Back to Journals » International Journal of Nanomedicine » Volume 18
Disulfide Bond-Based SN38 Prodrug Nanoassemblies with High Drug Loading and Reduction-Triggered Drug Release for Pancreatic Cancer Therapy
Authors Zhong ZX, Li XZ, Liu JT, Qin N, Duan HQ, Duan XC ![]()
Received 25 January 2023
Accepted for publication 11 March 2023
Published 15 March 2023 Volume 2023:18 Pages 1281—1298
DOI https://doi.org/10.2147/IJN.S404848
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lijie Grace Zhang
Zhi-Xin Zhong,1,* Xu-Zhao Li,1,* Jin-Tao Liu,1 Nan Qin,1 Hong-Quan Duan,1– 3 Xiao-Chuan Duan1,4
1School of Pharmacy, Tianjin Medical University, Tianjin, 300070, People’s Republic of China; 2Research Center of Basic Medical Sciences, Tianjin Medical University, Tianjin, 300070, People’s Republic of China; 3Key Laboratory of Immune Microenvironment and Disease (Ministry of Education), Tianjin Medical University, Tianjin, 300070, People’s Republic of China; 4School of Biomedical Engineering and Technology, Tianjin Medical University, Tianjin, 300070, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hong-Quan Duan; Xiao-Chuan Duan, School of Pharmacy, School of Biomedical Engineering and Technology, Tianjin Medical University, 22, Qi Xiang Tai Road, Tianjin, 300070, People’s Republic of China, Tel +86-22-83336680, Fax +86-22-83336560, Email [email protected]; [email protected]
Purpose: Chemotherapy is a significant and effective therapeutic strategy that is frequently utilized in the treatment of cancer. Small molecular prodrug-based nanoassemblies (SMPDNAs) combine the benefits of both prodrugs and nanomedicine into a single nanoassembly with high drug loading, increased stability, and improved biocompatibility.
Methods: In this study, a disulfide bond inserted 7-ethyl-10-hydroxycamptothecin (SN38) prodrug was rationally designed and then used to prepare nanoassemblies (SNSS NAs) that were selectively activated by rich glutathione (GSH) in the tumor site. The characterization of SNSS NAs and the in vitro and in vivo evaluation of their antitumor effect on a pancreatic cancer model were performed.
Results: In vitro findings demonstrated that SNSS NAs exhibited GSH-induced SN38 release and cytotoxicity. SNSS NAs have demonstrated a passive targeting effect on tumor tissues, a superior antitumor effect compared to irinotecan (CPT-11), and satisfactory biocompatibility with double dosage treatment.
Conclusion: The SNSS NAs developed in this study provide a new method for the preparation of SN38-based nano-delivery systems with improved antitumor effect and biosafety.
Keywords: 7-ethyl-10-hydroxycamptothecin, prodrug, nanoassemblies, glutathione responsive, pancreatic cancer therapy
Graphical Abstract:

Introduction
According to the GLOBOCAN database, there have been an estimated 19.3 million new cancer cases and nearly 10 million cancer deaths in 2020, and the global cancer burden is predicted to reach 28.4 million cases in 2040, a 47% increase from 2020.1 Surgery, radiation therapy, and systemic therapy including chemotherapy, targeted therapy, hormonal therapy, and immunotherapy are reported as cancer treatment modalities.2 Among them, chemotherapy is a crucial therapeutic approach and has been used extensively for the treatment of cancer.3 The treatment options for pancreatic ductal adenocarcinoma (PDAC) are limited. The majority of patients diagnosed with advanced disease are ineligible for surgical treatment. In addition, the progression of pancreatic cancer to an advanced stage is characterized by a rapid decline in clinical status, so chemotherapy remained the first-line treatment.4
In clinical trials, the topoisomerase inhibitor irinotecan (CPT-11) was highly recommended for the treatment of various kinds of solid tumors.5 However, CPT-11 has not been successfully implemented as a second-line pancreatic cancer therapy.6 This was primarily due to the insufficient conversion ratio (2–8%) of CPT-11 to its active metabolite 7-ethyl-10-hydroxycamptothecin (SN38) by carboxylesterase (CE) on the body.7 It has been reported that SN38 is 100–1000 times more potent than CPT-11, which has been considered a potential anticancer agent. However, because of its poor solubility (11–38 μg/mL in water), instability (inactive at pH 7.4), and dose-limiting side effects (myelosuppression and diarrhea), its applications as an anticancer drug were severely limited.8,9 As a result, the development of effective SN38-drug delivery systems has significant clinical implications.
The delivery of therapeutic agents using a nanoparticulate drug delivery system (nano-DDS) has been thoroughly studied.10,11 Several nano-DDS have been investigated in clinical trials for SN38. For example, SN38 conjugates such as NK012 (a SN38 and PEG-PLGA conjugate) and EZN-2208 (a poly (ethylene glycol) and SN38 conjugate). In addition, liposomal formulations, such as liposome-encapsulated SN38 (LE-SN38) and liposomal irinotecan (Onivyde®), have been developed.6,12–14 In the Phase III NAPOLI-1 trial, it was found that the use of Onivyde in combination with leucovorin and 5-fluorouracil (5-FU/LV) could significantly improve outcomes for patients with PDAC.15 Traditional nano-DDS, on the other hand, has a number of drawbacks, including a low drug-loading efficiency (less than 10% in most cases), significant drug leakage during blood circulation, inadequate drug release at target sites, and biosafety issues related to carrier material.16,17 Small molecular prodrug-based nano-assemblies (SMPDNAs) can be considered to overcome certain drawbacks and have demonstrated enormous potential for use in the treatment of cancer in clinical settings.18,19 The SMPDNAs have the following characteristics:1) Nanometer-sized vehicles have the potential to selectively accumulate in close proximity to tumor tissue because of the enhanced permeability and retention (EPR) effect;20,21 2) small molecular prodrugs that can prevent the premature release of the parent drug; 3) a relatively simple synthesis that lessens the synthetic complexity of polymer-based carriers; 4) extremely high drug loading capacities (about 90%) without the incorporation of carriers that prevent potential side effects brought on by excipients.22–24
Glutathione (GSH), the principal cellular thiol involved in scavenging reactive oxygen species (ROS) and maintaining redox equilibrium, is widely distributed and highly concentrated.25 Recent studies have shown that many different kinds of cancer cells have increased levels of GSH.26–29 Intracellular GSH levels in tumors can reach 2–10 mM, which is 100–1000 times higher than the extracellular matrix (2–10 μM) and 7–10 times higher than normal tissues. Furthermore, higher levels of GSH may regulate oxidative stress and contribute to chemotherapy resistance in cancer cells.30–32 Designing GSH-responsive nanoplatforms for prodrug delivery and drug release was therefore important.
In a previous study, paclitaxel conjugate (NPPA-PTX) and NPPA-PTX nanoparticles for antitumor therapy were synthesized.33–35 The self-assembly of the prodrug was carried out following the prediction criterion described in a previous study by our lab.36,37 Therefore, in the present study, a disulfide bond-based SN38 prodrug, SN38-diSS, was rationally designed and its XlogP values and Hansen solubility parameters were calculated to determine whether they met the prediction criterion for forming stable nanoparticles. Then, the prodrug was synthesized and GSH-responsive prodrug nanoassemblies (SNSS NAs) with few DSPE-MPEG2000 molecules were prepared and characterized. The antitumor effect of SNSS NAs was further evaluated in vitro and in vivo on a pancreatic cancer model.
Materials and Methods
Reagents
Irinotecan hydrochloride (CPT-11), 7-Ethyl-10-hydroxycamptothecin (SN38), n-butanethiol, sodium hydride, trichloroisocyanuric acid, 2-mercaptoethanol, 4-(Dimethylamino) pyridine, triphosgene were provided by Heowns Biochem Technologies, LLC.(Tianjin, China). DSPE-MPEG2000 and DSPE-PEG2000-NH2 were purchased from AVT Pharmaceutical Tech Co., Ltd. (Shanghai, China). DL-Dithiothreitol (DTT) and Sephadex G-50 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sulforhodamine B (SRB) was acquired from Macklin Biochemical Co., Ltd (Shanghai, China). L-Buthionine-sulfoximine (BSO) was obtained from yuanye Bio-Technology Co., Ltd. (Shanghai, China). EIPA and Filipin complex were provided by MedChemExpres LLC (Monmouth Junction, NJ, USA). Methyl-β-cyclodextrin (MβCD), chlorpromazine hydrochloride (CPZ), and D- (+)-Sucrose were purchased from J&K Scientific Ltd. (Beijing, China). Nuclear Green LCS-1 was acquired from Abcam. 4% Paraformaldehyde, dimethyl sulfoxide (DMSO), Tween-80 and Triton X-100 were obtained from Solarbio Science&Technology Co., Ltd. (Beijing, China).
Cell Lines
The human pancreatic carcinoma cells Panc-1 and BxPC-3 were acquired from the American Type Culture Collection (ATCC, Rockefeller, MD, USA). Panc-1 and BxPC-3 cells were grown in DMEM (high glucose) medium and RPMI 1640 medium, respectively. All of the cell culture medium was supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 µg/mL streptomycin. The cultures were kept at 37℃ in a humidified atmosphere with 5% CO2.
Animals
BALB/c nude mice (20 ± 2 g) were provided by SPF (Beijing) Biotechnology Co., Ltd. (Beijing, China) and were acclimated for 7 days prior to the experiment. They were then given free access to a standard diet and water under the maintained conditions of 25℃ temperature and 50% relative humidity.
The Institutional Animal Care and Use Committee of Tianjin Medical University gave its approval for all procedures involving the care and handling of animals to be carried out following the “3R”-rule (“replacement”, “refinement”, “reduction”). Approval number: TMUaMEC 2021001. Animals were anesthetized with an intraperitoneal injection of 50 mg/kg of 1% pentobarbital sodium solution and sacrificed via cervical dislocation.
Synthesis of SN38 Prodrug
Compound 1 was synthesized by pouring n-butyl mercaptan (30 mmol, 3.213 mL) into a solution of sodium hydride (720 mg, 30 mmol) in tetrahydrofuran (30 mL) and stirring for 1 minute at −20℃ with N2 protection. The mixture was then treated with trichloroisocyanuric acid (TCCA) (10 mmol, 2.32 g) in acetonitrile (10 mL), followed by a rapid addition of 2-mercaptoethanol (10 mmol, 2.106 mL). The reaction mixture was stirred for 10 minutes, after which the organic solvent was evaporated under reduced pressure and neutralized with a 1 mM solution of hydrochloric acid. The crude product was extracted three times with dichloromethane from water, and the organic layer was dried with anhydrous magnesium sulfate, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography with petroleum ether and ethyl acetate in a ratio of 6:1, 4:1 and 2:1 in sequence to yield a white solid (2.02 g, 12.15 mmol, 40.5%).
Compound 2 was synthesized in the second step by dissolving compound 1 (10 mmol, 1.66 g) and 4-(dimethylamino) pyridine (22.5 mmol, 2.75 g) in anhydrous dichloromethane (10 mL) under N2 protection. The mixture was then treated with triphosgene (3.3 mmol, 979.2 mg) and stirred for 15 minutes at 0℃. A solution of SN38 (1 mmol, 392.4 mg) in anhydrous N,N-dimethylformamide (5 mL) was added dropwise to the mixture at 0℃ and stirred for 2 h at room temperature. The organic solvent was extracted from the reaction mixture by evaporation at reduced pressure, and the remaining residue was neutralized with 1 mM hydrochloric acid. The crude product was extracted three times with dichloromethane from water, and the organic layer was dried with anhydrous magnesium sulfate, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography (petroleum ether-dichloromethane, 2:8; dichloromethane-methanol, 1:1) to yield an off-white solid (0.848 mmol, 658.9mg, 84.8%).
Thin-layer chromatography (TLC), with pre-coated silica gel glass plates containing a fluorescence indicator, was employed for monitoring the progress of the reactions. The column chromatography was performed on silica gel (SiO2; 300–400 mesh). The NMR spectra of SNSS were obtained using a Bruker AVANCE III 400 instrument (Billerica, MA, USA). High-performance liquid chromatography (HPLC) with Agilent Technologies 1200 Series (Palo Alto, CA, USA) was utilized to determine the purity of SN38-diSS.
Theoretical Partition Coefficient (XlogP) and Hansen Solubility Parameters of SN38-diSS
The XlogP value of SN38-diSS was calculated using the Wang group’s XLOGP3 algorithm.38 The dispersion [δd], polar [δp], hydrogen bonding [δh], and total solubility parameter [δt] were determined using Beerbower’s group contribution method (GCM).39,40 The specific details of the calculation are presented in Table S1 and S2.
Preparation and Characterization of SNSS NAs
SN38-diSS and DSPE-MPEG2000 were dissolved in DMSO (0.2 mL) at a ratio of 1:0.1. The mixture was added dropwise to 5 mL of distilled water with vigorous stirring at room temperature, resulting in the spontaneous formation of SNSS NAs. The SNSS NAs were then ultrafiltered and purified utilizing Sephadex G-50. Cy7-labled SNSS NAs were prepared by using DSPE-PEG2000-Cy7, which was obtained by conjugating DSPE-PEG2000-NH2 with Cy7.
The particle size of SNSS NAs as well as their zeta potential were measured and analyzed by employing dynamic light scattering (DLS) on a Nano ZS90 (Malvern Instruments, Malvern, UK). For the stability study, SNSS NAs were stored at 4℃ for two weeks while their size and PDI were measured daily using DLS. Additionally, the dilution stability of SNSS NAs was assessed by DLS at various PBS dilution ratios. Transmission electron microscopy (TEM) (HT7700, Tokyo, Japan) was employed for investigating and characterizing the morphology of SNSS NAs.
HPLC was used to measure the drug-loading efficiency of SNSS NAs. The solution of SNSS NAs was freeze-dried using a freeze drier. The freeze-dried powder was then redissolved in methanol and analyzed at 372 nm using HPLC. The loading efficiency of SN38-diSS was determined as follows:
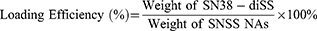
Hemocompatibility Analysis
The hemolysis test was performed as described previously.41 Red blood cells (RBCs) were separated from fresh mice blood by centrifugation. The collected RBCs were resuspended in PBS at a concentration of 2% (v/v). Following that, 0.2 mL of RBC suspension was incubated with SNSS NAs at concentrations of 25, 125, 250, and 500 µg/mL, respectively, in PBS, 0.9% saline (as a negative control), or distilled water (as a positive control). The samples were centrifuged at 3000 rpm for 10 min after being gently shaken at 37℃ for 3 h, and the supernatant was collected. A microplate reader (BioTek Synergy HTX, BioTek Instruments, Inc., Winooski, VT, USA) was used to measure the absorbance of the supernatant at 545 nm. The percentage of hemolysis was calculated using the following equation:
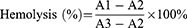
A1 = absorbance of sample; A2 = absorbance of negative control; and A3 = absorbance of positive control.
In vitro Drug Release Study
The in vitro drug release behavior of SNSS NAs was evaluated using the dialysis method. In brief, SNSS NAs (1 mL, 0.2 mg/mL) were added to dialysis bags (MWCO 7000 Da) and tightly sealed. The dialysis bags were then immersed in 40 mL of a 10mM phosphate-buffered saline solution (pH 7.4) containing 10mM GSH and 0.5% tween-80 and gently stirred at 37℃. 0.4 mL of dialysate was collected at various time intervals (1, 2, 4, 6, 8, 12, 24, 48, 72, and 96 h) and replaced with the same volume of fresh release medium to maintain sink conditions of the media. HPLC was used to ascertain the SN38 content.
In vitro Cytotoxicity Study
The SRB method was utilized to investigate the cytotoxicity of SNSS NAs in relation to the human pancreatic carcinoma cell lines Panc-1 and BxPC-3.34,42 Briefly, cells were seeded in 96-well plates at a density of 2×103 cells/well and incubated for 24 h. The cells were then treated with different concentrations of SNSS NAs, CPT-11, and SN38, respectively. The cells were washed, fixed, and stained with SRB after being exposed to an incubation period of 72 h. Then 10 mM Tris buffer was added, and the OD value of each well was measured at 540 nm using a microplate reader (BioTek Synergy HTX, BioTek Instruments, Inc., Winooski, VT, USA). GraphPad Prism 7 Software (GraphPad Software Inc., San Diego, CA, USA) was used to construct dose-effect curves and determine IC50 values.
Cells were pre-treated with 200 μM buthionine sulfoximine (BSO), an inhibitor of γ-glutamylcysteine synthetase (GCS), for 24 h to evaluate the GSH-responsive cytotoxicity. The medium was then replaced with different concentrations of drugs for an additional 72 h of incubation. An SRB assay was performed to evaluate the viability of the cells.
In vitro Cellular Uptake and Endocytosis Mechanism of SNSS NAs
The Panc-1 and BxPC-3 cells were seeded in 6-well plates at a density of 3.5×105 cells/well containing 2 mL of growth medium. After 24 h, the medium was discarded and replaced with an equal volume of fresh medium containing SNSS NAs (20 µM), which was then incubated at 37 ℃ for 2, 4, or 6 h. The 6-well plates were washed three times with cold PBS and lysed with SDS at a predetermined time. After that, samples of 5 µL were collected and analyzed using the PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The remaining lysate was then treated with methanol to precipitate proteins and extract drugs. The supernatant was collected and dried with nitrogen after centrifugation. The samples were redissolved in methanol and then examined using HPLC.
The fluorescence distribution of SNSS NAs in Panc-1 and BxPC-3 cells was observed using a confocal fluorescence microscope. Cells were seeded in confocal dishes at a density of 3×105 cells per dish for 24 h. The medium was then replaced with fresh medium containing SNSS NAs (20 µM) and the cells were incubated for 2, 4, or 6 h. Following incubation, the confocal dishes were washed three times with PBS and then fixed for 30 min with 4% paraformaldehyde. The cells were then stained with LCS-1 (2 µM, 200 µL) for 15 min. Drug distribution in cells was observed by CLSM (Olympus FV1000, Olympus Corporation, Tokyo, Japan).
Panc-1 and BxPC-3 cells were seeded in a 6-well plate (3.5×105 cells/well) for 24 ho and treated with various endocytosis inhibitors (listed in Table S3) for 60 min at 37 ℃ to investigate the endocytosis mechanism. The medium was then discarded and replaced with fresh medium containing SNSS NAs (20 µM) in combination with various inhibitors for an additional 5 h of incubation. HPLC was used to ascertain the amount of drug endocytosed, as previously mentioned.
In vivo Biodistribution of SNSS NAs
Nude mice bearing the Panc-1 tumor were used to evaluate the in vivo imaging of SNSS NAs. Mice were injected intravenously with Cy7-labeled SNSS NAs when the tumor volume reached approximately 400 mm3. In vivo fluorescence imaging was performed on an IVIS Spectrum (PerkinElmer Inc., Waltham, MA, USA). Mice were sacrificed, and tumor tissues were collected for further imaging after 24 h.
Evaluation of Antitumor Efficacy and Biosafety in vivo
To prepare the Panc-1 tumor-bearing nude mice, a 0.2 mL Panc-1 cell suspension (1×107 cells in PBS) was injected subcutaneously into the right armpit of the forelimb of female BALB/c nude mice. When the tumor volume reached approximately 100 mm3, Panc-1 tumor-bearing nude mice were randomly divided into four groups: saline; CPT-11 (15.88 mg/kg, equivalent to 10 mg/kg SN38); SNSS NAs (19.8 mg/kg, equivalent to 10 mg/kg SN38); and SNSS NAs (39.6 mg/kg, equivalent to 20 mg/kg SN38). Each group received a total of four intravenous injections at 3-day intervals. The volume and weight of the tumor were measured every other day. The volume of the tumor was calculated using the formula:
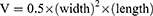
On day 16, all mice were sacrificed. Other organs (heart, liver, spleen, lung, and kidney) were also isolated for H&E staining in addition to the tumor tissues, which were then imaged. Beckman Coulter Chemistry Analyzer AU5800 Series (Beckman Coulter, Inc., Brea, CA, USA) was used to analyze the hematological and biochemical characteristics of blood samples.
Statistical Analysis
All data are displayed as the mean ± standard deviation (S.D.). GraphPad Prism 7.00 (GraphPad Software Inc., San Diego, CA, USA) was used to conduct statistical analysis. Differences were considered statistically significant when p < 0.05 (* p < 0.05, ** p < 0.01, and *** p < 0.001).
Results
Synthesis of SN38 Prodrug
Scheme 1 describes the synthesis of SN38-diSS, a GSH-responsive SN38 prodrug in this study. The mechanism of SN38 release from compound 2 is depicted in Scheme 2. The chemical structures of compound 1 and 2 were determined by 1H NMR, 13C NMR and HR-ESIMS as illustrated in Supplementary Materials (1.2, 1.3). Figure S1 depicts the purity as measured by HPLC.
 |
Scheme 1 Synthesis of compounds 1 and 2. a. NaH/n-butyl mercaptan/ TCCA/β-Mercaptoethanol/THF/CH3CN/-20 ℃/N2 b. DMAP/ Triphosgene/Anhydrous DCM/ Anhydrous DMF/r.t./N2. |
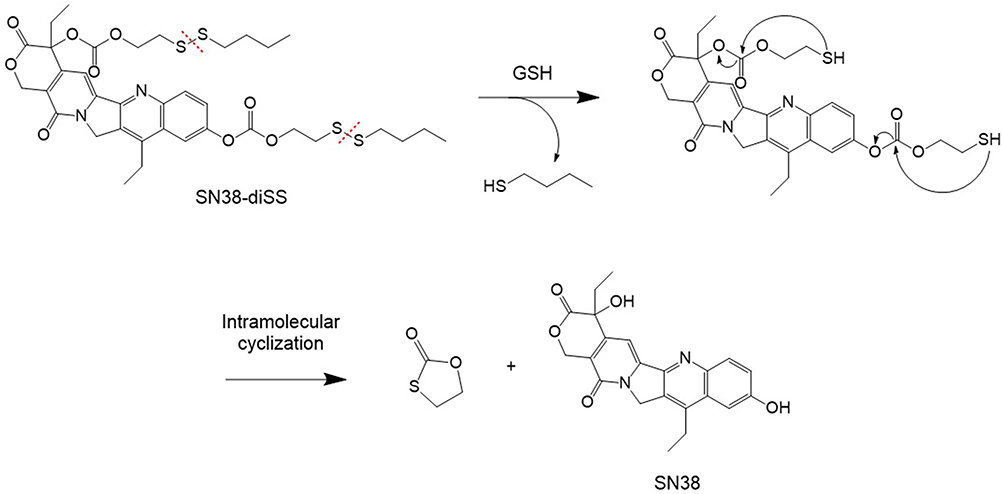 |
Scheme 2 The SN38 releasing mechanism from compound 2. |
XlogP and Hansen Solubility Parameters of SN38-diSS
According to Table 1, SN38-diSS had a 3.1-fold greater XlogP value than SN38, and its values for the Hansen solubility parameters (δp, δh, and δt) were all lower than those of SN38. SN38-diSS exhibited an ΔXlogP value of 2.1, while SN38-diSS exhibited a Δδh value of −42.2%. These findings were consistent with the proposed prediction criteria,36,37 which stated that conjugates should exhibit an increase in XlogP of greater than 1.0 relative to the parent drug, or a decrease in Hansen solubility of the polar solubility parameter (δp) and hydrogen bond solubility (δh) of greater than 10% relative to the parent drug. Therefore, it was proposed that SN38-diSS self-assemble into nanoassemblies.
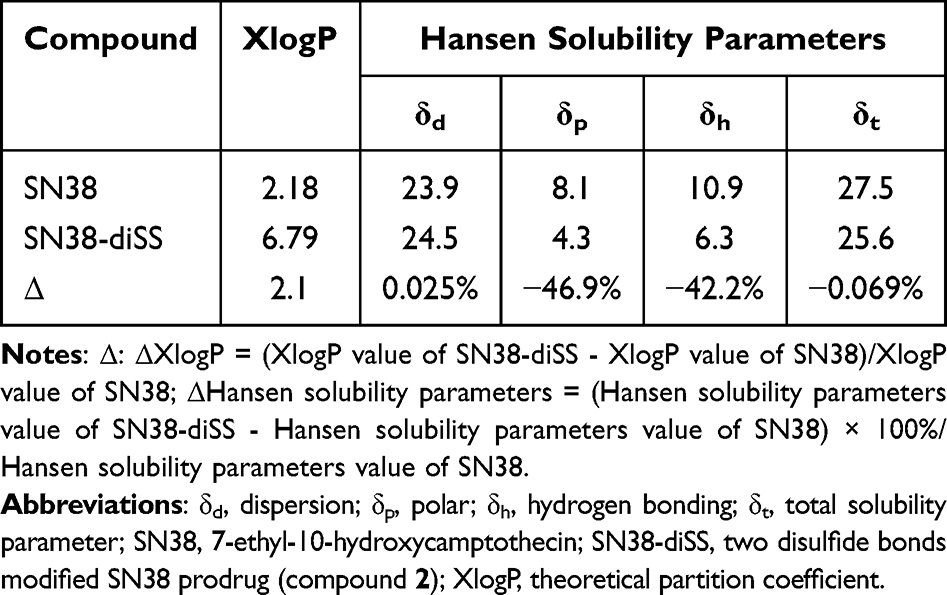 |
Table 1 XlogP Values and Hansen Solubility Parameters of SN38 and SN38-diSS |
Preparation and Characterization of SNSS NAs
SNSS NAs were produced through precipitation as described previously.34 The SNSS NAs were 88.8 ± 1.27 nm in size, with a polydispersity index value of 0.089 ± 0.016. (Figure 1a). Nanoassemblies had a zeta potential of −25.95 ± 0.32 mV (Figure 1b). The morphology of SNSS NAs was studied using TEM, which revealed a uniform spherical shape with a size of around 80 nm, which is equivalent to that measured by DLS. (Figure 1c). As depicted in Figure 1d, the cumulative release of SN38 from SNSS NAs was 58.4 ± 1.6% in 120 h and diffusion equilibrium was attained in 96 h. At 120 h, however, very little SN38 (1.5±0.49%) was found in SNSS NAs in PBS. As shown in Figure 2, the particle size of SNSS NAs remained stable after 14 days of storage and a 200-fold water dilution. Furthermore, SNSS NAs revealed less than 1.5% hemolysis, which is within the acceptable range for hemolysis (Table 2). The drug loading efficiency of SNSS NAs was determined to be 87.5% ± 4.8%.
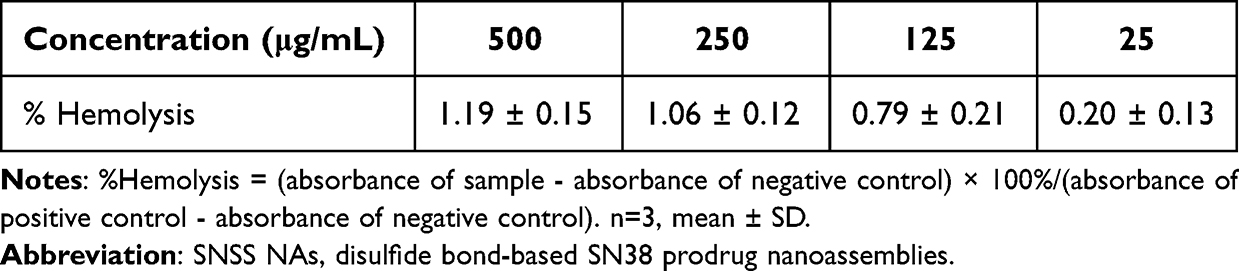 |
Table 2 % Hemolysis of Various SNSS NAs Concentrations |
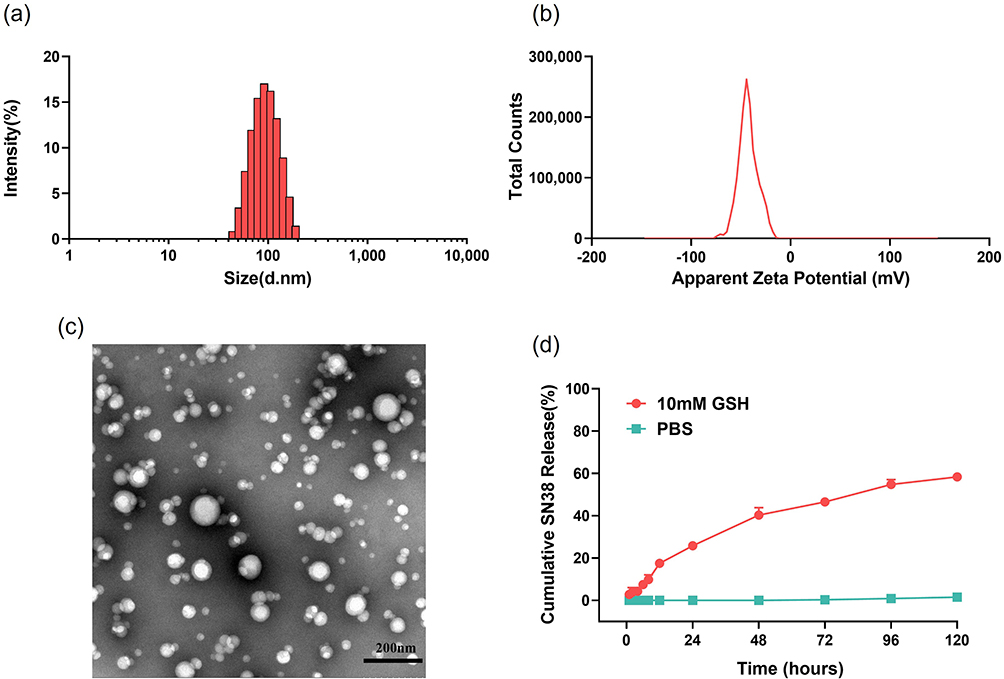 |
Figure 1 The preparation and characterization of SNSS NAs. (a and b) Particle size and zeta potential of SNSS NAs measured by DLS. (c) The TEM image of SNSS NAs was obtained using the HT7700 microscope (scale bar = 200 nm). (d) Cumulative SN38 release profiles of SNSS NAs in 10 mM PBS (pH 7.4) containing 10 mM GSH determined by HPLC (n=3, mean ± SEM). |
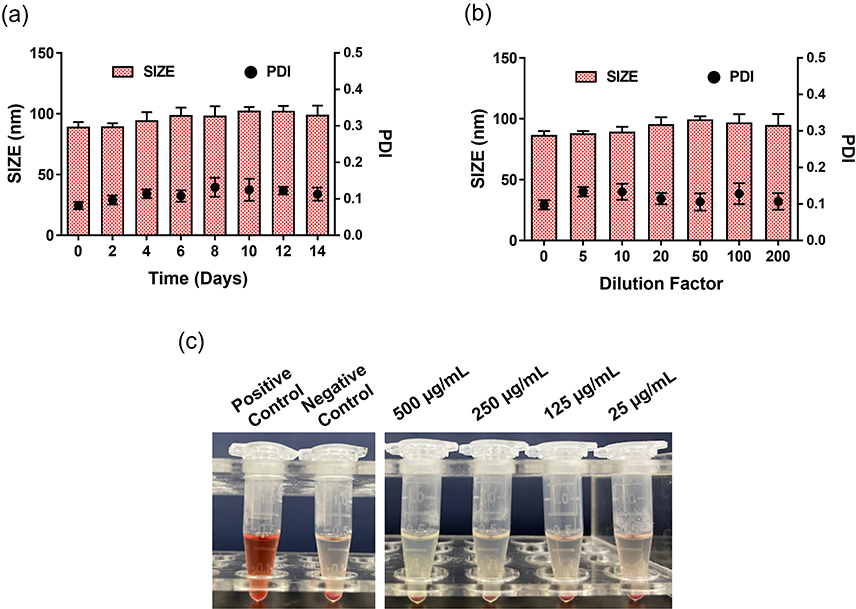 |
Figure 2 In vitro stability and blood compatibility of SNSS NAs. (a and b) The storage stability and dilution stability of the SNSS NAs were measured by DLS. (c) Image of SNSS NA hemolysis assay detected by a microplate reader at 545 nm (n=3, mean ± SEM). |
In vitro Cellular Uptake of SNSS NAs
Panc-1 and BxPC-3 cell lines were used to study the cellular uptake of SNSS NAs in vitro. As depicted in Figure 3a and b, two cell lines displayed improved uptake of SNSS NAs following incubation for 2, 4, or 6 h, indicating that nanoassemblies are taken up by cells via active endocytosis. Two cell lines showed time-dependent increases in both SN38 release and accumulation, hence ensuring cytotoxicity against pancreatic cancer cells. The intracellular behavior of SNSS NAs was then evaluated using CLSM. Cells treated with SNSS NAs exhibited noticeable intracellular green fluorescence through CLSM at 6 h, as shown in Figure 3c and d, and the fluorescence was maintained until 18 h post-treatment. In addition, several bright fluorescence spots were identified in the cytoplasm, indicating that cells uptake SNSS NAs via specific endocytosis pathways. Furthermore, nanoassemblies might dissolve within cells that exhibit dispersed green fluorescence throughout the cytoplasm.
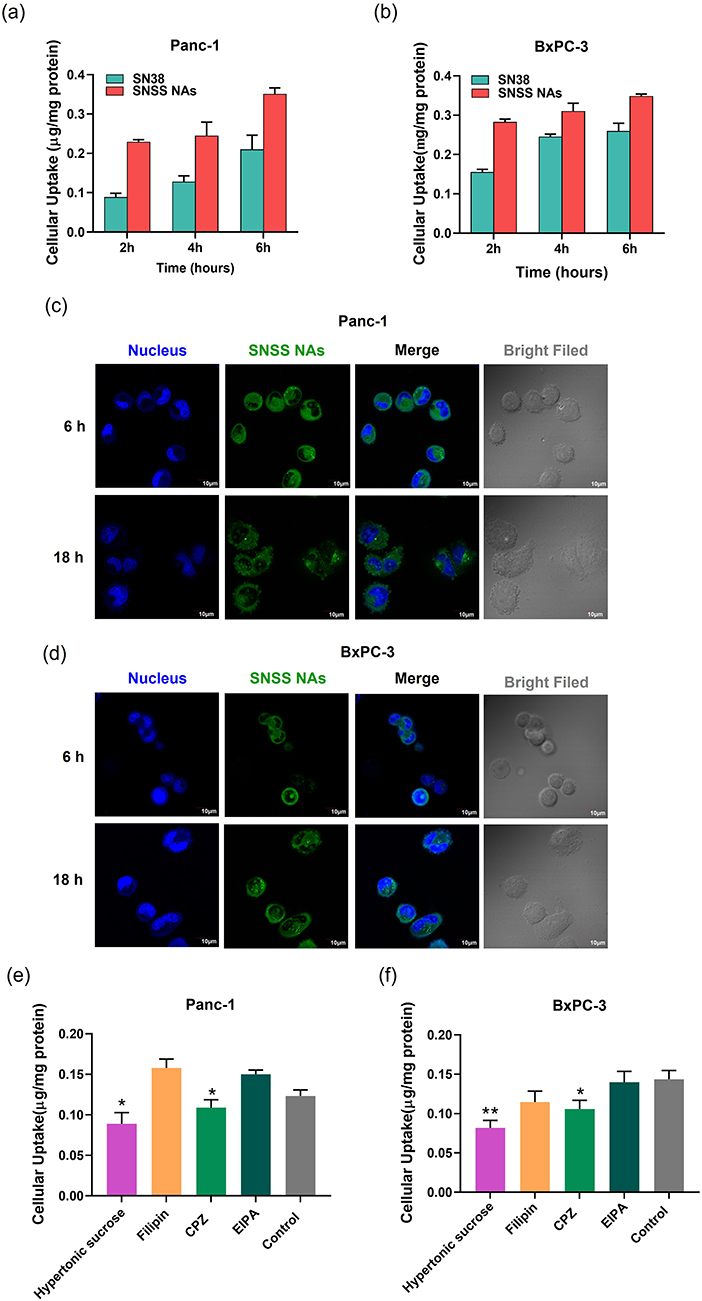 |
Figure 3 In vitro cellular uptake and endocytosis mechanism of SNSS NAs. (a and b) In vitro cellular uptake of SNSS NAs and degradation of SN38 in Panc-1 or BxPC-3 cells determined by HPLC. (c and d) Intracellular behavior of SNSS NAs in Panc-1 or BxPC-3 cells analyzed by CLSM. (e and f) The endocytosis mechanism of SNSS NAs in Panc-1 or BxPC-3 cells investigated by co-incubating with endocytosis inhibitors (n=3, mean ± SEM, *p < 0.05, compared with control). |
Endocytosis Pathway of SNSS NAs
As a result of the aforementioned findings, several pathway inhibitors were used to further identify the SNSS NAs’ endocytosis pathway (Figure 3e and f). In general, the endocytosis processes include phagocytosis, micropinocytosis, clathrin- and caveolin-independent endocytosis, caveolae-mediated endocytosis, and clathrin-mediated endocytosis.43 The inhibition of hypertonic sucrose and chlorpromazine (CPZ) in Panc-1 and BxPC-3 cells revealed that SNSS NAs were internalized via a clathrin-mediated endocytosis pathway (p < 0.01, p < 0.05).
In vitro Cytotoxicity of SNSS NAs
SRB assays were performed on Panc-1 and BxPC-3 cells incubated with CPT-11, SN38, and SNSS NAs to determine the cytotoxicity of GSH-responsive SNSS NAs (Figure 4a and b). The calculated half-maximal inhibitory concentrations (IC50) are presented in Table 3. In these cell lines, drugs and SNSS NAs exhibited dose-dependent toxicity, while free SN38 demonstrated the most potent cytotoxicity against all cell lines. SNSS NAs were less effective in reducing tumor cell proliferation, possibly due to the delayed release of active SN38 molecules. In contrast, CPT-11 showed the highest IC50 values in these cancer cells, which contributed to a reduced carboxylesterase level in tumor cells compared to liver cells.44,45
 |
Table 3 The IC50 Values (µm) of CPT-11, SN38, and SNSS NAs in Pancreatic Cancer Cells |
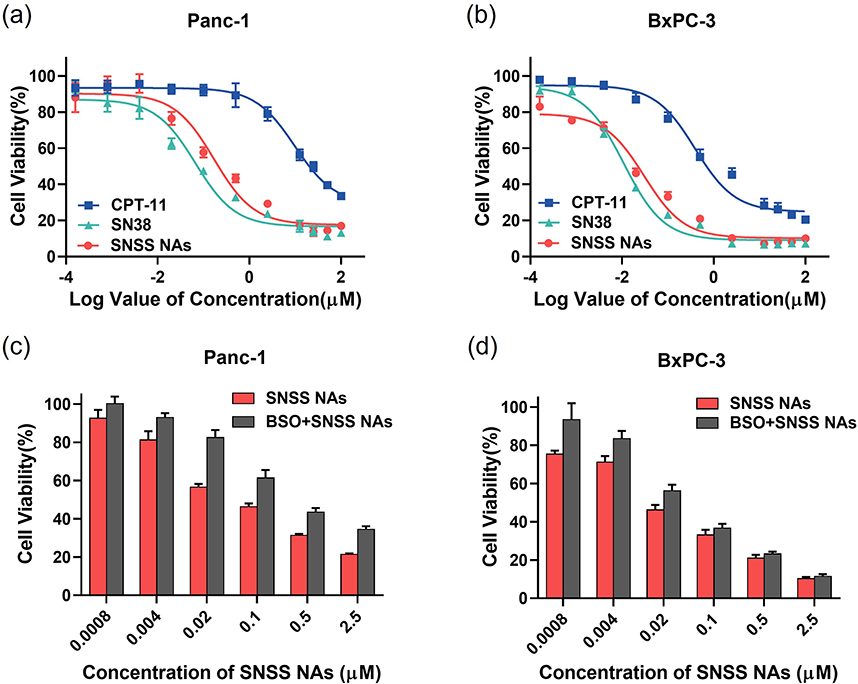 |
Figure 4 Cytotoxicity assay. (a and b) Cell viability of Panc-1 and BxPC-3 cells treated with various concentrations of CPT-11, SN38, and SNSS NAs. (c and d) Effect of BSO on the cytotoxic activity of SNSS NAs in Panc-1 and BxPC-3 cells (n=6, mean ± SEM). |
The cytotoxic activity of SNSS NAs was measured using the SRB assay in the presence of a GSH synthesis inhibitor, BSO, to further support the hypothesis that GSH triggers SN38 release (Figure 4c and d).46 To deplete GSH in Pnac-1 and BxPC-3 cells, BSO was added to the cell culture medium 24 h prior to the incorporation of nanoassemblies. The higher drug concentration resulted in the development of cell resistance to the antitumor drug. Additionally, the resistance effect was particularly potent at the high dose range. BSO reduced the cytotoxicity of SNSS NAs, demonstrating that SN38 release and cytotoxicity were dependent on GSH concentration.
In vivo Biodistribution of SNSS NAs
Cy7-labeled SNSS NAs were used to study the targeting effect of SNSS NAs in nude mice with the Panc-1 tumor. Figure 5 depicts the distribution and tumor accumulation of fluorescent Cy7. SNSS NAs-Cy7 produced a higher fluorescence signal in tumors compared to Cy7 dye injections used as a control. SNSS NAs-Cy7 accumulated at the tumor site with a slow clearance rate and was transported to the liver in part over the first 24 h after injection. In contrast, Cy7 dye was eliminated within 6 h through the bladder. In addition, tumor tissues removed for ex vivo analysis after 24 h demonstrate that Cy7-labeled SNSS NAs effectively targeted the tumor site.
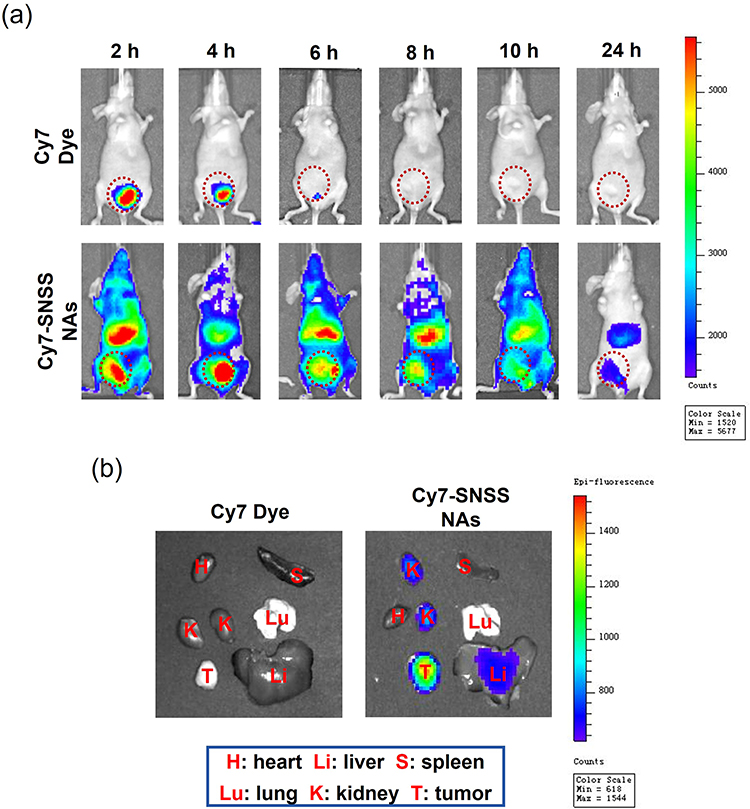 |
Figure 5 In vivo imaging of Cy7-labeled SNSS NAs in Panc-1 tumor-bearing nude mice. (a) The whole-body bioluminescence images of Panc-1 tumor-bearing nude mice after injection with Cy7 dye or Cy7-labeled SNSS NAs at 2, 4, 6, 8, 10, and 24 h, respectively. (b) The ex vivo bioluminescence images of tumors and organs of Panc-1 tumor-bearing nude mice sacrificed 24 h after Cy7 dye or Cy7-labeled SNSS NAs administration. |
In vivo Antitumor Effect of SNSS NAs
Nude mice bearing the Pnac-1 tumor were used to assess the in vivo antitumor effect of SNSS NAs. Figure 6a and b indicate that in comparison to the physiological saline treatment group, CPT-11 (15.88 mg/kg, corresponding to 10 mg/kg SN38) and SNSS NAs treatment groups (19.8 and 39.6 mg/kg, corresponding to 10 and 20 mg/kg SN38, respectively) significantly inhibited tumor growth in Panc-1 tumor-bearing nude mice (p < 0.001). It was noteworthy that the treatment group receiving SNSS NAs at 39.6 mg/kg exhibited more potent antitumor effect than the treatment group receiving CPT-11 (p < 0.001). The average tumor size at day 16 in the physiological saline, CPT-11 (15.88 mg/kg), SNSS NAs (19.8 mg/kg) and SNSS NAs (39.6 mg/kg) treatment groups were 1171±207, 733±47, 827±19 and 296±59 mm3, respectively. Additionally, the corresponding tumor inhibition rates for the different drug treatment groups were 37.4%, 29.3%, and 74.7%.
 |
Figure 6 In vivo antitumor effect and biosafety of SNSS NAs. (a) Average tumor growth curves of each treatment group in the Panc-1 tumor-bearing nude mice. (b) Tumor photograph from Panc-1 tumor-bearing nude mice after treatment for 16 days. (c) Curves showing the body weight change of mice during various treatments in the Panc-1 tumor model. (d) Levels of key indicators of liver and kidney functions in Panc-1 tumor-bearing nude mice. (e) H&E staining of main organs harvested from each treatment group in Panc-1 tumor-bearing nude mice at the experimental endpoint (scale bar = 200 μm) (n=6, mean ± SEM, ***p < 0.001, compared with saline, ###p < 0.001, compared with CPT-11 (15.88 mg/kg)). |
Biosafety Evaluation
Due to the high doses employed in this investigation, the toxicity of the SNSS NAs was also evaluated. Interestingly, despite receiving SNSS NAs at a drug dosage of 39.6 mg/kg (equal to 20 mg/kg SN38), the mice’s body weight did not decrease significantly throughout the experiment’s four in-vivo administrations (Figure 6c). In addition, a histological analysis was performed on all of the major organs (ie, the heart, liver, spleen, lung, and kidney of mice) of each treatment group. Upon administration of SNSS NAs at two doses to panc-1 tumor-bearing nude mice, H&E staining revealed no significant histological abnormalities (Figure 6e). The pathological images of the organs obtained from mice treated with CPT-11 and SNSS NAs were comparable to those obtained from mice treated with saline. Additionally, biochemical analysis (Figure 6d) of the serum at the end of the anticancer investigations in Panc-1 tumor-bearing nude mice revealed no damage to the liver and kidneys. These findings indicate that treatment with SNSS NAs, even at a higher dosage (ie, 39.6 mg/kg), resulted in minimal organ damage and systemic toxicity.
Discussion
Prodrug strategy is highly effective and fruitful in drug development, with around 20% of approved drugs from 2008 to 2020 being characterized as prodrugs.47 A successful prodrug method can effectively address the inferior properties of parent drugs, including poor water solubility, low permeability, chemical instability, inadequate pharmacokinetic characteristics, and high toxicity.48,49 Prodrug-driven self-assembled nanodelivery systems served as both carriers and payloads,50,51 combining the benefits of prodrug and nanodrug into a single nano-platform.
Different chemical conjugates of SN38 have been synthesized to self-assemble into nanostructures. The vast majority of moieties were macromolecular compounds, such as natural polysaccharides, linear polymers, hyperbranched polymers, dendrimers, albumin, and so on.52–56 However, because of the high-molecular-moiety prodrugs, the drug loading efficiency and drug release profiles of these nano-delivery systems are severely limited.57 Many small-molecular SN38 prodrugs with ultra-high drug loading efficiency have also been developed, including the SN38 phosphocholine prodrug (Di-SN38-PC), SN38-taxane heterodimeric prodrugs, SN38 homodimers, SN38-tocopherol oxyacetate conjugate, Artesunate-SN38 hybrid derivative, and Chol-SN38.58–63 However, these small molecular conjugates of SN38 were typically produced via an ester (succinic anhydride) or acetal linker, which degraded slowly and partially in the liver due to esterase.44,45 In addition, chemotherapeutic drugs having the capacity for temporal/spatial controlled release have always been an essential method for minimizing exposure to healthy cells and tissues.64
Based on the preceding, the chemical structure of the SN38 prodrug was rationally constructed based on various prerequisites: 1) The XlogP value of the prodrug must be greater than 1.0 and the Hansen solubility of the polar solubility parameter (δp) and hydrogen bond solubility (δh) must be less than 10% of that of SN38;36,37 2) Pro-moieties must be simple, biocompatible, and advantageous for molecular self-assembly. 3) a reversible linkage between SN38 and the pro-moiety that allows for stimuli-responsive, controlled release of SN38 without the production of any potentially cytotoxic byproducts.65 According to the chemical structure of SN38, two hydroxyl groups may be primarily responsible for the molecule’s polar solubility parameter (δp) and hydrogen bond solubility (δh). In addition, the rigid structure of camptothecin’s backbone has significant intermolecular forces due to the π-π stacking interaction among the planar aromatic ring,66,67 which leads to the formation of large aggregates that precipitate in water. Consequently, the two hydroxyl groups of SN38 should be modified with lipophilic flexible chains, which improved the XlogP value of the parent drug and decreased the δp and δh (Table 1). Furthermore, tumor microenvironment sensitive bonds should be implemented for simultaneous controlled drug release at cancerous sites.
In the current study, the two hydroxyl groups (at C10 and C20) of SN38 were modified with a GSH-responsive disulfide bond, which was then linked to n-butyl chains. The ethyl chain between SN38 and the disulfide bond was shown to have a higher bond cleavage and drug release rate than lengthy alkyl chains.68 In addition, alkyl (n-butyl) chains are the simplest lipid chains with good biocompatibility and little toxicity.69,70 This could potentially make the molecular structure less rigid and inhibit long-distance-ordered molecular packing. The disulfide bond is one of the most commonly employed flexible linkers,71,72 with almost vertical double bond angles and a single dihedral angle, which play a crucial role in enhancing structural flexibility and balancing intermolecular forces during molecule self-assembly.73 SN38-diSS contained two disulfide bonds, which increased structural flexibility and facilitated self-assembly.
In the absence of surfactant, the SNSS could self-assemble into nanoassemblies. As indicated in Table S4, size and PDI increased without the use of DSPE-MPEG2000 in the assembly procedure. Due to the potential for undesirable stability and in vivo drug behavior of non-PEGylated formulations,74,75 subsequent experiments utilized PEGylated SNSS nanoassemblies (SNSS NAs). PEGylated NAs displayed excellent stability and biocompatibility (Figure 2), indicating that SNSS NAs might be injected intravenously for in vivo cancer therapy.
Drug release behavior confirmed that GSH induced drug release (Figure 1d). The release of SN38 from NAs was stimulated by GSH and exhibited a profile of sustained drug release, which may contribute to the stabilizing effect of DSPE-MPEG2000. In addition, the cell viability analysis of NAs with BSO confirmed the controlled drug release and cytotoxicity among cancer cells (Figure 4).
Nanoassemblies are typically confined to a membrane-lined vesicle, such as an endosome,76 following endocytosis via a particular pathway. Therefore, drugs must be distributed to the cytoplasm for SN38 to bind to DNA topoisomerase I, thereby inhibiting cell proliferation and inducing apoptosis.77 SNSS NAs were taken up by pancreatic cancer cells via a clathrin-mediated endocytic pathway, according to the results (Figure 3e and f). The nanoassemblies were then distributed throughout the cytoplasm, accompanied by continual drug dissolution and degradation (Figure 3a and b). Finally, SN38 accumulation in cancer cells resulted in antitumor effect.
Analysis of in-vivo images showed that SNSS NAs could be passively targeted to the tumor site via the EPR effect (Figure 5). In contrast to CPT-11, SNSS NAs selectively accumulated and released the active chemical SN38 around tumor sites rather than in liver tissues, resulting in diarrhea and neutropenia.78 Therefore, double dosages of SSNA (39.6 mg/kg, corresponding to 20 mg/kg of SN38) were administered in animal studies, resulting in a greater antitumor effect in pancreatic cancer (Figure 6a and b). As anticipated, the systemic toxicity study revealed that a high dose of SNSS NAs did not result in any pathological alterations or organ dysfunction (Figure 6d and e). Finally, the disulfide-based SN38 prodrug delivery system demonstrated distinct benefits in anticancer efficacy and in vivo safety as a result of selective tumor accumulation and GSH-triggered drug release, and it demonstrated excellent future application potential.
Conclusion
In conclusion, a GSH-responsive SN38 prodrug containing disulfide bonds was synthesized using previously proposed prediction criteria. The SNSS NAs were then developed and characterized. In vitro experiments demonstrated that SNSS NAs had high drug loading efficiency and stability. The disulfide bonds endow the nanoassemblies with GSH-triggered release of SN38 and cytotoxicity. In vivo studies revealed that SNSS NAs exhibited higher tumor targeting accumulation, higher administration dosage, superior anticancer effect, and less adverse effects than CPT-11. The effort to incorporate a disulfide bond into the SN38 prodrug is crucial for the practical application of SN38-based drugs. SNSS NAs provide a new strategy for the synthesis of SN38-based nano-delivery systems, which offers a wide range of potential applications in the field of clinical antitumor therapy.
Acknowledgments
The present work was financially supported by the Natural Science Foundation of China (NSFC) (81673310).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Miller KD, Nogueira L, Devasia T, et al. Cancer treatment and survivorship statistics, 2022. CA Cancer J Clin. 2022;72(5):409–436.
3. Zhang X, Li N, Zhang S, et al. Emerging carrier-free nanosystems based on molecular self-assembly of pure drugs for cancer therapy. Med Res Rev. 2020;40(5):1754–1775.
4. Ducreux M, Cuhna AS, Caramella C, et al.; ESMO Guidelines Committee. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):56–68.
5. Si J, Zhao X, Gao S, Huang D, Sui M. Advances in delivery of Irinotecan (CPT-11) active metabolite 7-ethyl-10-hydroxycamptothecin. Int J Pharm. 2019;568:118499.
6. Milano G, Innocenti F, Minami H. Liposomal irinotecan (Onivyde): exemplifying the benefits of nanotherapeutic drugs. Cancer Sci. 2022;113(7):2224–2231.
7. Innocenti F, Kroetz DL, Schuetz E, et al. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J Clin Oncol. 2009;27(16):2604–2614.
8. Matsumura Y. Preclinical and clinical studies of NK012, an SN-38-incorporating polymeric micelles, which is designed based on EPR effect. Adv Drug Deliv Rev. 2011;63(3):184–192.
9. Huang Q, Liu X, Wang H, et al. A nanotherapeutic strategy to overcome chemoresistance to irinotecan/7-ethyl-10-hydroxy-camptothecin in colorectal cancer. Acta Biomater. 2022;137:262–275.
10. Musetti S, Huang L. Nanoparticle-mediated remodeling of the tumor microenvironment to enhance immunotherapy. ACS Nano. 2018;12(12):11740–11755. doi:10.1021/acsnano.8b05893
11. Chen Q, Liu G, Liu S, et al. Remodeling the tumor microenvironment with emerging nanotherapeutics. Trends Pharmacol Sci. 2018;39(1):59–74. doi:10.1016/j.tips.2017.10.009
12. Sapra P, Kraft P, Mehlig M, et al. Marked therapeutic efficacy of a novel polyethylene glycol-SN38 conjugate, EZN-2208, in xenograft models of B-cell non-Hodgkin’s lymphoma. Haematologica. 2009;94(10):1456–1459. doi:10.3324/haematol.2009.008276
13. Koizumi F, Kitagawa M, Negishi T, et al. Novel SN-38–incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor–secreting bulky tumors. Cancer Res. 2006;66(20):10048–10056. doi:10.1158/0008-5472.CAN-06-1605
14. Wang S, Ye T, Yang B, Yi X, Yao H. 7-Ethyl-10-hydroxycamptothecin proliposomes with a novel preparation method: optimized formulation, characterization and in-vivo evaluation. Drug Dev Ind Pharm. 2013;39(2):393–401. doi:10.3109/03639045.2012.683441
15. Wang-Gillam A, Li CP, Bodoky G, et al.; NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, Phase 3 trial. Lancet. 2016;387(10018):545–557.
16. Ganipineni LP, Danhier F, Préat V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J Control Release. 2018;281:42–57.
17. Li G, Sun B, Li Y, Luo C, He Z, Sun J. Small-molecule prodrug nanoassemblies: an emerging nanoplatform for anticancer drug delivery. Small. 2021;17(52):e2101460.
18. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37.
19. Xiao H, Guo Y, Liu H, et al. Structure-based design of charge-conversional drug self-delivery systems for better targeted cancer therapy. Biomaterials. 2020;232:119701.
20. Manzari MT, Shamay Y, Kiguchi H, Rosen N, Scaltriti M, Heller DA. Targeted drug delivery strategies for precision medicines. Nat Rev Mater. 2021;6(4):351–370.
21. Yu W, Liu R, Zhou Y, Gao H. Size-tunable strategies for a tumor targeted drug delivery system. ACS Cent Sci. 2020;6(2):100–116.
22. Li H, Zhao Y, Jia Y, Qu C, Li J. Covalently assembled dopamine nanoparticle as an intrinsic photosensitizer and pH-responsive nanocarrier for potential application in anticancer therapy. Chem Commun. 2019;55(100):15057–15060.
23. Dong S, He J, Sun Y, et al. Efficient click synthesis of a protonized and reduction-sensitive amphiphilic small-molecule prodrug containing camptothecin and gemcitabine for a drug self-delivery system. Mol Pharm. 2019;16(9):3770–3779.
24. Wang Y, Huang P, Hu M, Huang W, Zhu X, Yan D. Self-delivery nanoparticles of amphiphilic methotrexate-gemcitabine prodrug for synergistic combination chemotherapy via effect of deoxyribonucleotide pools. Bioconjug Chem. 2016;27(11):2722–2733.
25. Fu LH, Wan Y, Qi C, et al. Nanocatalytic theranostics with glutathione depletion and enhanced reactive oxygen species generation for efficient cancer therapy. Adv Mater. 2021;33(7):e2006892.
26. Yeh CC, Hou MF, Wu SH, et al. A study of glutathione status in the blood and tissues of patients with breast cancer. Cell Biochem Funct. 2006;24(6):555–559.
27. Grubben MJ, van den Braak CC, Nagengast FM, Peters WH. Low colonic glutathione detoxification capacity in patients at risk for colon cancer. Eur J Clin Invest. 2006;36(3):188–192.
28. Gupta A, Srivastava S, Prasad R, et al. Oxidative stress in non-small cell lung cancer patients after chemotherapy: association with treatment response. Respirology. 2010;15(2):349–356.
29. Kearns PR, Pieters R, Rottier MM, Pearson AD, Hall AG. Raised blast glutathione levels are associated with an increased risk of relapse in childhood acute lymphocytic leukemia. Blood. 2001;97(2):393–398.
30. Zhong X, Wang X, Cheng L, et al. GSH depleted PtCu3 nanocages for chemodynamicenhanced sonodynamic cancer therapy. Adv Funct Mater. 2019;30:1907954–1907966.
31. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591.
32. Noh J, Kwon B, Han E, et al. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat Commun. 2015;6:6907.
33. Song P, Yao X, Zhong T, et al. The anti-tumor efficacy of 3-(2-Nitrophenyl) propionic acid-paclitaxel (NPPA-PTX): a novel paclitaxel bioreductive prodrug. Oncotarget. 2016;7(30):48467–48480.
34. Duan XC, Yao X, Zhang S, et al. Antitumor activity of the bioreductive prodrug 3-(2-nitrophenyl) propionic acid-paclitaxel nanoparticles (NPPA-PTX NPs) on MDA-MB-231 cells: in vitro and in vivo. Int J Nanomedicine. 2018;14:195–204.
35. Duan XC, Peng LY, Yao X, et al. The synergistic antitumor activity of 3-(2-nitrophenyl) propionic acid-paclitaxel nanoparticles (NPPA-PTX NPs) and anti-PD-L1 antibody inducing immunogenic cell death. Drug Deliv. 2021;28(1):800–813.
36. Zhong T, Hao YL, Yao X, et al. Effect of XlogP and Hansen solubility parameters on small molecule modified paclitaxel anticancer drug conjugates self-assembled into nanoparticles. Bioconjug Chem. 2018;29(2):437–444.
37. Xu MQ, Zhong T, Yao X, et al. Effect of XlogP and Hansen solubility parameters on the prediction of small molecule modified docetaxel, doxorubicin and irinotecan conjugates forming stable nanoparticles. Drug Deliv. 2021;28(1):1603–1615.
38. Cheng T, Zhao Y, Li X, et al. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47(6):2140–2148.
39. Barton AFM. CRC Handbook of Solubility Parameters and Other Cohesion Parameters.
40. Hansen CM. Hansen Solubility Parameters: A User’s Handbook.
41. Zhang S, Li ZT, Liu M, et al. Anti-tumour activity of low molecular weight heparin doxorubicin nanoparticles for histone H1 high-expressive prostate cancer PC-3M cells. J Control Release. 2019;295:102–117.
42. Xu MQ, Hao YL, Wang JR, et al. Antitumor activity of α-linolenic acid-paclitaxel conjugate nanoparticles: in vitro and in vivo. Int J Nanomedicine. 2021;16:7269–7281.
43. Behzadi S, Serpooshan V, Tao W, et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev. 2017;46(14):4218–4244.
44. Taketani M, Shii M, Ohura K, Ninomiya S, Imai T. Carboxylesterase in the liver and small intestine of experimental animals and human. Life Sci. 2007;81(11):924–932.
45. Di L. The impact of carboxylesterases in drug metabolism and pharmacokinetics. Curr Drug Metab. 2019;20(2):91–102.
46. Dong Z, Feng L, Chao Y, et al. Amplification of tumor oxidative stresses with liposomal Fenton catalyst and glutathione inhibitor for enhanced cancer chemotherapy and radiotherapy. Nano Lett. 2019;19(2):805–815.
47. Rautio J, Meanwell NA, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov. 2018;17(8):559–587.
48. Bildstein L, Dubernet C, Couvreur P. Prodrug-based intracellular delivery of anticancer agents. Adv Drug Deliv Rev. 2011;63(1–2):3–23.
49. Huttunen KM, Raunio H, Rautio J. Prodrugs--from serendipity to rational design. Pharmacol Rev. 2011;63(3):750–771.
50. Bariwal J, Kumar V, Chen H, et al. Nanoparticulate delivery of potent microtubule inhibitor for metastatic melanoma treatment. J Control Release. 2019;309:231–243.
51. Liu Y, Shen G, Zhao L, Zou Q, Jiao T, Yan X. Robust photothermal nanodrugs based on covalent assembly of nonpigmented biomolecules for antitumor therapy. ACS Appl Mater Interfaces. 2019;11(45):41898–41905.
52. Vangara KK, Ali HI, Lu D, Liu JL, Kolluru S, Palakurthi S. SN-38-cyclodextrin complexation and its influence on the solubility, stability, and in vitro anticancer activity against ovarian cancer. AAPS Pharm Sci Tech. 2014;15(2):472–482.
53. Salmanpour M, Yousefi G, Samani SM, Mohammadi S, Anbardar MH, Tamaddon A. Nanoparticulate delivery of irinotecan active metabolite (SN38) in murine colorectal carcinoma through conjugation to poly (2-ethyl 2-oxazoline)-b-poly (L-glutamic acid) double hydrophilic copolymer. Eur J Pharm Sci. 2019;136:104941.
54. Liu B, Wang D, Liu Y, et al. Hydrogen peroxide-responsive anticancer hyperbranched polymer micelles for enhanced cell apoptosis. Polym Chem. 2015;6:3460–3471.
55. Vijayalakshmi N, Ray A, Malugin A, Ghandehari H. Carboxyl-terminated PAMAM-SN38 conjugates: synthesis, characterization, and in vitro evaluation. Bioconjug Chem. 2010;21(10):1804–1810.
56. Yao Y, Su X, Xie Y, et al. Synthesis, characterization, and antitumor evaluation of the albumin-SN38 conjugate. Anticancer Drugs. 2013;24(3):270–277.
57. Palakurthi S. Challenges in SN38 drug delivery: current success and future directions. Expert Opin Drug Deliv. 2015;12(12):1911–1921.
58. Du Y, Zhang W, He R, et al. 7-ethyl-10-hydroxycamptothecin conjugated phospholipid prodrug assembled liposomes with in vitro anticancer effects. Bioorg Med Chem. 2017;25(12):3247–3258.
59. Wang H, Chen J, Xu C, et al. Cancer nanomedicines stabilized by π-π stacking between heterodimeric prodrugs enable exceptionally high drug loading capacity and safer delivery of drug combinations. Theranostics. 2017;7(15):3638–3652.
60. Kasai H, Murakami T, Ikuta Y, et al. Creation of pure nanodrugs and their anticancer properties. Angew Chem Int Ed Engl. 2012;51(41):10315–10318.
61. Nguyen F, Alferiev I, Guan P, et al. Enhanced intratumoral delivery of SN38 as a tocopherol oxyacetate prodrug using nanoparticles in a neuroblastoma xenograft model. Clin Cancer Res. 2018;24(11):2585–2593.
62. Botta L, Filippi S, Zippilli C, et al. Artemisinin derivatives with antimelanoma activity show inhibitory effect against human DNA topoisomerase 1. ACS Med Chem Lett. 2020;11(5):1035–1040.
63. Jiang X, Lee M, Xia J, et al. Two-stage SN38 release from a core-shell nanoparticle enhances tumor deposition and antitumor efficacy for synergistic combination with immune checkpoint blockade. ACS Nano. 2022;16:1.
64. Dong X, Brahma RK, Fang C, Yao SQ. Stimulus-responsive self-assembled prodrugs in cancer therapy. Chem Sci. 2022;13(15):4239–4269.
65. Nguyen A, Böttger R, Li SD. Recent trends in bioresponsive linker technologies of prodrug-based self-assembling nanomaterials. Biomaterials. 2021;275:120955.
66. Wang Y, Wang X, Deng F, et al. The effect of linkers on the self-assembling and anti-tumor efficacy of disulfide-linked doxorubicin drug-drug conjugate nanoparticles. J Control Release. 2018;279:136–146.
67. Cai K, He X, Song Z, et al. Dimeric drug polymeric nanoparticles with exceptionally high drug loading and quantitative loading efficiency. J Am Chem Soc. 2015;137(10):3458–3461.
68. Muniesa C, Vicente V, Quesada M, et al. Glutathione-sensitive nanoplatform for monitored intracellular delivery and controlled release of Camptothecin. RSC Adv. 2013;3(35):15121–15131.
69. Yang C, Tu K, Gao H, et al. The novel platinum (IV) prodrug with self-assembly property and structure-transformable character against triple-negative breast cancer. Biomaterials. 2020;232:119751.
70. Dey J, Ghosh R, Das Mahapatra R. Self-assembly of unconventional low-molecular-mass amphiphiles containing a PEG chain. Langmuir. 2019;35(4):848–861.
71. Wang Y, Liu D, Zheng Q, et al. Disulfide bond bridge insertion turns hydrophobic anticancer prodrugs into self-assembled nanomedicines. Nano Lett. 2014;14(10):5577–5583.
72. Lin W, Sun T, Xie Z, Gu J, Jing X. A dual-responsive nanocapsule via disulfide-induced self-assembly for therapeutic agent delivery. Chem Sci. 2016;7(3):1846–1852.
73. Yang Y, Sun B, Zuo S, et al. Trisulfide bond-mediated doxorubicin dimeric prodrug nanoassemblies with high drug loading, high self-assembly stability, and high tumor selectivity. Sci Adv. 2020;6(45):eabc1725.
74. Luo C, Sun J, Sun B, et al. Facile fabrication of tumor redox-sensitive nanoassemblies of small-molecule oleate prodrug as potent chemotherapeutic nanomedicine. Small. 2016;12(46):6353–6362.
75. Wang J, Mao W, Lock LL, et al. The role of micelle size in tumor accumulation, penetration, and treatment. ACS Nano. 2015;9(7):7195–7206.
76. Kim CS, Li X, Jiang Y, et al. Cellular imaging of endosome entrapped small gold nanoparticles. MethodsX. 2015;2:306–315.
77. Li TK, Liu LF. Tumor cell death induced by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol. 2001;41:53–77.
78. Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8(3):641–661.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.