):681–692. doi:10.1042/BST20230549
37. Hu JJ, Liu X, Xia S, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21(7):736–745. doi:10.1038/s41590-020-0669-6
38. Okabe Y, Toda E, Urushiyama H, et al. Antifibrotic effect of disulfiram on bleomycin-induced lung fibrosis in mice and its impact on macrophage infiltration. Sci Rep. 2024;14(1):23653. doi:10.1038/s41598-024-71770-z
39. Heukels P, Moor CC, von der Thüsen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respir Med. 2019;147:79–91. doi:10.1016/j.rmed.2018.12.015
40. O’Sullivan KM, Vu AK, Lu L, Gottschalk TA, Lawlor KE. Disulfiram inhibits neutrophil extracellular traps and attenuates inflammation in autoimmune experimental myeloperoxidase anti-neutrophil cytoplasmic antibody-associated vasculitis 2780. J Immunol. 2025;214(Supplement_1). doi:10.1093/jimmun/vkaf283.662
41. Hastie R, Ye L, Hannan NJ, et al. Disulfiram inhibits placental soluble FMS-like tyrosine kinase-1 and soluble endoglin secretion independent of the proteasome. Pregnancy Hypertens. 2018;14:125–130. doi:10.1016/j.preghy.2018.09.005
42. Kolb P, Upagupta C, Vierhout M, et al. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur Respir J. 2020;55(6):1901105. doi:10.1183/13993003.01105-2019
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.