Back to Journals » Infection and Drug Resistance » Volume 16
Distribution Patterns of Pathogens Causing Lower Respiratory Tract Infection Based on Metagenomic Next-Generation Sequencing
Authors Chai S, Wang C, Liu Y, Xia J, Wang X, Shi J
Received 30 May 2023
Accepted for publication 14 September 2023
Published 10 October 2023 Volume 2023:16 Pages 6635—6645
DOI https://doi.org/10.2147/IDR.S421383
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Shukun Chai, Chuan Wang, Yi Liu, Jingrong Xia, Xiaolin Wang, Jinying Shi
Department of Respiratory Medicine, Shijiazhuang People’s Hospital, Shijiazhuang, Hebei, 050051, People’s Republic of China
Correspondence: Jinying Shi, Department of Respiratory Medicine, Shijiazhuang People’s Hospital, Shijiazhuang, Hebei, 050051, People’s Republic of China, Email [email protected]
Purpose: Lower Respiratory Tract Infection (LRTI) is a leading cause of morbidity and mortality worldwide. In this study, the distribution patterns of causative pathogens in LRTI were evaluated within a city-level hospital by combining conventional microbiological tests (CMT) with metagenomic next-generation sequencing (mNGS).
Patients and Methods: This retrospective cohort study involved 160 patients suspected of having LRTI in a single center. Specimens, including bronchoalveolar lavage fluid (BALF), blood, tissue, sputum, and pus were utilized to identify pathogens. The seasonal prevalence of pathogens and co-pathogens involved in multiple infections was analyzed.
Results: A total of 137 patients with 156 samples were included in this study. Pseudomonas aeruginosa, Corynebacterium striatum, Klebsiella pneumoniae, Candida, and human herpesvirus were the top prevalent pathogens. We observed seasonal dynamic variation in the top prevalent bacteria (Pseudomonas aeruginosa and Klebsiella pneumoniae) and herpesvirus (Epstein-Barr virus and Human herpesvirus-7). The majority of patients had single bacterial infections, followed by instances of bacterial-viral co-infections, as well as mixed infections involving bacteria, fungi, and viruses. Notably, the spectrum of co-infecting pathogens was broader among the elderly population, and positive Spearman correlations were observed among these co-infecting pathogens.
Conclusion: Co-infections were prevalent among patients with LRTI, and the pathogens displayed distinct seasonal distribution patterns. The findings underscored the significance of comprehending pathogen distribution and epidemic patterns, which can serve as a basis for early etiological identification.
Keywords: metagenomic next-generation sequencing, low respiratory tract infection, co-pathogens, seasonal prevalence
Graphical Abstract:

Introduction
Lower respiratory tract infection (LRTI) remains one of the leading causes of death worldwide.1 Several well-known pathogens, including Streptococcus pneumoniae, Pseudomonas aeruginosa, Klebsiella pneumoniae, Candida, Herpesvirus, and others, have been identified as significant causes of infection.2 Nonetheless, nearly half of the cases still have an undetermined etiology,3,4 despite the extensive application of clinical microbiology tests. In certain scenarios, LRTI can progress to severe pneumonia, multiple organ dysfunction syndrome, and septic shock.5
Rapid and precise microbiological diagnoses are essential for the appropriate use of antibiotics and reduce LRTI mortality.6 However, current clinical diagnosis methods face various challenges, including prolonged turnaround time and low sensitivity of cultures,6,7 as well as narrow spectrum of smear microscopy, serology tests, PCR tests and multiplex PCR.4 Recently, metagenomic next-generation sequencing (mNGS) is widely developed and applied as an effective pathogen detection technology due to its capacity to overcome complications from antibiotic exposure8 and to detect unexpected pathogens.9 The mNGS approach becomes a valuable supplement to conventional microbiological test (CMT).
mNGS allows for the comprehensive analysis of pathogen profile. Previous studies have demonstrated that the age of the patients as well as regional/seasonal features might affect the distribution of pathogens.10,11 For example, seasonal variation is often observed in human bacterial and viral infections.11–13 Pseudomonas aeruginosa, Enterobacter cloacae, Acinetobacter baumannii and Klebsiella pneumoniae were more prevalent during the warmer months of the year.13–15 Thus, a comprehensive analysis of the pathogens among LRTI patients at a city-level hospital can enhance our comprehension of these infections.
Therefore, we conducted a retrospective study at the Shijiazhuang People’s Hospital of Hebei Province between April 7, 2021, and November 13, 2022, which enrolled 160 hospitalized patients with suspected LRTI. The lower respiratory tract sample or blood was collected and tested by both mNGS and CMT methods. Pathogens identified by mNGS and CMT methods were compared, and their seasonal prevalence was further analyzed.
Materials and Methods
Ethics Statement
This study was carried out in accord with adherence to the principles of the Declaration of Helsinki and approved by the Ethics Review Committee of Shijiazhuang People’s Hospital of Hebei Province. Patients’ approval and informed consent were waived because of the retrospective design. Patient anonymity was preserved.
Patient Population and Study Design
This retrospective observational study enrolled patients with suspected LRTI who were hospitalized at the Shijiazhuang People’s Hospital of Hebei Province between 7 April 2021 and 13 November 2022. The enrollment criteria were (1) new-onset shadows on chest X-ray or computed tomography (CT) and (2) presence of at least one of the following typical symptoms: a) cough, sputum production, dyspnea, chest pain, or exacerbation of existing respiratory symptoms; b) fever; c) clinical signs of lung consolidation or moist rales; d) peripheral leukocytosis (> 10×109 /L) or leucopenia (< 4×109 /L). The exclusion criteria were (1) patients with incomplete clinical data and (2) patients with no confirmed clinical diagnosis results.
Bronchoalveolar lavage fluid (BALF), blood, tissue, sputum, and pus samples of patients were collected to identify pathogens by conventional microbiological tests (CMT) and mNGS simultaneously. The CMT methods included bacteria and fungi culturing, PCR of eight respiratory tract pathogens (including Mycoplasma pneumoniae, Chlamydia pneumoniae, Respiratory syncytial virus, Human adenovirus, Coxsackievirus B, Influenza A/B virus, and Parainfluenza virus) were performed using virus nucleic acid detection Kit (PCR-Fluorescence Probing) (HXD-01, Coyote, Beijing, China) and Mycoplasma pneumoniae and Chlamydia pneumoniae nucleic acid combined detection kit (Fluorescence PCR) (Liferiver, Shanghai, China). Serum 1, 3-beta-D-glucan level (G test) for fungi, as well as T-spot and GeneXpert MTB/RIF assay for Mycobacterium tuberculosis (TB) identification were also performed. A proportion of 97.4% (152/156) cases had performed culture to detect bacteria and fungi and patients with CMT test were detailed in Supplementary Table 1.
mNGS assays were performed within 24 h after sample collection, including body fluids (2–3 mL), the lung biopsy tissues (≥ 3 × 3 × 3 mm) and whole blood (≥ 4 mL) samples in EDTA tubes.
Sample Preparation and mNGS Sequencing
Blood samples were centrifuged at 1900 × g and 4°C for 10 min to get plasma for subsequent processing. Sputum samples underwent a liquefaction treatment, while tissues were initially fragmented into small pieces and subsequently homogenized. Plasma cell-free DNA (cfDNA) was extracted using PathoXtract® cell-free Nucleic Acid Kit (WYXM03010S, WillingMed Corp, Beijing, China). DNA from BALF, sputum, tissue, and pus were extracted using PathoXtract® Basic Pathogen Nucleic Acid Kit (WYXM03211S, WillingMed Corp, Beijing, China) according to the manufacturer’s protocol. RNA was extracted using PathoXtract® Virus DNA/RNA Isolation Kit (WYXM03009S, WillingMed Corp, Beijing, China). Extracted RNA was reverse transcribed to get the first strand cDNA using SuperScript® Double-Stranded cDNA Synthesis Kit (11917020, Invitrogen, United States).
cfDNA libraries were prepared using KAPA DNA HyperPrep Kit (KK8504, KAPA, Kapa Biosystems, Wilmington, MA, United States), and DNA libraries were constructed using the Illumina® DNA Prep, (M) Tagmentation (20018705, Illumina, San Diego, USA). The quality of the libraries was evaluated on an Agilent 2100 Bioanalyzer (Agilent Technologies), and qualified libraries were sequenced on the NextSeq™ 550Dx sequencer (Illumina, San Diego, USA) using a 75-bp single-end method. Nuclease free water was used as negative control (NTC) and was set for each sequencing run to control contaminating DNA.
Bioinformatic Analysis
The Trimmomatic v0.4016 was used to filter out low-quality sequences, contaminated adapters, duplicated reads and reads shorter than 36 bp. Then, the sequences were compared with the human reference genome GRCh37 (hg19) using Bowtie2 to remove human sequences.17 For taxonomic classification and identification of microbial reads, we utilized Kraken2 with non-redundant nucleotide sequence database of National Center for Biotechnology Information (NCBI).18
The following criteria were applied to report the positive pathogens. Reads per ten million (RPTM) was used to quantify pathogen abundance. Bacteria and fungi with RPTM ≥ 20, viruses with RPTM ≥ 3, and special pathogens (including Cryptococcus, Mycobacterium, Mycoplasma, Chlamydia, and parasites) with RPTM ≥ 1 were identified as positive.19,20
Statistical Analysis
The Wilcoxon-Mann-Whitney test was used for comparison between groups, and Fisher’s exact test was used for categorical variables. Statistical analyses were performed using Prism 9 (GraphPad, La Jolla, CA). P-values below 0.05 were considered statistical significance. The correlation between patient characteristics associated with the presence of multiple pathogen types and the representative microorganism was assessed using Spearman’s rank correlation coefficient.
Results
General Characteristics of the Patients
A total of 160 suspected LRTI patients were enrolled in this study. After excluding 21 patients who did not undergo CMT testing and 2 patients with undiagnosed disease, 156 samples from 137 patients were used for subsequent analysis (Figure 1). Seventeen patients provided ≥ 2 samples for mNGS testing, which from different locations and/or at different time. Among them, 15 patients provided 2 samples, 2 patients provided 3 samples (Supplementary Table 1). The samples analyzed in the study included 129 BALF, 10 blood, 9 sputum, 7 tissue, and 1 pus. The age of the patients ranged from 14 to 87 years, with a median of 63 years (60.1 ± 15.1). Males made up 62.8% of the patients (Table 1). The most common diagnoses were pneumonia (54.7%) and severe pneumonia (25.5%).
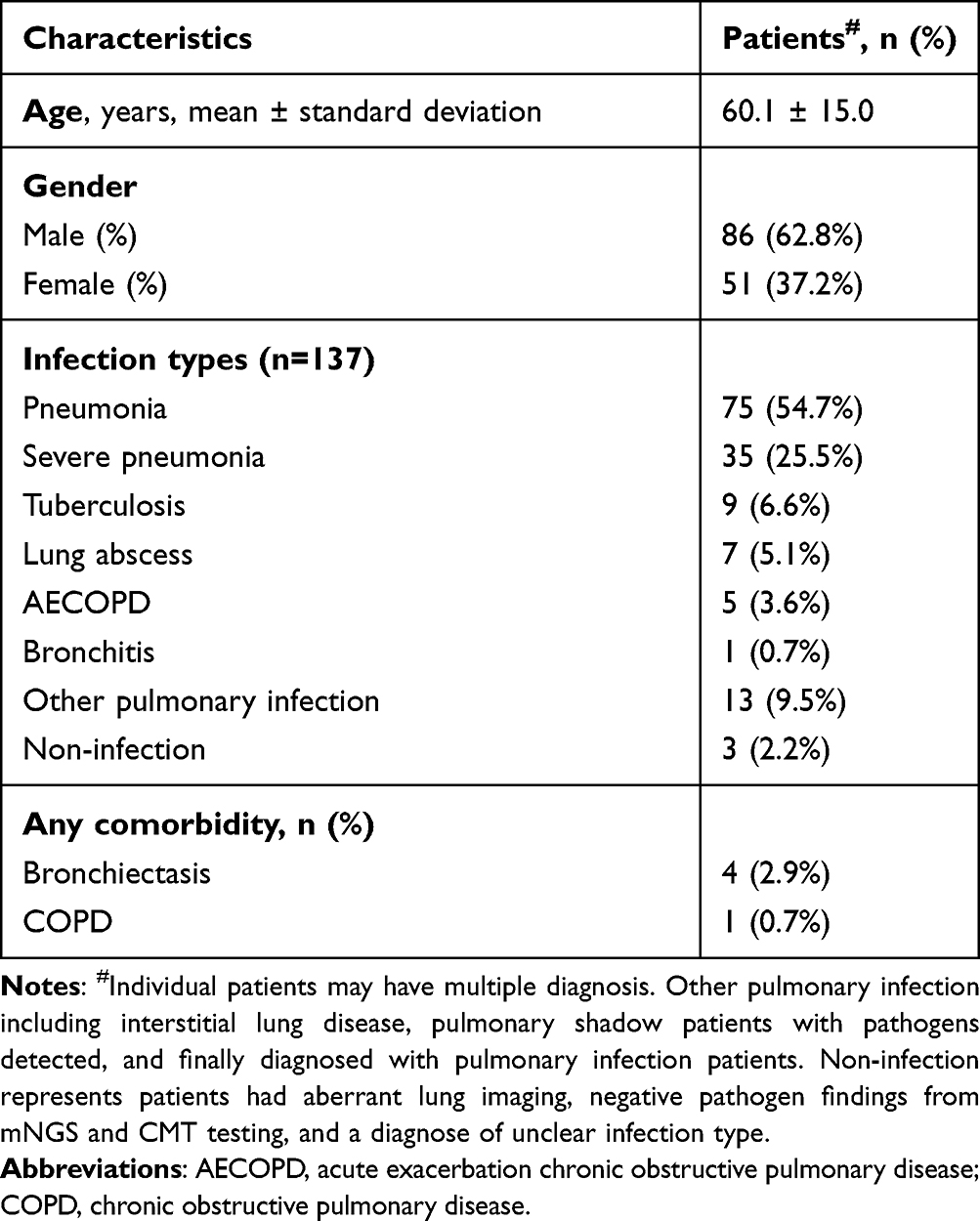 |
Table 1 Characteristics of the Patients |
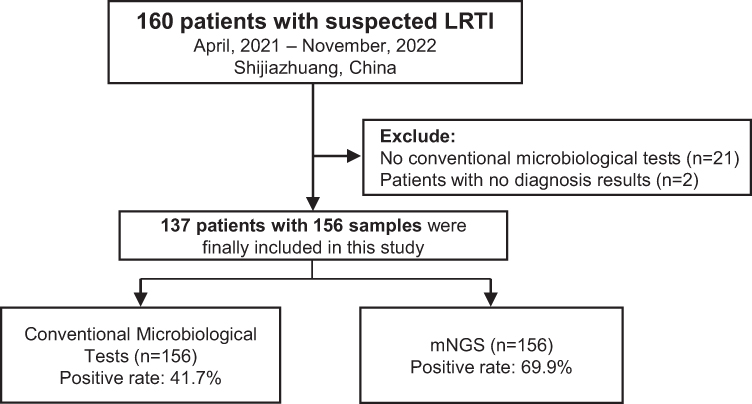 |
Figure 1 Flow diagram of patient’s inclusion in this study. |
Pathogens Detected by CMT and mNGS
An overall positive rate of 69.9% (109/156) was detected by mNGS. Among all sample types, the positive rate of sputum was 88.9% (8/9), BALF was 74.4% (96/129), tissue was 28.6% (2/7) and blood was 20% (2/10). The only one pus samples also showed positive result (Figure 2A). For the CMT results, 41.7% (65/156) samples showed positive results (Figure 2B). Further analysis showed that mNGS and CMT were both negative in 24% cases and both positive in 36% samples. Among the double-positive cases, 12.5% samples showed complete match pathogens, 53.6% showed partial match and the rest 33.9% demonstrated mismatch (Figure 2C).
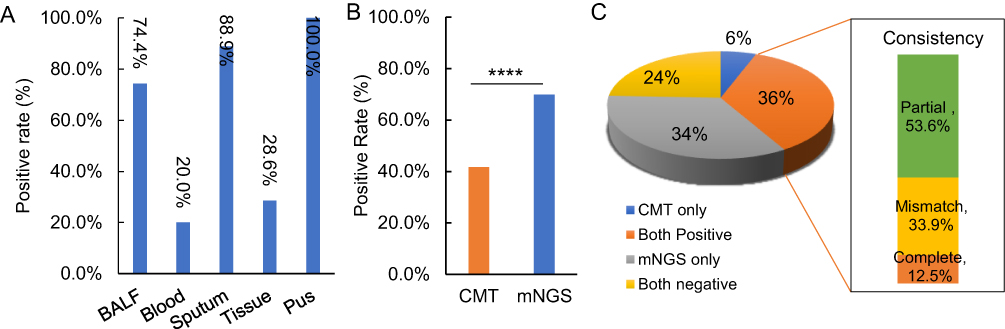 |
Figure 2 Pathogen detection rate and concordance between CMT and mNGS. (A) The positive rate of mNGS for different kinds of specimen. (B) The positive rate of CMT and mNGS results. P value present with ****Indicating less than 0.0001. (C) Concordance of detected pathogens between mNGS and CMT. |
Among the detected pathogens, there were 8 species of bacteria, 2 viruses and 5 fungi were identified by both mNGS and CMT methods. Additionally, 44 species of bacteria, 7 viruses and 12 fungi were only detected by mNGS, while another 1 species of bacteria, and 5 viruses were solely detected by CMT. Almost all of the bacteria and fungi detected by CMT were also detected by the mNGS technique (Figure 3A). Furthermore, 9 samples had positive results by CMT but negative by mNGS, including Candida albicans (n = 5), fungi (n = 2), Epstein-Barr virus (EBV, n = 1), and Pseudomonas aeruginosa (n = 1) in these samples (Table 2).
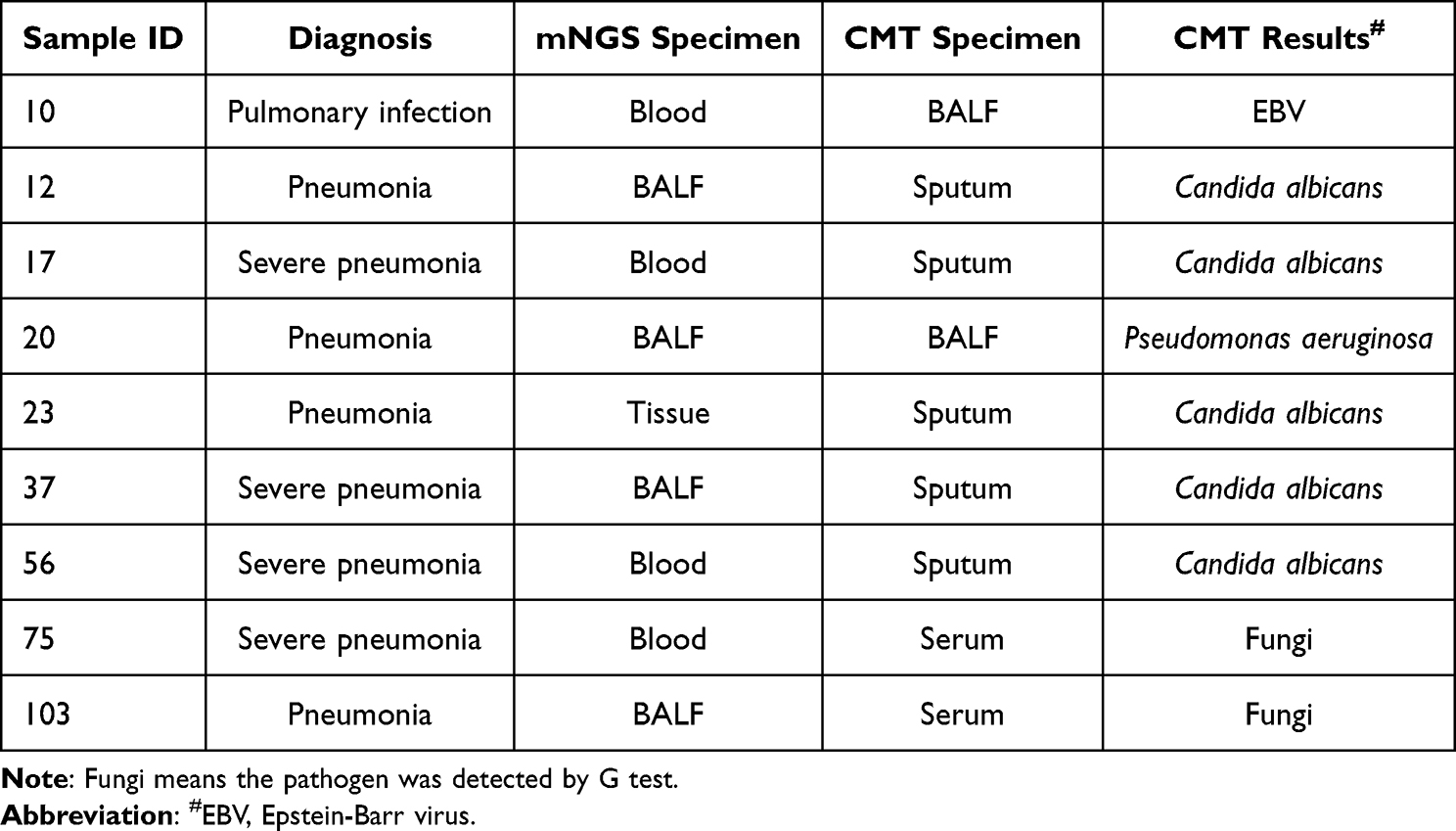 |
Table 2 The Samples with Only Positive Pathogen Results by CMT |
 |
Figure 3 Pathogens identified by CMT and mNGS. (A) Veen diagram showed the overlapped numbers of pathogen species detected by mNGS and CMT. (B) The type of infected pathogens for the patients based on the CMT and mNGS results. |
We classified the samples into two categories based on the types of pathogens identified: single type of pathogen infection and multiple types of pathogen infection. Among the mNGS findings, the proportion of the two categories were close (45% vs 55%, P > 0.05). Bacterial infection, bacteria and virus co-infections, and mixed bacteria-fungi-virus infections were the most common kinds of infection (Figure 3B). While in CMT results, only 18% of cases identified multiple types of pathogens. The top three prevalent infection types were single fungi infection, bacterial infection, and bacteria and fungi co-infections (Figure 3B).
Detailed pathogen profiles were shown in Figure 4. In both mNGS and CMT results, Pseudomonas aeruginosa (n = 24), Corynebacterium striatum (n = 16) and Klebsiella pneumoniae (n = 14) were the leading bacterial pathogens. Candida was the most frequently detected fungi, primarily Candida albicans (n = 31), Candida tropicalis (n = 10) and Candida glabrata (n = 7). Human herpesvirus was the predominantly virus detected, including EBV, Human herpesvirus-7 (HHV-7) and Cytomegalovirus (CMV). Additionally, the mNGS approach identified some special bacterial pathogens, including Mycoplasma pneumoniae (n = 6) and Chlamydia psittaci (n = 2). Among the mNGS results, only 9.6% (5/52) bacteria species were not detected from BALF samples, including Chroococcidiopsis thermalis (detected from tissue), Fusobacterium nucleatum (tissue), Filifactor alocis (Pus), Bilophila wadsworthia (sputum) and Mycolicibacterium smegmatis (sputum). Furthermore, herpesvirus has been found in both blood and sputum samples, and one instance of Candida tropicalis was identified in sputum samples.
 |
Figure 4 Pathogen profiles detected by mNGS and CMT. |
Seasonal Prevalence of the Pathogens
Pathogens found in both mNGS and CMT tests were used to investigate seasonal prevalence of infections. Of the 37 samples with consistent positive results (Figure 2C), 7, 14, 9 and 7 samples were obtained in spring, summer, autumn, and winter, respectively. TB was the most common pathogen in spring, while Pseudomonas aeruginosa and Klebsiella pneumoniae exhibited higher prevalence in the summer and autumn months. Moreover, Pseudomonas aeruginosa and Staphylococcus aureus were more likely to be identified in the winter. Candida albicans was consistently the most prevalent fungus across all seasons. Virus (Influenza B virus) was detected only in the winter season (Figure 5).
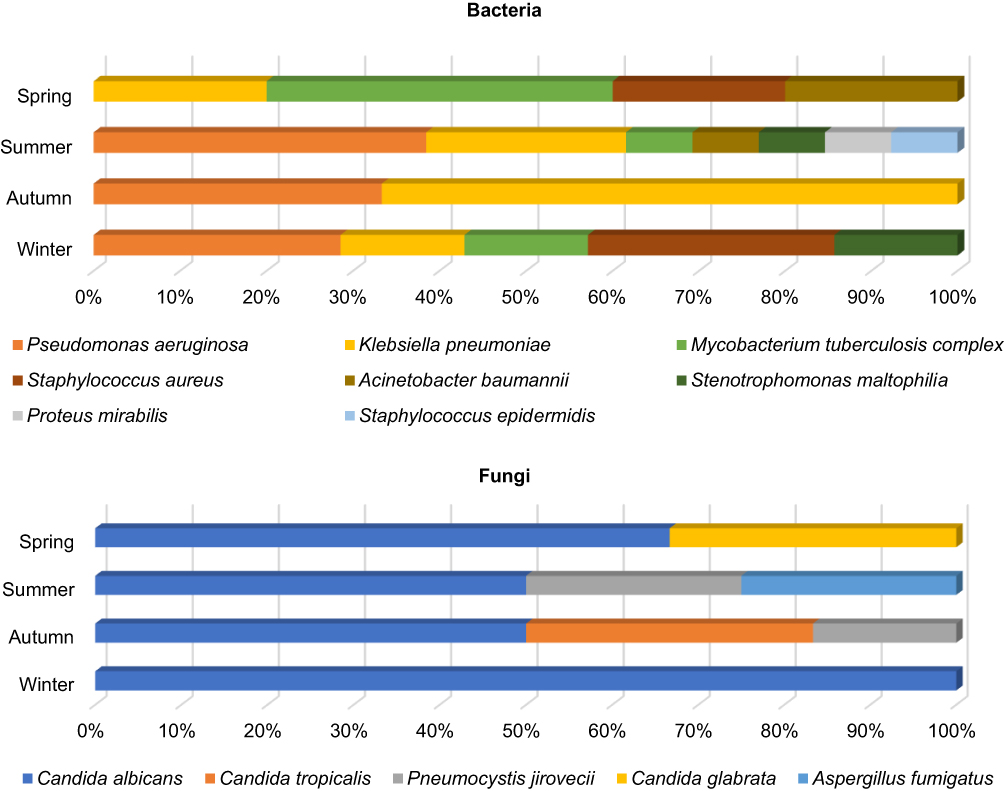 |
Figure 5 Seasonal prevalence of the pathogens detected by both mNGS and CMT. |
Furthermore, we analyzed the seasonal distribution of all the microorganisms identified by mNGS. There were 33, 50, 37 and 36 samples were collected in spring, summer, autumn, and winter, which achieved 63.6%, 70%, 75.7% and 69.4% positive rate, respectively. Bacteria that have been detected more than once are shown in Figure 6A and B. Gram-negative bacteria like Pseudomonas aeruginosa and Klebsiella pneumoniae, as well as gram-positive bacteria including Corynebacterium striatum and Enterococcus faecium, were detected more frequently during the spring seasons. The types of fungi detected in summer and winter were more diverse (Figure 6C). Additionally, the prevalence of EBV was highest during the spring and summer, while a higher detection of HHV-7 was observed in autumn and winter (Figure 6D).
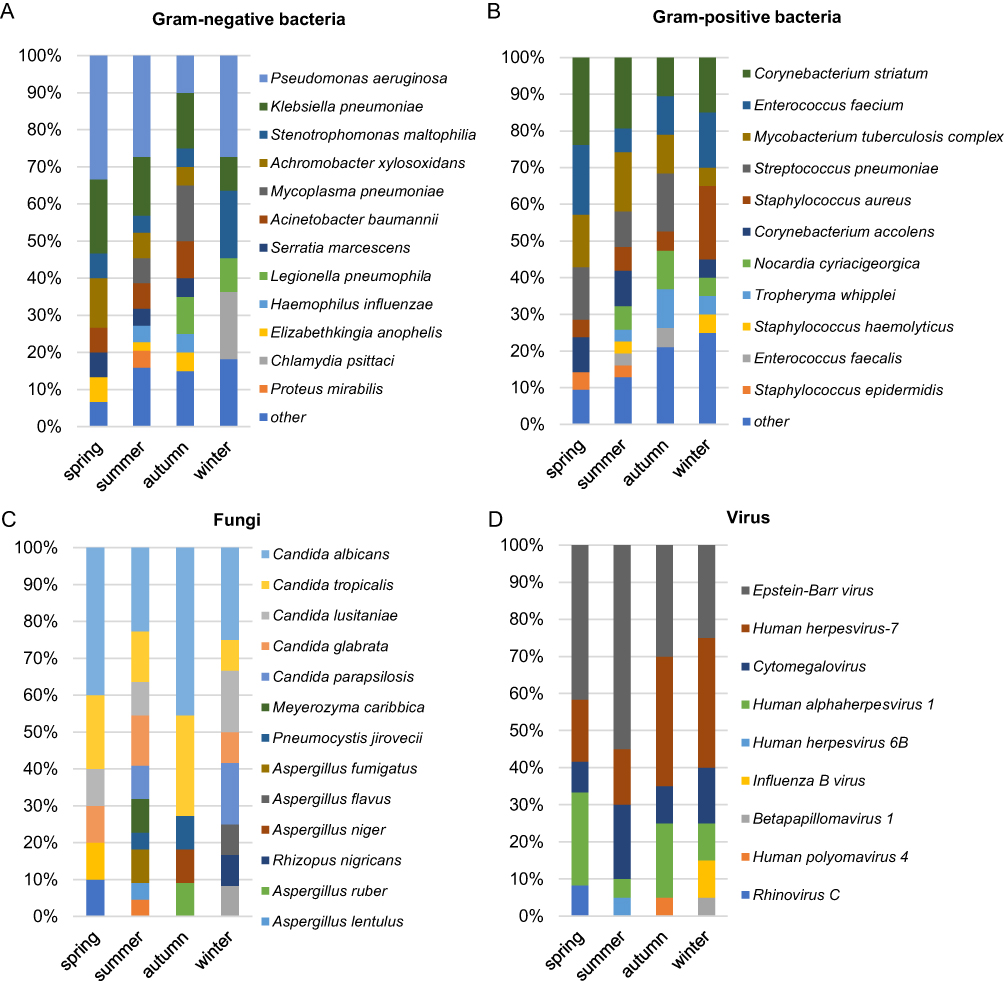 |
Figure 6 Seasonal prevalence of the pathogens detected by mNGS. (A) The distribution of gram-negative bacteria. (B) The distribution of gram-positive bacteria. (C) The seasonal prevalence of fungi. (D) Virus distribution based on seasons. “Other” represents the set of bacteria present in only one sample. |
Characteristics of Co-Infections with Multiple Types of Microorganisms
More than half of the samples had co-infection with multiple types of pathogens (Figure 3B). Only pathogens which have consistent results in both mNGS and CMT were applied to co-infection analysis. In total, 29 samples from 28 patients, comprising 27 species of bacteria, 12 species of fungi, and 6 viruses were used for the analysis. Male patients accounted for 64.3% of the total cohort, with a median age of 70 (range 37-87) years.
The findings revealed a broader range of co-infecting pathogens in elderly individuals, particularly involving gram-positive bacteria (Figure 7). Moreover, the prevalent forms of infection were attributed to a co-infection of bacteria, fungi, and viruses. No significant correlation was observed between the distribution of co-pathogens and the infection seasons of patients (P > 0.05, Supplementary Figure 1). Age exhibited a positive correlation only with Corynebacterium striatum, Stenotrophomonas maltophilia, and Candida lusitaniae (P < 0.05). Notably, all the correlations between the pathogens were positive. CMV and EBV were correlated with Aspergillus fumigatus (P < 0.05), and Aspergillus fumigatus also correlated with Pneumocystis jirovecii (P < 0.001). HHV-7 was correlated with Mycoplasma pneumoniae (P < 0.001). Human alphaherpesvirus 1 (HSV-1) was significantly correlated with two gram-negative bacteria (Achromobacter xylosoxidans and Elizabethkingia anopheles, P < 0.001). Candida glabrata and Candida tropicalis were more frequently co-infected with gram-negative bacteria, while other Candida species were more likely to coexist with gram-positive bacteria. Aspergillus and gram-negative bacteria were more typical co-pathogens (Supplementary Figure 1).
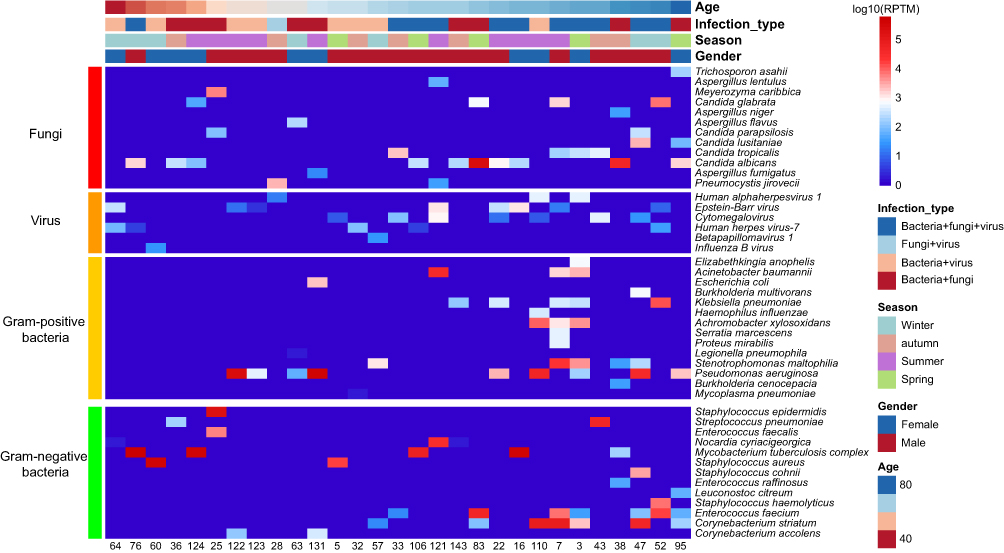 |
Figure 7 Heatmap of pathogens in samples with at least one consistent pathogen between mNGS and CMT. |
Discussion
Timely and accurately identification of pathogens is critical for treating LRTIs, particularly for critically ill patients or those infected with atypical pathogens. In this study, the seasonal prevalence of pathogens and potential co-pathogens in multiple infection events were analyzed.
Effective control and prevention of infectious diseases rely on a full understanding of their prevalence pattern and the factors that influence transmission.21 Bacterial infections showed seasonal fluctuation.11–13 The most prevalent bacterial pathogens discovered in this study were Pseudomonas aeruginosa, Corynebacterium striatum, and Klebsiella pneumoniae (Figure 4), which was consistent with previous studies.2,22 Additionally, Pseudomonas aeruginosa, and Klebsiella pneumoniae were more prevalent during the warmer months of the year.13–15 We found that Pseudomonas aeruginosa are most common in spring and summer, and Klebsiella pneumoniae was more prevalent in autumn (Figures 5 and 6). Tuberculosis is one of the most important infectious diseases in humans, and it is difficult to diagnose based on clinical signs and symptoms alone. Among the 9 TB patients diagnosed in this study, 4 cases were identified by both CMT and mNGS, and the remaining 5 were detected by mNGS only (Figure 4). TB was the most common pathogen in spring (Figure 5), which is consistent with the previous studies.23
Viral infections also demonstrated seasonal prevalence pattern.11–13 Human herpesviruses were the most frequently detected viruses, including EBV, HHV-7 and CMV (Figure 4). EBV was most frequently detected in spring and summer, while HHV-7 was more frequently detected in autumn and winter (Figure 6D). Herpesviruses are generally considered as noncausative pathogens. Herpesviruses are known to persist in the host in a latent state following infection, and the mechanisms governing their establishment, maintenance, and reactivation can vary among different subfamilies.24 Previous studies found that CMV, EBV and HSV-1 were the most commonly reactivated viruses, and their reactivation leading to an 2.052-fold increase in mortality.25 In addition, reactivation of CMV and EBV is also frequently seen in severe COVID-19 patients.26 Twenty samples from 18 patients underwent DNA and RNA mNGS for pathogen detection, and 2 of the cases identified Influenza B virus, 1 of which was further confirmed by PCR; another 2 cases tested by PCR have got negative results. Of the 136 remaining samples, only 4 RNA viruses were detected by PCR (Figure 3A). However, these samples were not performing RNA-sequencing by mNGS. Physicians did not perform RNA sequencing on all the samples using mNGS due to the low detection rate of RNA viruses in mNGS.27
Mixed infections were frequently observed among patients with LRTI (Figure 3), and there existed a noteworthy correlation between the pathogens in these instances (Supplementary Figure 1). The presence of polymicrobial pulmonary infection adds to the complexity of antibiotic spectrums and clinical manifestation. Conventional broad-spectrum antibiotics using may yield side effects before identifying the causative pathogens.28 However, research on mixed infections or co-pathogens is limited. The minimum inhibitory concentration test revealed that Acinetobacter baumannii cross-protects Klebsiella pneumoniae against cefotaxime, which helps elucidate the basis for their co-existence in polymicrobial infections and provides guidance for clinical medication.29 Competitive and cooperative interactions between Staphylococcus aureus and Pseudomonas aeruginosa influence the survival, antibiotic susceptibility, and persistence, consequently the disease progression of cystic fibrosis.30 The co-existing of Citrobacter freundii exacerbated the pathogenicity of Pseudomonas aeruginosa.31 Therefore, improving the understanding of the relationship between co-infectious pathogens and comparing the therapeutic effects of various antibiotics may provide a basis for reducing the use of antibiotics and promoting precision treatment.
This research has some limitations that should be acknowledged, such as the lack of in-depth analysis of the patients’ characteristics and clinical symptoms. Additionally, the data was obtained solely from a single hospital, which may have resulted in inherent bias.
Conclusion
In conclusion, this study indicates that mNGS holds great promise for detecting potential pathogens in the clinical samples of LRTI patients. Furthermore, the seasonal prevalence and co-pathogen patterns of the identified pathogens could facilitate the early and precise guidance of treatment procedures.
Acknowledgments
We would like to thank all the clinicians who contributed diagnostic data of patients to our study. Our thanks also go to Xiaojing Zhang, Yafeng Zheng and WillingMed Technology (Beijing) Co. for their technical support with mNGS.
Funding
This work was supported by Hebei Province Medical Science Research Project plan (grant number 20231610).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. WTO. WHO reveals leading causes of death and disability worldwide: 2000–2019; 2020. Available from: https://wwwwhoint/news/item/09-12-2020-who-reveals-leading-causes-of-death-and-disability-worldwide-2000-2019.
2. Diao Z, Han D, Zhang R, Li J. Metagenomics next-generation sequencing tests take the stage in the diagnosis of lower respiratory tract infections. J Adv Res. 2022;38:201–212. doi:10.1016/j.jare.2021.09.012
3. Zhu YG, Tang XD, Lu YT, Zhang J, Qu JM. Contemporary situation of community-acquired pneumonia in china: a systematic review. J Transl Int Med. 2018;6(1):26–31. doi:10.2478/jtim-2018-0006
4. Schlaberg R, Chiu CY, Miller S, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med. 2017;141(6):776–786. doi:10.5858/arpa.2016-0539-RA
5. Ljungstrom LR, Jacobsson G, Claesson BEB, Andersson R, Enroth H. Respiratory viral infections are underdiagnosed in patients with suspected sepsis. Eur J Clin Microbiol Infect Dis. 2017;36(10):1767–1776. doi:10.1007/s10096-017-2990-z
6. Simner PJ, Miller S, Carroll KC. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clin Infect Dis. 2018;66(5):778–788. doi:10.1093/cid/cix881
7. Chen S, Kang Y, Li D, Li Z. Diagnostic performance of metagenomic next-generation sequencing for the detection of pathogens in bronchoalveolar lavage fluid in patients with pulmonary infections: systematic review and meta-analysis. Int J Infect Dis. 2022;122:867–873. doi:10.1016/j.ijid.2022.07.054
8. Guo Y, Li H, Chen H, et al. Metagenomic next-generation sequencing to identify pathogens and cancer in lung biopsy tissue. EBioMedicine. 2021;73:103639. doi:10.1016/j.ebiom.2021.103639
9. Goldberg B, Sichtig H, Geyer C, Ledeboer N, Weinstock GM. Making the leap from research laboratory to clinic: challenges and opportunities for next-generation sequencing in infectious disease diagnostics. mBio. 2015;6(6):e01888. doi:10.1128/mBio.01888-15
10. Tsitsiklis A, Osborne CM, Kamm J, et al. Lower respiratory tract infections in children requiring mechanical ventilation: a multicentre prospective surveillance study incorporating airway metagenomics. Lancet Microbe. 2022;3(4):e284–e293. doi:10.1016/S2666-5247(21)00304-9
11. Qu J, Zhang J, Chen Y, et al. Aetiology of severe community acquired pneumonia in adults identified by combined detection methods: a multi-centre prospective study in China. Emerg Microbes Infect. 2022;11(1):556–566. doi:10.1080/22221751.2022.2035194
12. Richet H. Seasonality in Gram-negative and healthcare-associated infections. Clin Microbiol Infect. 2012;18(10):934–940. doi:10.1111/j.1469-0691.2012.03954.x
13. Eber MR, Shardell M, Schweizer ML, Laxminarayan R, Perencevich EN, Spellberg B. Seasonal and temperature-associated increases in gram-negative bacterial bloodstream infections among hospitalized patients. PLoS One. 2011;6(9):e25298. doi:10.1371/journal.pone.0025298
14. Perencevich EN, McGregor JC, Shardell M, et al. Summer peaks in the incidences of gram-negative bacterial infection among hospitalized patients. Infect Control Hosp Epidemiol. 2008;29(12):1124–1131. doi:10.1086/592698
15. Anderson DJ, Richet H, Chen LF, et al. Seasonal variation in Klebsiella pneumoniae bloodstream infection on 4 continents. J Infect Dis. 2008;197(5):752–756. doi:10.1086/527486
16. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
17. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi:10.1038/nmeth.1923
18. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20(1):257. doi:10.1186/s13059-019-1891-0
19. Langelier C, Kalantar KL, Moazed F, et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci U S A. 2018;115(52):E12353–E12362. doi:10.1073/pnas.1809700115
20. Chen H, Zhang Y, Zheng J, et al. Application of mNGS in the etiological diagnosis of thoracic and abdominal infection in patients with end-stage liver disease. Front Cell Infect Microbiol. 2021;11:741220. doi:10.3389/fcimb.2021.741220
21. Seventer JMV. Principles of infectious diseases: transmission, diagnosis, prevention, and control. Int Encyclop Public Health. 2017;2017:22–39.
22. Ping He JW, Rui K, Zhang W, et al. Comparison of metagenomic next-generation sequencing using cell-free DNA and whole-cell DNA for the diagnoses of pulmonary infections. Front Cell Infect Microbiol. 2022;12:1042945. doi:10.3389/fcimb.2022.1042945
23. Tedijanto C, Hermans S, Cobelens F, Wood R, Andrews JR. Drivers of seasonal variation in tuberculosis incidence: insights from a systematic review and mathematical model. Epidemiology. 2018;29(6):857–866. doi:10.1097/EDE.0000000000000877
24. Lieberman PM. Epigenetics and genetics of viral latency. Cell Host Microbe. 2016;19(5):619–628. doi:10.1016/j.chom.2016.04.008
25. Huang L, Zhang X, Pang L, et al. Viral reactivation in the lungs of patients with severe pneumonia is associated with increased mortality, a multicenter, retrospective study. J Med Virol. 2023;95(1):e28337. doi:10.1002/jmv.28337
26. Simonnet A, Engelmann I, Moreau AS, et al. High incidence of Epstein-Barr virus, cytomegalovirus, and human-herpes virus-6 reactivations in critically ill patients with COVID-19. Infect Dis Now. 2021;51(3):296–299. doi:10.1016/j.idnow.2021.01.005
27. Thorburn F, Bennett S, Modha S, Murdoch D, Gunson R, Murcia PR. The use of next generation sequencing in the diagnosis and typing of respiratory infections. J Clin Virol. 2015;69:96–100. doi:10.1016/j.jcv.2015.06.082
28. Hardak E, Avivi I, Berkun L, et al. Polymicrobial pulmonary infection in patients with hematological malignancies: prevalence, co-pathogens, course and outcome. Infection. 2016;44(4):491–497. doi:10.1007/s15010-016-0873-3
29. Semenec L, Cain AK, Dawson CJ, et al. Cross-protection and cross-feeding between Klebsiella pneumoniae and Acinetobacter baumannii promotes their co-existence. Nat Commun. 2023;14(1):702. doi:10.1038/s41467-023-36252-2
30. Biswas L, Gotz F. Molecular mechanisms of staphylococcus and pseudomonas interactions in cystic fibrosis. Front Cell Infect Microbiol. 2021;11:824042. doi:10.3389/fcimb.2021.824042
31. Peng Q, Chen L, Zhou S, et al. Co-existence of Citrobacter freundii exacerbated Pseudomonas aeruginosa infection in vivo. Int J Med Microbiol. 2020;310(1):151379. doi:10.1016/j.ijmm.2019.151379
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.