Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Disconnection of pulmonary and systemic arterial stiffness in COPD
Authors Weir-McCall JR ![]() , Liu-Shiu-Cheong PSK, Struthers AD, Lipworth BJ
, Liu-Shiu-Cheong PSK, Struthers AD, Lipworth BJ ![]() , Houston JG
, Houston JG ![]()
Received 16 December 2017
Accepted for publication 19 March 2018
Published 28 May 2018 Volume 2018:13 Pages 1755—1765
DOI https://doi.org/10.2147/COPD.S160077
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Jonathan R Weir-McCall,1 Patrick SK Liu-Shiu-Cheong,2 Allan D Struthers,1 Brian J Lipworth,2 J Graeme Houston1
1Division of Molecular and Clinical Medicine, Medical Research Institute, University of Dundee, Dundee, UK; 2Scottish Centre for Respiratory Research, Medical Research Institute, University of Dundee, Dundee, UK
Background: Both pulmonary arterial stiffening and systemic arterial stiffening have been described in COPD. The aim of the current study was to assess pulse wave velocity (PWV) within these two arterial beds to determine whether they are separate or linked processes.
Materials and methods: In total, 58 participants with COPD and 21 healthy volunteers (HVs) underwent cardiac magnetic resonance imaging (MRI) and were tested with a panel of relevant biomarkers. Cardiac MRI was used to quantify ventricular mass, volumes, and pulmonary (pulse wave velocity [pPWV] and systemic pulse wave velocity [sPWV]).
Results: Those with COPD had higher pPWV (COPD: 2.62 vs HV: 1.78 ms-1, p=0.006), higher right ventricular mass/volume ratio (RVMVR; COPD: 0.29 vs HV: 0.25 g/mL, p=0.012), higher left ventricular mass/volume ratio (LVMVR; COPD: 0.78 vs HV: 0.70 g/mL, p=0.009), and a trend toward a higher sPWV (COPD: 8.7 vs HV: 7.4 ms-1, p=0.06). Multiple biomarkers were elevated: interleukin-6 (COPD: 1.38 vs HV: 0.58 pg/mL, p=0.02), high-sensitivity C-reactive protein (COPD: 6.42 vs HV: 2.49 mg/L, p=0.002), surfactant protein D (COPD: 16.9 vs HV: 9.13 ng/mL, p=0.001), N-terminal pro-brain natriuretic peptide (COPD: 603 vs HV: 198 pg/mL, p=0.001), and high-sensitivity troponin I (COPD: 2.27 vs HV: 0.92 pg/mL, p<0.001). There was a significant relationship between sPWV and LVMVR (p=0.01) but not pPWV (p=0.97) nor between pPWV and RVMVR (p=0.27).
Conclusion: Pulmonary arterial stiffening and systemic arterial stiffening appear to be disconnected and should therefore be considered independent processes in COPD. Further work is warranted to determine whether both these cause an increased morbidity and mortality and whether both can be targeted by similar pharmacological therapy or whether different strategies are required for each.
Keywords: pulmonary vascular, cardiovascular, COPD, arterial compliance, ventricular function
Introduction
Despite being a disease of the pulmonary parenchyma, COPD is associated with significant cardiac morbidity and mortality, with up to 43% of mortality in COPD secondary to cardiovascular causes.1–3 Even in the absence of pulmonary hypertension, those with COPD demonstrate right ventricular (RV) dysfunction,4,5 and diastolic and systolic left heart failures are present in >20% of those with moderate-to-severe COPD.6,7
Arteriosclerosis is the stiffening of the arterial wall and is predominantly a result of aging and rising mean arterial pressure in both the systemic and pulmonary circulation.8–11 An increase in arterial stiffening leads to an increased afterload and an inefficient energy transfer from the ventricles to the vasculature, resulting in an increased cardiac workload.12,13 Increased stiffening of both the systemic and pulmonary circulation has been described in COPD, independent of traditional risk factors.14,15 Due to its adverse hemodynamic effects, arterial stiffening is a well-established risk factor for future cardiovascular mortality.16 Multiple studies have demonstrated systemic arterial stiffening in those with COPD when compared with matched smokers and for this stiffening to correlate with the percentage of emphysema.14,17 Several studies have also shown COPD therapy to result in an improvement in arterial stiffness, although this observation has not been consistent across all studies.18–20 Pulmonary arterial stiffness has also been shown to correlate with exercise capacity and right heart function in COPD.21 It has also been shown that those with COPD who are taking statins have lower pulmonary pressures on right heart catheterization compared with those not on statins and that statins are associated with improved functional capacity and lower mortality in COPD.22–24 Given that statins improve systemic cardiovascular disease and mediate adverse vascular remodeling in atherosclerosis,25,26 it may thus be posited that the disease process afflicting the pulmonary arteries and aorta may simply be the same pathological process resulting from a COPD-induced accelerated vascular degeneration.
Thus, the aim of the current study was to examine the association between aortic and pulmonary arterial stiffening using pulse wave velocity (PWV) and their effects on cardiac remodeling with the hypotheses that 1) aortic stiffening and pulmonary stiffening occur in parallel; 2) inflammatory markers would explain this interaction; and 3) elevated PWV would be associated with adaptive ventricular remodeling with an increased ventricular mass to end diastolic volume ratio.
Materials and methods
Between July 2014 and May 2016, participants were recruited from primary and secondary care clinics and from community-based spirometry and research databases. Inclusion criteria for the study were the following: 40–85 years of age with a diagnosis of COPD, based on the current Global Initiative for Chronic Obstructive Lung Disease guidelines of postbronchodilator forced expiratory volume in 1 second/forced vital capacity <0.7 with a history of smoking.27 Exclusion criteria were the following: history of cardiac condition, including but not limited to ischemic heart disease, valvular disease (mild functional regurgitation allowed), arrhythmia, cardiomyopathy, congestive cardiac failure, or congenital cardiac disease; previous cardiac or thoracic operation; other coexistent lung condition; connective tissue disease or systemic vasculitis; severe renal impairment (epidermal growth factor receptor <30 mL/min); or contraindication for magnetic resonance imaging (MRI). All recruited participants underwent a screening echocardiogram to exclude significant silent left ventricular systolic dysfunction (ejection fraction <45%).
In total, 104 participants with COPD were screened with 37 excluded due to either coexistent coronary artery disease, atrial fibrillation, prior thoracotomy, coexistent lung condition, left ventricular systolic dysfunction on echo, or no history of smoking. This left 67 participants in the cohort. Of these, 56 participants were included in the final analysis (6 participants were excluded due to incomplete scan secondary to claustrophobia, 1 due to a history of metal fragments in the orbits not picked up in screening, and 4 due to inadequate image quality for analysis). All the participants gave written informed consent for the study that was conducted in accordance with the Declaration of Helsinki and was approved by the East of Scotland Research Ethics Committee 1 (Reference Number: 14/ES/0034).
A healthy control (HC) group was recruited with the age and sex of them approximately matched to the COPD cohort, with no prior history of cardiac or pulmonary pathology.
All COPD participants underwent spirometry, single breath diffusion (diffusing capacity of the lungs for carbon monoxide [DLCO]), a 6-minute walk test (6MWT), and a cardiac MRI on the same day. All HCs underwent a cardiac MRI. Spirometry, DLCO, and 6MWT were performed as per European Respiratory Society/American Thoracic Society guidelines.28,29 The blood samples of all the participants were taken at the time of their attendance for the MRI scan.
MRI
Images were acquired on a 32–radiofrequency RF cardiac receiver channel, 3-Tesla MRI scanner (Prisma; Siemens Medical Solutions, Erlangen, Germany). A three-plane localizer was first obtained, following which 4-chamber, 2-chamber, and short-axis localizers of the heart were obtained. An axial half-Fourier acquisition turbo spin echo stack was acquired of the chest.
From these, a balanced steady-state free precession (bSSFP) stack was performed in breath-hold from the atrioventricular ring to the apex. To plan the main pulmonary artery (MPA) phase contrast sequence, a bSSFP sequence of the RV outflow tract was performed following which an orthogonal plane was acquired to optimally visualize the MPA and pulmonary valve. A bSSFP sequence was then performed through the MPA slice and was positioned midway between the valve and the bifurcation of the pulmonary artery in order to avoid both structures throughout the cardiac cycle. A free-breathing phase contrast sequence (slice thickness =6 mm, time to relaxation/time to echo (TR/TE) =12/4 ms, number of averages =1, phases =80, velocity encoding =150 cm/s, bandwidth/pixel =340 Hz, flip angle =15°, field of view =320×320 mm2, matrix =512×512) was then performed in the same position as previously described.30 For aortic PWV measurement, a two-dimensional gradient echo (fast low-angle shot [FLASH] was first acquired of the aorta in a “candy stick” double-oblique orientation (parameters: TR/TE =40/1.2 ms; flip angle =15°, slice thickness =8 mm, 23 cardiac phases, number of averages =1, a pixel size of 1.5×1.5 mm2, bandwidth =475 Hz/pixel). Two-phase contrast sequences were then obtained, the first positioned through the aortic arch at the level of the pulmonary bifurcation and the second slice placed through the proximal abdominal aorta just distal to the aortic hiatus. The same phase contrast sequence was used as for the pulmonary PWV measurement.
Image analysis
The images were exported with image analysis performed by using CVI 42 (Circle Cardiovascular Imaging Inc., Calgary, Alberta, Canada).
Ventricular quantification
Epicardial and endocardial contours were drawn around the right ventricle at end systole and end diastole. Trabeculae were included in the mass measurement and excluded from the volume calculation. The septum was treated as belonging to the left ventricle and was excluded from the RV mass. RV mass and volumes were normalized to height1.7.
Pulmonary PWV
For the time component, the phase and magnitude images were pulled up side by side. A contour was manually drawn around the perimeter of the vessel, then propagated throughout the cardiac cycle, and corrected as when automatic contouring led to erroneous boundaries. The program then automatically calculated area, flow, and velocity data, which were exported to Excel 2010 (Microsoft Corporation, Redmond, WA, USA). The area and flow were plotted against one another during early systole, which was defined as the time period in systole during which both the vessel area and flow were simultaneously increasing. The PWV was then calculated as described by Davies et al:31
|
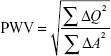
where Q =vessel flow and A=vessel area.
Aortic PWV
The distance was measured along the aorta between the two analysis planes (Δx value) using candy stick FLASH and the time delay calculated as the time delay between the arrival of the foot of the pulse wave at the ascending aorta and abdominal aorta. To calculate the latter, the flow curves from the ascending thoracic aorta and the abdominal aorta were plotted, and the time to the systolic upstroke of the waves was then calculated. The arrival time of the flow wave was identified as the intersection between the systolic upstroke and baseline flow. The systolic upstroke was calculated as the line through the data points lying between 20% and 80% of the maximum flow rate.32 The baseline was the horizontal line at minimum velocity before systole. Pulse wave was calculated using the following equation:
|
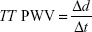
where TT stands for transit time, d stands for distance between the imaging planes, and t stands for time of systolic upstroke arrival.
Biomarkers
In order to better understand the links between COPD, vascular stiffening, and cardiac remodeling, several families of biomarkers were assessed: 1) connective tissue markers – carboxymethyllysine (CML), matrix metalloproteinase-9 (MMP-9), and fibroblast growth factor-23 (FGF-23); 2) inflammatory markers – high-sensitivity C-reactive protein (hsCRP), interleukin-6 (IL-6), and fibrinogen; 3) pulmonary markers – club cell secretory protein-16 (CC-16) and surfactant protein D (SPD); 4) cardiac markers – high-sensitivity troponin I (hsTrop I) and N-terminal pro-brain natriuretic peptide (NT-proBNP); and 5) lipid markers – total cholesterol (TC), high-density lipoprotein (HDL), low-density lipoprotein (LDL), and triglycerides.
For this analysis, blood samples were drawn into 3×4 mL EDTA tubes, which were then centrifuged at 2,000× g at room temperature for 15 minutes before the plasma from this was removed and then stored at −80°C. All the samples were stored until the study was completed and then analyzed en bloc. Table S1 presents the details of the manufacturers and interassay (plate-to-plate) and intra-assay coefficients of variations.
Statistics
Descriptive statistics were used for the analysis of the demographic and clinical features of the cohorts with data expressed as mean ± SD. Normality and equality of variances of the variables were tested. An independent sample t-test was used to compare the differences in continuous variables between the HCs and COPD cohort. χ2, Fisher’s exact, and Mann–Whitney U tests were used as appropriate to compare differences in ordinal and nominal data between the groups. Pearson or Spearman rank coefficients were used to assess the correlation between aortic and pulmonary PWV and to look at the correlates of both of these with baseline demographic, spirometric, and MRI factors in the COPD cohort. Multiple linear regression analysis was performed with systemic pulse wave velocity (sPWV) and pulmonary pulse wave velocity (pPWV) entered separately as the dependent variables, with those factors which were (p<0.1) in single variable analysis entered as independent variables. All data were analyzed using SPSS package (Version 21.0; IBM Corporation, Armonk, NY, USA). Significance was assumed when p<0.05.
Results
In total, 56 COPD patients (67.4±9.0 years, 55% male) and 20 HCs (60.4±5.1, 48% male) completed the study protocol. Despite approximate age and sex matching, those with COPD were significantly older (p<0.001) and had a higher body mass index (BMI; COPD: 26.8±5.3 kg/m2 vs HC: 24.6±2.5 kg/m2, p=0.02). While those with COPD had a significantly higher prevalence of hypertension, there was no significant difference in blood pressure or pulse pressure due to a higher prevalence of antihypertensive medication prescription. Table 1 presents the full baseline characteristics.
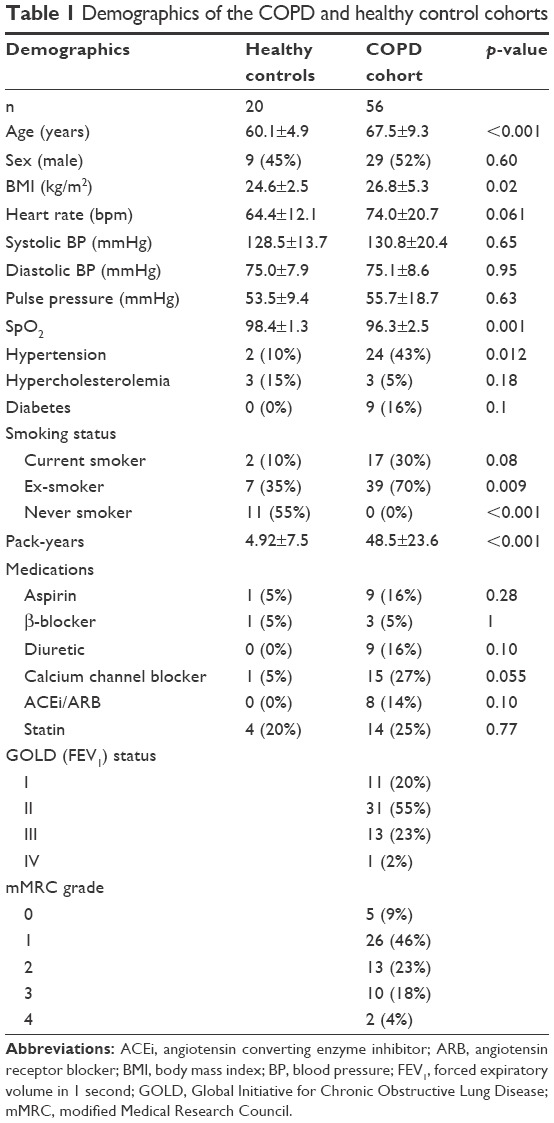 | Table 1 Demographics of the COPD and healthy control cohorts |
Those with COPD demonstrated a significantly higher pulmonary PWV (COPD: 2.6±1.3 ms−1 vs HC: 1.8±0.7 ms−1, p=0.006) and a trend toward a higher aortic PWV (COPD: 8.7±2.7 ms−1 vs HC: 7.4±2.1 ms−1, p=0.06). Compared with the HCs, those with COPD had a significantly lower RV stroke volume (COPD: 31.7±7.0 mL/m1.7 vs HC: 36.6±5.9 mL/m1.7, p=0.007) and a higher RV mass/volume ratio (RVMVR; COPD: 0.29±0.05 g/mL vs HC: 0.25±0.04 g/mL, p=0.012). Those with COPD also had a significantly higher left ventricular mass/volume ratio (LVMVR; COPD 0.78±0.13 g/mL vs HC: 0.70±0.09 g/mL, p=0.009; see Table 2 for full ventricular and pulmonary arterial parameters).
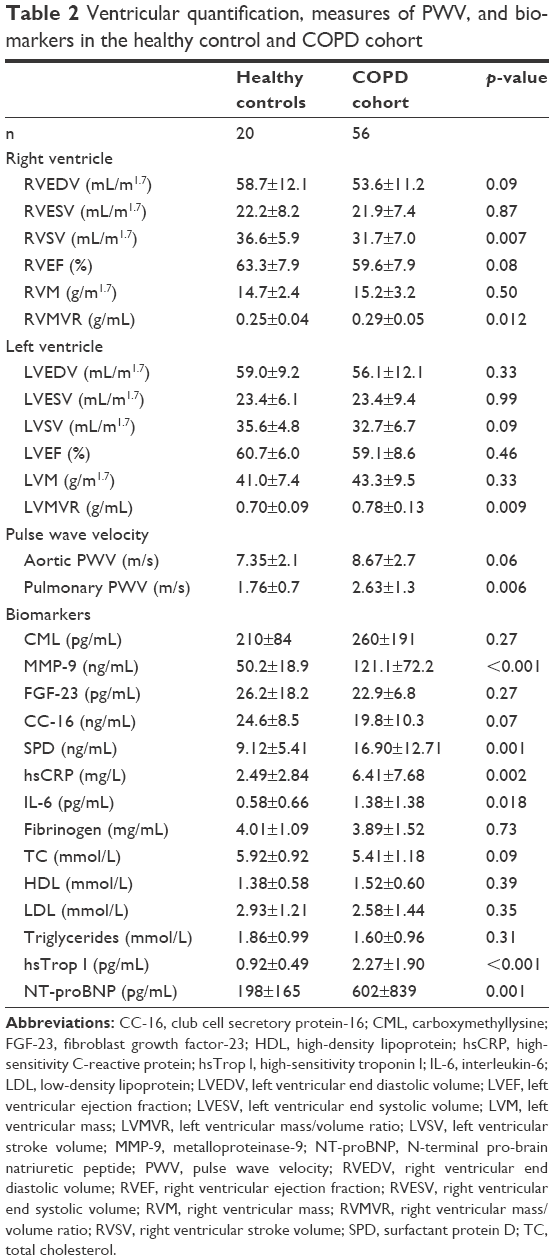 | Table 2 Ventricular quantification, measures of PWV, and biomarkers in the healthy control and COPD cohort |
In those with COPD, pulmonary PWV was correlated with percentage predicted total lung capacity (ρ=0.28, p=0.046), inversely correlated with BMI (ρ=−0.28, p=0.04), and correlated with increasing diastolic blood pressure (ρ=0.36, p=0.01) with a trend toward a positive correlation with percentage predicted residual lung volume (ρ=0.25, p=0.08; see Table S2 for full correlates). Applying a conservative Bonferroni correction for multiple comparisons would result in a significance level of p<0.0008, with none of the variables meeting this level. On multiple-variable backward linear regression, diastolic blood pressure (ß=0.28, p=0.035) and percentage predicted total lung capacity (ß=0.30, p=0.028) remained significantly associated with pPWV.
In comparison, aortic PWV showed a significant association with age (ρ=0.47, p<0.001), systolic blood pressure (ρ=0.32, p=0.02), and percentage predicted transfer coefficient for carbon monoxide (KCO) (ρ=0.43, p=0.001) and a trend toward a significant association with percentage predicted DLCO (ρ=0.27, p=0.050) and pulse pressure (ρ=0.25 p=0.067). Applying a conservative Bonferroni correction for multiple comparisons would result in a significance level of p<0.0008, as a result of which only age would remain a significant correlate of sPWV. On linear regression, age (ß=0.30, p=0.02), systolic blood pressure (ß=0.27, p=0.04), and percentage predicted KCO (ß=0.37, p=0.003) all remained significantly associated with aortic PWV.
sPWV was significantly associated with LVMVR (ρ=0.34, p=0.01), while no such correlation was found between pPWV and RVMVR (ρ=−0.15, p=0.27). Similarly, there was no significant association between aortic and pulmonary PWV (ρ=−0.004, p=0.97; Figure 1).
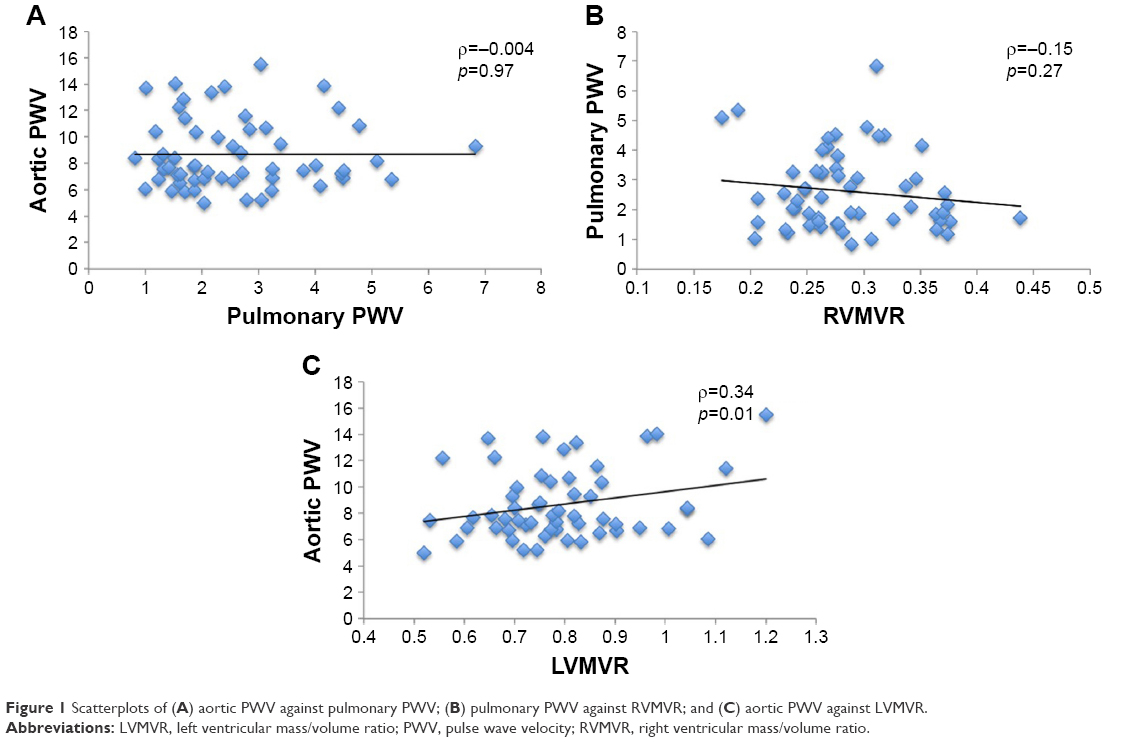 | Figure 1 Scatterplots of (A) aortic PWV against pulmonary PWV; (B) pulmonary PWV against RVMVR; and (C) aortic PWV against LVMVR. |
Those with COPD showed significantly higher levels of the following: inflammatory markers IL-6 (COPD: 1.38±1.38 pg/mL vs healthy volunteers [HVs]: 0.58±0.66 pg/mL, p=0.018) and hsCRP (COPD: 6.42±7.68 mg/L vs HV: 2.49±2.84 mg/L, p=0.002); connective tissue markers MMP-9 (COPD: 121±72 ng/mL vs HV: 50±19 ng/mL, p<0.001); lung tissue marker SPD (COPD: 16.9±12.7 ng/mL vs HV: 9.13±5.41 ng/mL, p=0.001); and cardiac markers NT-proBNP (COPD: 603±839 pg/mL vs HV: 198±165 pg/mL, p=0.001) and hsTrop I (COPD: 2.27±1.90 pg/mL vs HV: 0.92±0.49 pg/mL, p<0.001).
No significant difference was found in FGF-23, CC-16, CML, fibrinogen, cholesterol, HDL, or LDL (p>0.05 for all).
In those with COPD, there was a weak correlation between pPWV and hsCRP (ρ=−0.28, p=0.04), TC (ρ=0.28, p=0.044), and NT-proBNP (ρ=0.28, p=0.048). However, on linear regression accounting for diastolic blood pressure, the significance of these was lost. For sPWV, there was a weak correlation with CC-16 (ρ=0.31, p=0.028), TC (ρ=0.33, p=0.019), and LDL (ρ=0.32, p=0.023). However, on linear regression accounting for age and systolic blood pressure, these associations were no longer significant.
Discussion
In this study, we have seen that 1) both pulmonary and systemic arteries are stiffer in COPD compared with controls; 2) aortic PWV and pulmonary PWV are associated with different clinical parameters and biomarkers from one another; and 3) aortic but not pulmonary PWV is associated with ventricular hypertrophy.
Our finding of increased aortic stiffness is consistent with previous work demonstrating increased aortic PWV in those with COPD.33 In comparison with the previous studies that used carotid femoral PWV, the current study used MRI for the assessment of central aortic PWV. The two techniques have been shown to correlate well with one another, although carotid–femoral PWV significantly overestimates central aortic PWV due to the inclusion of the stiffer peripheral vessels in its calculation and inaccuracies in path length estimation.34–36 In addition, it excludes the ascending aorta from its measurement, with this playing a greater role in left ventricular remodeling compared with the rest of the aorta.37 By its direct assessment of central aortic PWV, the current study provides valuable insights into the interaction between central aortic stiffness and left ventricular remodeling. The underlying cause of the increased systemic arterial stiffness in COPD is still a topic of debate. While a previous association between aortic PWV and inflammatory markers has been described,38 other groups have found no such correlation.39,40 In the current study, we found that in those with COPD the aortic PWV was associated with age and blood pressure, but not with inflammatory markers. Our finding of a significant association between LVMVR – which is the earliest measure of concentric myocardial hypertrophic remodeling41 – and aortic PWV provides significant mechanistic insight into the interface between the pulmonary and cardiovascular disease evident in COPD. As with increased aortic PWV, multiple studies have documented left ventricular hypertrophy in COPD, demonstrating both a high prevalence and significant implications for mortality in this group.42–44 While the association between PWV and left ventricular mass has been described in the general population,45,46 to the best of the authors’ knowledge, no studies have examined the role of the aortic arterial stiffness as a linking mechanism in COPD. Indeed, a previous study ascribed the observed left ventricular hypertrophy to pulmonary hyperinflation, but did not measure or account for aortic stiffness.43 Given that LVMVR is a known risk factor for future cardiovascular events, this association may help explain both the increased cardiovascular risk in COPD and the beneficial effects in COPD of β-blockers – a known regressor of left ventricular mass.47–49
Increased pulmonary arterial stiffness in COPD has been previously reported both invasively on right heart catheterization and using pulsatility on MRI.4,15 Invasive pulmonary PWV has been demonstrated to correlate well with invasive pressure measurements in those with pulmonary hypertension.50 Pulmonary PWV as measured on MRI provides the benefit of not requiring an invasive procedure to obtain direct information on the arterial wall. It also provides additional information over pulmonary pulsatility as this latter metric is a flow-dependent measure of the relative change in the area of the pulmonary artery throughout the cardiac cycle. Thus, assuming a steady pulmonary arterial wall stiffness, a fall in stroke volume will result in a fall in pulmonary pulsatility. As both a reduced stroke volume and a reduced pulmonary pulsatility have been described in COPD,15,51 pulmonary PWV is a useful measure to better interrogate pulmonary arterial remodeling.
Despite those with COPD having significantly elevated hsCRP, IL-6, MMP-9, NT-proBNP, and hsTrop I, none of these markers showed any significant correlation with either aortic or pulmonary PWV. In the current study, we found no significant link between aortic and pulmonary PWV, with no overlap of clinical correlates or biomarkers. Thus, it would appear that the pulmonary vascular remodeling in COPD occurs separately to systemic arterial stiffening. Previous work has described pulmonary arterial stiffening to be significantly associated with exercise-induced elevations in pulmonary pressures;21,52 however, we found no link between this and RV remodeling; therefore, while the pressures may elevate in exercise, the clinical significance of this is uncertain. Longitudinal studies will be required to determine the long-term implications of pulmonary arterial stiffening.
Given the lack of significant correlation between pulmonic and arterial stiffening and biomarkers, particularly the lack of association with inflammation, another link between COPD and arterial stiffening must also be considered. It has been previously demonstrated that the two strongest predictors of aortic PWV are age and blood pressure.8 The former of these reflects a gross marker of the total pulsatile cycles the aorta has undergone, while the latter reflects the pressure at which the walls have been distended during these cycles. Thus, COPD, with its higher heart rate secondary to lower stroke volumes as found in this and other studies,51,53 may accelerate arterial stiffening without the need for an inflammatory link. A study in a population free from cardiovascular disease has shown an elevated resting heart rate to be associated with increased aortic stiffness.54 In those with COPD, resting heart rate is associated with cardiovascular mortality, but a direct link between heart rate and arterial stiffness in COPD has yet to be evaluated.53,55 While elevated heart rate would affect the aortic and pulmonary circulation in a similar pattern, the underlying distending pressures in the two systems vary independently of one another; thus the two circulations may remodel at a different rate even with the same accelerating factor. Another alternative is the role of autonomic nervous system activation, which has been described in COPD and is known to be associated with PWV.56–58
There are some limitations to the current study. Patients with known coronary artery disease were excluded; thus, the results may not be representative of the wider COPD cohort in whom cardiovascular disease is common. Despite attempted matching, the controls were younger than those with COPD and more likely to be on treatment for hypertension although the blood pressures were well matched. As age is a significant contributor to PWV, this may have contributed to the difference between the two groups although our observations are concordant with the current literature.8 Unfortunately, due to the limited group sizes, particularly of the controls, the data did not meet assumptions required for the analysis of covariance; therefore, multiple variable modeling to account for these could not be performed. We also used two different techniques for the MRI assessment of the aortic and pulmonary PWV, using a transit time method in the aorta and a flow area technique within the pulmonary artery. This was done as the former is more accurate in longer vessels such as the aorta;32 however, the short pulmonary artery is less amenable to this due to the rapid transit times, and prior work has shown the transit time within the pulmonary arteries to be less reproducible compared with the flow area technique.30 In addition, the two techniques demonstrate a reasonable agreement with one another minimizing the potential significance of this.59 Finally, if we had enrolled more severe hypoxic COPD patients in GOLD stages III/IV (ie, FEV<50%), then perhaps we would have seen more pronounced RV remodeling.
Conclusion
In conclusion, both aortic and pulmonary arterial stiffening occurs in COPD, but these processes are independent of one another with aortic but not pulmonary PWV, demonstrating a significant association with remodeling in the ventricle. Further work is warranted to determine whether this stiffening causes an increased morbidity and mortality and whether both can be targeted by similar pharmacological therapy or whether different strategies are required for each.
Acknowledgments
We would like to thank the support of Gwen Kennedy and Lesley McFarlane at the Biomarker and Immunoassay Biomarker Core Laboratory, University of Dundee, for their processing of all the biomarkers. The present study was funded by the Wellcome Trust through the Scottish Translational Medicine and Therapeutics Initiative (Grant Number: WT 085664) in the form of a Clinical Research Fellowship. They had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
Author contributions
JRWM and PSKLSC recruited the subjects, acquired the images, took the measurements and analyzed them, drafted the paper, and prepared the tables and figures. ADS, BJL, and JGH participated in the design of the study, helped in statistical analysis and the preparation of tables and figures, and worked on the text of the paper. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Anthonisen NR, Skeans MA, Wise RA, et al. The effects of a smoking cessation intervention on 14.5-year mortality: a randomized clinical trial. Ann Intern Med. 2005;142(4):233–239. | ||
McGarvey LP, John M, Anderson JA, Zvarich M, Wise RA; TORCH Clinical Endpoint Committee. Ascertainment of cause-specific mortality in COPD: operations of the TORCH Clinical Endpoint Committee. Thorax. 2007;62(5):411–415. | ||
Vestbo J, Anderson JA, Brook RD, et al. Fluticasone furoate and vilanterol and survival in chronic obstructive pulmonary disease with heightened cardiovascular risk ( SUMMIT ): a double-blind randomised controlled trial. Lancet. 2016;387(10030):1817–1826. | ||
Hilde JM, Skjørten I, Grøtta OJ, et al. Right ventricular dysfunction and remodeling in chronic obstructive pulmonary disease without pulmonary hypertension. J Am Coll Cardiol. 2013;62(12):1103–1111. | ||
Cuttica MJ, Shah SJ, Rosenberg SR, et al. Right heart structural changes are independently associated with exercise capacity in non-severe COPD. PLoS One. 2011;6(12):e29069. | ||
Rutten FH, Cramer JM, Grobbee DE, et al. Unrecognized heart failure in elderly patients with stable chronic obstructive pulmonary disease. Eur Heart J. 2005;26(18):1887–1894. | ||
Kubota Y, Asai K, Murai K, et al. COPD advances in left ventricular diastolic dysfunction. Int J Chron Obstruct Pulmon Dis. 2016;11:649–655. | ||
Cecelja M, Chowienczyk P. Dissociation of aortic pulse wave velocity with risk factors for cardiovascular disease other than hypertension: a systematic review. Hypertension. 2009;54(6):1328–1336. | ||
Dawes TJ, Gandhi A, de Marvao A, et al. Pulmonary artery stiffness is independently associated with right ventricular mass and function: a cardiac MR imaging study. Radiology. 2016;280(2):398–404. | ||
Milnor WR, Bergel DH, Bargainer JD. Hydraulic power associated with pulmonary blood flow and its relation to heart rate. Circ Res. 1966;19(3):467–480. | ||
Milnor WR, Conti CR, Lewis KB, O’Rourke MF. Pulmonary arterial pulse wave velocity and impedance in man. Circ Res. 1969;25(6):637–649. | ||
Fourie PR, Coetzee AR, Bolliger CT. Pulmonary artery compliance: its role in right ventricular-arterial coupling. Cardiovasc Res. 1992;26(9):839–844. | ||
O’Rourke MF. Vascular impedance in studies of arterial and cardiac function. Physiol Rev. 1982;62(2):570–623. | ||
Mills NL, Miller JJ, Anand A, et al. Increased arterial stiffness in patients with chronic obstructive pulmonary disease: a mechanism for increased cardiovascular risk. Thorax. 2008;63(4):306–311. | ||
Liu CY, Parikh M, Bluemke DA, et al. Pulmonary artery stiffness in chronic obstructive pulmonary disease (COPD) and emphysema: the Multi-Ethnic Study of Atherosclerosis (MESA) COPD Study. J Magn Reson Imaging. 2018;47(1):262–271. | ||
Ben-Shlomo Y, Spears M, Boustred C, et al. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta-analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol. 2014;63(7):636–646. | ||
McAllister DA, Maclay JD, Mills NL, et al. Arterial stiffness is independently associated with emphysema severity in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176(12):1208–1214. | ||
Iyer AS, Wells JM, Vishin S, Bhatt SP, Wille KM, Dransfield MT. CT scan-measured pulmonary artery to aorta ratio and echocardiography for detecting pulmonary hypertension in severe COPD. Chest. 2014;145(4):824–832. | ||
Pepin J, Cockcroft JR, Midwinter D, Sharma S, Rubin DB. Long-acting bronchodilators and arterial stiffness in patients with COPD: a comparison of fluticasone furoate/vilanterol with tiotropium. Chest. 2014;146(6):1521–1530. | ||
Bhatt SP, Dransfield MT, Cockcroft JR, et al. A randomized trial of once-daily fluticasone furoate/vilanterol or vilanterol versus placebo to determine effects on arterial stiffness in COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:351–365. | ||
Kubo K, Ge RL, Koizumi T, et al. Pulmonary artery remodeling modifies pulmonary hypertension during exercise in severe emphysema. Respir Physiol. 2000;120(1):71–79. | ||
Reed RM, Iacono A, DeFilippis A, et al. Statin therapy is associated with decreased pulmonary vascular pressures in severe COPD. COPD. 2011;8(2):96–102. | ||
Lawes CM, Thornley S, Young R, et al. Statin use in COPD patients is associated with a reduction in mortality: a national cohort study. Prim Care Respir J. 2012;21(1):35–40. | ||
Lee TM, Lin MS, Chang NC. Usefulness of C-reactive protein and interleukin-6 as predictors of outcomes in patients with chronic obstructive pulmonary disease receiving pravastatin. Am J Cardiol. 2008;101(4):530–535. | ||
Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;(1):CD004816. | ||
Kao MP, Ang DS, Gandy SJ, et al. Allopurinol benefits left ventricular mass and endothelial dysfunction in chronic kidney disease. J Am Soc Nephrol. 2011;22(7):1382–1389. | ||
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Am J Respir Crit Care Med. 2017;195:557–582. | ||
ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111–117. | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Weir-McCall JR, Kamalasanan A, Cassidy DB, Struthers AD, Lipworth BJ, Houston JG. Assessment of proximal pulmonary arterial stiffness using magnetic resonance imaging: effects of technique, age and exercise. BMJ Open Respir Res. 2016;3(1):e000149. | ||
Davies JE, Whinnett ZI, Francis DP, et al. Use of simultaneous pressure and velocity measurements to estimate arterial wave speed at a single site in humans. Am J Physiol Heart Circ Physiol. 2006;290(2):H878–H885. | ||
Ibrahim el-SH, Johnson KR, Miller AB, Shaffer JM, White RD. Measuring aortic pulse wave velocity using high-field cardiovascular magnetic resonance: comparison of techniques. J Cardiovasc Magn Reson. 2010;12:26. | ||
Vivodtzev I, Tamisier R, Baguet JP, Borel JC, Levy P, Pépin JL. Arterial Stiffness in COPD. Chest. 2014;145(4):861–875. | ||
Parikh JD, Hollingsworth KG, Kunadian V, Blamire A, MacGowan GA. Measurement of pulse wave velocity in normal ageing: comparison of Vicorder and magnetic resonance phase contrast imaging. BMC Cardiovasc Disord. 2016;16(1):50. | ||
Hickson SS, Butlin M, Graves M, et al. The relationship of age with regional aortic stiffness and diameter. JACC Cardiovasc Imaging. 2010;3(12):1247–1255. | ||
Weir-McCall JR, Khan F, Cassidy DB, et al. Effects of inaccuracies in arterial path length measurement on differences in MRI and tonometry measured pulse wave velocity. BMC Cardiovasc Disord. 2017;17(1):118. | ||
Quinaglia T, Bensalah MZ, Bollache E, et al. Differential impact of local and regional aortic stiffness on left ventricular remodeling. J Hypertens. 2018;36(3):552–559. | ||
Sabit R, Bolton CE, Edwards PH, et al. Arterial stiffness and osteoporosis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(12):1259–1265. | ||
Vanfleteren LE, Spruit MA, Groenen MT, et al. Arterial stiffness in patients with COPD: the role of systemic inflammation and the effects of pulmonary rehabilitation. Eur Respir J. 2014;43(5):1306–1315. | ||
Bhatt SP, Cole AG, Wells JM, et al. Determinants of arterial stiffness in COPD. BMC Pulm Med. 2014;14:1. | ||
Rodrigues JC, Amadu AM, Dastidar AG, et al. Comprehensive characterisation of hypertensive heart disease left ventricular phenotypes. Heart. 2016;102(20):1671–1679. | ||
Anderson WJ, Lipworth BJ, Rekhraj S, Struthers AD, George J. Left ventricular hypertrophy in COPD without hypoxemia: the elephant in the room? Chest. 2013;143(1):91–97. | ||
Smith BM, Kawut SM, Bluemke DA, et al. Pulmonary hyperinflation and left ventricular mass: the Multi-Ethnic Study of Atherosclerosis COPD Study. Circulation. 2013;127(14):1503–1511, 1511e1–e6. | ||
Short PM, Anderson WJ, Elder DHJ, Struthers AD, Lipworth BJ. Impact of left ventricular hypertrophy on survival in chronic obstructive pulmonary disease. Lung. 2015;193(4):487–495. | ||
Brandts A, van Elderen SG, Westenberg JJ, et al. Association of aortic arch pulse wave velocity with left ventricular mass and lacunar brain infarcts in hypertensive patients: assessment with MR imaging. Radiology. 2009;253(3):681–688. | ||
Schillaci G, Mannarino MR, Pucci G, et al. Age-specific relationship of aortic pulse wave velocity with left ventricular geometry and function in hypertension. Hypertension. 2007;49(2):317–321. | ||
Bluemke DA, Kronmal RA, Lima JA, et al. The relationship of left ventricular mass and geometry to incident cardiovascular events: the MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;52(25):2148–2155. | ||
Short PM, Lipworth SI, Elder DHJ, Schembri S, Lipworth BJ. Effect of beta blockers in treatment of chronic obstructive pulmonary disease: a retrospective cohort study. BMJ. 2011;342:d2549. | ||
Bhatt SP, Wells JM, Kinney GL, et al. β-Blockers are associated with a reduction in COPD exacerbations. Thorax. 2016;71(1):8–14. | ||
Kopeć G, Moertl D, Jankowski P, Tyrka A, Sobień B, Podolec P. Pulmonary artery pulse wave velocity in idiopathic pulmonary arterial hypertension. Can J Cardiol. 2013;29(6):683–690. | ||
Kawut SM, Poor HD, Parikh MA, et al. Cor pulmonale parvus in chronic obstructive pulmonary disease and emphysema: the MESA COPD study. J Am Coll Cardiol. 2014;64(19):2000–2009. | ||
Hilde JM, Skjørten I, Hansteen V, et al. Haemodynamic responses to exercise in patients with COPD. Eur Respir J. 2013;41(5):1031–1041. | ||
Jensen MT, Marott JL, Lange P, et al. Resting heart rate is a predictor of mortality in COPD. Eur Respir J. 2013;42(2):341–349. | ||
Whelton SP, Blankstein R, Al-Mallah MH, et al. Association of resting heart rate with carotid and aortic arterial stiffness: multi-ethnic study of atherosclerosis. Hypertension. 2013;62(3):477–484. | ||
Warnier MJ, Rutten FH, de Boer A, Hoes AW, De Bruin ML. Resting heart rate is a risk factor for mortality in chronic obstructive pulmonary disease, but not for exacerbations or pneumonia. PLoS One. 2014;9(8):e105152. | ||
Mäki-Petäjä KM, Barrett SML, Evans SV, Cheriyan J, McEniery CM, Wilkinson IB. The role of the autonomic nervous system in the regulation of aortic stiffness. Hypertension. 2016;68(5):1290–1297. | ||
van Gestel AJ, Steier J. Autonomic dysfunction in patients with chronic obstructive pulmonary disease (COPD). J Thorac Dis. 2010;2(4):215–222. | ||
Hulo S, Inamo J, Dehon A, Le Rouzic O, Edme JL, Neviere R. Chronotropic incompetence can limit exercise tolerance in COPD patients with lung hyperinflation. Int J COPD. 2016;11(1):2553–2561. | ||
Vulliémoz S, Stergiopulos N, Meuli R. Estimation of local aortic elastic properties with MRI. Magn Reson Med. 2002;47(4):649–654. |
Supplementary materials
 | Table S1 Intra-assay and interassay CoV for the biomarkers |
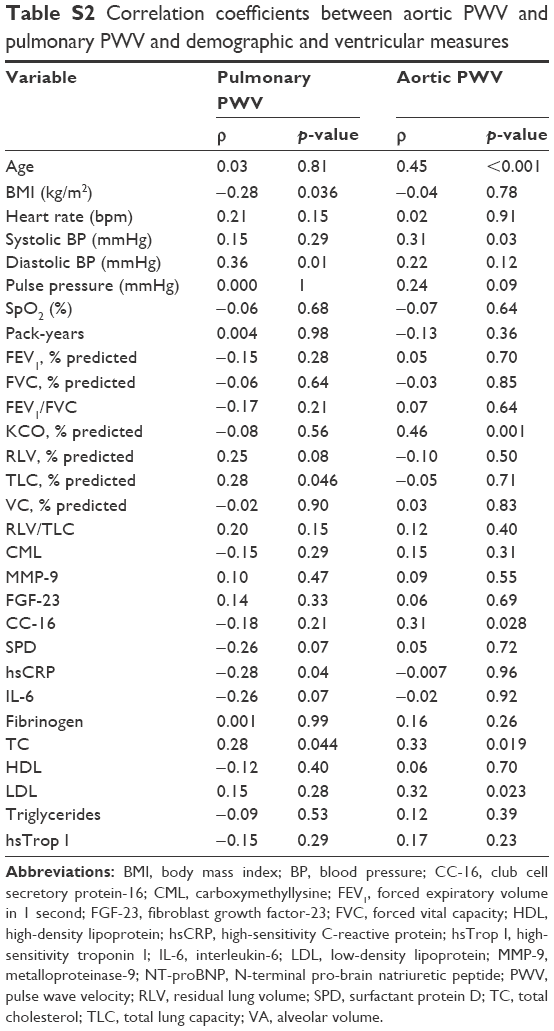 | Table S2 Correlation coefficients between aortic PWV and pulmonary PWV and demographic and ventricular measures |
 © 2018 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.
© 2018 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.