Back to Journals » OncoTargets and Therapy » Volume 13
Dioscin Inhibited Glycolysis and Induced Cell Apoptosis in Colorectal Cancer via Promoting c-myc Ubiquitination and Subsequent Hexokinase-2 Suppression
Authors Wu Z, Han X, Tan G, Zhu Q, Chen H, Xia Y, Gong J, Wang Z, Wang Y, Yan J
Received 22 July 2019
Accepted for publication 6 December 2019
Published 6 January 2020 Volume 2020:13 Pages 31—44
DOI https://doi.org/10.2147/OTT.S224062
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Zhenqian Wu, Xiaodong Han, Gewen Tan, Qingchao Zhu, Hongqi Chen, Yang Xia, Jianfeng Gong, Zhigang Wang, Yu Wang, Jun Yan
Department of General Surgery, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai 200233, People’s Republic of China
Correspondence: Jun Yan
Department of General Surgery, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, 600 Yishan Road, Shanghai 200233, People’s Republic of China
Tel +86-21-6436 9181
Email [email protected]
Purpose: Dioscin is a natural product isolated from traditional Chinese medicines and is reported to have antitumor activities against several cancers. In the present study, we aimed to investigate its potency against colorectal cancers, especially the effects on tumor glycolysis, and to elaborate related molecular mechanisms.
Methods: The antitumor activities of dioscin were evaluated by cell proliferation assays and colony formation assays in vitro and the mouse xenograft models in vivo. The effects of dioscin on tumor glycolysis were determined by measuring glucose absorption and lactate generation. Cell apoptosis was detected by cleaved PARP and the activity of caspase-3. Protein overexpression or gene knockdown was conducted to illustrate molecular mechanisms. Immunoprecipitation experiments were applied to identify the interaction between different proteins.
Results: Dioscin substantially inhibited colorectal cancer cell proliferation in vitro and suppressed the xenograft growth in nude mice. After dioscin treatment, with the suppression of hexokinase-2, the tumor glycolysis was significantly decreased. Dioscin substantially impaired the interaction between hexokinase-2 and VDAC-1, and induced cell apoptosis. Exogenous overexpression of hexokinase-2 significantly antagonized the glycolysis suppression and apoptosis induction by dioscin. Through enhancing the binding of E3 ligase FBW7 to c-myc, dioscin promoted the ubiquitination of c-myc and gave rise to c-myc degradation, which contributed to the inhibition of hexokinase-2.
Conclusion: Our studies revealed a novel mechanism by which dioscin exerted its antitumor activity in colorectal cancer, and verified that dioscin or its analog might have potentials for colorectal cancer therapy.
Keywords: colorectal cancer, tumor glycolysis, hexokinase-2, c-myc, ubiquitination
Introduction
As an important feature of tumor cells, aerobic glycolysis plays an essential role in tumor development. Unlike normal cells which metabolize glucose completely to CO2 and H2O, cancer cells prefer to acquire the energy by converting glucose into lactic acid even when sufficient oxygen is available.1 Although the efficiency of ATP generation in tumor glycolysis is low, the high speed of energy production is indispensable for rapid tumor growth.2 Moreover, the intermediate products generated in tumor glycolysis are useful for cancer cells to promote the biosynthesis to meet the requirement of cell proliferation.3 In addition, the product of tumor glycolysis can acidify the microenvironment around tumor tissue and help tumor cells escape the surveillance of the immune system and give rise to tumor cell invasion and metastasis.4 So far, the details involved in the transition from oxidative phosphorylation to aerobic glycolysis are not fully understood, and several mechanisms were considered to contribute to this metabolic reprogramming.5
In glucose metabolism, hexokinases are the crucial enzymes and responsible for catalyzing ATP-dependent phosphorylation of glucose to generate glucose-6-phosphate.6 So far, four different isoforms of hexokinases named hexokinase 1–4, respectively, have been characterized. Owing to the different affinities to glucose and distributions in cells, these four isoforms have different roles in glucose metabolism.7,8 Recently, increasing evidence suggested that hexokinase-2 is upregulated in cancer development and is associated with enhanced tumor glycolysis.9,10 By positioning itself on the outer membrane of mitochondria, hexokinase-2 not only can avoid the inhibitory effect of its own product, but also get easier access to newly generated ATP and increase tumor glycolysis.11 Except the role in glycolysis regulation, hexokinase-2 has been demonstrated to be also involved in the mediation of tumor cell survival. Hexokinase-2 is engaged in the maintenance of the integrity of outer membrane and protect cells from apoptosis.12 In some extreme conditions, such as glucose deprivation, hexokinase-2 can sense the glucose availability and function as a switcher from tumor glycolysis to autophagy.13
Dioscin is a kind of steroid saponins isolated from traditional Chinese medicines, such as Dioscoreae rhizome and Paridis rhizome. Pharmacology researches showed that dioscin possessed multiple biological functions, such as protection of chemical-induced liver damage, anti-inflammatory activity, lipid-lowering property.14 Recently, increasing studies reported that dioscin had potent activities against various cancers, including leukemia, non-small cell lung cancer, hepatocellular carcinoma, and gastric cancers.15,16 Different molecular mechanisms including DNA damage regulation, cell cycle arrest, apoptosis induction, reactive oxygen species elevation were suggested to be contributed to the antitumor activities of dioscin.17–19 Except that, by decreasing MDR1 levels and inhibiting the activity of the p-gp protein, dioscin can substantially reverse the multidrug resistance in several cancers.20,21 So far, the activity of dioscin against tumor glycolysis and the underlying mechanisms remain unclear, in this study, we mainly investigated the effect of dioscin on tumor glycolysis in colorectal cancers and the related molecular mechanisms.
Materials and Methods
Cell Lines and Reagents
Dioscin (≥95%) and MG132 were products of Sigma-Aldrich (St. Louis, MO). Normal colon epithelial cells FHC and CCD-18Co, and human colorectal cancer cells, including HCT116, HT29, DLD1, and SW620, were from the American Type Culture Collection (ATCC). All cells were cultured by following the standard protocols. Anti-HK2, anti-HK1, anti-VDAC1, anti-FBW7, anti-c-Myc, anti-cleaved-PARP, anti- cytochrome C, anti-cleaved-caspase-3, anti-Bax, anti-α-Tubulin, anti-β-actin, anti-mouse/rabbit secondary antibodies were from Cell Signaling Technology, Inc. (Danvers, MA). Anti-Ki67 and anti-c-Myc antibodies were purchased from Abcam (Cambridge, UK). Lipofectamine was from Invitrogen (Carlsbad, CA). HK2 overexpression plasmid was purchased from Addgene (Cat.: 23854). Control siRNA (Cat.: sc-37007) and FBW7 (Cat.: sc-37547) siRNAs were product of Santa Cruz.
Cell Proliferation Assay
Digested with trypsin, the CRC cells were resuspended with culture medium and adjusted to appropriate concentration, then 90μL cell suspensions were seeded into 96-well plate. After the cells were completely attached (usually overnight), 10μL solutions containing different concentrations of dioscin were added. At different time points, the examination of cell viability was performed with Cell Titer-Glo Luminescent Cell Viability Assay kit (Promega Madison, WI) by following manufacturer’s instructions.
Western Blotting
CRC cells treated with dioscin were lysed with RIPA solutions, the lysates were harvested and then centrifuged at 12,000 g for 10 mins, the supernatants were collected and protein concentrations were determined. 30 μg protein per lane was loaded into10% acrylamide gel and resolved by SDS-PAGE. After electrically transferring to the PVDF membrane, the unspecific sites on the membrane were blocked by 5% non-fat dry milk. After the incubation with primary and secondary antibodies, the membrane was visualized by ECL chemiluminescence reagents (Pierce Chemical Co., Rockford, Illinois, USA).
Tumor Glycolysis Measurement
The glycolytic levels in CRC levels were determined by glucose consumption and lactate generation in culture medium. Briefly, appropriate amount of CRC cells (5×105/well) were seeded into 6-well plate and cultured in the incubator until the cells were attached to the plate, then the fresh mediums with dioscin were added and incubated for 8 hrs. The culture mediums were collected and the glucose and lactate levels were examined with Glucose Assay Kit (ab65333, Abcam) and L-Lactate Assay Kit (ab65331, Abcam) respectively. The protein concentration per well was measured by Bradford assay (Bio-Rad, Philadelphia, PA, USA) and used to normalize glucose and lactate concentrations.
Caspase-3 Activity Measurement
The caspase-3 activity in CRC cells was measured by the Caspase-3 Assay Kit (ab39401, Abcam). Briefly, the CRC cells treated with/without dioscin were harvested and lysed on ice. After centrifugation, protein was measured and then adjusted to the concentration as suggested, then the caspase-3 activity was examined by following supplier’s protocols.
Soft Agar Colony Formation Assay
The soft agar assay was performed as described following, briefly, the cells were counted and diluted to the concentration of 10,000 cells/mL in a 5 mL tube, and mixed with the Eagle’s basal medium which containing 10% FBS, 0.3% agar, and dioscin at various concentrations. The mixture was loaded into the 6-well plates which coated with a 0.6% agar base layer. The plates were placed into cell incubator until obvious cell colonies were observed in the control group (generally about 2 weeks). The number of cell colonies (more than 50 cells/colony) was counted under the microscope.
Mitochondria Isolation
After digestion with trypsin, CRC cells from 100 mm dish were harvested and washed with ice-cold PBS. Cell pellets were obtained by centrifugation at 800 rpm for 5 min. The mitochondria in the pellets were extracted using the Mitochondria Isolation Kit (ab110170, Abcam) following the standard procedure.
Co-Immunoprecipitation (Co-IP) Assays
Briefly, after digestion, the tumor cells were harvested. The NP40 buffer (FNN0021, Thermo Fisher) was used for cell lysate extraction. After centrifugation, the supernatants from the cell lysates were incubated with agarose A/G beads and specific primary antibody at 4°C overnight. Washing three times with the lysis buffer, the interacted proteins were examined by immunoblotting.
In vivo Efficacy Study
The animal studies were performed with the approval of the Animal Ethics Committee of Shanghai Jiao Tong University. All operations were performed in accordance with the guideline of National Institute of Health Guide for the Care and Use of Laboratory Animals. HCT116 or HT29 xenograft model was established by subcutaneously injecting cell suspension (5×106 cell/mice) into the right flank of female nude mice. When the tumor was formed (about 100 mm3), the mice were randomly divided and designated as vehicle or treatment group, respectively (5 mice/group). The vehicle group received 0.5% sodium carboxymethylcellulose, and the treatment group was i.p. injected 5mg/kg dioscin every 2 days. Tumor volume was measured by microcalipers every 2 days and calculated as V= (length×width2)/2. Meanwhile, the weight of tumor-bearing mice was also examined. In the end of experiments, the mice were sacrificed with CO2, the tumors were stripped, photographed and weighed.
Immunohistochemical (IHC) Staining
The colorectal cancer tissue microarray (CO484a), which includes 45 malignant CRC tissue and matched adjacent tissue, was product of US Biomax, Inc. The tissue from mouse xenograft was embedded in paraffin and cut into 5μM slides. The whole IHC staining processes were performed as following. Briefly, after dewaxing and hydration, the antigen retrieval in the slides was carried out in boiled sodium citrate buffer (PH=6.0), then the slides were incubated with 3% H2O2 for 10mins and the endogenous peroxidase was blocked. The slides were blocked with the serum from goat and then incubated with the primary antibodies overnight at 4°C. After washing in PBS solution, the slides were treated with HRP-conjugated goat anti-rabbit polymer antibodies and visualized with DAB. By counterstaining with hematoxylin, the slides were dehydrated in alcohol and xylene and then mounted. The expression status was analyzed by the software Image-Pro PLUS (v.6).
Statistical Analysis
The quantitative data was calculated as mean±SD, and the statistical analysis was carried out by the SPSS software (version 13.0). Two-tailed student T test was used to analyze the statistical differences and p< 0.05 was considered to represent significant difference.
Results
Dioscin Inhibited CRC Proliferation and Colony Formation in vitro
Firstly, the antitumor activities of dioscin (Figure 1A) against CRC cells were evaluated by the cell proliferation assays. As shown in Figure 1B–D, in three CRC cells (HT-29, HCT-116, and SW480), after the treatment of dioscin, cell proliferation was significantly inhibited in a dose-dependent manner. At the top concentration 5μM, after the incubation for 72 hrs, cell proliferation was almost completely suppressed, and cell growth inhibition rate reached more than 90%. To further examine the antitumor potency of dioscin, we used anchorage-independent growth assay to measure the effect of dioscin on cell colony formation. As the results demonstrated (Figure 1E–G), in the cells with no dioscin, a number of cell clones were observed in the soft agar, however, with the treatment of dioscin, the number of clones formed was dramatically decreased, demonstrating that dioscin had a profound antitumor potency in CRC cells.
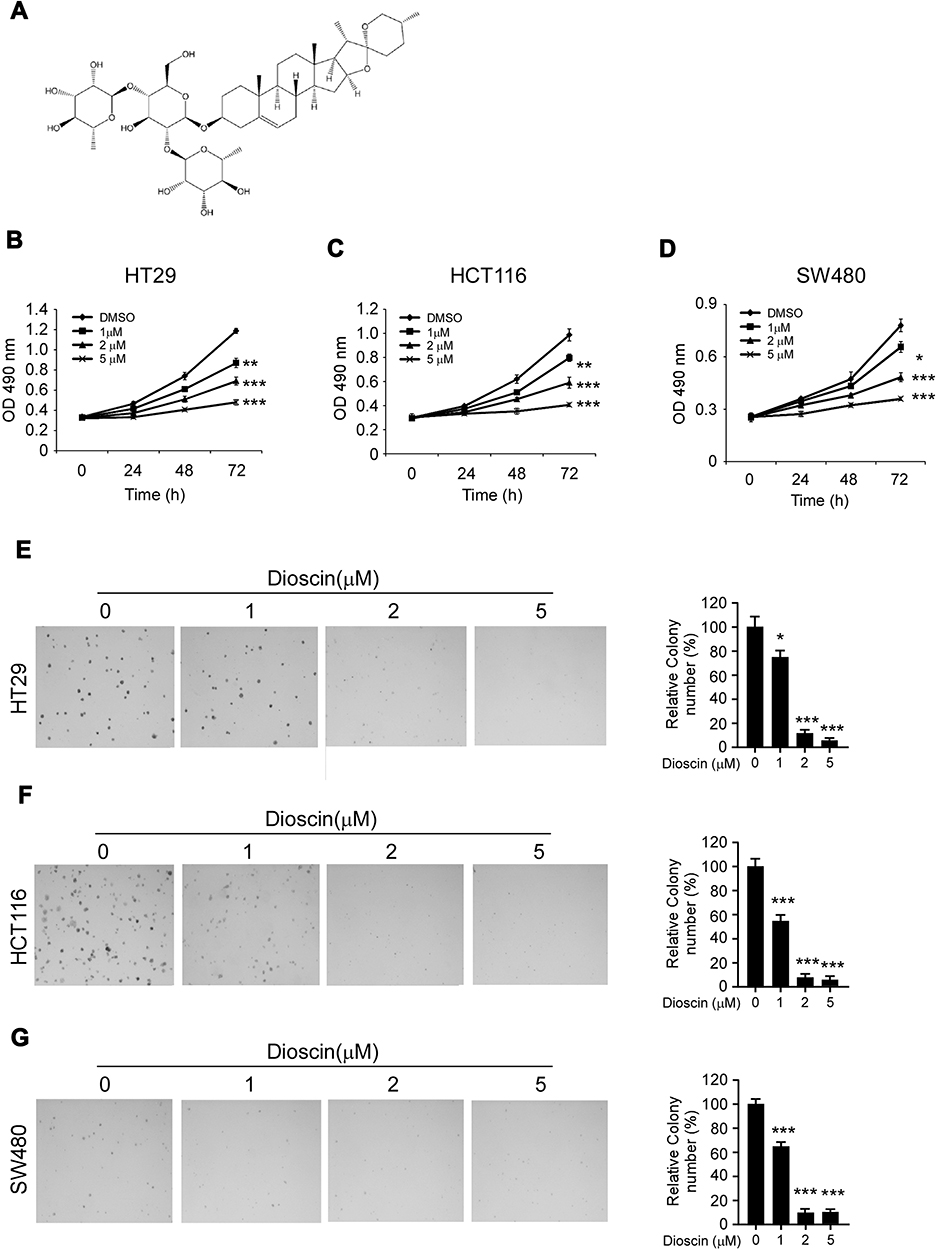 |
Figure 1 Dioscin inhibited colorectal cancer proliferation and colony formations. (A) The chemical structure of dioscin; (B–D) Dioscin inhibited colorectal cancer proliferation in vitro. HT-29 (B), HCT-116 (C) and SW-480 (D) cells were placed into 96-well plates and then treated with different concentrations of dioscin for 24, 48, 72 hrs, respectively, the cell viability was measured by the Cell Titer-Glo kit as described. (E–G) Dioscin inhibited the colony formation of colorectal cells. HT-29 (E), HCT-116 (F) and SW-480 (G) cell suspensions treated with dioscin were plated into 6-well plate, and the colony formation was examined as described in methods. Left, the representative images; right, quantitative statistics expressed as mean ± SD. *p<0.05, **p<0.01, ***p<0.001 versus the control. |
Dioscin Inhibited the Glycolysis in CRC Cells by Mediating Hexokinase-2
Hexokinase-2 has a critical role in the regulation of tumor glycolysis, so we examined the expression of hexokinase-2 in colorectal cancers. As shown in Figure 2A, in 45 paired tissue, the intensity of hexokinase-2 in tumor tissue was obviously higher than adjacent normal tissue. Moreover, the Western blotting results demonstrated compared with normal colonic cells FHC and CCD-18Co; the expression of hexokinase-2 was significantly increased in four detected colorectal cancer cells (Figure 2B). Next, we investigated the effect of dioscin on tumor glycolysis. As the results shown in Figure 2C–E, in CRC cells, the treatment of dioscin resulted in a substantial decrease in glucose consumption. With the decline of glucose absorption, the amount of the lactate generated by CRC cells was also decreased significantly, demonstrating the glycolysis in dioscin-treated CRC was suppressed. Moreover, the analysis of the crucial proteins in tumor glycolytic pathways showed that the expression of hexokinase-2, not hexokinase-1, was dose-dependently reduced. To further clarify the role of hexokinase-2 in dioscin-induced glycolysis inhibition, we exogenously expressed hexokinase-2 in HCT-116 and HT-29 cells, and the results showed hexokinase-2 overexpression significantly reversed the glycolysis inhibition (Figure 2F–G).
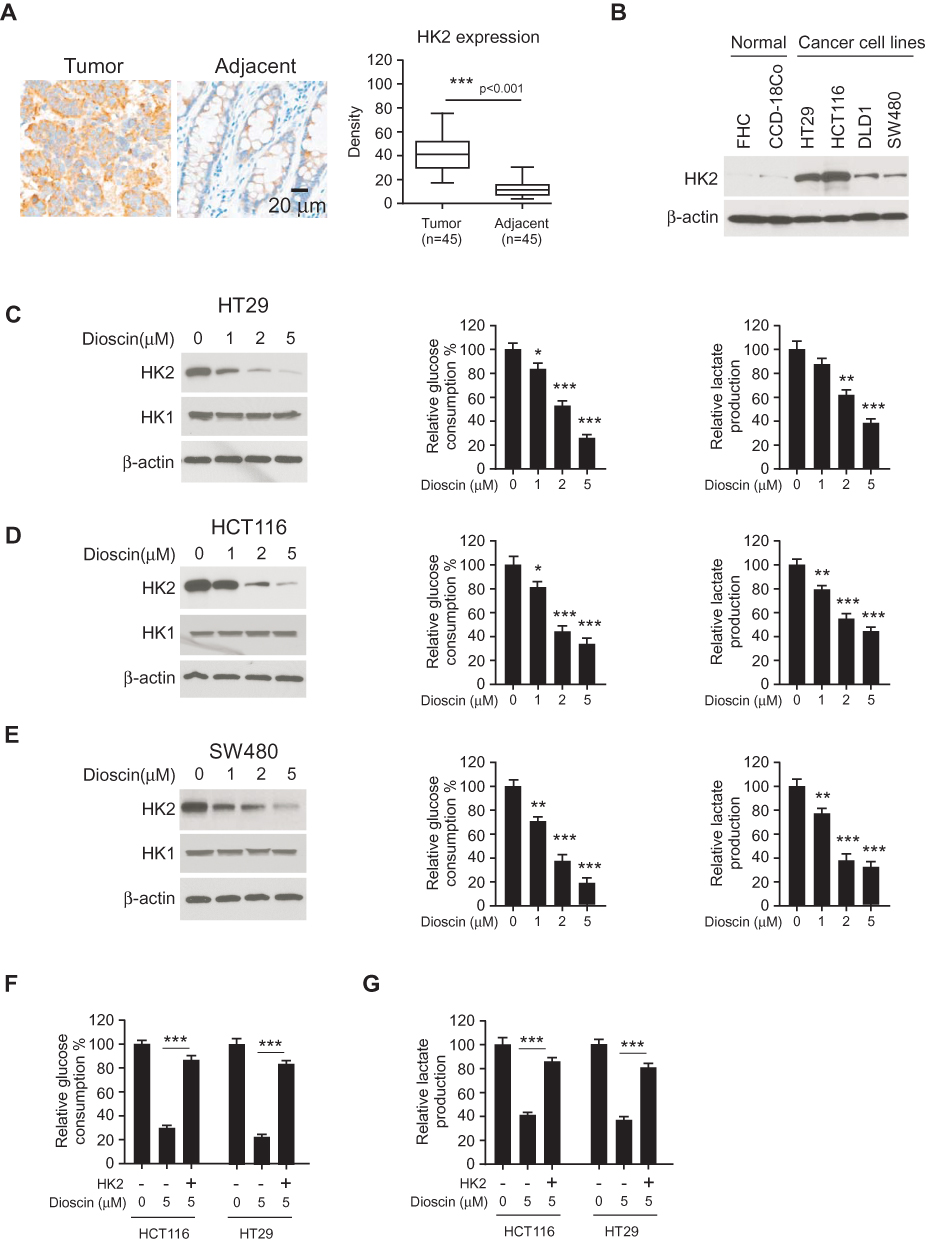 |
Figure 2 Dioscin inhibited tumor glycolysis in colorectal cancers by downregulating hexokinase-2. (A) The expression of hexokinase-2 in colorectal cancer tissue and paired adjacent tissue was examined by IHC staining. Left, the representative images; right, the statistics of hexokinase-2 expression. ***p<0.001 indicated a significant difference. (B) The expression of hexokinase-2 in normal colon cells and colorectal cancer cells was examined by Western blotting. (C–E) HT-29 (C), HCT-116 (D) and SW-480 (E) cells were treated with dioscin and the expression of hexokinase-2 (left), glucose consumption (middle) or lactate production (right) was measured, respectively. (F–G) Hexokinase-2 overexpression impaired dioscin-induced glycolysis inhibition. Ectopic overexpression of hexokinase-2 was performed in HCT-116 and HT-29 cells and then treated with 5 μM dioscin, the amount of glucose (F) and lactate (G) were measured as described. *p<0.05, **p<0.01, ***p<0.001 versus the control. |
Hexokinase-2 Suppression Contributed to Dioscin-Induced Cell Apoptosis
Generally, hexokinase-2 is bound to the VDAC-1 and maintains the integrity of outer membrane of mitochondria. Given the effects of dioscin on hexokinase-2 expression, we speculated that the interaction between hexokinase-2 and VDAC-1 would be affected by dioscin. We performed the co-immunoprecipitation experiments to detect the change of the binding of hexokinase-2 to VDAC-1. All results demonstrated that in dioscin-treated cells, the interaction between hexokinase-2 and VDAC-1 was significantly impaired (Figure 3A and B). Furthermore, we isolated the cytosolic and mitochondria fractions, respectively, and examined the expression of cytochrome C and Bax. As the results demonstrated in Figure 3C and D, in the cytosolic parts, the amount of cytochrome C was dose-dependently increased, whereas Bax was substantially decreased. However, in mitochondria, Bax expression was dramatically elevated. These results suggested that Bax was translocated from the cytoplasm to mitochondria after dioscin treatment, which gave rise to the release of cytochrome C. Cleaved PARP and Caspase-3 are important markers of cell apoptosis, as shown in Figure 3E, cleaved PARP and Caspase-3 were dose-dependently increased in dioscin-treated CRC cells, implying that substantial cells were induced apoptosis by dioscin. Meanwhile, we also examined the activities of Caspase-3 after dioscin treatment. The results showed that the activities of Caspase-3 were significantly increased (Figure 3F). To verify the role of hexokinase-2 suppression in dioscin-induced cell apoptosis, hexokinase-2 was ectopically overexpressed in HCT-116 and HT-29 cells and then measured the apoptosis induction by dioscin. As demonstrated in Figure 3G–H, dioscin-induced cell apoptosis in hexokinase-2 overexpression cells was dramatically attenuated, which was evidenced by the decrease of cleaved PARP and caspase-3 as well as the activity of caspase-3.
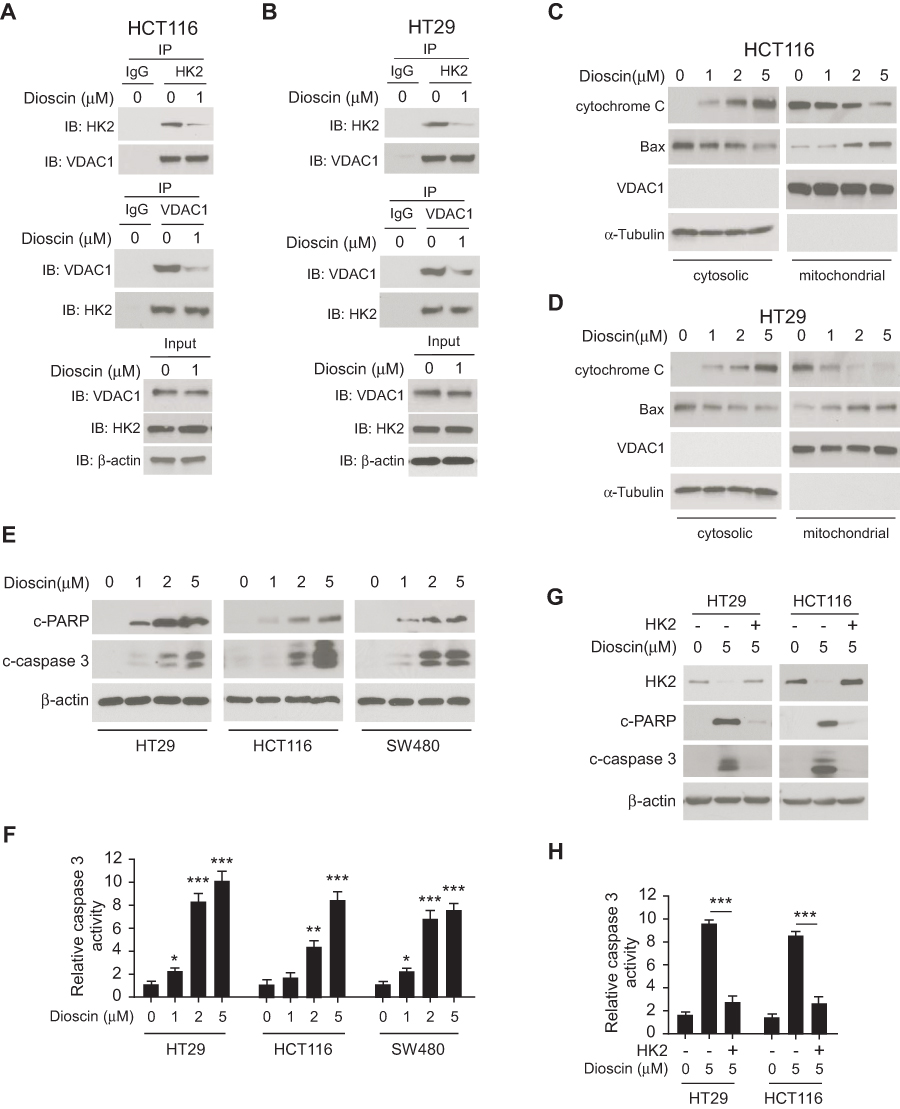 |
Figure 3 Dioscin-induced cell apoptosis in colorectal cancer via suppressing hexokinase-2. (A, B) Dioscin impaired the interaction between hexokinase-2 and VDAC-1. HCT-116 (A) or HT-29 (B) Cells were treated with 1μM dioscin for 24 hrs, and the cell lysates were harvested. The cell lysates were immunoprecipitated with anti-hexokinase-2 or anti-VDAC-1 antibodies, respectively, and then probed by Western blotting with indicated antibodies. (C, D), Dioscin induced the translocation of cytochrome C and Bax. HCT-116 (C) or HT-29 (D) cells were treated with the indicated concentration of dioscin, the cytosolic and mitochondria fractions were extracted as described, respectively, and then examined with indicated antibodies. (E, F) Dioscin-induced cell apoptosis in CRC cells. HT-29, HCT-116, or SW480 cells were treated with dioscin for 24 hrs, cleaved PARP and caspase-3 (E) or the activity of caspase-3 (F) was examined. (G, H) Ectopic overexpression attenuated dioscin-induced cell apoptosis. HT-29 or HCT-116 cells were transfected with plasmid overexpressing hexokinase-2 and then treated with 5 μM dioscin, and the expression of cleaved PARP and caspase-3 (G) or the activity of caspase-3 (H) was measured. *p<0.05, **p<0.01, ***p<0.001 represented significant differences. |
Dioscin-Induced Hexokinase-2 Suppression by Inhibiting c-myc
The transcriptional factor c-myc is reported to engage in hexokinase-2 regulation. In HCT-116 and HT-29 cell, we adopted shRNA to silence c-myc expression and examined the effects. As shown in Figure 4A–B, in c-Myc knockdown cells, the expression of hexokinase-2 was dramatically decreased, the glucose consumption and lactate production were significantly dropped accordingly. In dioscin-treated CRC cells, we examined the effect of dioscin on c-myc, the results revealed c-myc was dose-dependently downregulated (Figure 4C), suggesting the suppression of hexokinase-2 caused by dioscin maybe attribute to the inhibition of c-myc. To clear the importance of c-myc in dioscin-mediated hexokinase-2 suppression, we ectopically overexpressed c-myc in HCT-116 cells, and the results showed that dioscin-induced hexokinase-2 suppression was substantially attenuated (Figure 4D). With the recovery of HK-2, the expression of cleaved-PARP and caspase-3 was decreased in c-myc overexpression cells, and the activity of caspase-3 was also reduced accordingly, suggesting c-myc ectopic overexpression significantly impaired dioscin-induced cell apoptosis (Figure 4E–F).
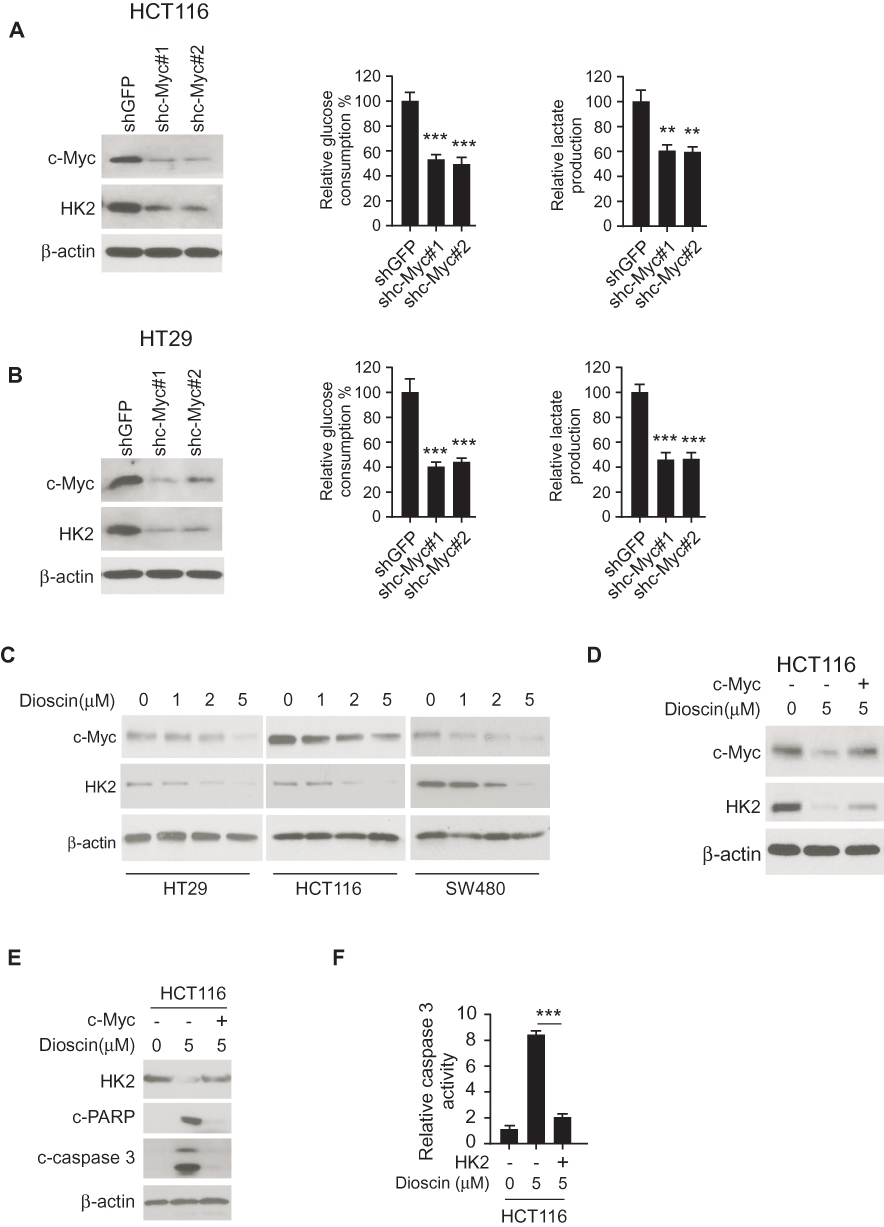 |
Figure 4 Dioscin inhibited hexokinase-2 expression by downregulating c-myc. (A, B) c-myc knockdown suppressed hexokinase-2 expression and tumor glycolysis. HCT-116 (A) or HT-29 (B) cells were transfected with c-myc shRNA, the expression of hexokinase-2 in transfected cells (left), glucose consumption (middle) or lactate production (right) was examined. (C) Dioscin reduced c-myc expression in CRC cells. CRC cells were treated with indicated concentrations of dioscin, and the expression of c-myc was detected. (D) Exogenous overexpression of c-myc attenuated hexokinase-2 suppression by dioscin. HCT-116 cells were transfected with plasmids overexpressing c-myc and then treated with 5μM dioscin, the expression of c-myc and hexokinase-2 was detected. (E, F) c-myc ectopic overexpression attenuated dioscin-induced cell apoptosis. HCT-116 cells were transfected with plasmid overexpressing c-myc and then treated with 5μM dioscin, and the expression of HK-2, cleaved PARP and caspase-3 (E) or the activity of caspase-3 (F) was measured. **p<0.01, ***p<0.001 represented significant differences. |
Dioscin Promoted FBW-7 Mediated c-myc Ubiquitination
To illustrate the mechanism by which c-myc was inhibited, we firstly examined the effects of dioscin on c-myc mRNA levels, and no difference was observed (data not shown). However, after CRC cells were treated with proteasome inhibitor MG132, the dioscin-mediated c-myc reduction was significantly recovered (Figure 5A). Furthermore, in HCT-116 cells, we examined the ubiquitination levels of c-myc. As the results are shown in Figure 5B, in MGC-132 pretreated HCT-116 cells, after the treatment of dioscin, the ubiquitin levels of c-myc were significantly increased. FBW-7 is the E3 ligase responsible for the ubiquitination of c-myc; therefore, in HCT-116 cells, we adopted siRNA to knockdown the expression of FBW7 and observed the effects on c-myc ubiquitination. As demonstrated, in FBW7-knockdown cells, dioscin-induced ubiquitination of c-myc was substantially decreased, the expression of c-myc was slightly increased (Figure 5C). Furthermore, we investigated whether dioscin affected the interaction between FBW7 and c-myc. In Figure 5D, the results showed that dioscin had no effects on the expression of FBW7, however, in dioscin-treated cells, the amount of c-myc bound to FBW7 was significantly increased, implying dioscin promoted the binding between FBW7 and c-myc.
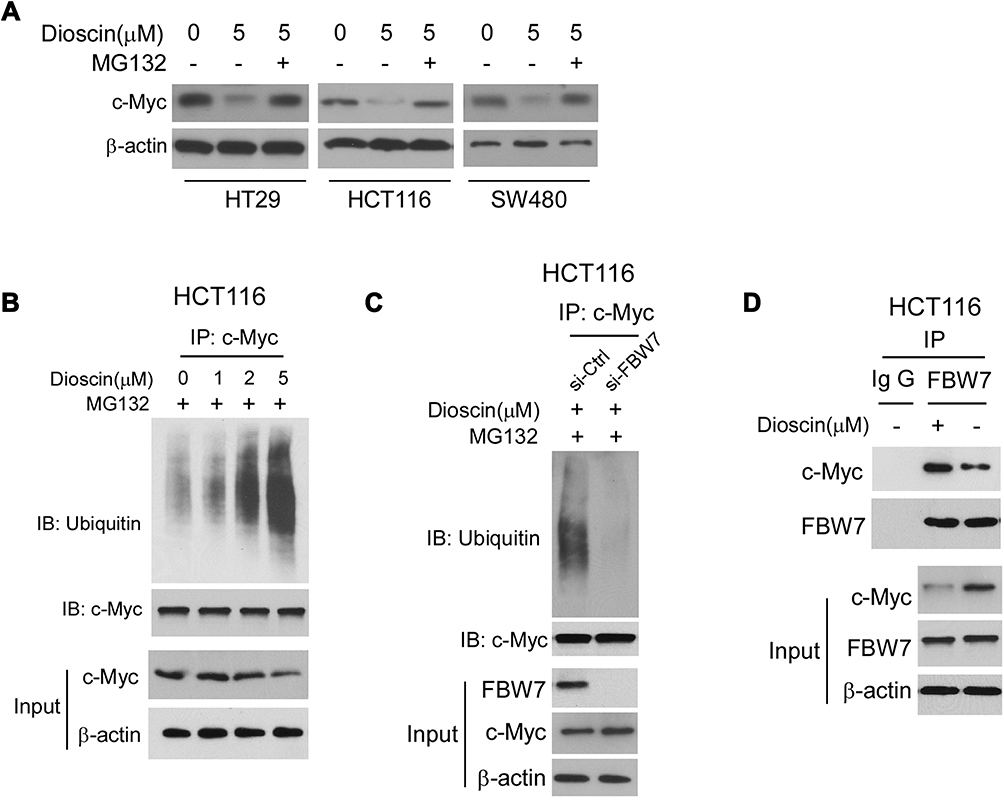 |
Figure 5 Dioscin promoted the ubiquitination of c-myc. (A) Proteasome inhibitor MG132 antagonized dioscin-mediated c-myc reduction. CRC cell was pretreated with 20 μM MG132 and then treated with 5μM dioscin; the expression of c-myc was examined by Western blotting. (B) Dioscin increases the ubiquitination levels of c-myc. HCT-116 cells were pretreated with 20 μM MG132 and then incubated with indicated dioscin, the cell lysates were immunoprecipitated with anti-c-myc antibody and then probed with anti-ubiquitin antibody. (C) FBW-7 was involved in dioscin-mediated ubiquitination of c-myc. HCT-116 were transfected with FBW7 siRNA and then treated with 5μM dioscin, the ubiquitination of c-myc was detected as described. (D) Dioscin enhanced the binding between c-myc and FBW-7. HCT-116 cells were treated with 5μM dioscin, the cell lysates were immunoprecipitated with anti-FBW7 antibody, and the precipitation was probed with the anti-c-Myc antibody. |
Dioscin Inhibited the Growth of CRC Xenografts in vivo
Finally, we have established HCT-116 and HT-29 xenograft models in nude mice and evaluated the antitumor potency of dioscin. As the results are shown in Figure 6A–D, in these two models, dioscin at the dose of 5mg/kg had displayed potent antitumor activities, and the tumor growth was remarkably restrained. In HCT-116 models, the tumor volume of vehicle and the dioscin-treated group were 825mm3 and 340mm3 respectively, and in HT-29 models, the volume was 605mm3 and 240mm3 respectively. In addition, during the experiment, the administration of dioscin did not cause significant weight loss in tumor-bearing mice, and no obvious side effects were observed, proving that dioscin was safe at the dose of 5mg/kg (Figure 6E–F). Consistent with the in vitro findings, the immunohistochemistry results revealed a significant decrease of the expression of c-myc and hexokinase-2 in tumor tissues treated with dioscin. Meanwhile, the expression level of ki67, an important marker of cell proliferation potentials, also decreased significantly. Conversely, in dioscin-treated tumor tissue, the expression of cleaved caspase-3 was substantially increased, suggesting an increase of cell apoptosis induction (Figure 6G).
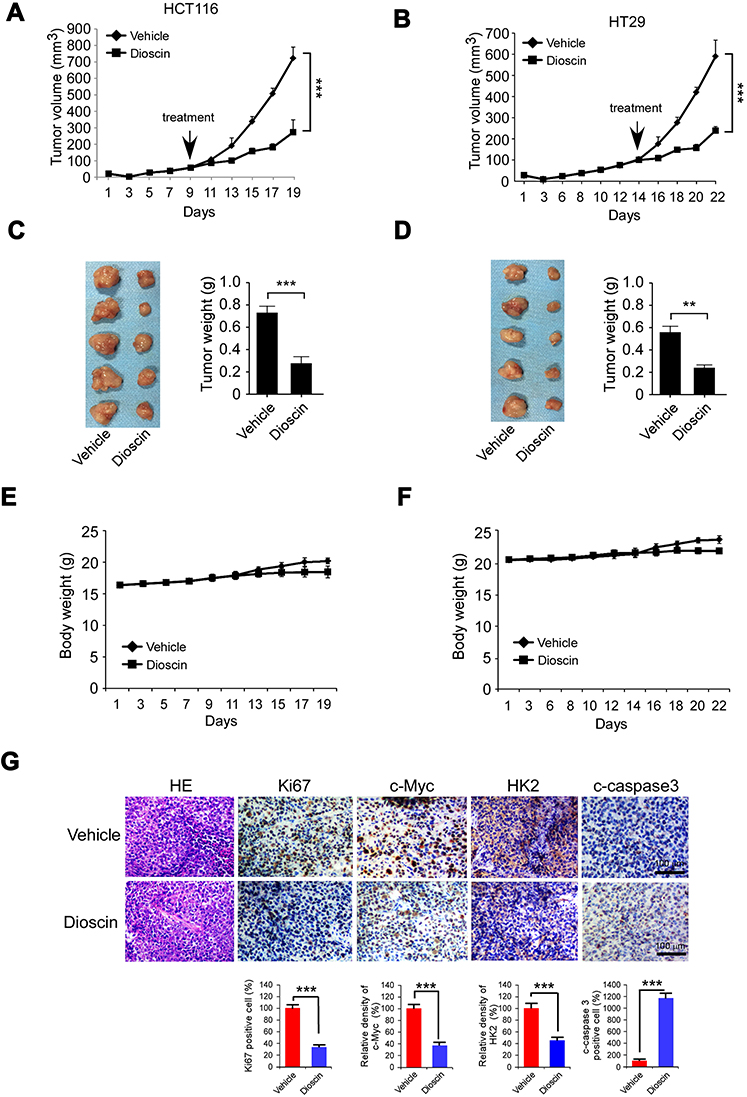 |
Figure 6 Disocin inhibited CRC xenograft growth in nude mice. (A–D) Dioscin restrained the growth of HCT-116 and HT-29 xenografts. The nude mice bearing HCT-116 or HT-29 xenografts were administrated with dioscin, and the growth rate of HCT-116 (A) or HT-29 (C) xenograft was measured. At the end of experiments, HCT-116 (B) or HT-29 (D) xenograft was isolated, photography, and weighed. Left: the images of tumor tissue; right: the weight of tumor tissue expressed as mean±SD. **p<0.01, ***p<0.001 versus the vehicle. (E–F) The curve of the body weight of nude mice bearing HCT-116 (E) or HT-29 (F) tumors during the experiments. (G) The expression of Ki67, c-myc, hexokinase-2 and cleaved-caspase-3 was decreased after dioscin treatment. In the end of in vivo experiments, tumor tissue was collected and stained with Ki67, c-myc, hexokinase-2 and cleaved caspase-3 antibodies, respectively. Upper, the representative photograph. Below, the expression was quantified, ***p<0.001 indicated a significant difference. |
Discussion
Colorectal cancer is a significant threat to the health of the public in worldwide. With the improvement of drug development, more and more advanced therapeutics such as targeted therapy, immunotherapy, CART were applied to the clinical treatment of colorectal cancer. However, some serious adverse effects of these therapeutics prevented patients from clinical benefits.22 Therefore, seeking potential drugs with high efficacy and low toxicity is in urgent needs. In contrast with the compounds obtained through synthetic chemistry, natural products, especially those extracted from traditional Chinese medicines with long clinical use, have more advantages in structural diversity and safety profiles. Our experiments demonstrated that dioscin had exerted profound antitumor activities against colorectal cancer. In vitro, dioscin dose-dependently inhibited colorectal cancer proliferation and anchorage-independent growth. In vivo, in HT-29 and HCT-116 xenograft models, dioscin at the dose of 5mg/kg substantially inhibited xenograft growth. As reported by Xu et al in Sprague-Dawley rats, the no observed adverse effect level (NOAEL) dose is estimated 300mg/kg, which was significantly higher than the effective dose required for dioscin to provide antitumor activity, suggesting dioscin had an excellent therapy margin.23
As an important hallmark of tumor cells, tumor glycolysis played a vital role to support rapid growth. Owing to the importance of tumor glycolysis, the deregulation of hexokinase-2 is often associated with high glycolytic activity. Hexokinase-2 overexpression is detected in multiple cancers, including hepatocellular carcinoma, lung cancer, gastric cancer, ovarian cancer, and breast cancer.24–26 In colorectal cancer, we also found that compared to the adjacent normal tissue and cells, hexokinase-2 expression was significantly increased (Figure 2A and B), implying that hexokinase-2 was involved in the regulation of colorectal cancer glycolysis and contributed to the tumor growth. In dioscin-treated cells, because of hexokinase-2 suppression, glucose uptake and lactate generation were substantially decreased, indicating the glycolysis activity in colorectal cancer cells was significantly inhibited by dioscin. On the other hand, in exogenously overexpressed hexokinase-2 cells, the impairment of glycolysis suppression further validated that the reduction of hexokinase-2 played an essential role in dioscin-induced glycolysis inhibition (Figure 2C–G). According to previous studies, different mechanisms, for example, activation of estrogen receptor-β, inhibition of ROS-mediated PI3K/Akt signaling, demethylation of DAPK-1 and RASSF-1α genes, activation of apoptosis-inducing factor (AIF) is reported to be contributed to the induction of cell apoptosis by dioscin.27 In our studies, as shown in Figure 3G and H, exogenous overexpression of hexokinase-2 significantly reverses the apoptosis induced by dioscin, indicating hexokinase-2 was also engaged in dioscin-induced cell apoptosis. As reported by Pastorino et al the pro-apoptotic protein Bax could compete with hexokinase-2 for combination with VDAC-1, and the separation of hexokinase-2 from the membrane enhanced the binding between Bax and VDAC-1, which led to the substantial increase of the permeability of the membrane.28 In dioscin-treated cells, the results of immunoprecipitation indicated that the interaction between hexokinase-2 and VDAC-1 was impaired, and the amount of Bax in mitochondria was dose-dependently increased (Figure 3A–D). Therefore, we disclosed a novel mechanism by which dioscin-induced CRC cells apoptosis. That is, owing to hexokinase-2 inhibition after dioscin treatment, Bax could bind to the VDAC-1 on the membrane more efficiently, which led to the increase of permeability and the release of cytochrome C and subsequent cell apoptosis.
Because of the importance of hexokinase-2 in cellular metabolism and survival, the regulation of hexokinase-2 is complicated in tumor cells. Sequence analysis of hexokinase-2 promoter region demonstrated that there were multiple functional response elements for transcriptional factors such as HIF-1, p53.29,30 As a transcription factor that plays a crucial role in cancer development, c-myc was also involved in hexokinase-2 regulation. In lung adenocarcinoma induced by c-myc, the expression of hexokinase-2 in mitochondrial was up-regulated near 5-fold and contributed significantly to the apoptosis inhibition.31 In Burkitt’s lymphoma, it was reported by Kim et al that dysregulated c-myc collaborated with HIF-1α to induce hexokinase-2 overexpression and resulted in the metabolic switches to tumor glycolysis.32 In colon cancers, c-myc-mediated upregulation of hexokinase-2 contributed significantly to interleukin-22-induced aerobic glycolysis.33 In the present studies, we also clarified that c-myc inhibition was closely associated with dioscin-mediated hexokinase-2 suppression, which was evidenced by the recovery of hexokinase-2 in c-myc ectopic-overexpression cells (Figure 4D). Posttranslational modification is an important way to regulate the activity and stability of c-myc. With the addition of the proteasome inhibitor, dioscin-induced c-myc decrease was substantially recovered, suggesting the proteasome system participated in the regulation of c-myc. FBW-7 is a well-known E3 ligase responsible for c-myc ubiquitination.34 In FBW-7 deficient cells, dioscin-mediated c-myc ubiquitination was almost completely diminished, verifying that FBW-7 was involved in the regulation of c-myc by dioscin. Although dioscin had no effects on FBW-7 expression, the binding between FBW-7 and c-myc was substantially enhanced in dioscin-treated cells. Based on the results above, we proposed that through the enhancement of the interaction between FBW7 and c-myc, dioscin promoted the ubiquitination of c-myc, which resulted in the degradation of c-myc and hexokinase-2 suppression.
Conclusion
In the present study, we demonstrated that through suppressing tumor glycolysis and inducing cell apoptosis, dioscin exerted potent antitumor activities against colorectal cancer. Furthermore, we revealed a novel antitumor mechanism of dioscin. By reinforcing FBW-7 mediated ubiquitination of c-myc and promoting c-myc degradation, dioscin substantially inhibited the activity of hexokinase-2, which gave rise to the glycolysis inhibition and apoptosis induction. In summary, our data provided the preclinical evidence for the application of dioscin or its derivatives in the treatment or prevention of colorectal cancers.
Acknowledgment
This work was supported by the grants from the National Natural Science Foundation of China (No. 81500401), Shanghai Pujiang Program (16PJD034) and Shanghai Jiao Tong University Interdisciplinary (Biomedical Engineering) Research Fund (No. YG2015MS12).
Author Contributions
ZQW, YW, and JY conceived and designed the experiments. ZQW, XDH and GT had carried out the most of the experimental work. QCZ and HQC performed the animal studies. YX and JFG have done the IHC experiments. ZQW, ZGW and YW analyzed and interpreted the results. ZQW and JY wrote and revised the manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
All authors declared no conflicts of interest to be disclosed in this study.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
2. Danhier P, Banski P, Payen VL, et al. Cancer metabolism in space and time: beyond the Warburg effect. Biochim Biophys Acta Bioenerg. 2017;1858(8):556–572. doi:10.1016/j.bbabio.2017.02.001
3. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi:10.1126/sciadv.1600200
4. Pillai SR, Damaghi M, Marunaka Y, Spugnini EP, Fais S, Gillies RJ. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019;38:205–222. doi:10.1007/s10555-019-09792-7
5. Dai C, Arceo J, Arnold J, et al. Metabolomics of oncogene-specific metabolic reprogramming during breast cancer. Cancer Metab. 2018;6:5. doi:10.1186/s40170-018-0175-6
6. Garcia SN, Guedes RC, Marques MM. Unlocking the potential of HK2 in cancer metabolism and therapeutics. Curr Med Chem. 2018;26. doi:10.2174/0929867326666181213092652
7. Li W, Ma X, Li N, et al. Resveratrol inhibits Hexokinases II mediated glycolysis in non-small cell lung cancer via targeting Akt signaling pathway. Exp Cell Res. 2016;349(2):320–327. doi:10.1016/j.yexcr.2016.11.002
8. McDonald AJ, Curt KM, Patel RP, et al. Targeting mitochondrial hexokinases increases efficacy of histone deacetylase inhibitors in solid tumor models. Exp Cell Res. 2019;375(2):106–112. doi:10.1016/j.yexcr.2018.12.012
9. DeWaal D, Nogueira V, Terry AR, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9(1):446. doi:10.1038/s41467-017-02733-4
10. Chen G, Zhang Y, Liang J, et al. Deregulation of hexokinase II is associated with glycolysis, autophagy, and the epithelial-mesenchymal transition in tongue squamous cell carcinoma under hypoxia. Biomed Res Int. 2018;2018:8480762.
11. John S, Weiss JN, Ribalet B, Rodrigues-Lima F. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE. 2011;6(3):e17674. doi:10.1371/journal.pone.0017674
12. Yao J, Liu J, Zhao W. By blocking hexokinase-2 phosphorylation, limonin suppresses tumor glycolysis and induces cell apoptosis in hepatocellular carcinoma. Onco Targets Ther. 2018;11:3793–3803. doi:10.2147/OTT
13. Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell. 2014;53(4):521–533. doi:10.1016/j.molcel.2013.12.019
14. Zhang W, Yin L, Tao X, et al. Dioscin alleviates dimethylnitrosamine-induced acute liver injury through regulating apoptosis, oxidative stress and inflammation. Environ Toxicol Pharmacol. 2016;45:193–201. doi:10.1016/j.etap.2016.06.002
15. Wang Y, He QY, Chiu JF. Dioscin induced activation of p38 MAPK and JNK via mitochondrial pathway in HL-60 cell line. Eur J Pharmacol. 2014;735:52–58. doi:10.1016/j.ejphar.2014.04.018
16. Mao Z, Han X, Chen D, et al. Potent effects of dioscin against hepatocellular carcinoma through regulating TP53-induced glycolysis and apoptosis regulator (TIGAR)-mediated apoptosis, autophagy, and DNA damage. Br J Pharmacol. 2019;176(7):919–937. doi:10.1111/bph.v176.7
17. Zhang G, Zeng X, Zhang R, et al. Dioscin suppresses hepatocellular carcinoma tumor growth by inducing apoptosis and regulation of TP53, BAX, BCL2 and cleaved CASP3. Phytomedicine. 2016;23(12):1329–1336. doi:10.1016/j.phymed.2016.07.003
18. Si L, Zheng L, Xu L, et al. Dioscin suppresses human laryngeal cancer cells growth via induction of cell-cycle arrest and MAPK-mediated mitochondrial-derived apoptosis and inhibition of tumor invasion. Eur J Pharmacol. 2016;774:105–117. doi:10.1016/j.ejphar.2016.02.009
19. Li S, Cheng B, Hou L, et al. Dioscin inhibits colon cancer cells’ growth by reactive oxygen species-mediated mitochondrial dysfunction and p38 and JNK pathways. Anticancer Drugs. 2018;29(3):234–242. doi:10.1097/CAD.0000000000000590
20. Wang L, Wang C, Peng J, et al. Dioscin enhances methotrexate absorption by down-regulating MDR1 in vitro and in vivo. Toxicol Appl Pharmacol. 2014;277(2):146–154. doi:10.1016/j.taap.2014.03.013
21. Wang C, Huo X, Wang L, et al. Dioscin strengthens the efficiency of adriamycin in MCF-7 and MCF-7/ADR cells through autophagy induction: more than just down-regulation of MDR1. Sci Rep. 2016;6:28403. doi:10.1038/srep28403
22. Sun X, Suo J, Yan J. Immunotherapy in human colorectal cancer: challenges and prospective. World J Gastroenterol. 2016;22(28):6362–6372. doi:10.3748/wjg.v22.i28.6362
23. Xu T, Zhang S, Zheng L, Yin L, Xu L, Peng J. A 90-day subchronic toxicological assessment of dioscin, a natural steroid saponin, in Sprague-Dawley rats. Food Chem Toxicol. 2012;50(5):1279–1287. doi:10.1016/j.fct.2012.02.027
24. Liu Y, Wu K, Shi L, Xiang F, Tao K, Wang G. Prognostic significance of the metabolic marker hexokinase-2 in various solid tumors: a meta-analysis. PLoS ONE. 2016;11(11):e0166230. doi:10.1371/journal.pone.0166230
25. Zhou L, Li M, Yu X, Gao F, Li W. Repression of hexokinases II-mediated glycolysis contributes to piperlongumine-induced tumor suppression in non-small cell lung cancer cells. Int J Biol Sci. 2019;15(4):826–837. doi:10.7150/ijbs.31749
26. Yu M, Chen S, Hong W, et al. Prognostic role of glycolysis for cancer outcome: evidence from 86 studies. J Cancer Res Clin Oncol. 2019;145(4):967–999. doi:10.1007/s00432-019-02847-w
27. Tao X, Yin L, Xu L, Peng J. Dioscin: a diverse acting natural compound with therapeutic potential in metabolic diseases, cancer, inflammation and infections. Pharmacol Res. 2018;137:259–269. doi:10.1016/j.phrs.2018.09.022
28. Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277(9):7610–7618. doi:10.1074/jbc.M109950200
29. Masoud GN, Li W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015;5(5):378–389. doi:10.1016/j.apsb.2015.05.007
30. Shen L, Sun X, Fu Z, Yang G, Li J, Yao L. The fundamental role of the p53 pathway in tumor metabolism and its implication in tumor therapy. Clin Cancer Res. 2012;18(6):1561–1567. doi:10.1158/1078-0432.CCR-11-3040
31. Ciribilli Y, Singh P, Inga A, Borlak J. c-Myc targeted regulators of cell metabolism in a transgenic mouse model of papillary lung adenocarcinoma. Oncotarget. 2016;7(40):65514–65539. doi:10.18632/oncotarget.v7i40
32. Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27(21):7381–7393. doi:10.1128/MCB.00440-07
33. Liu Y, Xiang F, Huang Y, et al. Interleukin-22 promotes aerobic glycolysis associated with tumor progression via targeting hexokinase-2 in human colon cancer cells. Oncotarget. 2017;8(15):25372–25383. doi:10.18632/oncotarget.15913
34. Farrell AS, Sears RC. MYC degradation. Cold Spring Harb Perspect Med. 2014;4:3. doi:10.1101/cshperspect.a014365
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.