Back to Journals » Drug Design, Development and Therapy » Volume 20
Discharge Readiness After Propofol with or without Esketamine for Flexible Bronchoscopy: A Randomized Non-Inferiority Trial
Authors Zhang X, Chen L, Chen Z, Yang J ![]() , Zheng T, Lin W, Guan J, Yao Y
, Zheng T, Lin W, Guan J, Yao Y ![]()
Received 26 March 2026
Accepted for publication 15 June 2026
Published 22 June 2026 Volume 2026:20 612153
DOI https://doi.org/10.2147/DDDT.S612153
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Solomon Tadesse Zeleke
Xiaohong Zhang,1,* Lei Chen,2,* Zaizhi Chen,3,* Jiahan Yang,1 Ting Zheng,1 Wenjun Lin,1 Jinsheng Guan,4 Yusheng Yao1
1Department of Anesthesiology, Shengli Clinical Medical College of Fujian Medical University, Fujian Provincial Hospital, Fuzhou, People’s Republic of China; 2Department of Anesthesiology, People’s Hospital Affiliated to Fujian University of Traditional Chinese Medicine, Fuzhou, People’s Republic of China; 3Department of Anesthesiology, The Third Hospital of Xiamen, Xiamen, People’s Republic of China; 4Department of Anesthesiology, Fujian Maternity and Child Health Hospital, College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fuzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jinsheng Guan, Department of Anesthesiology, Fujian Maternity and Child Health Hospital, College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, No. 18 Daoshan Road, Fuzhou, 350001, People’s Republic of China, Email [email protected] Yusheng Yao, Department of Anesthesiology, Shengli Clinical Medical College of Fujian Medical University, Fujian Provincial Hospital, 134 Dongjie Street, Fuzhou, 350001, People’s Republic of China, Email [email protected]
Purpose: To determine whether low-dose esketamine combined with propofol is non-inferior to propofol alone for discharge readiness after outpatient flexible bronchoscopy.
Patients and Methods: In this single-center, randomized, double-blind, non-inferiority trial, 246 adults aged 18 to 75 years with ASA physical status I to III scheduled for elective outpatient flexible bronchoscopy were randomly assigned (1:1:1) to propofol alone (propofol plus 0.9% saline; n = 82), propofol plus esketamine 0.1 mg/kg (n = 82), or propofol plus esketamine 0.2 mg/kg (n = 82). The primary endpoint was the proportion achieving discharge readiness (Modified Post-Anesthetic Discharge Scoring System [MPADSS] score ≥ 9) within 30 minutes of procedure completion; the non-inferiority margin was − 18 percentage points. Secondary endpoints included propofol consumption, intraoperative cough severity, adverse events, and patient-reported recovery quality.
Results: Both esketamine doses were non-inferior to propofol alone for discharge readiness: the risk difference was − 3.0 percentage points (95% CI, − 13.8 to 7.8; Pnon-inferiority = 0.003) at 0.1 mg/kg and − 5.1 percentage points (95% CI, − 16.2 to 6.1; Pnon-inferiority = 0.012) at 0.2 mg/kg. In exploratory secondary analyses, both doses reduced propofol consumption (by 20% and 28%; both P < 0.001) and intraoperative cough severity (P = 0.005 and P = 0.002). Hypotension occurred in 42 (51%), 24 (29%), and 10 (12%) participants in the propofol-alone, propofol + esketamine 0.1, and propofol + esketamine 0.2 groups, respectively, and hypoxemia in 17 (21%), 10 (12%), and 3 (4%). Recovery quality was comparable across the three groups.
Conclusion: Adding low-dose esketamine to propofol maintained timely discharge readiness during outpatient flexible bronchoscopy. Among the secondary outcomes, esketamine at 0.2 mg/kg was associated with lower rates of hypotension and hypoxemia, although these exploratory findings warrant confirmation.
Registration: ClinicalTrials.gov Identifier: NCT05643066. A trial examined esketamine′s effect on propofol sedation for discharge readiness post-bronchoscopy. Participants: 246 adults, aged 18-75, with ASA status I-III, at Fujian Provincial Hospital, China. Groups: 82 received propofol alone, 82 received propofol plus 0.1 mg/kg esketamine and 82 received propofol plus 0.2 mg/kg esketamine. Goal: Achieve a sedation score below 3. Primary outcome: Discharge readiness within 30 minutes, scoring 9+ on the Postanesthetic Discharge Scoring System. Results: Low-dose esketamine with propofol ensures quick recovery and fewer cardiorespiratory issues. Propofol plus 0.1 mg/kg esketamine vs. propofol alone showed a -3.0% difference (95% CI: -13.8% to 7.8%), P=0.003. Propofol plus 0.2 mg/kg esketamine vs. propofol alone showed a -5.1% difference (95% CI: -16.2% to 6.1%), P=0.012.Infographic on esketamine and propofol sedation trial for bronchoscopy discharge readiness.
Keywords: bronchoscopy, discharge readiness, esketamine, non-inferiority, procedural sedation, propofol
Introduction
Lung cancer accounts for approximately 1.8 million deaths annually, while chronic respiratory diseases rank as the third leading cause of death worldwide.1 Flexible bronchoscopy remains the principal diagnostic and therapeutic tool for these conditions, enabling airway inspection, tissue sampling, and staging assessment.2 However, topical anesthesia alone often causes discomfort and anxiety that may compromise examination quality and diagnostic yield.3 Procedural sedation is therefore standard practice during flexible bronchoscopy.
Propofol is the most widely used sedative for flexible bronchoscopy owing to its rapid onset and short duration of action, which facilitates efficient patient throughput in outpatient settings.4,5 Nevertheless, it carries well-documented adverse effects, including injection-site pain, respiratory depression, and dose-dependent hypotension.6,7 To mitigate these effects, clinicians have sought adjunctive agents that reduce propofol dose requirements without compromising sedation quality.8–10
Esketamine, the S-enantiomer of ketamine, is an attractive adjunct to propofol in this context. It provides approximately twice the analgesic potency of racemic ketamine with fewer psychomimetic effects,11,12 while preserving respiratory drive, producing bronchodilation, and maintaining cardiovascular stability — properties that directly counteract the principal hemodynamic and respiratory limitations of propofol.13,14 In bidirectional gastrointestinal endoscopy, esketamine added to propofol reduced hypotension and desaturation,15 and a previous bronchoscopy trial reported lower transient hypoxemia with propofol–esketamine than with propofol–remifentanil.16 However, neither study used an inactive-adjunct comparator, assessed discharge readiness, or compared multiple esketamine doses. The effect of esketamine on discharge readiness — an operational priority and key quality metric in outpatient settings — therefore remains undefined.
We hypothesized that combining low-dose esketamine with propofol would not delay discharge readiness compared with propofol alone. We further explored whether esketamine would reduce adverse events, including hemodynamic instability and respiratory depression. To test this hypothesis, we conducted a randomized, double-blind, non-inferiority trial comparing propofol plus normal saline with propofol plus esketamine at 0.1 or 0.2 mg/kg. The primary endpoint was the proportion of participants achieving discharge readiness within 30 minutes of procedure completion, defined as a Modified Post-Anesthetic Discharge Scoring System (MPADSS) score ≥ 9.17 Secondary endpoints included propofol consumption, intraoperative cough severity, adverse events, and patient-reported recovery quality.
Materials and Methods
Study Design and Ethics
We conducted a single-center, randomized, double-blind, non-inferiority trial at Fujian Provincial Hospital (Fuzhou, People’s Republic of China) between December 7, 2022, and December 18, 2023. The Institutional Review Board of Fujian Provincial Hospital approved the study protocol (approval number K2021-12-067) on May 10, 2022, and the trial was prospectively registered at ClinicalTrials.gov (NCT05643066; https://clinicaltrials.gov/study/NCT05643066) before enrollment commenced. All participants provided written informed consent after a detailed explanation of the study procedures. The trial was conducted in accordance with the Declaration of Helsinki and reported in accordance with the CONSORT extension for non-inferiority trials.18 No protocol modifications were made after trial commencement.
Patient Selection
Eligible participants were adults aged 18 to 75 years with an American Society of Anesthesiologists (ASA) physical status of I to III and who were scheduled for elective outpatient flexible bronchoscopy. Exclusion criteria were hypersensitivity to any study medication, obstructive sleep apnea–hypopnea syndrome, psychiatric or neurological disease, cognitive impairment, pregnancy, substance abuse, current use of centrally acting medications, and inability to provide informed consent or to complete the study assessments owing to a language barrier or illiteracy.
Randomization and Allocation
A total of 246 participants were randomly assigned in a 1:1:1 ratio to receive propofol plus 0.9% saline (propofol-alone group), propofol plus esketamine at 0.1 mg/kg (propofol + esketamine 0.1 group), or propofol plus esketamine at 0.2 mg/kg (propofol + esketamine 0.2 group). An independent statistician generated the randomization sequence using permuted blocks of 6 and 9 with R software, version 4.0.5 (R Foundation for Statistical Computing, Vienna, Austria); block sizes were concealed from all enrolling investigators. Allocation concealment was achieved using sequentially numbered, opaque, sealed envelopes that were opened only after participant enrollment. An unblinded pharmacist prepared visually identical 10-mL syringes daily, containing esketamine at 1 mg/mL (propofol + esketamine 0.1 group), esketamine at 2 mg/mL (propofol + esketamine 0.2 group), or 0.9% saline (propofol-alone group). All participants, bronchoscopists, anesthesiologists, nurses, and outcome assessors remained blinded throughout the study.
Study Procedures
Participants fasted from solid foods for ≥ 6 hours and from clear liquids for ≥ 2 hours before the procedure. Standard monitoring included continuous electrocardiography, noninvasive blood pressure measurement, pulse oximetry, and continuous capnography. Topical anesthesia was administered according to a standardized protocol consisting of nebulized 2% lidocaine (10 mL) delivered over 15 minutes, followed by intranasal application of 2% lidocaine gel (3 mL). During bronchoscopy, supplemental 1% lidocaine was instilled in 2-mL aliquots through the working channel at key anatomical landmarks; the total lidocaine dose was limited to < 7 mg/kg to avoid systemic toxicity.19
After baseline measurements were obtained, the attending anesthesiologist administered the assigned study solution (0.1 mL/kg) intravenously over 30 seconds, followed immediately by propofol (1 mg/kg) over 30 seconds. Once the participant achieved adequate sedation — defined as a Modified Observer’s Assessment of Alertness/Sedation (MOAA/S) score of 2 or lower20 — a nasopharyngeal airway was inserted and oxygen was delivered at 3 L·min–1 (100% FiO2). The bronchoscopist performed the examination through the contralateral nostril (Figure S1).
If a participant required additional sedation after 2 minutes (MOAA/S score ≥ 3), rescue propofol (0.5 mg/kg) was administered at intervals of ≥ 1 minute until adequate sedation was re-established. Spontaneous ventilation was maintained throughout, with continuous hemodynamic monitoring. Following the procedure, participants recovered under direct observation. Discharge readiness was assessed at 5-minute intervals beginning when the participant achieved full alertness (MOAA/S score of 5). Discharge was permitted when the MPADSS score reached 9 or higher.17
Study Outcomes
The primary endpoint was the proportion of participants who achieved discharge readiness within 30 minutes of procedure completion, defined as an MPADSS score ≥ 9 on a 10-point scale assessing five domains: vital signs, ambulation, nausea and vomiting, pain, and surgical bleeding.17 Discharge readiness was assessed by a blinded nurse at 5-minute intervals from the time of full alertness (MOAA/S score of 5) until discharge criteria were met.
Secondary endpoints comprised recovery quality, procedural efficiency, patient experience, and safety. Recovery quality was assessed using the Postoperative Quality of Recovery Scale (PostopQRS) at baseline and at 30 minutes, 24 hours, and 72 hours after the procedure.21 This instrument evaluates five domains: physiological, nociceptive, emotive, activities of daily living, and cognitive. Recovery at each time point was defined as a score meeting or exceeding the baseline value in the corresponding domain. PostopQRS assessments at baseline and 30 minutes were performed in person by a blinded assessor, and the 24-hour and 72-hour assessments were conducted by telephone by the same assessor using a standardized interview script to ensure consistency.
Procedural efficiency endpoints included induction time (defined as the interval from drug administration to a MOAA/S score ≤ 2), total propofol consumption, and emergence time (defined as the interval from bronchoscope withdrawal to a MOAA/S score of 5). Injection-site pain was assessed immediately after study drug administration with a numerical rating scale (0–10). Patient and bronchoscopist satisfaction were evaluated immediately after the procedure with 5-point Likert scales (1 = very dissatisfied to 5 = very satisfied).22 At 24 hours, participants were asked to report their willingness to undergo repeat bronchoscopy and their likelihood of recommending the procedure to others (response options: yes, no, or unsure).
Adverse events were defined a priori and categorized as follows: cardiovascular disturbances (heart rate < 50 or > 100 beats·min–1; mean arterial pressure < 65 mmHg or a decrease of > 30% from baseline; systolic blood pressure > 140 mmHg or an increase of > 30% from baseline), respiratory events (oxygen saturation < 90% despite supplemental oxygen), gastrointestinal events (nausea and vomiting), and neuropsychiatric effects (nightmares or visual disturbances).
Sample Size Calculation
The sample size was calculated to demonstrate non-inferiority of esketamine-supplemented propofol sedation relative to propofol alone for the primary endpoint. A retrospective audit at our institution showed that 88% of patients achieved discharge readiness within 30 minutes of procedure completion when sedated with propofol alone; we therefore set a non-inferiority margin of −18 percentage points, corresponding to a minimum acceptable rate of 70% in the esketamine groups. This threshold was considered the lowest proportion compatible with maintaining acceptable outpatient throughput and is consistent with the margin adopted for the same discharge-readiness endpoint in a previous randomized non-inferiority trial of ambulatory sedation.23 To control the familywise type I error rate across the two pairwise comparisons, we used a prespecified fixed-sequence testing strategy: the propofol + esketamine 0.1 mg/kg group was compared with the propofol-alone group first (one-sided α = 0.025), and the propofol + esketamine 0.2 mg/kg group was tested only if non-inferiority was established for the lower dose. Assuming a discharge readiness rate of 88% in both arms of each comparison, 90% power, a one-sided α of 0.025, and an anticipated 15% attrition rate, the required sample size was 82 participants per group (246 in total). The calculation was performed using PASS software (version 2021; NCSS, LLC, Kaysville, UT, USA).
Statistical Analysis
All randomized participants (n = 246) were analyzed according to their originally allocated groups (intention-to-treat principle). Missing primary outcome data were addressed using multiple imputation with chained equations, generating 20 imputed datasets combined using Rubin’s rules.24 Per-protocol sensitivity analyses (n = 232) were conducted to assess the robustness of the primary findings. For secondary outcomes with missing data (< 6%), missing values were imputed using group-specific means for continuous variables and within-group modal values for categorical variables. Complete-case analyses, which included only participants with fully observed data, were performed as sensitivity analyses to verify the robustness of the imputed results.
For the primary endpoint, risk differences with two-sided 95% confidence intervals (CIs) were calculated using the Farrington–Manning method; non-inferiority was declared for each comparison if the lower bound of the 95% CI exceeded −18 percentage points. Time to discharge readiness was additionally analyzed using Kaplan–Meier estimates and Cox proportional hazards models; hazard ratios with 95% CIs were reported as the measure of effect.
Secondary outcomes were tested for superiority (two-sided; P < 0.05). Longitudinal recovery data from the PostopQRS were analyzed using a generalized linear mixed model with a binomial distribution and a logit link function, with all domain outcomes dichotomized as “recovered” (postoperative score meeting or exceeding the baseline value) or “not recovered.” Group allocation, time (modeled as a categorical variable), and the group-by-time interaction were included as fixed effects, and a random intercept was specified for each participant to account for within-participant correlation across repeated measures. The group-by-time interaction was the prespecified test for between-group differences in recovery trajectories; where a significant interaction was detected, pairwise comparisons at each time point were performed with Bonferroni correction. Exploratory subgroup analyses examined potential effect modification by sex, age, ASA physical status, and procedure indication; results are presented as forest plots with interaction P values. All P values for secondary and exploratory outcomes are descriptive and were not adjusted for multiple comparisons. All analyses were performed by a statistician blinded to group allocation using R software, version 4.3.1 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Between December 7, 2022, and December 18, 2023, a total of 265 individuals were screened for eligibility, of whom 246 were randomly assigned to the propofol-alone group (n = 82), the propofol + esketamine 0.1 group (n = 82), or the propofol + esketamine 0.2 group (n = 82) (Figure 1). Baseline demographic and clinical characteristics were well balanced across the three groups (Table 1). Of the 246 randomized participants, 9 did not receive their assigned intervention (8 because of procedure cancellation and 1 because of withdrawal of consent); 5 additional participants were subsequently lost to follow-up, yielding a per-protocol population of 232 (94.3%) (Table S1).
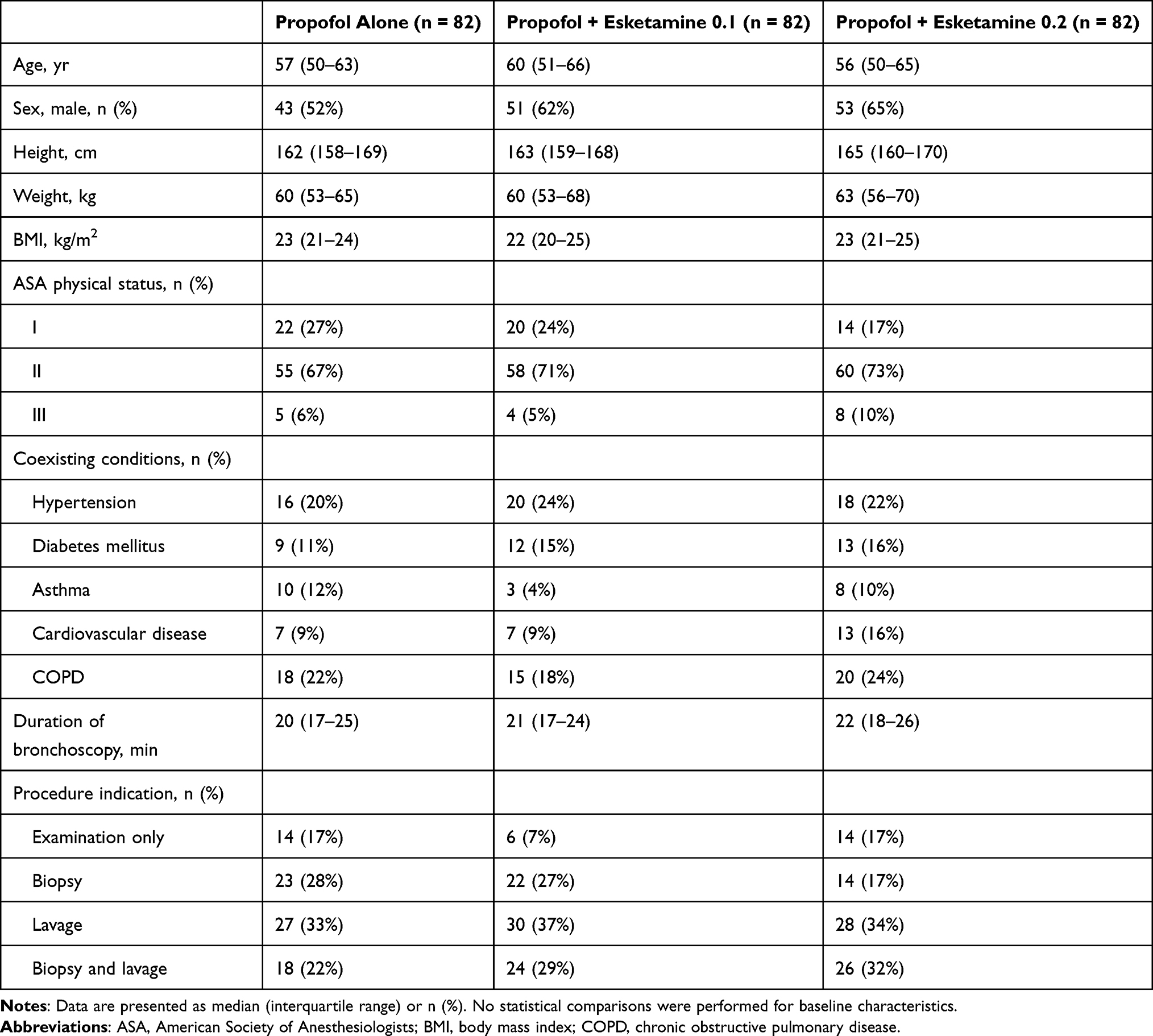 |
Table 1 Baseline Demographic and Clinical Characteristics |
 |
Figure 1 CONSORT flow diagram showing participant enrollment, allocation, follow-up, and analysis. |
At 30 minutes, discharge readiness (MPADSS score ≥ 9) met the prespecified non-inferiority criterion for both esketamine doses compared with the propofol-alone group (Figure 2). In the intention-to-treat population, the risk difference compared with the propofol-alone group was −3.0 percentage points (95% CI, −13.8 to 7.8; Pnon-inferiority = 0.003) for the propofol + esketamine 0.1 group and −5.1 percentage points (95% CI, −16.2 to 6.1; Pnon-inferiority = 0.012) for the propofol + esketamine 0.2 group. All 232 per-protocol participants met discharge criteria within 40 minutes. Per-protocol results were consistent: the risk difference was −2.8 percentage points (95% CI, −14.0 to 8.2; Pnon-inferiority = 0.005) for the propofol + esketamine 0.1 group and −4.8 percentage points (95% CI, −16.1 to 6.4; Pnon-inferiority = 0.011) for the propofol + esketamine 0.2 group. In both the ITT and the per-protocol analyses, the lower bounds of the 95% CIs for all risk differences remained above the prespecified −18 percentage point non-inferiority margin.
 |
Figure 2 Non-inferiority analysis of discharge readiness within 30 minutes of procedure completion. Notes: Forest plots depict risk differences (squares) with 95% CIs (horizontal lines) for the proportion of participants achieving discharge readiness (MPADSS score ≥ 9). (A) Intention-to-treat population (n = 246). (B) Per-protocol population (n = 232). In both analyses, the lower bounds of the 95% CIs exceeded the prespecified non-inferiority margin of −18 percentage points, confirming non-inferiority for both esketamine doses relative to the propofol-alone group. |
Kaplan–Meier analysis demonstrated comparable time to discharge readiness across the three groups (Figure 3), with Cox hazard ratios of 0.93 (95% CI, 0.69–1.27; P = 0.685) for the propofol + esketamine 0.1 group and 0.84 (95% CI, 0.62–1.14; P = 0.117) for the propofol + esketamine 0.2 group versus the propofol-alone group. Exploratory subgroup analyses showed no evidence of effect modification by sex, age, ASA physical status, or procedure indication (all Pinteraction > 0.05) (Figure 4).
 |
Figure 3 Kaplan–Meier analysis of time to discharge readiness. Notes: Cumulative probability of achieving discharge readiness (MPADSS score ≥ 9) in the intention-to-treat population (n = 246). Cox hazard ratios with 95% CIs and log-rank P values are presented for each esketamine dose versus the propofol-alone group. The numbers at risk are displayed below the curves. |
 |
Figure 4 Subgroup analysis of discharge readiness within 30 minutes of procedure completion. Notes: Forest plots depict relative risks with 95% CIs for discharge readiness stratified by sex, age category, American Society of Anesthesiologists physical status, and procedure indication. (A) Propofol + esketamine 0.1 mg/kg versus propofol alone; (B) Propofol + esketamine 0.2 mg/kg versus propofol alone. Pinteraction values test for effect modification across subgroups. |
Generalized linear mixed model analysis demonstrated that recovery rates across all PostopQRS domains improved progressively over the 72-hour assessment period in all three groups. There were no significant group-by-time interactions for either the propofol + esketamine 0.1 group (Pinteraction = 0.835) or the propofol + esketamine 0.2 group (Pinteraction = 0.837) relative to the propofol-alone group, indicating comparable recovery trajectories across the treatment arms (Figure S2; Tables S2 and S3).
Total propofol consumption was significantly lower in both esketamine groups than in the propofol-alone group: 200 [160–240] mg (propofol + esketamine 0.1) and 180 [150–210] mg (propofol + esketamine 0.2) versus 250 [215–300] mg (propofol alone) (both P < 0.001), representing relative reductions of 20% and 28%, respectively (Table 2). Induction time was shorter with both esketamine doses than in the propofol-alone group (P = 0.021 and P = 0.009, respectively), and injection-site pain scores were lower with both esketamine doses (P = 0.006 and P < 0.001, respectively). Intraoperative cough severity was also reduced in both esketamine groups (P = 0.005 and P = 0.002 versus the propofol-alone group, respectively). However, severe coughing (grade 3) did not differ significantly across groups, occurring in 8 (10%), 4 (5%), and 3 (4%) participants in the propofol-alone, propofol + esketamine 0.1, and propofol + esketamine 0.2 groups, respectively (P = 0.230 and P = 0.119 versus the propofol-alone group). Patient satisfaction was significantly higher in the propofol + esketamine 0.2 group than in the propofol-alone group (P = 0.014). No significant differences were observed in emergence time, bronchoscopist satisfaction, or willingness to undergo repeat bronchoscopy among the three groups.
 |
Table 2 Secondary Outcomes and Adverse Events |
Hypotension occurred in 42 (51%), 24 (29%), and 10 (12%) participants in the propofol-alone, propofol + esketamine 0.1, and propofol + esketamine 0.2 groups, respectively (P = 0.007 and P < 0.001 versus the propofol-alone group) (Table 2). The incidence of hypoxemia was significantly lower in the propofol + esketamine 0.2 group than in the propofol-alone group: 3 (4%) versus 17 (21%) (P = 0.002); the propofol + esketamine 0.1 group showed an intermediate incidence of 10 (12%) (P = 0.206 versus the propofol-alone group). The rates of hypertension, bradycardia, tachycardia, postoperative nausea and vomiting, and neuropsychiatric effects did not differ significantly between groups. Complete-case analyses of secondary outcomes yielded consistent effect estimates (Table S4).
Discussion
In this randomized non-inferiority trial, supplementing propofol with low-dose esketamine preserved timely discharge readiness and was associated with favorable intraprocedural cardiorespiratory stability, indicating that the analgesic and sympathomimetic benefits of esketamine can be obtained without the recovery delay historically associated with racemic ketamine.
The 30-minute discharge-readiness threshold was selected a priori as an operational benchmark reflecting recovery-bay capacity rather than a physiological recovery boundary. That all per-protocol participants achieved readiness within 40 minutes, and that the time-to-readiness curves largely overlapped, indicate that esketamine did not meaningfully delay recovery and support the interpretation of preserved operational efficiency.
Our results contrast with those of Singh et al,25 who reported longer discharge times with ketamine–propofol than with propofol alone during endoscopic ultrasonography (46 versus 33 minutes). This discrepancy likely reflects methodological differences: Singh et al used racemic ketamine at a substantially higher dose (0.5 mg/kg), which has lower NMDA receptor selectivity and more pronounced psychomimetic and sedative effects that delay recovery.26 Esketamine has approximately twice the NMDA receptor affinity of the racemate,27 allowing the lower doses used in our study (0.1 and 0.2 mg/kg) to provide effective analgesia while minimizing recovery-delaying effects.28 This pharmacological profile supports the suitability of esketamine as an adjunct for time-sensitive outpatient procedures.
These findings extend previous research on esketamine as a procedural sedation adjunct.29 Song et al15 reported improved hemodynamic and respiratory profiles with esketamine–propofol during bidirectional endoscopy. Our results corroborated these observations and suggested dose-related differences: esketamine at 0.2 mg/kg was associated with greater cardiovascular stability than the 0.1 mg/kg dose, without a higher incidence of tachycardia, hypertension, postoperative nausea and vomiting, or neuropsychiatric effects. These associations may reflect the sympathomimetic and bronchodilatory properties of esketamine, which counteract propofol-induced vasodilation, negative inotropy, and respiratory depression.30 Because these dose-related differences derived from exploratory analyses that were not powered for formal comparison or adjusted for multiplicity, they should be regarded as hypothesis-generating.
Recovery quality, including cognitive function, was equivalent across the three groups, allaying concerns about the potential psychoactive effects of esketamine at these doses.31 The reduction in injection-site pain is consistent with previous findings, with esketamine at 0.2 mg/kg demonstrating greater analgesic efficacy than the 0.1 mg/kg dose. This benefit likely arises from both peripheral and central mechanisms: NMDA receptor antagonism and local anesthetic-like effects attenuate nociceptor activation at the venous endothelium, while enhancement of descending inhibitory pathways raises pain thresholds.32,33
Both esketamine doses reduced intraoperative cough severity compared with the propofol-alone group. This antitussive effect is mechanistically plausible: NMDA receptors on airway sensory neurons mediate central cough reflex processing in the nucleus tractus solitarius,34 and antagonism of these receptors by esketamine, combined with its direct bronchodilatory effects,35 likely attenuates both afferent signaling and the motor cough response. The absence of a clear dose–response pattern may reflect an antitussive ceiling effect at 0.1 mg/kg, beyond which additional receptor blockade confers limited incremental benefit in the context of standardized lidocaine topicalization. Reduced coughing may in turn have contributed to the favorable hemodynamic stability observed in the esketamine groups, as vigorous coughing is a recognized trigger of transient hypertension and desaturation during flexible bronchoscopy. Although randomization produced a balanced distribution of procedure types across groups, which limits confounding, the influence of procedure type on the secondary outcomes cannot be fully excluded and warrants dedicated investigation.
This study has three principal implications for procedural sedation practice. First, low-dose esketamine preserved discharge readiness and operational efficiency without prolonging recovery, supporting its further evaluation for outpatient flexible bronchoscopy sedation. Second, the reduction in intraoperative cough severity is consistent with the recognized antitussive properties of NMDA-receptor antagonists and supports a role for esketamine in modulating airway reflexes during airway instrumentation. Third, the observed dose-related differences in hemodynamic and respiratory outcomes may inform individualized optimization of sedation regimens. However, P values for secondary outcomes were not adjusted for multiple comparisons, and all findings beyond the primary endpoint of discharge readiness should therefore be regarded as hypothesis-generating rather than confirmatory.
Several limitations warrant consideration. First, the single-center design and the inclusion of predominantly low-risk to moderate-risk patients (ASA physical status I–III) limit generalizability to higher-risk populations and to more complex bronchoscopic interventions; enrollment was confined to standard diagnostic flexible bronchoscopy and did not include endobronchial ultrasound–guided or other advanced interventional procedures, in which sedation requirements and airway stimulation may differ. Second, several higher-risk groups were excluded, including patients with obstructive sleep apnea–hypopnea syndrome, significant neurological disease, or ASA physical status of IV or higher. This is particularly relevant because the respiratory-sparing and hemodynamic-stabilizing properties of esketamine may be of greatest benefit in these groups; our findings therefore cannot be extrapolated to them, and dedicated trials in higher-risk populations are warranted. Third, the sample size was calculated for the primary endpoint of discharge readiness, potentially leaving the study underpowered for some secondary outcomes; the safety and cough findings, although clinically coherent, require confirmation in larger, adequately powered trials. Fourth, we did not measure plasma esketamine concentrations, which would have enabled characterization of the pharmacokinetic–pharmacodynamic relationship between drug exposure and clinical effects. Fifth, the 72-hour follow-up may have been insufficient to capture delayed neurocognitive effects, although none have been reported with similar low-dose regimens in previous studies.
Future research should examine esketamine–propofol combinations during more complex procedures, such as endobronchial ultrasound–guided transbronchial needle aspiration, in which sedation requirements and airway stimulation differ from those of standard diagnostic bronchoscopy. Head-to-head trials comparing esketamine with other adjuncts such as remifentanil or dexmedetomidine would further clarify the position of esketamine in the procedural sedation armamentarium. Such studies should be adequately powered for safety endpoints and incorporate longer-term neurocognitive follow-up to address the limitations identified in the present trial.
Conclusion
In this randomized, double-blind, non-inferiority trial, adding low-dose esketamine to propofol maintained rapid discharge readiness and did not compromise operational efficiency during outpatient flexible bronchoscopy. Among the secondary outcomes, both esketamine doses were associated with lower propofol consumption and lower intraoperative cough severity, and esketamine at 0.2 mg/kg was associated with lower rates of hypotension and hypoxemia. Because these secondary analyses were exploratory and unadjusted for multiplicity, these observations should be regarded as hypothesis-generating. These findings support further evaluation of low-dose esketamine as an adjunct to propofol-based sedation for outpatient flexible bronchoscopy, with confirmation of the secondary outcomes in adequately powered trials.
Data Sharing Statement
De-identified individual participant data underlying the results reported in this article will be made available upon reasonable request to the corresponding authors, beginning 3 months and ending 3 years after publication.
Acknowledgments
The authors gratefully acknowledge the contributions of Dr. Guiyang Lin and Dr. Changjian Lin from the Bronchoscopy Center, Fujian Provincial Hospital, for their clinical support and cooperation during the conduct of this study.
Author Contributions
All authors made substantial contributions to the conception and design, data acquisition, or data analysis and interpretation; participated in drafting the manuscript or revising it critically for important intellectual content; approved the final version for publication; agreed to submit to the current journal; and accept accountability for all aspects of the work.
Funding
This study was supported by the Joint Funds for the Innovation of Science and Technology, Fujian Province (grant number 2023Y9275), and the Natural Science Foundation of Fujian Province (grant number 2025J01071). The funders had no role in the study design, data collection, analysis, interpretation, manuscript preparation, or the decision to submit for publication.
Disclosure
The authors declare no conflicts of interest.
References
1. Wang L, Wang M, Xu R, et al. Comparative analysis of guidelines and recommendations for sedation during flexible bronchoscopy: a narrative review. Eur Respir Rev. 2025;34(177):240045. doi:10.1183/16000617.0045-2025
2. American Thoracic Society. Flexible bronchoscopy. Am J Respir Crit Care Med. 2015;191:P7–13. doi:10.1164/rccm.1919P7
3. Magazine R, Sisupalan KN, Surendra VU, et al. Effect of bronchoscopist-directed sedation and other factors on patient comfort during diagnostic flexible bronchoscopy. Scientifica. 2022;2022:8643844. doi:10.1155/2022/8643844
4. José RJ, Shaefi S, Navani N. Sedation for flexible bronchoscopy: current and emerging evidence. Eur Respir Rev. 2013;22:106–116. doi:10.1183/09059180.00006412
5. Grendelmeier P, Tamm M, Pflimlin E, et al. Propofol sedation for flexible bronchoscopy: a randomised, noninferiority trial. Eur Respir J. 2014;43:591–601. doi:10.1183/09031936.00200412
6. Hemphill S, McMenamin L, Bellamy MC, et al. Propofol infusion syndrome: a structured literature review and analysis of published case reports. Br J Anaesth. 2019;122:448–459. doi:10.1016/j.bja.2018.12.025
7. Sahinovic MM, Struys MMRF, Absalom AR. Clinical pharmacokinetics and pharmacodynamics of propofol. Clin Pharmacokinet. 2018;57:1539–1558. doi:10.1007/s40262-018-0672-3
8. Heybati K, Zhou F, Ali S, et al. Outcomes of dexmedetomidine versus propofol sedation in critically ill adults requiring mechanical ventilation: a systematic review and meta-analysis of randomised controlled trials. Br J Anaesth. 2022;129:515–526. doi:10.1016/j.bja.2022.06.020
9. Li J, Liu Y, Chen S, et al. Pharmacological agents for procedural sedation and analgesia in patients undergoing gastrointestinal endoscopy: a systematic review and network meta-analysis. EClinicalMedicine. 2025;85:103307. doi:10.1016/j.eclinm.2025.103307
10. Ghojazadeh M, Sanaie S, Paknezhad SP, et al. Using ketamine and propofol for procedural sedation of adults in the emergency department: a systematic review and meta-analysis. Adv Pharm Bull. 2019;9:5–11. doi:10.15171/apb.2019.002
11. Mion G, Himmelseher S. Esketamine: less drowsiness, more analgesia. Anesth Analg. 2024;139:78–91. doi:10.1213/ANE.0000000000006851
12. Jelen LA, Young AH, Stone JM. Ketamine: a tale of two enantiomers. J Psychopharmacol. 2021;35:109–123. doi:10.1177/0269881120959644
13. Eikermann M, Grosse-Sundrup M, Zaremba S, et al. Ketamine activates breathing and abolishes the coupling between loss of consciousness and upper airway dilator muscle dysfunction. Anesthesiology. 2012;116:35–46. doi:10.1097/ALN.0b013e31823d010a
14. Jonkman K, van Rijnsoever E, Olofsen E, et al. Esketamine counters opioid-induced respiratory depression. Br J Anaesth. 2018;120:1117–1127. doi:10.1016/j.bja.2018.02.021
15. Song N, Yang Y, Zheng Z, et al. Effect of esketamine added to propofol sedation on desaturation and hypotension in bidirectional endoscopy: a randomized clinical trial. JAMA Network Open. 2023;6:e2347886. doi:10.1001/jamanetworkopen.2023.47886
16. Nie J, Chen W, Jia Y, et al. Comparison of remifentanil and esketamine in combination with propofol for patient sedation during fiberoptic bronchoscopy. BMC Pulm Med. 2023;23:254. doi:10.1186/s12890-023-02517-1
17. Chung F. Discharge criteria—a new trend. Can J Anaesth. 1995;42:1056–1058. doi:10.1007/BF03011083
18. Piaggio G, Elbourne DR, Pocock SJ, et al. Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA. 2012;308:2594–2604. doi:10.1001/jama.2012.87802
19. Wahidi MM, Jain P, Jantz M, et al. American College of Chest Physicians consensus statement on the use of topical anesthesia, analgesia, and sedation during flexible bronchoscopy in adult patients. Chest. 2011;140:1342–1350. doi:10.1378/chest.10-3361
20. Chernik DA, Gillings D, Laine H, et al. Validity and reliability of the Observer’s Assessment of Alertness/Sedation Scale: study with intravenous midazolam. J Clin Psychopharmacol. 1990;10:244–251.
21. Royse CF, Newman S, Chung F, et al. Development and feasibility of a scale to assess postoperative recovery: the post-operative quality recovery scale. Anesthesiology. 2010;113:892–905. doi:10.1097/ALN.0b013e3181d960a9
22. Dreher M, Ekkernkamp E, Storre JH, et al. Sedation during flexible bronchoscopy in patients with pre-existing respiratory failure: midazolam versus midazolam plus alfentanil. Respiration. 2010;79:307–314. doi:10.1159/000267227
23. Edokpolo LU, Mastriano DJ, Serafin J, et al. Discharge readiness after propofol with or without dexmedetomidine for colonoscopy: a randomized controlled trial. Anesthesiology. 2019;131:279–286. doi:10.1097/ALN.0000000000002809
24. Sterne JAC, White IR, Carlin JB, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ. 2009;338:b2393. doi:10.1136/bmj.b2393
25. Singh SA, Prakash K, Sharma S, et al. Comparison of propofol alone and in combination with ketamine or fentanyl for sedation in endoscopic ultrasonography. Korean J Anesthesiol. 2018;71:43–47. doi:10.4097/kjae.2018.71.1.43
26. Lee YS, Kim WY, Choi JH, et al. The effect of ketamine on the incidence of emergence agitation in children undergoing tonsillectomy and adenoidectomy under sevoflurane general anesthesia. Korean J Anesthesiol. 2010;58:440–445. doi:10.4097/kjae.2010.58.5.440
27. Eberl S, Koers L, van Hooft J, et al. The effectiveness of a low-dose esketamine versus an alfentanil adjunct to propofol sedation during endoscopic retrograde cholangiopancreatography: a randomised controlled multicentre trial. Eur J Anaesthesiol. 2020;37:394–401. doi:10.1097/EJA.0000000000001134
28. Zheng L, Wang Y, Ma Q, et al. Efficacy and safety of a subanesthetic dose of esketamine combined with propofol in patients with obesity undergoing painless gastroscopy: a prospective, double-blind, randomized controlled trial. Drug Des Devel Ther. 2023;17:1347–1356. doi:10.2147/DDDT.S408076
29. Huang X, Lin F, Chen Q, et al. Safety and efficacy of the combination of esketamine and propofol in procedural sedation/analgesia: a systematic review and meta-analysis. Minerva Anestesiol. 2023;89:680–689. doi:10.23736/S0375-9393.23.17100-8
30. Jansen SC, van Velzen M, Sarton E, et al. Acute effects of esketamine on hypoxic ventilatory response, haemodynamics, and brain function in healthy volunteers. Br J Anaesth. 2025;134:557–563. doi:10.1016/j.bja.2024.08.040
31. Xu C, Wei X, Zhang C, et al. Esketamine prevents propofol-induced injection pain: randomized controlled trial. Front Pharmacol. 2022;13:991559. doi:10.3389/fphar.2022.991559
32. Wagner II LE, Gingrich KJ, Kulli JC, et al. Ketamine blockade of voltage-gated sodium channels: evidence for a shared receptor site with local anesthetics. Anesthesiology. 2001;95:1406–1413. doi:10.1097/00000542-200112000-00020
33. Niesters M, Dahan A, Swartjes M, et al. Effect of ketamine on endogenous pain modulation in healthy volunteers. Pain. 2011;152:656–663. doi:10.1016/j.pain.2010.12.015
34. Mazzone SB, Undem BJ. Vagal afferent innervation of the airways in health and disease. Physiol Rev. 2016;96:975–1024. doi:10.1152/physrev.00039.2015
35. Gateau O, Bourgain JL, Gaudy JH, et al. Effects of ketamine on isolated human bronchial preparations. Br J Anaesth. 1989;63:692–695. doi:10.1093/bja/63.6.692
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Low-Dose Esketamine as an Adjuvant to Propofol Sedation for Same-Visit Bidirectional Endoscopy: Protocol for a Multicenter Randomized Controlled Trial
Song N, Shan XS, Yang Y, Zheng Z, Shi WC, Yang XY, Li Y, Tan AP, Liu H, Peng K, Ji FH
International Journal of General Medicine 2022, 15:4733-4740
Published Date: 6 May 2022
A Comparative Study of Esketamine-Propofol and Sufentanil-Propofol for Analgesia and Sedation During Breast Minimally Invasive Rotary Resection with Local Anesthesia: A Randomized Double-Blind Clinical Trial
Li N, Qi X, Bao J, Gu Y, Zhou X, Wang T, Jiang N, Wang Y, Ye Q
Drug Design, Development and Therapy 2024, 18:5397-5407
Published Date: 25 November 2024
The Efficacy and Safety Profile of Balanced Propofol Sedation for Bronchoscopy
Wu X, Zhang L, Zhou Z, Qi L, Liu Y, Du X, Ma L, Ji X
Therapeutics and Clinical Risk Management 2024, 20:849-860
Published Date: 12 December 2024
Comparison of Esketamine/Propofol and Sufentanil/Propofol on Intraoperative Hypoxemia During Bronchoscopy: A Randomized Trial
Huang X, Li X, Sun Y, Wu A, Ai P
Drug Design, Development and Therapy 2025, 19:4429-4436
Published Date: 27 May 2025
Efficacy of Different Doses of Remimazolam Tosilate Combined with Esketamine in Painless Abortion Patients: A Prospective, Double-Blind, Randomized Controlled Trial
Chen J, Zhang J, Zhang M, Zou X, Hu B, Yang Y, Li H
Drug Design, Development and Therapy 2025, 19:9117-9126
Published Date: 8 October 2025