")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Dihydromyricetin Attenuates Cerebral Ischemia Reperfusion Injury by Inhibiting SPHK1/mTOR Signaling and Targeting Ferroptosis
Authors Xie J, Zhang T, Li P, Wang D, Liu T, Xu S
Received 16 June 2022
Accepted for publication 31 August 2022
Published 11 September 2022 Volume 2022:16 Pages 3071—3085
DOI https://doi.org/10.2147/DDDT.S378786
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Jiangbo Xie,1,2 Tingting Zhang,3 Peichun Li,3 Dong Wang,2 Tao Liu,2 Shunliang Xu1
1Department of Neurology, The Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 2Department of Neurology, Weifang Traditional Chinese Hospital, Weifang, People’s Republic of China; 3Department of Rehabilitation Medicine, Weifang Traditional Chinese Hospital, Weifang, People’s Republic of China
Correspondence: Shunliang Xu, Department of Neurology, The Second Hospital, Cheeloo College of Medicine, Shandong University, 247 Beiyuan Road, Jinan, Shandong, 250033, People’s Republic of China, Tel +86 15153169998, Email [email protected]
Background: Dihydromyricetin (DHM) exerts protective effects in various brain diseases. The aim of this research was to investigate the biological role of DHM in cerebral ischemia reperfusion (I/R) injury.
Methods: We generated a rat model of cerebral I/R injury by performing middle cerebral artery occlusion/reperfusion (MCAO/R). The neurological score and brain water content of the experimental rats was then evaluated. The infarct volume and extent of apoptosis in brain tissues was then assessed by 2,3,5-triphenyltetrazolium (TTC) and TdT-mediated dUTP nick end labeling (TUNEL) staining. Hippocampal neuronal cells (HT22) were subjected to oxygen-glucose deprivation/reperfusion (OGD/R) and cell counting kit-8 (CCK-8) assays and flow cytometry were performed to detect cell viability and apoptosis. The levels of lipid reactive oxygen species (ROS) and iron were detected and the expression levels of key proteins were assessed by Western blotting.
Results: DHM obviously reduced neurological deficits, brain water content, infarct volume and cell apoptosis in the brain tissues of MCAO/R rats. DHM repressed ferroptosis and inhibited the sphingosine kinase 1 (SPHK1)/mammalian target of rapamycin (mTOR) pathway in MCAO/R rats. In addition, DHM promoted cell viability and repressed apoptosis in OGD/R-treated HT22 cells. DHM also suppressed the levels of lipid ROS and intracellular iron in OGD/R-treated HT22 cells. The expression levels of glutathione peroxidase 4 (GPX4) was enhanced while the levels of acyl-CoA synthetase long-chain family member 4 (ACSL4) and phosphatidylethanolamine binding protein 1 (PEBP1) were reduced in OGD/R-treated HT22 cells in the presence of DHM. Moreover, the influence conferred by DHM was abrogated by the overexpression of SPHK1 or treatment with MHY1485 (an activator of mTOR).
Conclusion: This research demonstrated that DHM repressed ferroptosis by inhibiting the SPHK1/mTOR signaling pathway, thereby alleviating cerebral I/R injury. Our findings suggest that DHM may be a candidate drug for cerebral I/R injury treatment.
Keywords: dihydromyricetin, ferroptosis, SPHK1, mTOR, cerebral ischemia reperfusion injury
Graphical Abstract:
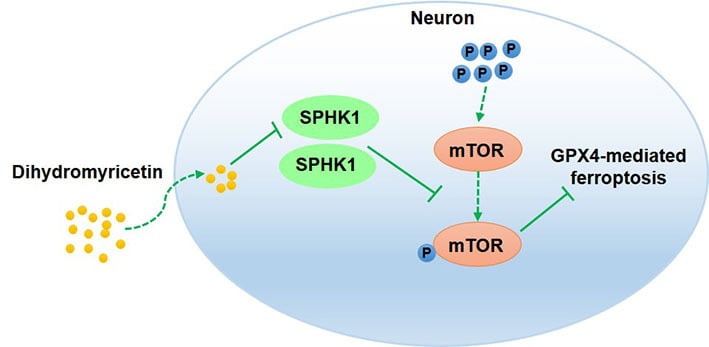
Introduction
Stroke is the second leading cause of disability and death worldwide1 and includes both ischemic stroke and hemorrhagic stroke. Ischemic stroke, defined as brain, retina or spinal cord infarction, develops in more than 85% of all strokes worldwide.2 As reported previously, the incidence of stroke is positively associated with patient age; estimates show that approximately 13.7 million individuals suffer from stroke every year globally.3,4 Stroke creates a significant burden on the physical and mental health of patients and also the health-care system. The rapid restoration of blood flow (reperfusion) via mechanical thrombectomy or thrombolysis is the primary method used to reduce or prevent neuronal damage in patients with ischemia stroke. Paradoxically, reperfusion may aggravate neurological deficits or result in additional damage; this condition is defined as ischemia reperfusion (I/R) injury.5 Previous research showed that cerebral I/R injury is the main pathophysiological mechanism of ischemic stroke.6 The duration and severity of ischemic injury determines the reversibility of the injury response and the ultimate survival rate of the tissues.7 Thus, the development of effective methods is particularly important for the treatment of cerebral I/R injury.
Ferroptosis, a cell death pathway caused by excessive iron-dependent lipid peroxidation, is dominated by antioxidant systems and integrated oxidation.8 The concept of ferroptosis was first described in 2012 and refers to cell death caused by erastin. Erastin-induced ferroptosis occurs mainly by inhibiting the cystine/glutamate antiporter (known as system XC-) and a loss in the activity of glutathione peroxidase 4 (GPX4). System XC- can exchange intracellular glutamate and extracellular cystine. Intracellular cystine is essential for the synthesis of glutathione (GSH).9 Solute carrier family 7 member 11 (SLC7A11) is the light chain of the XC- system. It was previously reported that the downregulation of SLC7A11 ultimately causes a reduction in intracellular cystine and the subsequent depletion of GSH synthesis.10 The reduction of GSH synthesis and the loss of GPX4 activity result in damage to the cellular antioxidant defense system and leads to the accumulation of reactive oxygen species (ROS), eventually causing the activation of ferroptosis.9,11 Accumulating studies have confirmed that ferroptosis is closely related to diseases of the nervous system, including Parkinson’s disease, Alzheimer’s disease, neurotrauma and ischemic stroke.12,13 Ferroptosis has also been detected in the brain tissues of rat models with ischemic stroke; the suppression of ferroptosis by carthamin yellow led to an obvious reduction in the size of the stroke area and improved neurological deficit in a rat model of I/R, thus suggesting that the inhibition of ferroptosis may effectively alleviate I/R-induced brain damage.14
Dihydromyricetin (DHM), a flavonoid compound widely found in Vine tea (Ampelopsis grossedentata), has antioxidant, anti-inflammatory, anti-hyperlipidemia and various other useful properties.15 A previous study found that DHM could improve brain damage in ischemic stroke.16 DHM was also shown to protect mouse hippocampal neuron HT22 cells from oxygen-glucose deprivation/reperfusion (OGD/R)-induced oxidative stress and apoptosis by activating the nuclear factor E2-associated factor 2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathway.16 However, whether DHM can improve cerebral I/R injury in an animal model has yet to be elucidated. Bioinformatics analysis of the potential targets of DHM found that sphingosine kinase 1 (SPHK1) and mammalian target of rapamycin (mTOR) may represent targets of DHM. DHM may play a protective role in cerebral I/R injury by targeting SPHK1 and mTOR. Based on published research relating to cerebral I/R injury, we previously found that suppressing the expression of SPHK1 and enhancing the inactivation of mTOR signaling helped to attenuate I/R-induced brain injury.17 In diabetic nephropathy, DHM was shown to alleviate renal fibrosis and inhibit the mTOR signaling pathway.18 In addition, the overexpression of SPHK1 promotes the phosphorylation of mTOR.19 Eugenol alleviates cerebral I/R injury by inhibiting the mTOR signaling pathway, thus indicating that inhibition of the mTOR signaling pathway may reduce cerebral I/R injury.20 Therefore, in the present study, we investigated whether DHM can repress ferroptosis by inhibiting the SPHK1/mTOR signaling pathway and thus alleviate I/R-induced brain damage.
Materials and Methods
Animals
Sprague Dawley (SD) rats (male, 280 ± 20 g) were provided by the Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). Rats were housed under specific pathogen-free conditions with controlled temperature (26 ± 2°C) and humidity (55 ± 10%). All protocols were authorized by the Ethics Committee of The Second Hospital, Cheeloo College of Medicine, Shandong University (Reference: 2021SDL547). Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research, National Research Council, Washington, DC: National Academy Press, 1996).
Animal Groups
Rats were subjected to middle cerebral artery occlusion/reperfusion (MCAO/R) to induce I/R injury as reported previously.21 Rats were anesthetized by pentobarbital sodium at a dosage of 40 mg/kg by intraperitoneal injection. Rats were first anchored on to an operating table in the supine position. The fur around the incision was shaved and then disinfected. Subsequently, the neck of each rat was incised in the middle to expose the right common carotid artery (CCA), external carotid artery (ECA) and internal carotid artery (ICA). The proximal end of the CCA and ECA were ligated and severed using a 0.285 mm nylon suture. The suture was inserted from the ECA stump through the ICA to reach the MCA. The MCA was then occluded for 2 h to create ischemic conditions. Next, the nylon suture was slowly pulled out to restore blood flow and simulate reperfusion condition. Rats were randomly divided into 6 groups (n = 5): control, sham, MCAO/R, L-DHM (low-dose DHM), M-DHM (medium-dose DHM) and H-DHM (high-dose DHM). The MCAO/R, L-DHM, M-DHM and H-DHM groups all received MCAO/R surgery; the control group did not receive any surgical intervention. Rats in the sham group received the same treatment and exposure in the area of the common carotid artery but without MCAO/R. The control, sham, and MCAO/R groups all received normal saline. The remaining intervention groups received L-DHM (150 mg/kg), M-DHM (200 mg/kg) and H-DHM (250 mg/kg) for 7 days. Rats were administered with DHM by gavage prior to MCAO/R. After modeling, the neurological score of each rat was determined. Then, the rats were euthanized by cervical dislocation. The brain tissues were separated from rats and frozen in liquid nitrogen to await subsequent analysis.
Neurological Score Measurement
After modeling, the neurological deficit of each rat was evaluated at a point 24 hours after the last drug treatment. The neurological score was assessed by the Zea Longa scoring method in a blinded manner.22 Zea Longa scores are classified into five grades, including normal performance, no neurological deficits (0), contralateral forepaws cannot fully extend (1); circling to the opposite side when walking (2); falling to the opposite side when walking (3), and no spontaneous walking and loss of consciousness (4). Rats with a score of 1 to 3 were used for follow-up experiments. Rats with a score of 0 or 4 were removed from analysis (along with those that died).
Brain Water Content
At a point 24 hours after the last treatment, we collected brain tissue from each rat. The brain water content was then determined by the dry weight method.20 Blood stains on the surface of the brain tissues were carefully cleaned with filter paper. Then, the wet weight of the brain tissues was determined with an analytical balance. After that, the brain tissues were placed in an oven and dried at 105°C for 24 h; the dry weight of the brain tissues was then determined. Brain water content (%) = (wet weight - dry weight)/wet weight ×100.
2,3,5-Triphenyltetrazolium Chloride (TTC) Staining
At a point 24 hours after the last drug treatment, TTC staining was carried out to estimate the area of infarction in each rat.23 Brain tissues were embedded in optimal cutting temperature compound (OTC) and frozen at −20°C for 15 min; then, the tissues were cut into 2 mm coronal sections. The coronal sections were incubated with 2% TTC dye reagent at 37°C in darkness. After 20 min of incubation, the sections were washed with PBS to terminate the dyeing and then fixed with 4% paraformaldehyde for 6 h. Non-infarct areas stained red while infarct areas stained off-white. The sections were photographed with a digital camera and the infarct volume and brain volume were determined by Image-Pro Plus 6.0 software. The sum of the infarct area from each section was equal to the total infarct volume. To minimize the error induced by edema, the infarct volume was calculated as follows: Infarct volume = contralateral hemisphere region - non-infarcted region in the ipsilateral hemisphere. Infarct percentage (%) = infarct volume/contralateral hemisphere volume × 100.
TUNEL Staining
Paraffin-embedded brain tissues were sectioned at a thickness of 5 µm. The sections were stained with Biotin-dUTP and Streptavidin-HRP following dewaxing and hydration in accordance with the Colorimetric TdT-mediated dUTP Nick-End Labeling (TUNEL) Apoptosis Assay Kit (Beyotime, Shanghai, China).20 The sections were then stained with diaminobenzidine. TUNEL-positive cells (apoptotic cells) displayed cytoplasmic staining and brown nuclei. The apoptotic cells were assessed with Image J software.
Cell Culture and Treatments
Hippocampal neuronal cells (HT22; Zhong Qiao Xin Zhou Biotechnology Co., Ltd, Shanghai, China) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Hyclone, Logan, UT, USA) at 37°C and 5% CO2 in an incubator. The medium was supplemented with 10% fetal bovine serum (FBS, Hyclone) and 1% penicillin/streptomycin (Solarbio, Beijing, China).20
HT22 cells were administered with OGD/R to mimic cerebral I/R injury in vitro. In brief, HT22 cells were grown in glucose and FBS-free DMEM under specific conditions (37°C, 0.5% O2, and 5% CO2). Then, 12 hours later, the cells were grown in normal culture conditions for another 24 h. HT22 cells were pre-treated with different concentrations of DHM (1, 10, 20 and 30 μM) for 24 h before OGD/R treatment. To induce the activation of mTOR, HT22 cells were treated with 10 μM MHY1485 (an activator of mTOR; MedChem Express, Monmouth Junction, NJ, USA) for 24 h.24
Cell Transfection
For SPHK1 overexpression, the full length of the SPHK1 gene was sub-cloned into the pcDNA3.1 vector to establish a recombinant pcDNA3.1-SPHK1. Specific siRNA targeting GPX4 (si-GPX4) was utilized to silence the GPX4 gene. Empty vector and scramble siRNA (si-NC) served as negative controls (NCs). All plasmids were purchased from GeneChem (Shanghai, China). Cells were transfected with these vectors or siRNAs utilizing Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA).25 Successfully transfected cells were then collected for further use.
Western Blotting
Relative expression levels of proteins were determined by using Western blotting.26 Total protein was extracted from HT22 cells and brain tissues with a Total Protein Extraction Kit (Solarbio) and a Bicinchoninic Acid (BCA) Protein Assay Kit (Solarbio) was used to determine the concentration of proteins. Equal concentrations and volumes of each protein sample were then separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Subsequently, the separated proteins were transferred onto nitrocellulose membranes. The membranes were then incubated at 4°C overnight with a range of primary antibodies, including anti-SPHK1 (1:2000; Thermo Fisher Scientific, Waltham, MA, US), anti-mTOR (1:1000; Thermo Fisher Scientific), anti-p-mTOR (phosphorylated mTOR; 1:2000; Thermo Fisher Scientific), anti-GPX4 (1:5000; Abcam, Cambridge, MA, USA), anti-acyl-CoA synthetase long-chain family member 4 (ACSL4) (1:10000; Abcam) or anti-phosphatidylethanolamine binding protein 1 (PEBP1) (1:1000; Abcam). Subsequently, the membranes were incubated with goat-anti rabbit horseradish peroxidase-conjugated antibody (1:2000; Abcam) at room temperature for 2 h. β-actin antibody (1:5000; Abcam) was used as reference protein for normalization. The protein bands were developed by enhanced chemiluminescence reagent (Beyotime) and data were analyzed by Image J software.
CCK-8 Assay
Cell Counting Kit-8 assays (Beyotime) were used to assess the viability of HT22 cells.27 The HT22 cells were seeded into 96-well plates at a density of 2000 cells/100 μL and were then incubated with 10 μL of CCK-8 reagent at 37°C for 2 h. Finally, the absorbance of the cells at 450 nm was detected using a microplate reader (Thermo Fisher Scientific).
Flow Cytometry
We used an Annexin V-Fluorescein 5-isothiocyanate (Annexin V-FITC) Apoptosis Detection Kit (Beyotime) to detect apoptosis in HT22 cells. The HT22 cells were washed with phosphate buffered saline (PBS) several times. Then, cells were incubated with 5 μL of Annexin V-FITC and 10 μL propidium iodide (PI) in darkness at room temperature for 20 min. Apoptotic cells were then detected using a flow cytometer (ACEA Biosciences, San Diego, CA, USA).
Lipid ROS and Intracellular Iron Measurement
The levels of lipid ROS and intracellular iron were detected with a Lipid Peroxidation Malondialdehyde Assay Kit (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) and an Iron Assay Kit (Sigma-Aldrich) in accordance with the manufacturer’s instructions.28 The absorbance at 523 nm (for lipid ROS) or 593 nm (for intracellular iron) was then detected using a spectrophotometer.
Statistical Analysis
Each assay was performed in triplicate. Data are expressed as mean ± standard deviation and GraphPad Prism 8.0 software (La Jolla, CA, USA) was used for statistical analysis. The Kruskal–Wallis test followed by Dunn’s test was used to compare the differences in neurological score among groups. One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used to analyze statistical differences among multiple groups. P < 0.05 was considered as a significant difference.
Results
DHM Alleviated Cerebral I/R Injury in Rats
Rats in the control and sham groups did not exhibit any neurological deficits. In contrast to the rats in control and sham groups, those in the MCAO/R group exhibited obvious neurological deficits (**P < 0.01), thus indicating a severe neurological injury. The therapeutic effects of DHM on the neurological deficits of MCAO/R rats were dose-dependent (Figure 1A) (#P < 0.05 and ##P < 0.01 vs MCAO/R group). Moreover, compared with sham rats, the brain water content and infarct volume were significantly increased in rats following MCAO/R (**P < 0.01). L-DHM, M-DHM and H-DHM treatment effectively reduced brain water content and cerebral infarct volume in MCAO/R rats in a dose-dependent manner (Figure 1B and C) (#P < 0.05 and ##P < 0.01 vs MCAO/R group). Furthermore, TUNEL staining showed that cellular apoptosis was exacerbated in the brain tissues of MCAO/R rats when compared with sham rats (**P < 0.01). The number of apoptotic cells in MCAO/R rats was notably reduced following treatment with L-DHM, M-DHM and H-DHM in a dose-dependent manner (Figure 1D) (#P < 0.05 and ##P < 0.01 vs MCAO/R group). These findings demonstrated that DHM treatment alleviated cerebral I/R injury in rats.
 |
Figure 1 Dihydromyricetin treatment alleviated cerebral I/R injury in rats. Rats were subjected to MCAO/R to induce cerebral I/R injury and then administered DHM treatment in increasing doses. Sham-operated rats served as controls. (A) The neurological scores of rats. (B) Brain water content of rats. (C) Infarct volume of brain tissues as assessed by TTC staining. (D) Apoptotic cells in brain tissues were examined by TUNEL staining. **P < 0.01, compared with the sham group. #P < 0.05, ##P < 0.01, compared with the MCAO/R group. |
DHM Regulated the SPHK1/mTOR Pathway and Suppressed Ferroptosis in Rats with Cerebral I/R Injury
Figure 2A and B show that SPHK1 was highly expressed in MCAO/R rats with respect to sham rats (**P < 0.01 vs sham group). DHM treatment caused a down-regulation of SPHK1 in MCAO/R rats in a dose-dependent manner (##P < 0.01 vs MCAO/R group). The expression of mTOR did not change in these groups, while the phosphorylation level of mTOR was decreased in MCAO/R rats following DHM treatment in a dose-dependent manner (Figure 2A and C) (**P < 0.01 vs sham group, and #P < 0.05 and ##P < 0.01 vs MCAO/R group). Moreover, the expression levels of ferroptosis-related proteins and GPX4 was significantly decreased while the levels of ACSL4 and PEBP1 were increased in MCAO/R rats (**P < 0.01 vs sham group). DHM treatment enhanced GPX4 expression and reduced ACSL4 and PEBP1 expression in a dose-dependent manner (Figure 2A and D–F) (#P < 0.05 and ##P < 0.01 vs MCAO/R group). These data showed that DHM suppressed the SPHK1/mTOR pathway and ferroptosis in rats with cerebral I/R injury.
 |
Figure 2 Dihydromyricetin repressed SPHK1 and p-mTOR expression and reduced ferroptosis in rats with cerebral I/R injury. (A–F) Rats were subjected to MCAO/R to induce I/R injury or followed by treatment with L-DHM, M-DHM, H-DHM. Sham-operated rats served as controls. Western blotting analysis of SPHK1, p-mTOR/mTOR, GPX4, ACSL4 and PEBP1 in the brain tissues of rats. **P < 0.01, compared with the sham group. #P < 0.05, ##P < 0.01, compared with the MCAO/R group. |
DHM Enhanced Cell Viability and Suppressed Apoptosis in OGD/R-Treated HT22 Cells
The cell viability of HT22 cells was significantly suppressed following OGD/R treatment (**P < 0.01 vs control group). DHM treatment at doses of 10, 20 and 30 μM enhanced the cell viability of OGD/R-treated HT22 cells, especially at a dose of 30 μM (Figure 3A) (#P < 0.05, and ##P < 0.01 vs OGD/R group, and @P < 0.05 vs 20-DHM group). Furthermore, OGD/R treatment notably accelerated apoptosis in HT22 cells; this was effectively rescued by DHM treatment in a dose-dependent manner (Figure 3B) (**P < 0.01 vs control group, and ##P < 0.01 vs OGD/R group, and @@P < 0.01 vs 20-DHM group). Because 30 μM of DHM exhibited the best inhibitory effect on OGD/R-induced apoptosis in HT22 cells, this dose was used to treat HT22 cells in all subsequent experiments. These results showed that DHM enhanced cell viability and suppressed apoptosis in OGD/R-treated HT22 cells. Subsequently, we analyzed the extent of ferroptosis in OGD/R-treated HT22 cells.
 |
Figure 3 Dihydromyricetin enhanced cell viability and repressed apoptosis in OGD/R-treated HT22 cells. HT22 cells were subjected to OGD/R or pretreated with 1, 10, 20 or 30 μM of DHM. CCK-8 assay (A) and flow cytometry (B) were performed to detect viability and apoptosis in HT22 cells. **P < 0.01, compared with the control group. #P < 0.05, ##P < 0.01, compared with the OGD/R group. @P < 0.05, @@P < 0.01, compared with the 20-DHM group. |
DHM Regulated the SPHK1/mTOR Pathway and Reduced Ferroptosis in OGD/R-Treated HT22 Cells
OGD/R treatment caused an excessive accumulation of lipid ROS and increased intracellular iron levels in HT22 cells (**P < 0.01 vs control group). DHM treatment limited the OGD/R-mediated accumulation of lipid ROS and intracellular iron in HT22 cells (#P < 0.05 vs OGD/R group) (Figure 4A and B). Western blotting results showed that SPHK1 expression was notably increased in OGD/R-treated HT22 cells, while SPHK1 expression was rescued in the presence of DHM (**P < 0.01 vs control group, and ##P < 0.05 vs OGD/R group) (Figure 4C and D). The expression of mTOR did not change in HT22 cells following OGD/R or DHM treatment. The phosphorylation of p-mTOR was significantly reduced in OGD/R-treated HT22 cells; this was further suppressed by DHM treatment (**P < 0.01 vs control group, and #P < 0.05 vs OGD/R group) (Figure 4C and E). Moreover, the expression levels of GPX4 were decreased, whereas the levels of ACSL4 and PEBP1 were enhanced in OGD/R-treated HT22 cells. DHM treatment led to an up-regulation of GPX4 and a down-regulation of ACSL4 and PEBP1 in OGD/R-treated HT22 cells (**P < 0.01 vs control group, and #P < 0.05 vs OGD/R group) (Figure 4C and F–H). Collectively, these data show that DHM effectively suppressed the SPHK1/mTOR pathway and ferroptosis in OGD/R-treated HT22 cells.
 |
Figure 4 Dihydromyricetin suppressed SPHK1 and p-mTOR expression and reduced ferroptosis in OGD/R-treated HT22 cells. HT22 cells were subjected to OGD/R or pretreated with 30 μM of DHM. The levels of lipid ROS (A) and intracellular iron (B) were detected. (C–H) Western blotting analysis of SPHK1, p-mTOR/mTOR, GPX4, ACSL4 and PEBP1 in HT22 cells. **P < 0.01, compared with the control group. #P < 0.05, ##P < 0.01, compared with the OGD/R group. |
DHM Reduced Ferroptosis in OGD/R-Treated HT22 Cells by Regulating the Expression of GPX4
OGD/R treatment notably suppressed cell viability and enhanced apoptosis in HT22 cells; this was rescued by DHM treatment (**P < 0.01 vs control group, and ##P < 0.01 vs OGD/R group. The DHM treatment-mediated enhancement of cell viability and the inhibition of apoptosis was suppressed by GPX4 knockdown (@P < 0.05 vs OGD/R+30-DHM+si-NC group) (Figure 5A and B). Moreover, the levels of lipid ROS and intracellular iron were significantly increased in HT22 cells following OGD/R treatment (**P < 0.01 vs control group). DHM treatment reduced the levels of lipid ROS and intracellular iron in OGD/R-treated HT22 cells while these effects were abrogated by GPX4 knockdown (##P < 0.01 vs OGD/R group, and @P < 0.05 vs OGD/R+30-DHM+si-NC group) (Figure 5C and D). In addition, GPX4 expression was down-regulated while ACSL4 and PEBP1 expression was up-regulated in OGD/R-treated HT22 cells (**P < 0.01 vs control group). GPX4 deficiency impaired the DHM-mediated up-regulation of GPX4 and the down-regulation of ACSL4 and PEBP1 in OGD/R-treated HT22 cells (##P < 0.01 vs OGD/R group, and @P < 0.05 vs OGD/R+30-DHM+si-NC group) (Figure 5E–H). Collectively, these data indicated that DHM reduced ferroptosis in OGD/R-treated HT22 cells by regulating the expression of GPX4.
 |
Figure 5 Dihydromyricetin reduced ferroptosis in OGD/R-treated HT22 cells by regulating GPX4 expression. HT22 cells were transfected with si-GPX4 or si-NC, followed by OGD/R or combined with 30 μM of DHM treatment. CCK-8 assay (A) and flow cytometry (B) were performed to detect cell viability and apoptosis in HT22 cells. The levels of lipid ROS (C) and intracellular iron (D) were detected. (E–H) Western blotting analysis of GPX4, ACSL4 and PEBP1 in HT22 cells. **P < 0.01, compared with the control group. ##P < 0.01, compared with the OGD/R group. @P < 0.05, @@P < 0.01, compared with the OGD/R+30-DHM+si-NC group. |
DHM Reduced Ferroptosis in OGD/R-Treated HT22 Cells by Inhibiting the SPHK1/mTOR Signaling Pathway
DHM treatment enhanced cell viability and suppressed apoptosis in OGD/R-treated HT22 cells, although SPHK1 overexpression and MHY1485 treatment weakened the effects of DHM (**P < 0.01 vs control group, ##P < 0.01 vs OGD/R group, and @P < 0.05 vs OGD/R+30-DHM+vector group) (Figure 6A and B). DHM treatment also reduced the levels of lipid ROS and intracellular iron in OGD/R-treated HT22 cells; these effects were abolished by SPHK1 overexpression or MHY1485 treatment (**P < 0.01 vs control group, ##P < 0.01 vs OGD/R group, and @P < 0.05 and @@P < 0.01 vs OGD/R+30-DHM+vector group) (Figure 6C and D). Moreover, Western blotting revealed that the expression of SPHK1 was elevated while the expression of p-mTOR was decreased in OGD/R-treated HT22 cells. DHM treatment caused a reduction of SPHK1 expression and a further reduction of p-mTOR expression in OGD/R-treated HT22 cells (*P < 0.05 and **P < 0.01 vs control group). The impacts of DHM treatment on SPHK1 and p-mTOR expression were rescued by SPHK1 overexpression or MHY1485 treatment (##P < 0.01 vs OGD/R group, and @@P < 0.01 vs OGD/R+30-DHM+vector group) (Figure 6E–G). DHM suppressed OGD/R-induced ferroptosis in HT22 cells by inhibiting the SPHK1/mTOR signaling pathway.
 |
Figure 6 Dihydromyricetin limited OGD/R-induced ferroptosis by inhibiting SPHK1/mTOR signaling pathway. HT22 cells were transfected with pcDNA3.1-SPHK1/Vector or treated with MHY1485, followed by OGD/R or combined with 30 μM of DHM treatment. CCK-8 assay (A) and flow cytometry (B) were performed to detect cell viability and apoptosis in HT22 cells. The levels of lipid ROS (C) and intracellular iron (D) were detected. (E–G) Western blotting analysis of SPHK1, p-mTOR/ mTOR in HT22 cells. **P < 0.01, compared with the control group. ##P < 0.01, compared with the OGD/R group. @P < 0.05, @@P < 0.01, compared with the OGD/R+30-DHM group. |
Discussion
Cerebral I/R injury is one of the main pathophysiological mechanisms of ischemic stroke. In the current study, we verified the functions and molecular mechanism of DHM in cerebral I/R injury. The results of in vivo experiments revealed that DHM effectively reduced the neurological deficits and brain injury of MCAO/R rats. In cellular experiments, DHM promoted cell viability and suppressed the cellular apoptosis of OGD/R-treated HT22 cells. The activity of the SPHK1/mTOR signaling pathway and the extent of ferroptosis in MCAO/R rats and in OGD/R-treated HT22 cells was inhibited by DHM treatment; these effects were partly rescued by GPX4 knockdown, SPHK1 overexpression or MHY1485 treatment. Therefore, these data demonstrated that DHM reduced ferroptosis and alleviated cerebral I/R injury by inhibiting the SPHK1/mTOR signaling pathway.
DHM is one of the main effective ingredients in vine tea. Due to its anti-oxidative stress, anti-inflammatory activity, anti-cancer, antimicrobial, anti-apoptosis and neuroprotective activities, DHM exerts protective effects on I/R injury, diabetic cardiomyopathy, myocardial remodeling, neurodegenerative diseases, atherosclerosis and other multiple disorders.15,29,30 Huang et al reported that DHM represses palmitic acid-induced oxidative stress in hepatocytes by activating autophagy, thus contributing to the development of non-alcoholic steatohepatitis.31 DHM was previously shown to induce cardioprotective effects in rats with myocardial I/R injury-induced acute myocardial infarction through the PI3K/Akt and HIF-1α signaling pathways.32 In addition, a previous study confirmed that DHM inhibits oxidative stress and apoptosis in OGD/R‑induced HT22 cells, thus indicating the neuroprotective role of DHM.16 In current study, we attempted to determine the functions of DHM in cerebral I/R injury by establishing a rat model of typical cerebral I/R injury through MCAO/R. The rat models were subsequently treated with different doses of DHM. HT22 cells were subjected to OGD/R to mimic cerebral I/R injury in vitro. Our present work demonstrated that DHM alleviated neurological injury, brain water content, cerebral infarction and cellular apoptosis in MCAO/R rats. DHM was also shown to promote cell viability and repress apoptosis in OGD/R-treated HT22 cells. Collectively, these data indicated that DHM may exert protective effects on cerebral I/R injury.
Ferroptosis is a newly discovered form of regulatory cell death that is usually accompanied by a large amount of iron accumulation during the process of cell death. Ferroptosis is differ from traditional cell death processes such as necrosis, apoptosis, and pyroptosis.33,34 The deposition of lipid ROS is a hallmark event for ferroptosis.34 In the current study, the accumulation of ROS and iron in OGD/R-treated HT22 cells was partly reversed by DHM treatment at a dose of 30 mM. A previous study suggested that amyloid-β deposition and hippocampal dendritic spine loss could be improved by ferrostatin-1 (a well-known inhibitor of ferroptosis), thereby alleviating brain damage caused by cardiac I/R injury.35 Mitochondrial ferritin could reduce lipid peroxidation and stabilize GSH to inhibit ferroptosis, thus attenuating brain injury and neurological deficits in rats with cerebral I/R.36 Previous studies suggested that the inhibition of ferroptosis may exert effects against cerebral I/R injury. In the present study, we found that the expression of ferroptosis-related proteins (ACSL4 and PEBP1) was increased while that of GPX4 expression was decreased in the brain tissues of MCAO/R rats and in OGD/R-treated HT22 cells. The loss of GPX4 activity is a crucial hallmark of ferroptosis. As an antioxidant enzyme, GSH is a necessary co-factor for the normal function of GPX4. The suppression of GSH-GPX4-dependent antioxidant defense could result in the accumulation of lipid hydroperoxides and then induce the canonical pathway of ferroptosis.8,37 During the process of ferroptosis, ACSL4 and PEBP1 participate in the activation of lipid hydroperoxides and thus facilitate ferroptosis.37 Our results demonstrated that DHM treatment effectively suppressed ferroptosis; this was reflected by the increased GPX4 expression and decreased expression of ACSL4 and PEBP1. In particular, the DHM-induced increase in cell viability and decrease in ferroptosis was partly limited by GPX4 silencing. Thus, DHM can alleviate cerebral I/R injury by inhibiting ferroptosis.
To further investigate the regulatory mechanism underlying the ability of DHM to inhibit ferroptosis and improve cerebral I/R injury, we predicted the potential molecular targets of DHM using the TCMID database (http://119.3.41.228:8000/tcmid/ingredient/32748/) and found that SPHK1 and mTOR were two candidate targets of DHM. Previous research confirmed that miR-19a/b-3p could enhance inflammation and cell death during cerebral I/R by regulating the SPHK1-mediated signaling pathway.38 SPHK1 was also found to be expressed at high levels in the microglial cells of mice with cerebral I/R injury; the inhibition of SPHK1 reduced the inflammatory response and therefore hindered the progression of cerebral I/R injury.39 The overexpression of SPHK1 can aggravate cerebral I/R injury. In addition, annexin A1 exerts potent anti-inflammatory effects and modulates the polarization of microglia/macrophages by repressing the AMPK-mTOR signaling pathway, thereby ameliorating cerebral I/R injury.40 During cancer progression, SPHK1 overexpression was shown to increase the levels of mTOR phosphorylation;19 however, whether SPHK1 can regulate mTOR phosphorylation during cerebral I/R injury remain unclear. In the present research, our data showed that the expression of SPHK1 increased while the levels of mTOR phosphorylation declined in MCAO/R rats and OGD/R-treated HT22 cells; these results were consistent with previous studies. Some previous studies also indicated that the level of mTOR phosphorylation was lower in an animal model of MCAO/R or OGD/R-induced neurons than that in controls.41,42 In the present study, we found that DHM treatment suppressed the expression of SPHK1 and further reduced the level of mTOR phosphorylation. We noted that the continuing decline of mTOR phosphorylation was not incidental. In a previous study, Sun et al also showed that the reduced levels of mTOR phosphorylation were further decreased following drug administration in MCAO/R rats and OGD/R-treated neurons.20 Based on these results, we hypothesized that DHM attenuates cerebral I/R injury by suppressing the SPHK1/mTOR signaling pathway. To validate this hypothesis, SPHK1 was overexpressed in OGD/R-treated HT22 cells and MHY1485 (an activator of mTOR) was used to treat the cells. The improvement of DHM with regards to OGD/R-induced cell apoptosis and ferroptosis was partly rescued by SPHK1/mTOR signaling activation.
In conclusion, this research demonstrated that DHM repressed ferroptosis by inhibiting the SPHK1/mTOR signaling pathway, thereby alleviating cerebral I/R-induced brain damage. Thus, our data suggests that DHM may be a candidate drug for cerebral I/R injury treatment.
Funding
This study was financially supported by the Shandong Provincial Natural Science Foundation, China (ZR2015HM024 and 2019GSF108066), IIFSDU, and SFR for ROCS, SEM.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Prust ML, Saylor D, Zimba S, et al. Inpatient management of acute stroke of unknown type in resource-limited settings. Stroke. 2022;53(3):e108–e117. doi:10.1161/STROKEAHA.121.037297
2. Mollet I, Marto JP, Mendonça M, Baptista MV, Vieira HLA. Remote but not distant: a review on experimental models and clinical trials in remote ischemic conditioning as potential therapy in ischemic stroke. Mol Neurobiol. 2022;59(1):294–325. doi:10.1007/s12035-021-02585-6
3. Liao S, Apaijai N, Chattipakorn N, Chattipakorn SC. The possible roles of necroptosis during cerebral ischemia and ischemia/reperfusion injury. Arch Biochem Biophys. 2020;695:108629. doi:10.1016/j.abb.2020.108629
4. Campbell BCV, De Silva DA, Macleod MR, et al. Ischaemic stroke. Nat Rev Dis Primers. 2019;5(1):70. doi:10.1038/s41572-019-0118-8
5. Stegner D, Klaus V, Nieswandt B. Platelets as modulators of cerebral ischemia/reperfusion injury. Front Immunol. 2019;10:2505. doi:10.3389/fimmu.2019.02505
6. Sun M, Jin H, Sun X, et al. Free radical damage in ischemia-reperfusion injury: an obstacle in acute ischemic stroke after revascularization therapy. Oxid Med Cell Longev. 2018;2018:3804979. doi:10.1155/2018/3804979
7. Vadlamani A, Albrecht J. Severity of traumatic brain injury in older adults and risk of ischemic stroke and depression. J Head Trauma Rehabil. 2020;35(5):E436–E440. doi:10.1097/HTR.0000000000000561
8. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–2081. doi:10.1080/15548627.2020.1810918
9. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
10. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. doi:10.1038/cdd.2015.158
11. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi:10.1016/j.tcb.2015.10.014
12. Weiland A, Wang Y, Wu W, et al. Ferroptosis and its role in diverse brain diseases. Mol Neurobiol. 2019;56(7):4880–4893. doi:10.1007/s12035-018-1403-3
13. Ren J, Sun X, Yan X, Guo Z, Yang Y. Ferroptosis in Neurological Diseases. Front Cell Neurosci. 2020;14:218. doi:10.3389/fncel.2020.00218
14. Guo H, Zhu L, Tang P, et al. Carthamin yellow improves cerebral ischemia‑reperfusion injury by attenuating inflammation and ferroptosis in rats. Int J Mol Med. 2021;47(4):52. doi:10.3892/ijmm.2021.4885
15. Zhang J, Chen Y, Luo H, et al. Recent update on the pharmacological effects and mechanisms of dihydromyricetin. Front Pharmacol. 2018;9:1204. doi:10.3389/fphar.2018.01204
16. Zhang Q, Wang J, Zhang H, Zeng T. Dihydromyricetin inhibits oxidative stress and apoptosis in oxygen and glucose deprivation/reoxygenation‑induced HT22 cells by activating the Nrf2/HO‑1 pathway. Mol Med Rep. 2021;23(6):397. doi:10.3892/mmr.2021.12036
17. Zhang M, Zhou D, Ouyang Z, Yu M, Jiang Y. Sphingosine kinase 1 promotes cerebral ischemia-reperfusion injury through inducing ER stress and activating the NF-κB signaling pathway. J Cell Physiol. 2020;235(10):6605–6614. doi:10.1002/jcp.29546
18. Guo L, Tan K, Luo Q, Bai X. Dihydromyricetin promotes autophagy and attenuates renal interstitial fibrosis by regulating miR-155-5p/PTEN signaling in diabetic nephropathy. Bosn J Basic Med Sci. 2020;20(3):372–380. doi:10.17305/bjbms.2019.4410
19. Kim M, Park J, Lee E, et al. C16‑ceramide and sphingosine 1‑phosphate/S1PR2 have opposite effects on cell growth through mTOR signaling pathway regulation. Oncol Rep. 2018;40(5):2977–2987. doi:10.3892/or.2018.6689
20. Sun X, Wang D, Zhang T, et al. Eugenol attenuates cerebral ischemia-reperfusion injury by enhancing autophagy AMPK-mTOR-P70S6K pathway. Front Pharmacol. 2020;11:84. doi:10.3389/fphar.2020.00084
21. Zhang Y, Zhang Y, Jin X, et al. The role of astragaloside IV against cerebral ischemia/reperfusion injury: suppression of apoptosis via promotion of P62-LC3-autophagy. Molecules. 2019;24(9):1838. doi:10.3390/molecules24091838
22. Zhang Y, Shan Z, Zhao Y, Ai Y. Sevoflurane prevents miR-181a-induced cerebral ischemia/reperfusion injury. Chem Biol Interact. 2019;308:332–338. doi:10.1016/j.cbi.2019.06.008
23. Yin D, Zhou C, Kusaka I, et al. Inhibition of apoptosis by hyperbaric oxygen in a rat focal cerebral ischemic model. J Cereb Blood Flow Metab. 2003;23(7):855–864. doi:10.1097/01.WCB.0000073946.29308.55
24. Rakhmanova V, Jin M, Shin J. Inhibition of mast cell function and proliferation by mTOR activator MHY1485. Immune Netw. 2018;18(3):e18. doi:10.4110/in.2018.18.e18
25. Liu P, Zou D, Chen K, et al. Dihydromyricetin improves hypobaric hypoxia-induced memory impairment via modulation of SIRT3 signaling. Mol Neurobiol. 2016;53(10):7200–7212. doi:10.1007/s12035-015-9627-y
26. Qiu P, Dong Y, Li B, et al. Dihydromyricetin modulates p62 and autophagy crosstalk with the Keap-1/Nrf2 pathway to alleviate ethanol-induced hepatic injury. Toxicol Lett. 2017;274:31–41. doi:10.1016/j.toxlet.2017.04.009
27. Ma H, Wang X, Zhang W, et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxid Med Cell Longev. 2020;2020:9067610. doi:10.1155/2020/9067610
28. Li L, Hao Y, Zhao Y, et al. Ferroptosis is associated with oxygen-glucose deprivation/reoxygenation-induced Sertoli cell death. Int J Mol Med. 2018;41(5):3051–3062. doi:10.3892/ijmm.2018.3469
29. Li H, Li Q, Liu Z, et al. The versatile effects of dihydromyricetin in health. Evid Based Complement Alternat Med. 2017;2017:1053617. doi:10.1155/2017/1053617
30. Martínez-Coria H, Mendoza-Rojas MX, Arrieta-Cruz I, López-Valdés HE. Preclinical research of dihydromyricetin for brain aging and neurodegenerative diseases. Front Pharmacol. 2019;10:1334. doi:10.3389/fphar.2019.01334
31. Huang L, Zeng X, Li B, et al. Dihydromyricetin attenuates palmitic acid-induced oxidative stress by promoting autophagy via SIRT3-ATG4B signaling in hepatocytes. Nutr Metab. 2021;18(1):83. doi:10.1186/s12986-021-00612-w
32. Liu S, Ai Q, Feng K, Li Y, Liu X. The cardioprotective effect of dihydromyricetin prevents ischemia-reperfusion-induced apoptosis in vivo and in vitro via the PI3K/Akt and HIF-1α signaling pathways. Apoptosis. 2016;21(12):1366–1385. doi:10.1007/s10495-016-1306-6
33. Hirschhorn T, Stockwell B. The development of the concept of ferroptosis. Free Radic Biol Med. 2019;133:130–143. doi:10.1016/j.freeradbiomed.2018.09.043
34. Li J, Cao F, Yin HL, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. doi:10.1038/s41419-020-2298-2
35. Liao S, Apaijai N, Luo Y, et al. Cell death inhibitors protect against brain damage caused by cardiac ischemia/reperfusion injury. Cell Death Discov. 2021;7(1):312. doi:10.1038/s41420-021-00698-4
36. Wang P, Cui Y, Ren Q, et al. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021;12(5):447. doi:10.1038/s41419-021-03725-5
37. Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and cancer: mitochondria meet the ”Iron Maiden” cell death. Cells. 2020;9(6):1505. doi:10.3390/cells9061505
38. Zhou F, Wang Y, Zhang C, Wu B. miR-19a/b-3p promotes inflammation during cerebral ischemia/reperfusion injury via SIRT1/FoxO3/SPHK1 pathway. J Neuroinflammation. 2021;18(1):122. doi:10.1186/s12974-021-02172-5
39. Sun W, Ding Z, Xu S, Su Z, Li H. Crosstalk between TLR2 and Sphk1 in microglia in the cerebral ischemia/reperfusion-induced inflammatory response. Int J Mol Med. 2017;40(6):1750–1758. doi:10.3892/ijmm.2017.3165
40. Xu X, Gao W, Li L, et al. Annexin A1 protects against cerebral ischemia-reperfusion injury by modulating microglia/macrophage polarization via FPR2/ALX-dependent AMPK-mTOR pathway. J Neuroinflammation. 2021;18(1):119. doi:10.1186/s12974-021-02174-3
41. Liu T, Han S, Dai Q, et al. IL-17A-mediated excessive autophagy aggravated neuronal ischemic injuries via Src-PP2B-mTOR pathway. Front Immunol. 2019;10:2952. doi:10.3389/fimmu.2019.02952
42. Zhang P, Guo ZF, Xu YM, Li YS, Song JG. N-Butylphthalide (NBP) ameliorated cerebral ischemia reperfusion-induced brain injury via HGF-regulated TLR4/NF-κB signaling pathway. Biomed Pharmacother. 2016;83:658–666. doi:10.1016/j.biopha.2016.07.040
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.