Back to Journals » OncoTargets and Therapy » Volume 8
Differential regulation of human Eag1 channel expression by serum and epidermal growth factor in lung and breast cancer cells
Authors Acuña-Macías I, Vera E, Vázquez-Sánchez AY, Mendoza-Garrido ME, Camacho J ![]()
Received 26 March 2015
Accepted for publication 17 July 2015
Published 16 October 2015 Volume 2015:8 Pages 2959—2965
DOI https://doi.org/10.2147/OTT.S85504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Isabel Acuña-Macías,1 Eunice Vera,1 Alma Yolanda Vázquez-Sánchez,1 María Eugenia Mendoza-Garrido,2 Javier Camacho1
1Department of Pharmacology, 2Department of Physiology, Biophysics and Neurosciences, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Mexico City, Mexico
Abstract: Oncogenic ether à-go-go-1 (Eag1) potassium channels are overexpressed in most primary human solid tumors. Low oxygen and nutrient/growth factor concentrations play critical roles in tumorigenesis. However, the mechanisms by which tumor cells survive and proliferate under growth factor-depleted conditions remain elusive. Here, we investigated whether serum-deprived conditions and epidermal growth factor (EGF) regulate Eag1 expression in human lung and breast cancer cells. The human cancer cell lines A549 and MCF-7 (from the lungs and breast, respectively) were obtained from the American Type Culture Collection and cultured following the manufacturer’s recommendations. Eag1 gene and protein expression were studied by real-time PCR and immunocytochemistry, respectively. Cell proliferation was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, and ERK1/2 phosphorylation was investigated by Western blot. Serum-deprived conditions increased Eag1 mRNA and protein expression in both cell lines. This Eag1 upregulation was prevented by EGF and the ERK1/2 inhibitor U0126 in only lung cancer cells; vascular endothelial growth factor did not prevent Eag1 upregulation. Our results suggest that Eag1 may act as a survival and mitogenic factor under low-serum and nutrient conditions and may be a clinical target during the early stages of tumor development.
Keywords: lung cancer, serum deprivation, ether à-go-go, potassium channels, EGF, epidermal growth factor, ERK 1/2
Introduction
Many solid tumors display poorly developed blood vessels during progression to advanced stage disease. This deficiency in blood supply deprives tumor cells of oxygen and growth factors,1–3 which might trigger responses that promote survival, neoplastic progression, and malignancy.4–9 However, the mechanisms by which tumor cells survive and proliferate under growth factor-depleted conditions remain elusive. Transfection of the ether à-go-go-1 (Eag1, KCNH1, Kv10.1) potassium channel into mammalian cells confers the ability to grow in low-serum conditions.10 Accordingly, Eag1 expression favors tumor progression when transfected cells are injected into immunosuppressed mice.10 The expression of Eag1 under normal conditions is restricted to a few tissues, but Eag1 is aberrantly expressed in several cell lines derived from human tumors as well as in more than 75% of primary solid tumors, including lung tumors.11–18 In addition, Eag1 enhances tumor cell proliferation, is involved in tumor progression and metastasis, and is associated with poor prognosis.10,14–16,18–24 Chemical carcinogens, human papilloma virus oncogenes, and hormones regulate Eag1 expression.19,25–27 However, Eag1 regulation by serum-deprived conditions is poorly understood. Here, we investigated the effect of serum deprivation, epidermal and vascular endothelial growth factors (EGF and VEGF, respectively), and the involvement of the ERK1/2 pathway on Eag1 expression in cancer cells. Our results suggest that Eag1 may act as a survival and mitogenic factor under serum-deprived conditions, favoring the proliferation of solid tumors. Thus, targeting Eag1 expression during the early stages of tumor development may have therapeutic benefits.
Materials and methods
Reagents and cell culture
The MEK inhibitor U0126 and VEGF were purchased from Sigma-Aldrich, St Louis, MO, USA; EGF was purchased from Roche Diagnostic GmbH (Mannheim, Germany). The cancer cell lines A549 and MCF-7 (from human alveolar adenocarcinoma and breast, respectively) were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured according to the manufacturer’s instructions. The studies using these commercially available human cell lines followed the principles of the Declaration of Helsinki (following the recommendation and guidelines of the journal, we state that at our institution this type of in vitro experiments do not need a special Review Board approval to be performed).
Metabolic activity
A549 cells were plated in triplicate in 96-well plates at 3,500 cells/well (n=12) in Dulbecco’s modified Eagle’s medium/F12-K culture medium + 10% fetal bovine serum (FBS) and allowed to proliferate for 24 hours. Then, the cells were incubated for 24 hours under serum-deprived conditions (0.5% FBS) followed by incubation in either 10% FBS or 0.5% FBS in the absence or presence of EGF (10 ng/mL) and/or U0126 (10 μM) for 48 hours. Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell proliferation assay kit I (Boehringer Mannheim GmbH, Mannheim, Germany) as described.26
Western blot
Cells were prepared for standard Western blotting. The membranes were incubated overnight at 4°C with primary mouse monoclonal antibodies against p-ERK1/2 (Thr202/Tyr204; E-10; 1:2,000, Cell Signaling Technology, Danvers, MA, USA) or β-actin (1:2,000, Cell Signaling Technology). The membranes were incubated with a peroxidase-conjugated donkey anti-mouse IgG secondary antibody (1:17,000) for p-ERK1/2 (Thr202/Tyr204; E-10) and β-actin. The membranes were visualized using enhanced chemiluminescence (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA), followed by exposure onto photographic films (Hyperfilm, Amersham, Pittsburgh, PA, USA) to analyze peroxidase activity. Finally, the densitometric quantification of immunoblots was performed using Kodak Molecular Imaging Software (Carestream Health, Rochester, NY, USA). β-Actin was used as a loading control.
Real-time RT-PCR
Total RNA was isolated with TRIzol reagent (Sigma-Aldrich). cDNA was obtained from RNA (5 μg) previously treated with DNAse I and M-MuLV reverse transcriptase (New England Biolabs, Ipswich, MA, USA). Eag1 gene expression was determined by real-time reverse transcription polymerase chain reaction (RT-PCR) using the TaqMan™ detection system (Thermo Fisher Scientific, Waltham, MA, USA). Hypoxanthine-guanine phosphoribosyl transferase was used as the internal standard. Data were analyzed by the 2−ΔΔCt method and by the threshold cycle.
Immunocytochemistry
A549 and MCF-7 cells were prepared for standard immunochemistry experiments as described previously in Reagents and cell culture using a primary antibody against Eag1 (rabbit polyclonal, 1:600) from Novus Biologicals (Littleton, CO, USA) and a horseradish peroxidase polymer (Biocare Medical, Concord, CA, USA).27 Brown immunostaining indicated Eag1 expression. Negative controls were obtained in the absence of the primary antibody.
Statistical analysis
Analysis of variance followed by the Tukey–Kramer test or Student’s t-test was performed using GraphPad Prism software version 5.0 (San Diego, CA, USA). P-values <0.05 were considered statistically significant.
Results
Serum deprivation and EGF regulate Eag1 expression in lung cancer cells
A549 lung cancer cells were cultured in 10% FBS for 24 hours. Then, the cells were incubated for 24 hours in serum-deprived conditions (0.5% FBS) followed by incubation for 48 hours in different concentrations of FBS. Figure 1A and C shows that decreasing serum concentrations led to increasing Eag1 mRNA levels. Eag1 protein expression was also higher at low-serum concentrations (Figure 1B). Whereas, Eag1 protein expression (brown immunostaining) was exclusively detected in the nucleus of cells grown in 10% FBS, and it was also clearly observed in the cytoplasm of cells in low-serum conditions. Because EGF and VEGF are present in the serum and relevant in cancer, we hypothesized that these factors missing in the low-serum conditions might regulate the expression of Eag1. While EGF prevented the increase in Eag1 expression induced by serum deprivation (Figure 1C), treatment with VEGF did not affect Eag1 expression.
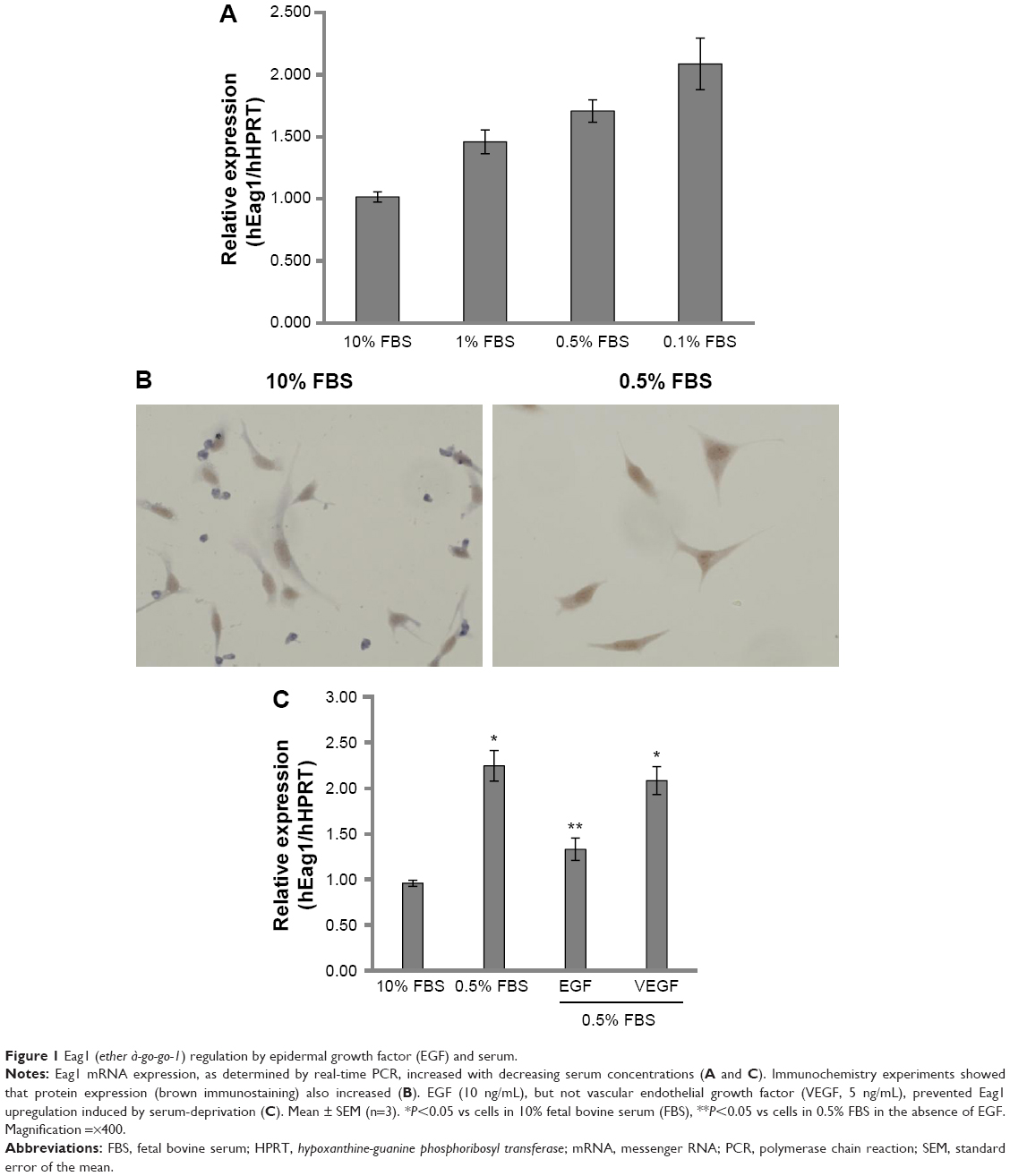 | Figure 1 Eag1 (ether à-go-go-1) regulation by epidermal growth factor (EGF) and serum. |
Potential participation of the canonical ERK1/2 pathway in Eag1 regulation
We investigated whether the canonical EGF receptor signaling pathway was involved in Eag1 regulation. Western blot experiments showed that ERK1/2 phosphorylation was lower in low-serum conditions compared with those with 10% FBS (Figure 2A), suggesting that ERK1/2 phosphorylation is inversely involved in the upregulation of Eag1 by serum deprivation. Accordingly, ERK1/2 phosphorylation was clearly increased by EGF in low-serum conditions (Figure 2A). The ERK1/2 pathway inhibitor U0126 decreased ERK1/2 phosphorylation in all conditions as expected (Figure 2A). Low Eag1 expression was observed in the presence of EGF and U0126, despite the cells being deprived of serum (Figure 2B).
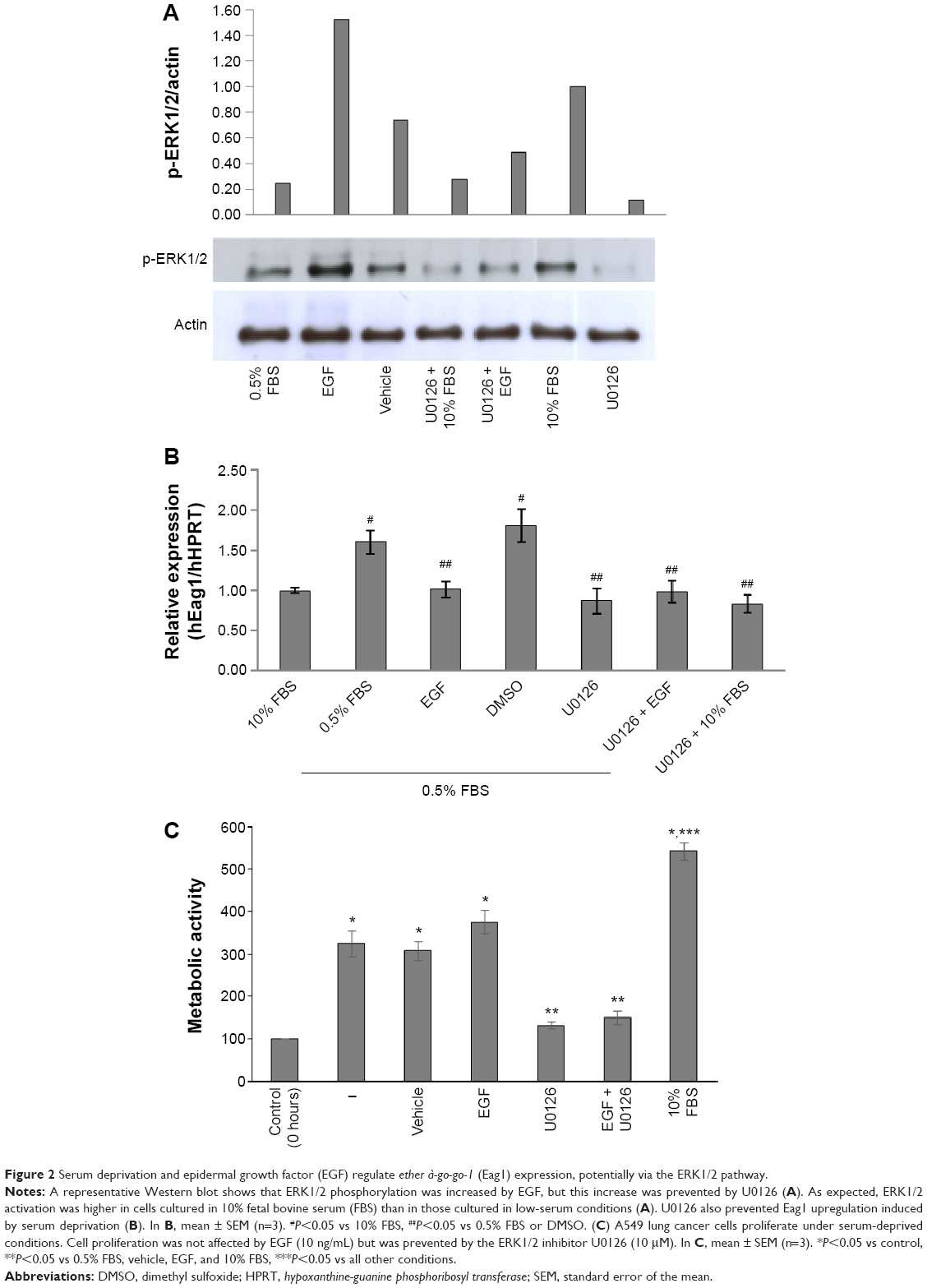 | Figure 2 Serum deprivation and epidermal growth factor (EGF) regulate ether à-go-go-1 (Eag1) expression, potentially via the ERK1/2 pathway. |
Cell proliferation in low-serum conditions is inhibited by U0126
In accordance with the Eag1 upregulation, A549 cells not only survived under serum-deprived conditions but also proliferated (Figure 2C). EGF was unable to increase such proliferation, most likely due to its inhibitory effect on Eag1 expression. As expected, the ERK1/2 inhibitor U0126 decreased cell proliferation.
Eag1 expression is upregulated by serum deprivation in breast cancer cells
Because the EGF pathway is also very important for the proliferation of breast cancer cells, we investigated whether the same serum- and EGF-mediated regulation of Eag1 occurred in MCF-7 cells. Eag1 mRNA expression was upregulated under low-serum conditions (Figure 3A). Eag1 protein expression (brown immunostaining) was higher in serum-deprived cells in comparison to cells grown in 10% FBS (Figure 3B). However, EGF did not prevent the Eag1 upregulation induced by serum deprivation.
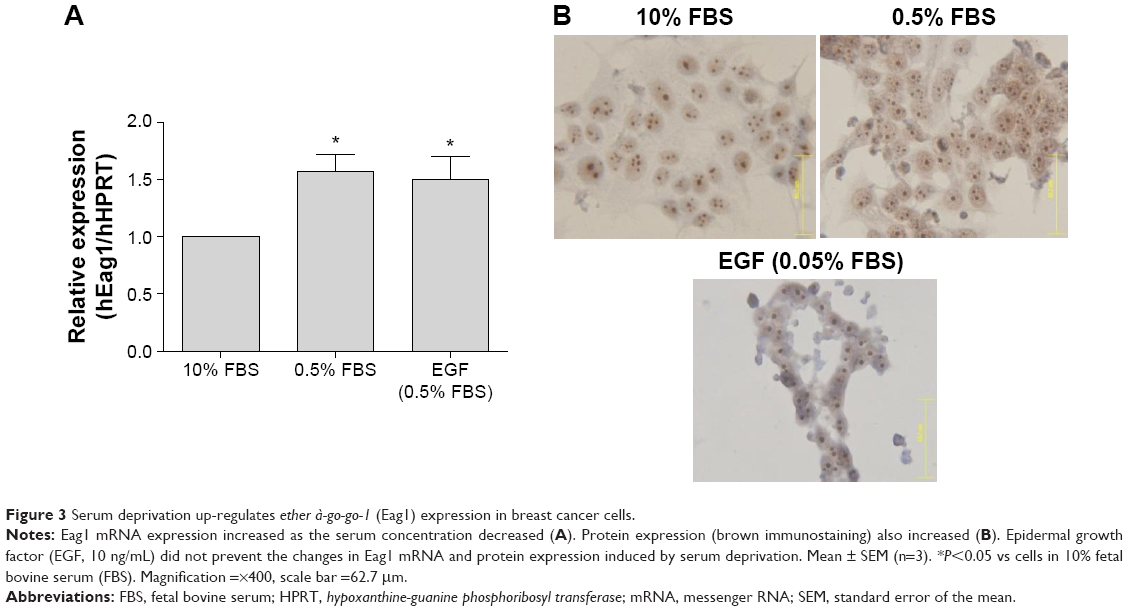 | Figure 3 Serum deprivation up-regulates ether à-go-go-1 (Eag1) expression in breast cancer cells. |
Discussion
Hypoxia and nutrient deprivation are the features of most solid tumors, especially during the early stages.1 Eag1 potassium channels display oncogenic properties, confer cell growth capability under serum-deprived condition, and favor tumor progression.10 In this study, we show that serum-deprived conditions increased Eag1 expression in A549 lung cancer cells. This Eag1 upregulation was prevented by EGF and inversely correlated with the phosphorylation of ERK1/2, suggesting that phosphorylated ERK1/2 inhibits Eag1 expression. However, when serum-deprived cells were treated with U0126, Eag1 upregulation was prevented. The inhibitory effect of U0126 may be associated with other kinases/pathways that are affected by this compound, including stress-activated protein kinase 2a/p38, p38-regulated/activated kinase, protein kinase Bα (also called Akt), and ERK5.28,29 Our results suggest that the MAPK pathway might not be the only one involved in Eag1 regulation by EGF.30
Our data suggest that Eag1 may also be regulated by other pathways associated with stress. For instance, NOX1-dependent reactive oxygen species (ROS) are involved in cell cycle progression under low-serum conditions in lung epithelial cells via ERK1/2 phosphorylation and cyclin D1 expression.31 Moreover, FaO rat hepatoma and human hepatocellular carcinoma cell lines proliferate in the absence of serum, in contrast with non-tumoral hepatocytes.32–34 Upon serum withdrawal, early ROS production by NOX1 activates the Src/ERK pathway, promoting a positive feedback loop that leads to NOX1 upregulation. In parallel, NOX1-induced ROS stimulate p38 and Akt activation, which in turn induce TGF-α and EGFR expression and consequently activate the EGFR pathway to induce cell growth.34 Wu et al showed that high Eag1 expression is regulated by p38 MAPK in osteosarcoma cells.35 Thus, Eag1 upregulation in response to serum-deprived conditions can also be mediated by p38 activation, ROS and NOX1. It is important to study additional pathways that regulate Eag1 expression in lung cancer cells.
Eag1 mRNA and protein expression was also upregulated in breast cancer cells by serum deprivation. These results suggest that this channel upregulation may be a general response in different cancer cell types. Nevertheless, because EGF did not prevent Eag1 upregulation in these cells, different pathways should be studied to elucidate the potential mechanisms that regulate Eag1 expression.
Conclusion
Our results suggest that Eag1 expression may act as a survival and mitogenic factor under serum-deprived conditions, favoring the proliferation of solid tumors. These results provide a strong rational for targeting Eag1 expression at early stages of tumor development.
Acknowledgment
The authors thank Beatriz Alcántara for secretarial assistance and Osbaldo Ríos and Guadalupe Montiel for technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Symonds H, Krall L, Remington L, et al. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–711. | ||
Harrington EA, Fanidi A, Evan GI. Oncogenes and cell death. Curr Opin Genet. 1994;4:120–129. | ||
Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72. | ||
Towle HC. Metabolic regulation of gene transcription in mammals. J Biol Chem. 1995;270:23235–23238. | ||
Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumors. Nature. 1996;379:88–91. | ||
Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–23763. | ||
Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509–4515. | ||
Bae SK, Baek JH, Lee YM, Lee OH, Kim KW. Hypoxia-induced apoptosis in human hepatocellular carcinoma cells: a possible involvement of the 6-TG-sensitive protein kinase(s)-dependent signaling pathway. Cancer Lett. 1998;126:97–104. | ||
Boix J, Fibla J, Yuste V, Piulats JM, Llecha N, Comella JX. Serum deprivation and protein synthesis inhibition induce two different apoptotic processes in N18 neuroblastoma cells. Exp Cell Res. 1998;238:422–429. | ||
Pardo LA, del Camino D, Sánchez A, et al. Oncogenic potential of EAG K(+) channels. EMBO J. 1999;18:5540–5547. | ||
Martin S, Lino de Oliveira C, Mello de Queiroz F, Pardo LA, Stühmer W, Del Bel E. Eag1 potassium channel immunohistochemistry in the CNS of adult rat and selected regions of human brain. Neuroscience. 2008;155:833–844. | ||
Meyer R, Heinemann SH. Characterization of an eag-like potassium channel in human neuroblastoma cells. J Physiol. 1998;508:49–56. | ||
Meyer R, Schönherr R, Gavrilova-Ruch O, Wohlrab W, Heinemann SH. Identification of ether à go-go and calcium-activated potassium channels in human melanoma cells. J Membr Biol. 1999;171:107–115. | ||
Ouadid-Ahidouch H, Le Bourhis X, Roudbaraki M, Toillon RA, Delcourt P, Prevarskaya N. Changes in the K+ current-density of MCF-7 cells during progression through the cell cycle: possible involvement of a h-ether.a-go-go K+ channel. Receptors Channels. 2001;7:345–356. | ||
Mello de Queiroz F, Suarez-Kurtz G, Stühmer W, Pardo LA. Ether à go-go potassium channel expression in soft tissue sarcoma patients. Mol Cancer. 2006;5:42. | ||
Hemmerlein B, Weseloh RM, Mello de Queiroz F, et al. Overexpression of Eag1 potassium channels in clinical tumors. Mol Cancer. 2006;5:41. | ||
Farias LM, Ocaña DB, Díaz L, et al. Ether à-go-go potassium channels as human cervical cancer markers. Cancer Res. 2004;64:6996–7001. | ||
Pardo LA, Contreras-Jurado C, Zientkowska M, Alves F, Stühmer W. Role of voltage-gated potassium channels in cancer. J Membr Biol. 2005;205:115–124. | ||
Ousingsawat J, Spitzner M, Puntheeranurak S, et al. Expression of voltage-gated potassium channels in human and mouse colonic carcinoma. Clin Cancer Res. 2007;13:824–831. | ||
Weber C, Mello de Queiroz F, Downie BR, Suckow A, Stühmer W, Pardo LA. Silencing the activity and proliferative properties of the human EagI potassium channel by RNA interference. J Biol Chem. 2006;281:13033–13037. | ||
Gavrilova-Ruch O, Schönherr K, Gessner G, et al. Effects of imipramine on ion channels and proliferation of IGR1 melanoma cells. J Membr Biol. 2002;188:137–149. | ||
García-Ferreiro RE, Kerschensteiner D, Major F, Monje F, Stühmer W, Pardo LA. Mechanism of block of hEag1 K+ channels by imipramine and astemizole. J Gen Physiol. 2004;124:301–307. | ||
Camacho J. Ether à go-go potassium channels and cancer. Cancer Lett. 2006;233:1–9. | ||
Ding XW, Luo HS, Jin X, Yan JJ, Ai YW. Aberrant expression of Eag1 potassium channels in gastric cancer patients and cell lines. Med Oncol. 2007;24:345–350. | ||
Rodríguez-Rasgado JA, Acuña-Macías I, Camacho J. Eag1 channels as potential cancer biomarkers. Sensors. 2012;12:5986–5995. | ||
Díaz L, Ceja-Ochoa I, Restrepo-Angulo I, et al. Estrogens and human papilloma virus oncogenes regulate human ether-à-go-go-1 potassium channel expression. Cancer Res. 2009;69:3300–3307. | ||
Ortiz CS, Montante-Montes D, Saqui-Salces M, et al. Eag1 potassium channels as markers of cervical dysplasia. Oncol Rep. 2011;26:1377–1383. | ||
Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. | ||
Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. | ||
Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. | ||
Ranjan P, Anathy V, Burch PM, Weirather K, Lambeth JD, Heintz NH. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid Redox Signal. 2006;8:1447–1459. | ||
Hisaka T, Yano H, Haramaki M, Utsunomiya I, Kajiro M. Expressions of epidermal growth factor family and its receptor in hepatocellular carcinoma cell lines: relationship to cell proliferation. Int J Oncol. 1999;14:453–460. | ||
Ortiz C, Caja L, Sancho P, Bertran E, Fabregat I. Inhibition of the EGF receptor blocks autocrine growth and increases the cytotoxic effects of doxorubicin in rat hepatoma cells: role of reactive oxygen species production and glutathione depletion. Biochem Pharmacol. 2008;15:1935–1945. | ||
Sancho P, Fabregat I. NADPH oxidase NOX1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J Biol Chem. 2010;285:24815–24824. | ||
Wu X, Zhong D, Lin B, Zhai W, Ding Z, Wu J. p38 MAPK regulates the expression of ether à go-go potassium channel in human osteosarcoma cells. Radiol Oncol. 2013;47:42–49. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.