")
Back to Journals » OncoTargets and Therapy » Volume 13
Dihydroartemisinin Inhibits the Proliferation, Colony Formation and Induces Ferroptosis of Lung Cancer Cells by Inhibiting PRIM2/SLC7A11 Axis
Authors Yuan B, Liao F, Shi ZZ, Ren Y, Deng XL, Yang TT, Li DY, Li RF, Pu DD, Wang YJ, Tan Y, Yang Z, Zhang YH
Received 5 February 2020
Accepted for publication 31 August 2020
Published 27 October 2020 Volume 2020:13 Pages 10829—10840
DOI https://doi.org/10.2147/OTT.S248492
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Bing Yuan,1,* Feng Liao,1,* Zhi-Zhou Shi,2 Yuan Ren,1 Xiao-Li Deng,1 Ting-Ting Yang,1 Deng-Yuan Li,1 Ru-Fang Li,1 Dan-Dan Pu,1 Yu-Jue Wang,1 Yan Tan,1 Zhen Yang,1 Yun-Hui Zhang1
1Department of Pulmonary and Critical Care Medicine, The First People’s Hospital of Yunnan Province, The Affiliated Hospital of Kunming University of Science and Technology, Kunming, Yunnan, People’s Republic of China; 2Medical School, Kunming University of Science and Technology, Kunming, Yunnan, People’s Republic of China
*These authors contributed equally to this work.
Correspondence: Zhen Yang
Department of Pulmonary and Critical Care Medicine, The First People’s Hospital of Yunnan Province, The Affiliated Hospital of Kunming University of Science and Technology, Kunming, Yunnan, People’s Republic of China
Email [email protected]
Yun-Hui Zhang
Department of Pulmonary and Critical Care Medicine, The First People’s Hospital of Yunnan Province, The Affiliated Hospital of Kunming University of Science and Technology, Kunming, Yunnan, People’s Republic of China
Email [email protected]
Objective: Lung cancer is the first leading cause of cancer-related deaths both worldwide and in China and threatens human health and quality of life. New drugs and therapeutic methods are urgently needed. Our study evaluated the roles of dihydroartemisinin (DHA) in lung cancer and further explored its underlying mechanisms.
Methods: CCK-8, colony formation and trypan blue exclusion assays were used to detect the cell viability, colony formation ability and cell death. qRT-PCR and Western blotting assays were applied to analyze the expressions of key molecules.
Results: DHA inhibited the proliferation and colony formation abilities and enhanced the cell death and induced ferroptosis of lung NCI-H23 and XWLC-05 cancer cells. DHA reduced PRIM2 expression and silencing PRIM2 mimicked the inhibitory roles on proliferation and colony formation and promotive roles on cell death and ferroptosis of DHA in lung NCI-H23 and XWLC-05 cancer cells. We further found that DHA treatment and loss of PRIM2 reduced the GSH level and increased the cellular lipid ROS and mitochondrial MDA levels, and further downregulated the expressions of SLC7A11 and β-catenin in lung cancer cells, respectively. Exogenetic overexpression of PRIM2 recovered the inhibitory effects of DHA on proliferation and colony formation in lung NCI-H23 cancer cells, meanwhile loss of PRIM2 sensitizes NCI-H23 cells to DHA therapy. In vivo experiment further showed that DHA treatment significantly suppressed the tumor growth and downregulated PRIM2 and SLC7A11.
Conclusion: Our study suggested that DHA inhibited the proliferation, colony formation and enhanced cell death and induced ferroptosis of lung cancer cells by inactivating PRIM2/SLC7A11 axis. Loss of PRIM2 induced ferroptosis might developed to be a novel therapeutic method in lung cancer therapy.
Keywords: dihydroartemisinin, lung cancer, proliferation, ferroptosis, PRIM2, SLC7A11
Introduction
Lung cancer is the first leading cause of cancer-related deaths both worldwide and in China1,2 and threatens human health and quality of life. Every year, 1.8 million people are diagnosed with lung cancer, 1.6 million people die, and 5-year survival rates vary from 4% to 18% depending on stage and regional differences.3,4 Xuanwei City, located in the northeast of Yunnan Province, is one of the regions with the highest incidence and mortality of lung cancer in China, which seriously endangers the health of local people and causes serious social and economic burden. From 2011 to 2013, the number of lung cancer deaths in Xuanwei accounted for 63.03% of all cancer deaths, the age standardized mortality rate for men was 82.53/100,000 and for women was 62.62/100,000, three times and six times higher than that in other regions of the country, respectively.5 The incidence of non-smoking women in Xuanwei is the highest in China. In some villages, the mortality rate of female patients is as high as 400/100,000, which is significantly higher than the national average (19.84/100,000).6 On the basis of traditional treatment, it is urgently necessary to develop traditional Chinese medicine and find new drugs and therapeutic methods.
Dihydroartemisinin (DHA) is the active compound of Artemisia annua and one of metabolites of Artemisinin.7 During the clinical application of artemisinin and its analogues, it was found that DHA showed good anti-tumor ability in many types of cancers include lung cancer, in addition to the traditional anti-malarial effect. The anti-tumor effect of DHA may result in tumor cell growth inhibition and apoptosis by regulating genes and proteins related to growth signal, apoptosis, proliferation, angiogenesis, tissue invasion and metastasis through different signal pathways.8 For example, DHA combined with gefitinib can significantly down regulate the expression level of G2/M regulatory protein (including cyclin B1 and CDK1) in NSCLC (NCI-H1975) cells and inhibit the formation of cdk1-cyclinb1 complex, which is essential for the initiation of mitosis in some organisms and leads to cell cycle arrest in G2/M phase stagnation, inhibition of cell proliferation.9 Apoptosis is a process mediated by the balance between Bax and Bcl-2 family genes. DHA induces apoptosis by regulating Bax/Bcl-2 ratio.8 Tumor angiogenesis is a sign of tumor malignant transformation. Inhibition of neovascularization can reduce the oxygen and nutrition supply of tumor cells, thus preventing tumor growth. DHA can significantly reduce the expression of many angiogenesis genes in cancer cells, so as to reduce angiogenesis and vascular density.10–12
Several studies have shown that another important anti-tumor mechanism of DHA is closely related to the iron content in tumor cells, mainly Fe2+,13,14 and its mechanism mainly includes the following three aspects: a. Oxidative stress reaction: tumor cells are vulnerable to damage by free radicals (ROS), high oxidative stress is a common anti-tumor characteristic of anti-tumor drugs.15 The divalent iron in tumor cells can activate and catalyze the cleavage of DHA molecular oxygen bridge, which produce a large number of highly alkylated carbon centered free radicals and reactive oxygen species, and the reactive oxygen species and other active intermediates can damage DNA of tumor cells.16 Hydrogen peroxide, a common oxidant, can enhance the antitumor effect of DHA, while antioxidant vitamin E can weaken the antitumor effect of DHA.17 N-tert-butyl-a-phenylnitrone (PBN), an oxygen free radical scavenger, can reduce the antitumor activity of DHA.18 b. Disturbed the balance of iron ions in cells: DHA can decreased the Levels of cancer cell-surface Transferrin receptor 1 (TFR1), leading to the decline of TFR1 mediated iron uptake and deficiency of cellular iron stores, which indicate that DHA can lead to the deficiency of Fe2+ in cancer cells and affect the proliferation of tumor cells.19 The antitumor effect of DHA was obviously weakened when iron ion was chelated by desferrioxamine.18 C. Ferroptosis: Ferroptosis is a new form of programmed cell death with characteristic accumulation of reactive oxygen species (ROS) which are generated by lipid peroxidation and iron accumulation. DHA can induce lysosomal degradation of ferritin in an autophagy-independent manner, increasing the cellular free iron level and causing cells to become more sensitive to ferroptosis.20
PRIM2, a large subunit of DNA primer enzyme, is located in 6p11.1 – p12 of human chromosome. It encodes 58 kDa protein (P58) containing 4Fe-4S cofactor, which can form the heterodimeric DNA primase enzyme (P49 · P58) with PRIM1 (P49), a small subunit of DNA primer enzyme. The DNA primase plays a key role in both the initiation of DNA replication and the synthesis of Okazaki fragments for lagging strand synthesis,21,22 previous study reported that PRIM2 was upregulated by SIX1 (sine oculis homeobox homolog 1) in cervical cancer, which enhanced DNA synthesis, accelerated G1 to S phase progression, and promoted the proliferation of cervical cancer cells and the growth of cervical cancer.23 In addition, some studies have confirmed that PRIM2 plays a critical role in DNA damage repair, transcription and other cell functions. After DNA damage of HeLa cells, PRIM2 can combine with hXRCC1, inhibit G1/S checkpoint, delay S-phase transformation, and promote DNA damage repair.24 These results indicate that PRIM2 is the main regulator of primer enzyme, which directly determines the start and end of primer synthesis and acts as a “molecular brake” in DNA replication.25 But up to present the role of PRIM2 in carcinogenesis especially lung cancer was largely unknown.
Previous studies have shown that DHA treatment can down regulate the expression of cyclin D in NSCLC (A549 and H1299) cells, resulting in G1 phase cell cycle arrest.26 In addition, DHA can inhibit the growth of tumor cells in an iron ion-dependent manner, and ferritin may be the drug target of DHA. As a key member of DNA replicase group, PRIM2, in line with DHA, also targets G1/S checkpoint; At the same time, PRIM2 is rich in Fe-S clusters and is the key molecule to initiate DNA replication. Therefore, PRIM2 may be the core target of DHA and other artemisinins to interfere with DNA replication. Based on the above theoretical and experimental evidence, we hypothesize that DHA may inhibit the proliferation of lung cancer by interfering with PRIM2 signaling. In order to test the hypothesis, the Xuanwei specific lung cancer cell line XWLC-05 and lung cancer cell line NCI-H23 with high expression of PRIM2 were used as the research objects. The aim of this study is to confirm that PRIM2 is a possible target gene of DHA and the possible molecular mechanism of DHA inhibiting the proliferation of lung cancer cells.
Materials and Methods
Cell Culture and Treatments
The human lung cancer cell lines including NCI-H23 which was purchased from the American Type Culture Collection (Rockville, MD) and XWLC-05 which was obtained from Yunnan Cancer Institute were cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS, Gibco, Carlsbad, USA), penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37 °C with 5% CO2. The use of XWLC-05 cell line was approved by the ethics committee of the Yunnan Cancer Institute. All above cell lines were authenticated by short tandem repeat profiling and were tested for mycoplasma contamination routinely.
The PRIM2 siRNAs and negative control sequences were designed and synthesized by GeneParma Co., Ltd (Shanghai, China). Transfection of 50 nM PRIM2 siRNAs and negative control siRNA was performed using lipofectamine 3000 (Thermo scientific, USA) according to manufacturer’s instruction. The PRIM2 shRNA vectors (sh1 and sh2) and negative control vector were obtained from GeneParma Co., Ltd (Shanghai, China) and the transfected cells were selected by G418 for 4 weeks. We conducted the pcDNA3.1-PRIM2 vector (OE vector) and the transfected cells were selected by G418 for 4 weeks.
Total RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from lung cancer cells and tumor tissues collected from in vivo experiment using the RNeasy Mini Kit (Qiagen, Hiden, Germany) according to the manufacturer’s instruction.
The expression levels of PRIM2 were examined by qRT-PCR using Power SYBRTM Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA). The PCR conditions are as follows: 1 cycle of 2 min at 50 °C and 2 min at 95 °C, 40 cycles of 15 sec at 95 °C and 1 min at 60 °C. The relative gene expressions were normalized to an endogenous reference glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and those relative to the calibrator were given by the formula 2−ΔΔCt. The following primer pairs are used for qRT-PCR: PRIM2 forward primer, 5ʹ-CGGCTTGCTTATTGCCAGTCT-3ʹ, reverse primer, 5ʹ-CAATCTCCTGTTCTCGAAGAGTC-3ʹ; GAPDH forward primer, 5ʹ-ACAACTTTGGTATCGTGGAAGG-3ʹ, reverse primer, 5ʹ-GCCATCACGCCACAGTTTC-3ʹ.
Western Blotting Analysis
Total proteins were extracted using a RIPA protein extraction buffer (Beyotime, Shanghai, China), and the concentration was detected using the BCA Protein Assay Kit (Beyotime, Shanghai, China). Equal amounts (7–10 μg) of proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore Corporation, Billerica, MA, USA). After blocking in 5% skimmed milk (in PBS) for 1 h at room temperature, the membrane was incubated with primary antibodies at 4 ºC overnight. Detection was done by peroxidase-conjugated secondary antibodies (KPL, Gaithersburg, MD, USA) and chemiluminescence (Millipore Corporation, Temecula, CA, USA). Anti-PRIM2 antibody (1:2000, ab241990), anti-SLC7A11 antibody (1:1000, ab175186), anti-β-catenin antibody (1:5000, ab32572) and anti-β-actin antibody (1:1000, ab8226) were purchased from Abcam, Inc.
Cell Proliferation and Colony Formation Assays
Cell proliferation was detected by using Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instruction. In colony formation assay, the 103 cells were seeded into 6-well plates and cultivated for 14 days in RPMI 1640 with 10% FBS. After removing the medium, cells were washed using PBS and stained using crystal violet (Sigma-Aldrich, MO, USA) for 30 min at room temperature. Colonies were visualized and quantitated.
Cell Death Assay
Cell death was detected by staining with trypan blue as described previously.27 Trypan blue could penetrate into a dead cell but could not stain the live cell.
GSH and Lipid ROS Assays
Cellular GSH levels in lung cancer cells were detected using a GSH colorimetric detection kit (BioVision Inc., Milpitas, CA, USA) according to the manufacturer’s instruction. Lipid ROS levels in lung cancer cells were assessed by using BODIPY 581/591 C11 (lipid peroxidation sensor, Thermo scientific, USA) according to the manufacturer’s instruction. Mitochondria were prepared by using the mitochondrial isolation kit (Thermo scientific, USA). Malondialdehyde (MDA) is an end product of lipid peroxidation and evaluated by using lipid peroxidation (MDA) assay kit (Sigma-Aldrich, USA) according to the manufacturer’s instruction.
In vivo Experiment
The Animal Ethics Committee of Kunming University of Science and Technology approved the animal experiments. Eight 3-4-week-old female nude mice (18–20g) were used in our study. 5×106 NCI-H23 cells were subcutaneously injected. When tumors reached around 25 mm3 in size (about 1 week), the mice were randomly divided into two groups: NC group (n = 4, saline) and DHA group (n = 4, 30mg/kg). The mice were closely monitored. Tumor size was detected every week. About 4 weeks later, the mice were euthanized, and the tumors were collected for analysis.
Statistical Analysis
Statistical analyses are performed using GraphPad Prism 6 (La Jolla, CA, USA). All quantitative data are presented as the mean ± SD. The Student’s t-test and ANOVA are used to evaluate the data. P < 0.05 was considered statistically significant.
Results
DHA Inhibits the Proliferation and Colony Formation, and Induces Ferroptosis of Lung Cancer Cells
First, we evaluated the effects of DHA on the proliferation and colony formation of human lung cancer cell lines NCI-H23 and XWLC-05 (Xuanwei female lung cancer cell line) using CCK-8 and colony formation assays. Both 40 μM and 60 μM DHA significantly reduced the cell viabilities of NCI-H23 and XWLC-05 cells at 24 h and 48 h, but the 20 μM DHA had no significant effect (Figure 1A and B). In colony formation assay, both 40 μM and 60 μM DHA significantly inhibited the colony formation abilities of NCI-H23 and XWLC-05 cells, but the 20 μM DHA had no significant effect (Figure 1C and D). We further found that 40 μM and 60 μM DHA treatment significantly enhanced the cell deaths of NCI-H23 and XWLC-05 cells (Figure 1E). Apoptosis and ferroptosis are the important forms of cell death and play important roles in cancer cell survival,28 therefore we further analyzed whether DHA induced the apoptosis or ferroptosis of lung cancer cells. Importantly, the ferroptosis inhibitor ferrostatin-1 significantly recovered the DHA-induced cell survival reduction and cell death both in NCI-H23 and XWLC-05 cells, however the apoptosis inhibitor Z-VAD-FMK could not reverse the DHA-induced cell survival reduction and cell death (Figure 1F and G). These results suggested that DHA inhibited the lung cancer cell proliferation and colony formation mainly by inducing ferroptosis.
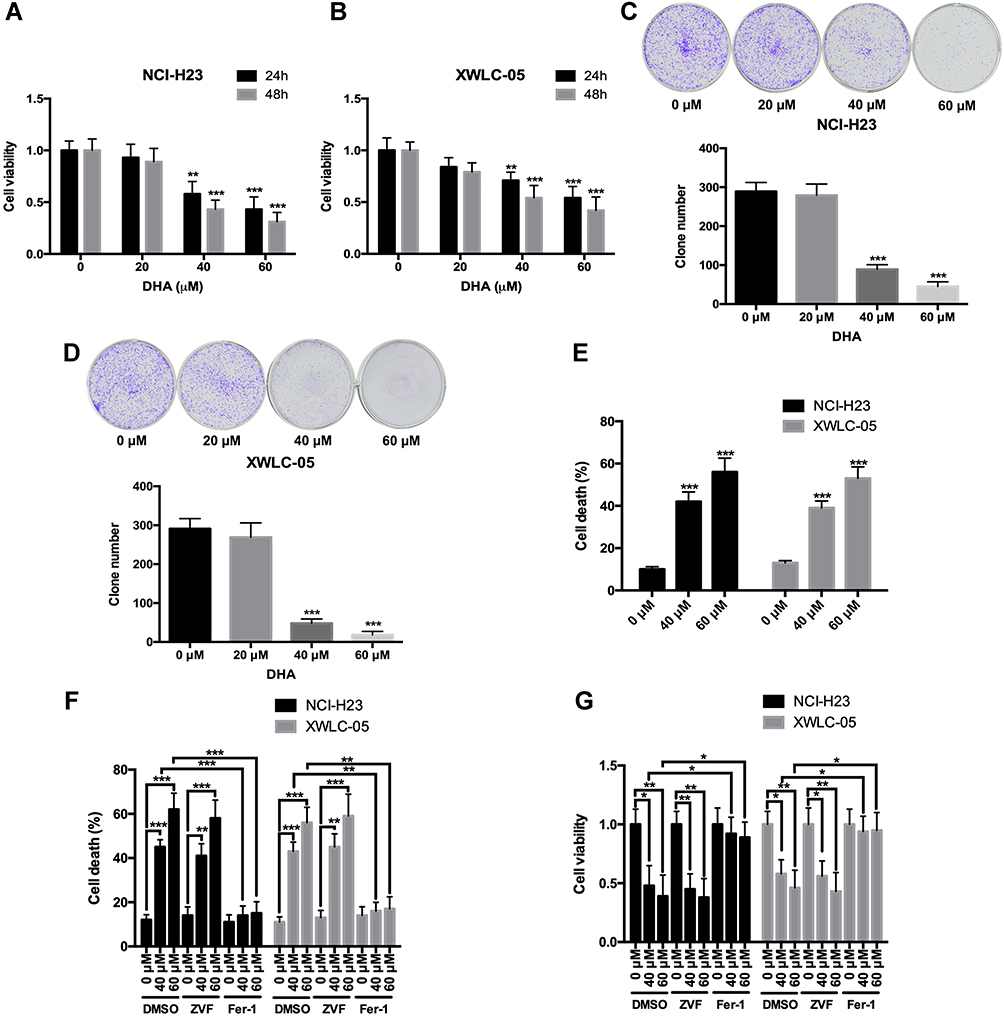 |
Figure 1 DHA inhibits the proliferation and colony formation, and induces ferroptosis of lung cancer cells. (A and B) The cell viabilities of NCI-H23 and XWLC-05 cells which were treated by DHA (0 μM, 20 μM, 40 μM and 60 μM) were detected by CCK-8 assay. (C and D) The colony formation abilities of NCI-H23 and XWLC-05 cells which were treated by DHA (0 μM, 20 μM, 40 μM and 60 μM) were detected by colony formation assay. (E) The cell deaths of NCI-H23 and XWLC-05 cells which were treated by DHA (0 μM, 40 μM and 60 μM) were detected by trypan blue exclusion assay. (F and G) The cell deaths and viabilities of NCI-H23 and XWLC-05 cells which were treated with DHA and ZVF (Z-VAD-FMK)/Fer-1 (Ferrostatin-1) were analyzed by trypan blue exclusion and CCK-8 assays. The experiments were repeated for three times. Compared with 0 μM DHA treated cells: *p < 0.05, **p < 0.01, ***p < 0.001. |
DHA Downregulates the PRIM2 Expression and Silencing PRIM2 Inhibits the Proliferation and Colony Formation and Induces Ferroptosis of Lung Cancer Cells
Our results found that 40 μM and 60 μM DHA significantly decreased the mRNA and protein levels of PRIM2 both in NCI-H23 and XWLC-05 cells (Figure 2A–C). CCLE database and our results showed that the expression levels of PRIM2 in XWLC-05 and NCI-H23 were higher than those in NCI-H1229 and A549 cells (Figure 2D and E). In order to reveal the role of PRIM2 in the DHA-induced proliferation and colony formation inhibition, we silenced the PRIM2 and evaluated its effects on cancer cell phenotypes. Interestingly, we found that knockdown of PRIM2 decreased the cell viabilities of NCI-H23 and XWLC-05 cells (Figure 2F–H), and enhanced the cell deaths and inhibited the colony formation abilities of NCI-H23 and XWLC-05 cells (Figure 2I and J).
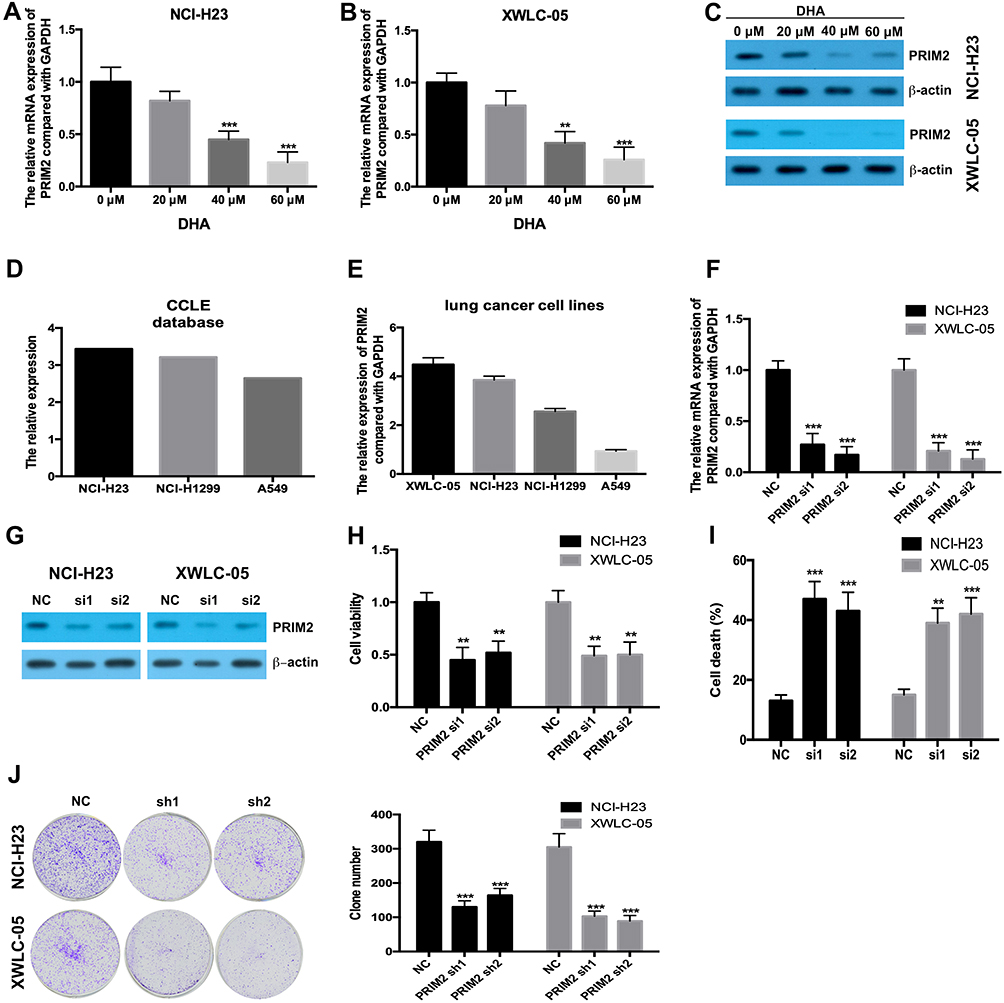 |
Figure 2 DHA downregulates the PRIM2 expression and silencing PRIM2 inhibits the proliferation and colony formation and induces ferroptosis of lung cancer cells. (A and B) The mRNA expression of PRIM2 was detected by qRT-PCR. (C) The protein expression of PRIM2 was detected by Western blotting assay. (D and E) The expression level of PRIM2 in lung cancer cells was analyzed by using CCLE database and detected by qRT-PCR. (F and G) The efficiency of PRIM2 silencing was analyzed by qRT-PCR and Western blotting assays. (H) The cell viabilities of NCI-H23 and XWLC-05 cells which were transfected with PRIM2 siRNAs were detected by CCK-8 assay. (I) The cell deaths of NCI-H23 and XWLC-05 cells were analyzed by trypan blue exclusion assay. (J) The colony formation abilities of NCI-H23 and XWLC-05 cells were analyzed by colony formation assay. The experiments were repeated for three times. Compared with 0 μM DHA treated cells or Negative control group (NC): **p < 0.01, ***p < 0.001. |
DHA and Loss of PRIM2 Reduce the GSH Level and Increase the Cellular Lipid ROS and Mitochondrial MDA Levels, and Downregulate the Expressions of SLC7A11 and β-Catenin in Lung Cancer Cells
Both 40 μM and 60 μM DHA significantly decreased the GSH level and also increased the cellular lipid ROS and mitochondrial MDA levels in both NCI-H23 and XWLC-05 cells after 48 h treatment (Figure 3A–C). Meanwhile, 20 μM DHA treatment had no effect on the GSH, cellular lipid ROS and mitochondrial MDA levels (Figure 3A–C). Knockdown of PRIM2 had the similar effects with DHA and also significantly reduced the GSH level and increased the cellular lipid ROS and mitochondrial MDA levels in both NCI-H23 and XWLC-05 cells (Figure 3D–F). SLC7A11 is the key regulator of ferroptosis and transported extracellular cystine to cells, and finally participated in the biosynthesis of glutathione.29 Wnt/β-catenin signaling pathway is one of the key signaling pathway in carcinogenesis including lung cancer.30 Therefore, we further evaluated whether DHA affected the SLC7A11 and β-catenin expressions. Importantly, our results showed that 40 μM and 60 μM DHA significantly decreased the protein expression levels of SLC7A11 and β-catenin, and loss of PRIM2 also downregulated SLC7A11 and β-catenin both in NCI-H23 and XWLC-05 cells (Figure 3G and H).
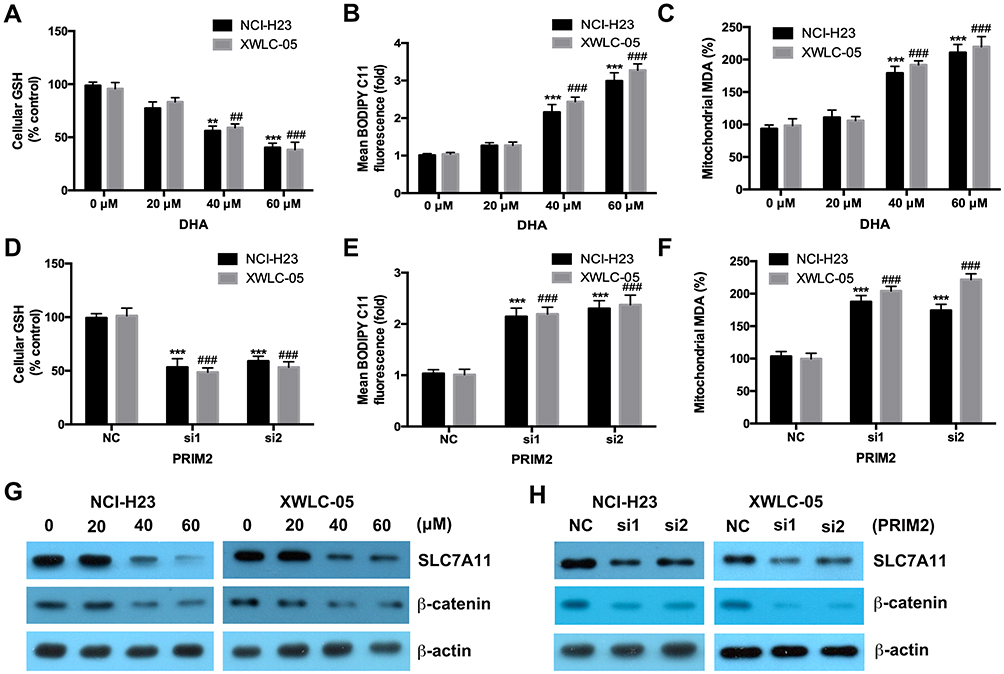 |
Figure 3 DHA and loss of PRIM2 reduce the GSH level and increase the cellular lipid ROS and mitochondrial MDA levels, and downregulate the expressions of SLC7A11 and β-catenin in lung cancer cells. (A–C) Measurement of cellular GSH, cellular ROS and mitochondrial MDA in DHA treated NCI-H23 and XWLC-05 cells. (D–F) Measurement of cellular GSH, cellular ROS and mitochondrial MDA in PRIM2 siRNAs transfected NCI-H23 and XWLC-05 cells. (G and H) The protein expressions of SLC7A11 and β-catenin were analyzed by Western blotting assay. The experiments were repeated for three times. Compared with 0 μM DHA treated cells or Negative control group (NC): **p < 0.01, ***p < 0.001; Compared with 40 μM/60 μM DHA treated cells or only PRIM2 siRNAs transfected cells, ##p < 0.01, ###p < 0.001. |
Exogenetic Overexpression of PRIM2 Recovered the Inhibitory Effects of DHA on Proliferation and Colony Formation of Lung Cancer Cells
In order to confirm the role of PRIM2 in the inhibitory role of DHA in lung cancer cells, we overexpressed PRIM2 in NCI-H23 cells and evaluated its effects on tumorigenic phenotypes (Figure 4A). Overexpression of PRIM2 significantly restored the cell viability and colony formation ability of NCI-H23 cells which were inhibited by 60 μM DHA treatment (Figure 4B–D). Exogenetic overexpression of PRIM2 also increased the GSH and decreased the cellular lipid ROS and mitochondrial MDA levels which were regulated by 60 μM DHA treatment (Figure 4E–G). Exogenetic overexpression of PRIM2 recovered the expressions of SLC7A11 and β-catenin which were downregulated by 60 μM DHA treatment (Figure 4H). These results further confirmed that DHA inhibited the malignant phenotypes of lung cancer cells by reducing PRIM2.
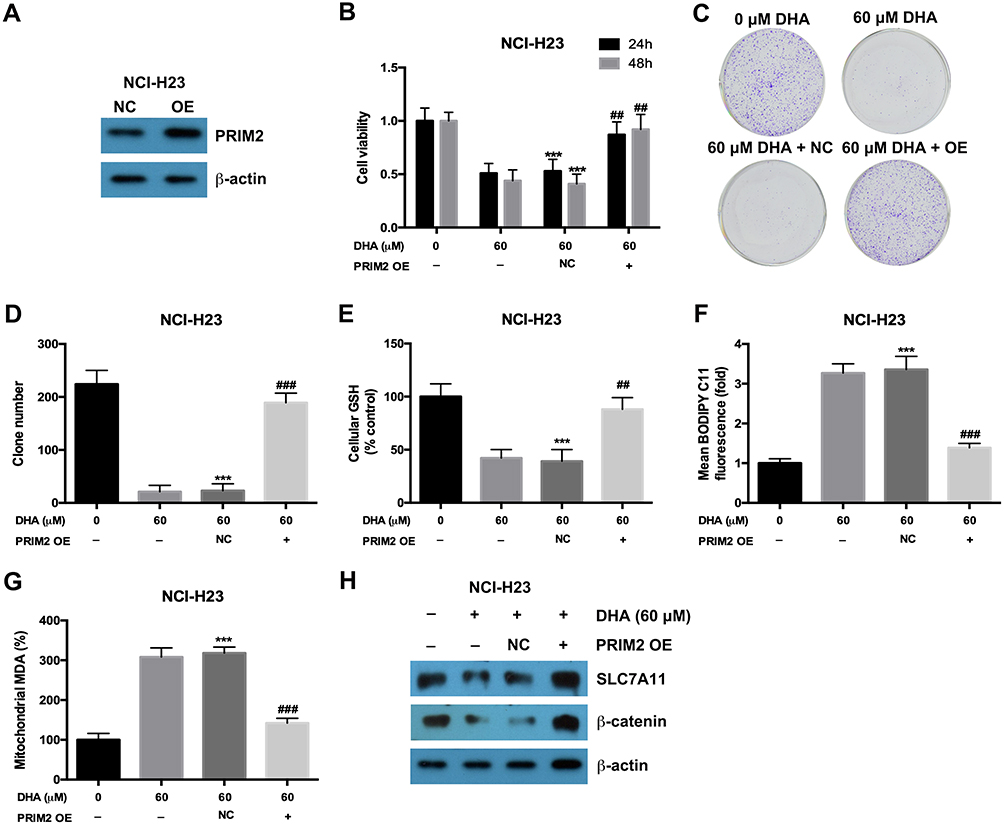 |
Figure 4 Exogenetic overexpression of PRIM2 recovered the inhibitory effects of DHA on proliferation and colony formation in lung cancer cells. (A) The overexpression of PRIM2 was confirmed by Western blotting assay. (B–D) The cell viability and colony formation ability of NCI-H23 cells were detected by CCK-8 assay and colony formation assay. (E–G) Measurement of cellular GSH, cellular ROS and mitochondrial MDA in NCI-H23 cells which were treated with 60 μM DHA and transfected with PRIM2 OE plasmid. (H) The protein expressions of SLC7A11 and β-catenin were analyzed by Western blotting assay. The experiments were repeated for three times. Compared with 0 μM DHA treated cells: ***p < 0.001; Compared with 60 μM DHA treated and empty vector transfected cells, ##p < 0.01, ###p < 0.001. |
Loss of PRIM2 Sensitizes NCI-H23 Cells to DHA Therapy
Our results further showed that silencing PRIM2 significantly decreased the cell viability and colony formation ability under the condition of 40 μM DHA treatment (Figure 5A–C). Loss of PRIM2 also decreased the GSH level and increased the cellular lipid ROS and mitochondrial MDA levels in NCI-H23 cells under the condition of 40 μM DHA treatment (Figure 5D–F). Comparing with 40 μM DHA treated NCI-H23 cells, combination of PRIM2 silencing and 40 μM DHA treatment significantly downregulated the expressions of SLC7A11 and β-catenin (Figure 5G). By analyzing TCGA data using starBase database,31 we further found that PRIM2 was significantly overexpressed in lung adenocarcinoma (Fold change = 2.09, p < 0.001, Figure 5H).
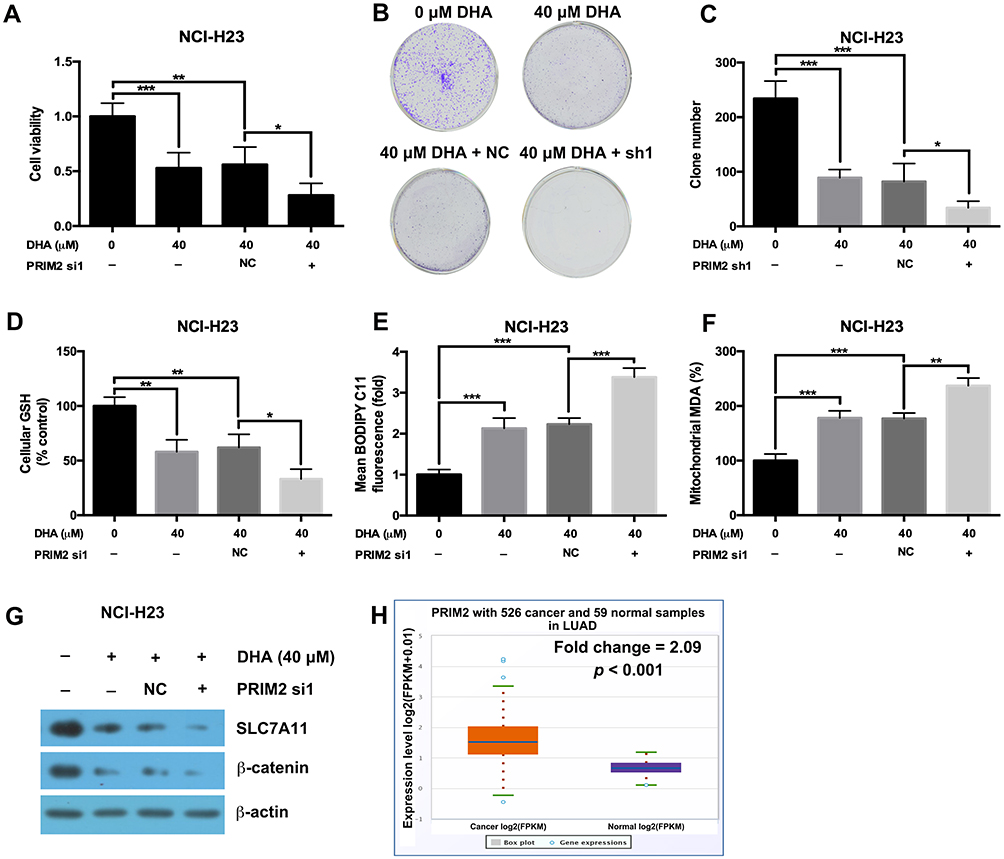 |
Figure 5 Loss of PRIM2 sensitizes NCI-H23 cells to DHA therapy. (A–C) The cell viability and colony formation ability of NCI-H23 cells were detected by CCK-8 assay and colony formation assay. (D–F) Measurement of cellular GSH, cellular ROS and mitochondrial MDA in NCI-H23 cells. (G) The protein expressions of SLC7A11 and β-catenin were analyzed by Western blotting assay. (H) The expression of PRIM2 in LUAD was analyzed by TCGA data using starBase database. The experiments were repeated for three times. Compared with 40 μM DHA treated and Negative control shRNA transfected cells: *p < 0.05, **p < 0.01, ***p < 0.001. |
DHA Inhibits the Tumor Growth in vivo
DHA treatment clearly inhibited the tumor growth compared with the control group (Figure 6A). By using Western blotting assay, we further found that in the tumor tissues, the expressions of PRIM2, SLC7A11 and β-catenin were significantly lower in DHA treatment group than those in control group (Figure 6B).
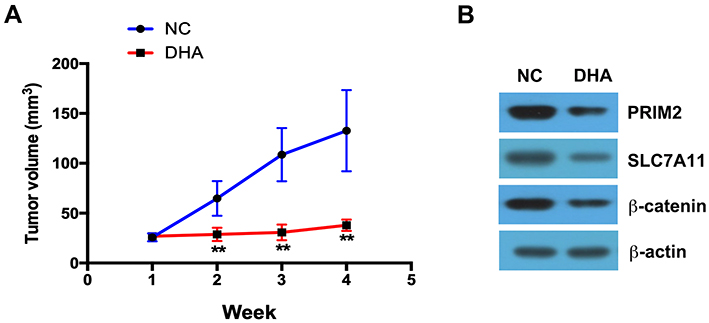 |
Figure 6 DHA inhibits the tumor growth in vivo. (A) DHA treatment clearly inhibited the tumor growth compared with the control group. (B) The expressions of PRIM2, SLC7A11 and β-catenin were analyzed by Western blotting assay. Compared with Negative control group (NC): **p < 0.01. |
Discussion
Lung cancer is the leading cause of cancer-related deaths both worldwide and in China.1,2 Although the treatment options have improved, the 5-year survival rate of lung cancer is only 18%.4 Serious indoor soot air pollution makes Xuanwei area of Yunnan Province a high incidence area of lung cancer in the world.32 At present, a large number of studies focus on the epidemiology and oncogenes of lung cancer in Xuanwei.6,33 However, there is no progress in the study of Xuanwei lung cancer drugs. Recent studies have found that DHA has significant antitumor effect and low toxicity, so it is a potential new antitumor drug. In gallbladder cancer, DHA could inhibit TCTP-dependent cell migration and invasion by decreasing Cdc42 (cell division control protein 42 homolog) activation.34 In lung cancer, DHA showed remarkable antitumor ability on A549 and H1299 cells by inhibiting Wnt/β-catenin signaling pathway.26 DHA could also suppress the migration and invasion of lung cancer A549 and H1975 cells in vitro by inhibiting NF-kB signaling pathway and GLUT1 translocation.35 DHA could sensitize cancer cells to many antitumor drugs. In NSCLC and malignant mesothelioma, the In vitro and in vivo results revealed that combination of DHA and onconase synergistically inhibited the tumor growth and angiogenesis.36 DHA sensitized ATO caused A549 lung cancer cells apoptosis by increasing ROS level.37 Combination of DHA and doxorubicin (DOX) showed significant antitumor ability in many types of cancer cells including A549, OVCAR-3, PC-3, HeLa and MCF-7 cells.38 Our study revealed that DHA significantly decreased the cell viability and the colony formation ability, and importantly we further found that DHA led to the cell death and induced ferroptosis of lung cancer NCI-H23 and XWLC-05 cells. XWLC-05 was a Xuanwei lung adenocarcinoma cell line and established in 2007 from a woman of Xuanwei lung adenocarcinoma. Whether the inhibitory roles of DHA relies on the sex or hormone is needed to be studied in future.
Ferroptosis is a new form of programmed cell death with characteristic accumulation of reactive oxygen species (ROS) which are generated by lipid peroxidation and iron accumulation.39 Ferroptosis might be developed to be a novel therapeutic method for lung cancer therapy.40 In cisplatin-resistant NSCLC cells, erastin and sorafenib could inhibit the Nrf2/SLC7A11 signaling pathway and further induce ferroptosis of cancer cells.41 Inducing ferroptosis by erastin could attenuate radioresistance of NSCLC cells.42 Meanwhile, radiation could promote lipid peroxidation and synergize with ferroptosis inducers to enhance the antitumor effect in a murine xenograft model.43 Acetaminophen was reported to promote erastin caused ferroptosis of NSCLC cancer cells by regulating Nrf2/heme oxygenase-1 signaling pathway.44 Our study revealed that DHA could induce ferroptosis of lung cancer cells.
PRIM2 is a subunit of DNA primase which is involved in the regulation of DNA replication. Although previous study reported that PRIM2 was upregulated by SIX1 (sine oculis homeobox homolog 1) in cervical cancer,23 its role in carcinogenesis especially lung cancer was largely unknown. Our findings showed that DHA reduced the PRIM2 expression and silencing PRIM2 had the similar antitumor effect to DHA. Importantly, we found that exogenetic overexpression of PRIM2 rescued DHA-induced ferroptosis. These results suggested that DHA-induced ferroptosis of lung cancer cells by regulating PRIM2.
We further elucidated that DHA and loss of PRIM2 downregulated SLC7A11 and β-catenin protein expressions. SLC7A11 is the key regulator of ferroptosis, and it could regulate cellular GSH biosynthesis.45 SLC7A11 was overexpressed in patients with KRAS-mutant lung adenocarcinoma (LUAD) and positively associated with tumor progression. And blocking SLC7A11 showed selective cytotoxicity on KRAS-mutant LUAD.46 In lung cancer A549 cells, knockdown of SLC7A11 improved the sensitivity of cancer cells to cisplatin.47 Previous study reported that silencing SLC7A11 increased caveolin-1 and promoted β-catenin recruitment to plasma membrane and finally inhibited the β-catenin transcriptional activity.48 Our findings revealed that DHA decreased PRIM2 and further reduced β-catenin and SLC7A11 levels. Considering DHA could not completely abolish the SLC7A11 expression, whether DHA/PRIM2 combination with other drugs had better anti-tumor effects need to be studied in future.
Taken together, our study suggested that DHA inhibited the proliferation, colony formation and induced ferroptosis of lung cancer cells by inhibiting PRIM2/SLC7A11 axis. Loss of PRIM2 induced ferroptosis might developed to be a novel therapeutic method in cancer therapy.
Funding
This work was supported by the Joint Special Fund Project of Yunnan Provincial Science and Technology Department-Kunming Medical University (No.2017FE468 (−239)), Foundation for Medical Discipline Leader in Health and Family Planning Commission of Yunnan Province (D-2017050) and Yunnan respiratory disease clinical medical center project (ZX20190103).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi:10.3322/caac.21338
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
3. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311. doi:10.1016/S0140-6736(16)30958-8
4. Conway EM, Pikor LA, Kung SH, et al. Macrophages, Inflammation, and Lung Cancer. Am J Respir Crit Care Med. 2016;193(2):116–130. doi:10.1164/rccm.201508-1545CI
5. Chen G, Sun X, Ren H, et al. The mortality patterns of lung cancer between 1990 and 2013 in Xuanwei, China. Lung Cancer. 2015;90(2):155–160. doi:10.1016/j.lungcan.2015.08.006
6. Wu H, Meng S, Xu Q, et al. Gene expression profiling of lung adenocarcinoma in Xuanwei, China. Eur J Cancer Prev. 2016;25(6):508–517. doi:10.1097/CEJ.0000000000000214
7. Crespo-Ortiz MP, Wei MQ. Antitumor activity of artemisinin and its derivatives: from a well-known antimalarial agent to a potential anticancer drug. J Biomed Biotechnol. 2012;2012:247597. doi:10.1155/2012/247597
8. Das AK. Anticancer Effect of AntiMalarial Artemisinin Compounds. Ann Med Health Sci Res. 2015;5(2):93–102. doi:10.4103/2141-9248.153609
9. Jin H, Jiang AY, Wang H, Cao Y, Wu Y, Jiang XF. Dihydroartemisinin and gefitinib synergistically inhibit NSCLC cell growth and promote apoptosis via the Akt/mTOR/STAT3 pathway. Mol Med Rep. 2017;16(3):3475–3481. doi:10.3892/mmr.2017.6989
10. Wu L, Cheng Y, Deng J, Tao W, Ye J. Dihydroartemisinin Inhibits Proliferation and Induces Apoptosis of Human Hepatocellular Carcinoma Cell by Upregulating Tumor Necrosis Factor via JNK/NF-kappaB Pathways. Evid Based Complement Alternat Med. 2019;2019:9581327. doi:10.1155/2019/9581327
11. Wang SJ, Sun B, Cheng ZX, et al. Dihydroartemisinin inhibits angiogenesis in pancreatic cancer by targeting the NF-kappaB pathway. Cancer Chemother Pharmacol. 2011;68(6):1421–1430. doi:10.1007/s00280-011-1643-7
12. Zhang JL, Wang Z, Hu W, Chen SS, Lou XE, Zhou HJ. DHA regulates angiogenesis and improves the efficiency of CDDP for the treatment of lung carcinoma. Microvasc Res. 2013;87:14–24. doi:10.1016/j.mvr.2013.02.006
13. Efferth T, Benakis A, Romero MR, et al. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radic Biol Med. 2004;37(7):998–1009. doi:10.1016/j.freeradbiomed.2004.06.023
14. Lai H, Sasaki T, Singh NP. Targeted treatment of cancer with artemisinin and artemisinin-tagged iron-carrying compounds. Expert Opin Ther Targets. 2005;9:995–1007. doi:10.1517/14728222.9.5.995
15. May WS, Cuatrecasas P. Transferrin receptor: its biological significance. J Membr Biol. 1985;88(3):205–215. doi:10.1007/bf01871086
16. Mercer AE, Maggs JL, Sun XM, et al. Evidence for the involvement of carbon-centered radicals in the induction of apoptotic cell death by artemisinin compounds. J Biol Chem. 2007;282(13):9372–9382. doi:10.1074/jbc.M610375200
17. Gerhardt T, Jones R, Park J, et al. Effects of antioxidants and pro-oxidants on cytotoxicity of dihydroartemisinin to Molt-4 human leukemia cells. Anticancer Res. 2015;35:1867–1871.
18. Chan HW, Singh NP, Lai HC. Cytotoxicity of dihydroartemisinin toward Molt-4 cells attenuated by N-tert-butyl-alpha-phenylnitrone and deferoxamine. Anticancer Res. 2013;33:4389–4393.
19. Ba Q, Zhou N, Duan J, et al. Dihydroartemisinin exerts its anticancer activity through depleting cellular iron via transferrin receptor-1. PLoS One. 2012;7(8):e42703. doi:10.1371/journal.pone.0042703
20. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27(1):242–254. doi:10.1038/s41418-019-0352-3
21. Shiratori A, Okumura K, Nogami M, et al. Assignment of the 49-kDa (PRIM1) and 58-kDa (PRIM2A and PRIM2B) subunit genes of the human DNA primase to chromosome bands 1q44 and 6p11.1-p12. Genomics. 1995;28(2):350–353. doi:10.1006/geno.1995.1155
22. Yatsula B, Galvao C, McCrann M, Perkins AS. Assessment of F-MuLV-induced tumorigenesis reveals new candidate tumor genes including Pecam1, St7, and Prim2. Leukemia. 2006;20(1):162–165. doi:10.1038/sj.leu.2404034
23. Liu D, Zhang XX, Xi BX, et al. Sine oculis homeobox homolog 1 promotes DNA replication and cell proliferation in cervical cancer. Int J Oncol. 2014;45(3):1232–1240. doi:10.3892/ijo.2014.2510
24. Levy N, Oehlmann M, Delalande F, et al. XRCC1 interacts with the p58 subunit of DNA Pol alpha-primase and may coordinate DNA repair and replication during S phase. Nucleic Acids Res. 2009;37(10):3177–3188. doi:10.1093/nar/gkp144
25. Lee JB, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM. DNA primase acts as a molecular brake in DNA replication. Nature. 2006;439(7076):621–624. doi:10.1038/nature04317
26. Tong Y, Liu Y, Zheng H, et al. Artemisinin and its derivatives can significantly inhibit lung tumorigenesis and tumor metastasis through Wnt/beta-catenin signaling. Oncotarget. 2016;7(21):31413–31428. doi:10.18632/oncotarget.8920
27. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7(7):e2307. doi:10.1038/cddis.2016.208
28. Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-Induced Endoplasmic Reticulum Stress: cross-talk between Ferroptosis and Apoptosis. Mol Cancer Res. 2018;16(7):1073–1076. doi:10.1158/1541-7786.MCR-18-0055
29. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
30. Lecarpentier Y, Schussler O, Hebert JL, Vallee A. Multiple Targets of the Canonical WNT/beta-Catenin Signaling in Cancers. Front Oncol. 2019;9:1248. doi:10.3389/fonc.2019.01248
31. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(D1):D92–D97. doi:10.1093/nar/gkt1248
32. Kim C, Chapman RS, Hu W, et al. Smoky coal, tobacco smoking, and lung cancer risk in Xuanwei, China. Lung Cancer. 2014;84(1):31–35. doi:10.1016/j.lungcan.2014.01.004
33. Seow WJ, Downward GS, Wei H, et al. Indoor concentrations of nitrogen dioxide and sulfur dioxide from burning solid fuels for cooking and heating in Yunnan Province, China. Indoor Air. 2016;26(5):776–783. doi:10.1111/ina.12251
34. Zhang F, Ma Q, Xu Z, et al. Dihydroartemisinin inhibits TCTP-dependent metastasis in gallbladder cancer. J Exp Clin Cancer Res. 2017;36(1):68. doi:10.1186/s13046-017-0531-3
35. Jiang J, Geng G, Yu X, et al. Repurposing the anti-malarial drug dihydroartemisinin suppresses metastasis of non-small-cell lung cancer via inhibiting NF-kappaB/GLUT1 axis. Oncotarget. 2016;7(52):87271–87283. doi:10.18632/oncotarget.13536
36. Shen R, Li J, Ye D, Wang Q, Fei J. Combination of onconase and dihydroartemisinin synergistically suppresses growth and angiogenesis of non-small-cell lung carcinoma and malignant mesothelioma. Acta Biochim Biophys Sin (Shanghai). 2016;48(10):894–901. doi:10.1093/abbs/gmw082
37. Chen H, Gu S, Dai H, Li X, Zhang Z. Dihydroartemisinin Sensitizes Human Lung Adenocarcinoma A549 Cells to Arsenic Trioxide via Apoptosis. Biol Trace Elem Res. 2017;179(2):203–212. doi:10.1007/s12011-017-0975-5
38. Tai X, Cai XB, Zhang Z, Wei R. In vitro and in vivo inhibition of tumor cell viability by combined dihydroartemisinin and doxorubicin treatment, and the underlying mechanism. Oncol Lett. 2016;12(5):3701–3706. doi:10.3892/ol.2016.5187
39. Shi ZZ, Fan ZW, Chen YX, et al. Ferroptosis in Carcinoma: regulatory Mechanisms and New Method for Cancer Therapy. Onco Targets Ther. 2019;12:11291–11304. doi:10.2147/OTT.S232852
40. Kuang Y, Wang Q. Iron and lung cancer. Cancer Lett. 2019;464:56–61. doi:10.1016/j.canlet.2019.08.007
41. Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol Lett. 2020;19:323–333. doi:10.3892/ol.2019.11066
42. Pan X, Lin Z, Jiang D, et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol Lett. 2019;17:3001–3008. doi:10.3892/ol.2019.9888
43. Ye LF, Chaudhary KR, Zandkarimi F, et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol. 2020;15(2):469–484. doi:10.1021/acschembio.9b00939
44. Gai C, Yu M, Li Z, et al. Acetaminophen sensitizing erastin-induced ferroptosis via modulation of Nrf2/heme oxygenase-1 signaling pathway in non-small-cell lung cancer. J Cell Physiol. 2020;235(4):3329–3339. doi:10.1002/jcp.29221
45. Lang X, Green MD, Wang W, et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019;9(12):1673–1685. doi:10.1158/2159-8290.CD-19-0338
46. Hu K, Li K, Lv J, et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest. 2019. doi:10.1172/JCI124049
47. Horibe S, Kawauchi S, Tanahashi T, Sasaki N, Mizuno S, Rikitake Y. CD44v-dependent upregulation of xCT is involved in the acquisition of cisplatin-resistance in human lung cancer A549cells. Biochem Biophys Res Commun. 2018;507(1–4):426–432. doi:10.1016/j.bbrc.2018.11.055
48. Chen RS, Song YM, Zhou ZY, et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene. 2009;28:599–609. doi:10.1038/onc.2008.414
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.