Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Differential effects of dopamine-directed treatments on cognition
Authors Ashby FG, Valentin V, von Meer S
Received 1 May 2015
Accepted for publication 9 June 2015
Published 29 July 2015 Volume 2015:11 Pages 1859—1875
DOI https://doi.org/10.2147/NDT.S65875
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Video abstract presented by Gregory Ashby
Views: 964
F Gregory Ashby, Vivian V Valentin, Stella S von Meer
Department of Psychological and Brain Sciences, University of California, Santa Barbara, CA, USA
Abstract: Dopamine, a prominent neuromodulator, is implicated in many neuropsychiatric disorders. It has wide-ranging effects on both cortical and subcortical brain regions and on many types of cognitive tasks that rely on a variety of different learning and memory systems. As neuroscience and behavioral evidence for the existence of multiple memory systems and their corresponding neural networks accumulated, so did the notion that dopamine’s role is markedly different depending on which memory system is engaged. As a result, dopamine-directed treatments will have different effects on different types of cognitive behaviors. To predict what these effects will be, it is critical to understand: which memory system is mediating the behavior; the neural basis of the mediating memory system; the nature of the dopamine projections into that system; and the time course of dopamine after its release into the relevant brain regions. Consideration of these questions leads to different predictions for how changes in brain dopamine levels will affect automatic behaviors and behaviors mediated by declarative, procedural, and perceptual representation memory systems.
Keywords: dopamine, cognition, memory systems, learning
Introduction
In the half a century since its discovery, dopamine (DA) has been among the most widely studied neurotransmitters in the brain.1 Dopaminergic systems have been studied in the context of neuropsychiatric and other disorders (eg, schizophrenia, attention deficit hyperactivity disorder (ADHD), Parkinson’s disease [PD], and drug addiction), as well as from the perspectives of cognitive development and aging. This broad interest in DA primarily reflects its important neuromodulatory function in many different subcortical and cortical neural networks, and the wide range of motor control, motivational, and cognitive functions that recruit these networks.
Not surprisingly, brain DA levels have been shown to affect performance of many different behaviors.2 Even so, DA can have qualitatively different effects on some behaviors and on other behaviors it can have little if any effect. There are several reasons for this. First, DA neurons project to much, but not all, of the brain, so behaviors mediated within brain regions that are not DA targets will be relatively unaffected by DA-related medications compared with behaviors mediated within brain regions that receive rich DA projections. Second, the effects of phasic bursts of DA neurons have a vastly different time course in the cortex3,4 and basal ganglia,5,6 and thus, the effects of DA-related medications on cortical-mediated behaviors will be much different than on behaviors mediated by the basal ganglia. Third, there is now overwhelming evidence that humans have multiple learning and memory systems that for the most part are neuroanatomically and functionally distinct.7,8 For example, declarative memory systems are mediated largely within frontal cortical areas that receive a dense DA projection and where DA release has a slow time course.3,4 Procedural memory systems are mediated largely within the basal ganglia, which receive a dense DA projection that has a fast time course,5,6 and the perceptual representation memory system is mediated largely within posterior cortical regions that receive almost no DA projection, at least compared with the heavily innervated frontal cortex.9,10 Thus, to make accurate predictions of how DA-related medications will affect a behavior, it is critical to understand: which memory system is mediating that behavior, the neural basis of that system, the DA projections into the relevant brain regions, and the time course of DA release in those regions.
This article reviews the current literature on each of these four questions. We begin, in the next section, with a brief overview of some of the most common memory systems that have been identified by memory researchers. The following section reviews the neuroanatomy and neurophysiology of the DA system, and subsequent sections highlight the differing roles of DA in different memory systems and examines some of the reasons why brain DA levels can vary so much across individuals. The final section closes with some general comments and conclusions.
Memory systems
The existence of multiple memory systems is supported by a rich armamentarium of behavioral neuroscience studies in non-human animals (eg, single-unit recordings, and lesion and pharmacological studies) and cognitive neuroscience and neuropsychology studies in humans (neuropsychiatric patient studies and neuroimaging studies). A common taxonomy is to distinguish between declarative and nondeclarative systems.8 Briefly, declarative memories are those accessible to conscious awareness, whereas nondeclarative memories are stored and retrieved without conscious awareness.
Declarative memory
Declarative memory is a phylogenetically advanced system that allows conscious manipulation of information. It is typically thought to include three interrelated subsystems, ie, working memory, episodic memory, and semantic memory. Working memory is a temporary, limited capacity memory system that can store information for brief time periods (up to ~1 minute). Episodic memory is the long-term memory of specific events or episodes and semantic memory is the long-term memory of facts.
Working memory
Working memory is a limited capacity system that temporarily stores and maintains roughly 3–5 chunks of information11 for online manipulation and organization, thereby supporting the human thought process by providing an interface between perception, long-term memory, and action.12 The term “working memory” has largely superseded the term “short-term memory”, since the latter traditionally only describes retention, whereas the former also includes the ability to manipulate information.
Based on experiments that demonstrate low levels of interference during the maintenance of information acquired via different sensory modalities, it appears that working memory reflects a multidimensional process supported by different brain structures that are controlled by a central executive located in the frontal lobes.12 Executive-attention and working memory circuits dependent on the prefrontal cortex (PFC) and the anterior cingulate cortex (ACC) are thought to select and maintain newly acquired stimulus representations from different sensory modalities, integrate these sensory inputs with long-term memory traces, action plans, and goal representations to form strategies that guide behavior.12–14 To accommodate this higher-order manipulation of information, working memory processes are subject to prefrontal executive control, such as inhibition of non-relevant information, attentional guidance, and associative monitoring.12,15,16 Thus, working memory processes may be grossly divided into two parts, ie, maintenance of information and the dynamic integration and manipulation of this information, which is heavily dependent on the PFC.16–18
Working memory maintenance appears to be a volatile phenomenon that is reflected in short-term electrical/synaptic activity, in contrast with long-term memory, which is encoded in stable structural changes that depend on genetic transcription and are preserved over long periods of time until information is recalled and structural traces become unstable.19,20 By this, we mean that working memories seem to be encoded via the sustained firing of neurons during the maintenance period. When the firing stops, the memories are lost, unless they have been transferred to some long-term structural store. In support of this hypothesis, many single-unit recording studies have found neurons that show sustained activity during delay periods during which an animal is motivated to maintain the memory of the location of some reward. Neurons of this type have been reported in a wide variety of different brain areas, including the PFC,21 the posterior parietal cortex,22 the medial dorsal nucleus of the thalamus,23 the caudate nucleus,24,25 and the globus pallidus.26 Ashby et al27 proposed a computational model in which sustained activation in all these structures contributes to working memory maintenance.
As part of the declarative system, working memory is an important conscious gate in the creation of declarative long-term memories. The transition from short-term to long-term memory is a dynamic process that includes acquisition, working memory maintenance, transition to intermediate-term memory, consolidation to long-term memory, and destabilization and restabilization of existing long-term memory traces during retrieval and integration of new information.19
Episodic and semantic memory
Declarative long-term memory is typically subdivided into the episodic memory of events and the semantic memory of facts.28 Episodic and semantic memories are characterized by high specificity, capacity for partial retrieval, conscious awareness, capacity limitations, and slow access. The term “remember” is commonly used to refer to retrieval from these memory systems.29 Declarative memory formation is subject to motivational and emotional control30 and is potentiated by attention.31
The encoding of declarative information depends on conceptually driven, top-down processing of stimulus associations and experiences. Declarative memories are flexible by nature; flexibility arises because memories are stored in a relational network in which semantic information is shared among multiple episodes.32 The processing sequences that were implemented during encoding also regulate recall. When retrieving information from episodic and semantic memory, a major distinction is made between recollection, which is a slow controlled search process, and familiarity, which is a fast perceptual process.33 The recall of an episodic or semantic memory requires recollection, but recognition can occur via either process.
Episodic and semantic memory systems depend on a widespread neural network. The hippocampus is critical for the consolidation of episodic memories,34 whereas the consolidation of semantic memories also depends on surrounding medial temporal lobe structures, including the entorhinal, perirhinal, and parahippocampal cortices.35 Long-term storage of the memories is distributed across much of the neocortex,32 whereas recall depends on the PFC, medial temporal lobe structures, and posterior midline regions.17
At the cellular level, long-term consolidation depends on long-term potentiation (LTP) at synapses within the hippocampus.36 Hippocampal LTP begins with activation of post-synaptic N-methyl-D-aspartate (NMDA) receptors. A complex chemical cascade is then initiated, which eventually leads to gene transcription, causing structural changes that strengthen the synapse.20
Nondeclarative memory
Nondeclarative memories, which are often called implicit memories, are expressed through performance rather than conscious recollection. In contrast with declarative memories that describe the “what” dimension of experience, implicit memories are concerned with the mechanism of “how” the organism interacts with its environment. Memories of this type mature early, appear to be highly specific, perceptually based, habitual, and inflexible. Non-declarative memory is an umbrella term with several subcategories tied to specific brain regions. Here we will focus on two subcategories, ie, the procedural and perceptual representation systems.
Procedural memory
Procedural memory systems store memories of habits and skills that are learned via repeated practice.37 Compared with declarative memories, the major distinguishing characteristics of procedural memories are: little conscious recollection or even awareness of what was learned; slow and incremental learning; learning that requires immediate and consistent feedback;37,38 and learning that includes a motor component.39,40 Traditionally, this type of memory refers to motor skills, such as playing a musical instrument, riding a motorcycle, or tying a shoe (known colloquially as muscle memories).
More recently, however, it has been discovered that many more purely cognitive behaviors also recruit procedural learning and memory systems. The best evidence for this comes from information-integration category learning tasks. In category learning tasks, two or more categories of unfamiliar visual images are presented to the subject one at a time and the subject’s task is to use trial-by-trial feedback about response accuracy to learn to assign each image to its correct category. Information-integration category learning tasks are those in which accuracy is maximized only if information from two or more non-commensurable stimulus dimensions is integrated at some predecisional stage.41 Typically, the optimal strategy in information-integration tasks is difficult or impossible to describe verbally, so it is unlikely to be discovered via logical reasoning or by a process of explicit hypothesis generation and testing. Tasks in which these latter explicit strategies succeed are called rule-based category learning tasks. Figure 1 shows examples of rule-based and information-integration tasks that are frequently used in laboratory studies. A real life example of an information-integration task might be learning to discriminate between German shepherds and wolves. Much evidence suggests that success in information-integration tasks depends on procedural learning. For example, switching the locations of the response keys interferes with information-integration categorization, but not with rule-based categorization.39,42
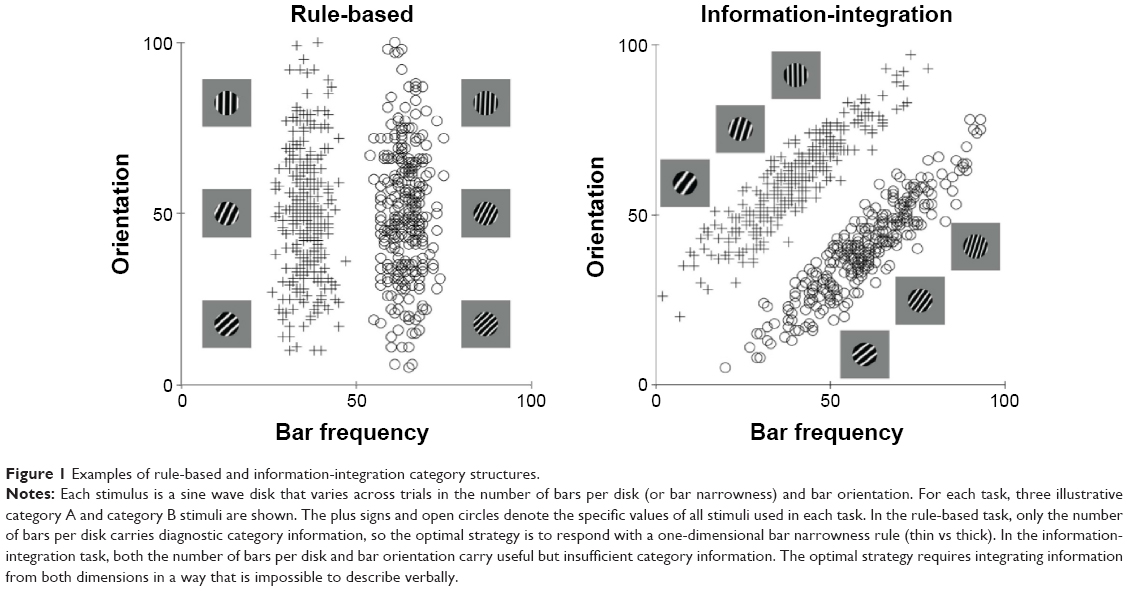 | Figure 1 Examples of rule-based and information-integration category structures. |
Another difference between the declarative and procedural systems is that learning in tasks that depend on declarative memory is flexible with regards to feedback timing, in the sense that long timing delays often have no detrimental effect on learning. In contrast, for procedural learning, the timing of feedback is critical. Learning is best when feedback immediately follows the behavior. Several studies have shown that rule-based category learning is unaffected by feedback delays as long as 10 seconds,38,43 supporting the notion that declarative and especially working memory systems are recruited. In contrast, information-integration category learning is best with a feedback delay of 500 milliseconds, slightly worse with delays of 0 or 1,000 milliseconds,44 and highly impaired with feedback delays of 2.5 seconds or longer.38,43 This complex pattern of results suggests that there is an optimal time for feedback to arrive after a response.
There is now overwhelming evidence that procedural learning depends on the basal ganglia, and especially on the striatum. This evidence comes from single-unit recording studies in non-human animals,45,46 animal lesion experiments,47–50 neuropsychological patient studies,51–53 and human neuroimaging studies.54–56
Perceptual representation memory
The perceptual representation memory system mediates “improvement in identifying or processing a stimulus as the result of its having been observed previously”.57 This type of learning is often referred to as repetition priming and it occurs even in the absence of feedback.58 Phenomenologically, perceptual representation memory can mediate the feeling of visual familiarity that occurs when one sees a visual image that has been seen before, but perceptual representation memory is an implicit system that can operate without conscious awareness.59 Behavioral effects of this type of memory can be observed after only a single stimulus repetition60 and can persist for months.61
Neuroimaging data and neuropsychological patient studies suggest a locus within the visual cortex. For example, perceptual priming is associated with a repetition-dependent suppression of activity in extrastriate regions of the visual cortex, such as the extrastriate occipital cortex.17,62 Similarly, animal studies show reduced cellular activity in the inferior temporal cortex that is proportional to stimulus repetition.63 However, these data do not provide a clear cellular mechanism for perceptual learning within the perceptual representation system.
Memory of automatic behaviors
Many actions performed by adults are automatic. When we brush our teeth, make our morning coffee, or ride a bicycle, our actions are typically automatic. The evidence is now good that automatic behaviors are not stored via any of the declarative or nondeclarative memory systems discussed so far.64 For example, PD patients have impaired declarative and nondeclarative memories. Even so, some PD patients are nevertheless able to emit an automatic motor response when presented with a familiar visual cue (eg, kicking a ball), despite difficulties in initiating novel voluntary movements.65
Ashby et al66 hypothesized that after practicing a behavior that was initially acquired via procedural learning long enough for it to become automatic, its representation is entirely cortical. According to this view, the development of automaticity is associated with a gradual transfer of control from the striatum to cortical–cortical projections from the relevant sensory areas directly to the premotor areas that initiate the behavior.
Dopamine systems
Neuroanatomy
Although DA cells are found in a number of central nervous system sites, for the purposes of understanding cognitive behaviors, the most important DA neurons originate in the midbrain regions of the substantia nigra pars compacta (SNpc) and the ventral tegmental area (VTA). DA neurons in these areas widely innervate various subcortical and cortical regions via the nigrostriatal, mesolimbic, mesocortical, and thalamic ascending DA projection pathways. The cell bodies of the nigrostriatal DA system, which are located in the SNpc, send dense projections to the striatum. The mesolimbic DA system originates from a more diffuse collection of neurons in the VTA. One portion of these neurons projects to limbic regions such as the nucleus accumbens, the amygdala, the hippocampus, and the ACC. A third pathway, referred to as the mesocortical DA system, also originates from the VTA and projects to virtually all of the frontal cortex. A fourth pathway, recently identified in the primate brain, projects to the thalamus and may be independent from the other systems.67
It is important to note that DA neurons in the VTA and SNpc projecting to striatal, limbic, and cortical areas are at least partly intermixed, suggesting that ascending DA systems interact.68 For example, it is true that most cortical DA comes from the VTA, but there are notable exceptions, such as a group of neurons in the dorsolateral SNpc that project to the dorsolateral PFC,69 and another group in the ventromedial SNpc that projects to the ventromedial PFC, ACC, and orbitofrontal cortex.70,71
Within the basal ganglia, an additional organizational scheme is that striatal neurons (within patch compartments) send direct inhibitory projections to those DA neurons from which they receive DA input and also to other DA neurons that in turn project to a different striatal subregion.72 Thus, ascending DA pathways interact and enable functional interactions via a series of parallel cortical-striatal-cortical loops.73 This spiral organization allows for information flow from ventral parts of the basal ganglia important in emotional learning to dorsal parts related more to cognitive and motor function.74
Through these pathways, the DA systems contribute to a wide variety of cognitive behaviors. Not surprisingly, DA dysfunction in neurological and neuropsychiatric disorders such as PD, schizophrenia, depression, ADHD, anorexia, autism spectrum disorders, and drug addiction is associated with multiple types of impairments, including of motor control, cognition, and motivation.75–79
Neurophysiology
DA neuron firing includes both phasic and tonic phases.80 The phasic bursts are brief, on the time scale of milliseconds, while the tonic mode includes spontaneous single spike activity that provides a baseline firing rate, which can briefly pause under certain conditions. Spike bursts trigger a high amplitude phasic DA release into targeted synapses,81,82 whereas the tonic firing of DA neurons is responsible for the baseline DA concentration.
After its release, free DA is either reabsorbed into the presynaptic terminal for reuse or broken down by enzymes. DA finely tunes its own synthesis and release with an inhibitory feedback loop mediated by presynaptic (D2) autoreceptors. Dopamine active transporter (DAT) is a membrane-bound, presynaptic protein that serves as a regulator of the synaptic concentration of DA at nerve terminals.83 DAT provides a rapid and efficient mechanism for reuptake of synaptic DA and is essential for the regulation of DA neurotransmission.84
The concentration of DAT serves as a marker of the homeostatic tone of the DA system.85 The highest concentrations of DAT are found in the striatum, with much lower concentrations in the brain stem, thalamus, basolateral amygdala, ACC, and neocortex, including the PFC.86,87 The high striatal concentrations of DAT cause exceptionally fast DA reuptake and therefore rapid clearing of DA from striatal synapses.5 This is an extremely important feature that makes the function of striatal DA unique, where a single phasic DA burst has the ability for temporal coding of the event that generated it. In contrast, low concentrations of cortical DAT cause DA to persist for much longer in cortical synapses.3,88 For example, the delivery of a single food pellet to a hungry rat elevates DA levels in the PFC for approximately 30 minutes.89 In the cortex, DA must either diffuse out of the synapse or be degraded by enzymes such as catechol-o-methyltransferase (COMT).90
Most DA receptors are located on post-synaptic neurons. Five receptor subtypes (D1–D5) are currently identified. The DA receptor subtypes have distinct anatomical distributions in the brain91 and can be viewed as markers for different clusters of DA-related functions. The five subtypes are grouped into two families on the basis of structural homology and biochemical characteristics. The family of D1-like receptors includes the D1 and D5 subtypes, and the family of D2-like receptors includes the D2, D3, and D4 subtypes. Of all DA receptors, the D1 and D2 receptors are, by far, the most common. Among these two, D1 receptors are more abundant than D2 receptors, reflecting high concentrations not only in the striatum but also throughout the neocortex.92 For example, in the frontal cortex, there are approximately ten times as many D1 as D2 receptors.93 D2 receptors are highly concentrated in the striatum, lower concentrations are expressed in the brainstem and thalamus, and concentrations are minute in the neocortex.86,94
Receptors of the D1 family are expressed throughout the brain. In contrast, receptors in the D2 family exhibit more regional specificity: D2 receptors are expressed primarily in the dorsal striatum, D3 receptors in the ventral striatum, including the nucleus accumbens but less so in the dorsal striatum, and D4 receptors in the frontal cortex and limbic regions.95 These differences have important consequences for cognition and behavior.96,97
The D1 and D2 receptors are slow-acting metabotropic receptors that are linked to G proteins. Activation of D1 receptors leads to an increase in intracellular levels of cyclic AMP, whereas activation of D2 receptors leads to decreases in the levels of intracellular cyclic AMP. In addition, phasic vs tonic DA release may have differential effects on D1 and D2 receptors.80
Post-synaptic DA receptors are located on neurons that are the targets of the most common excitatory neurotransmitter in the brain, namely glutamate, and there is now substantial evidence that DA modulates the postsynaptic effects of glutamate.98,99 There are two primary mechanisms via which DA exerts these neuromodulatory effects, ie, fast and slow. The fast effect of increases in synaptic DA levels is to increase the signal-to-noise ratio of the postsynaptic glutamate response,100–102 resulting in immediate effects on behavior that persist until DA levels decline. The slower effect is to potentiate LTP and long-term depression (LTD), leading to increased levels of synaptic plasticity103,104 which causes long-term changes in learned behaviors.
Neurons select meaningful signals out of noise by suppressing relatively weaker signals, while amplifying stronger ones. To achieve this dual effect, glutamate exerts different effects on two classes of receptors, ie, NMDA and non-NMDA. A common member of the non-NMDA class is the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor. AMPA and NMDA are both ionotropic receptors, but NMDA requires higher levels of presynaptic glutamate release for activation than AMPA. Therefore, at low levels of stimulation, the postsynaptic glutamate response is dominated by activity at AMPA (and other non-NMDA) receptors, whereas at high levels of stimulation, the glutamate response is dominated by activity at NMDA receptors. Evidence suggests that DA potentiates the glutamate response through the NMDA receptor102,105,106 and depresses the glutamate response through the AMPA receptor.100,101 The net effect of these modulations is to weaken the postsynaptic response when presynaptic stimulation is weak and to potentiate the postsynaptic response when presynaptic stimulation is strong. This increases the overall signal-to-noise ratio because noise should cause weak presynaptic stimulation, whereas signal should cause the presynaptic stimulation to be strong. Several mathematical models of this modulatory effect have been proposed.107,108
DA is known to have pronounced effects on synaptic plasticity. This has been observed in hippocampal,109 striatal,110,111 and PFC neurons.112 Evidence suggests that presynaptic glutamate release initiates several postsynaptic intracellular cascades that alter the strength of these synapses, and that DA plays an important role in these processes. Much evidence suggests that, when presynaptic and postsynaptic activation are both strong, DA levels above baseline promote LTP.104 Furthermore, within the striatum at least, if strong presynaptic and postsynaptic activation are present, but DA release is below baseline, then LTD is observed.103
A large literature shows that DA neurons in the VTA and SNpc increase their firing above baseline following unexpected rewards and decrease their firing below baseline following failure to receive an expected reward.113–115 Thus, DA-enhanced LTP should be in effect following an unexpected reward in any brain region that is a target of VTA or SNpc DA neurons.
The mechanisms via which DA potentiates LTP are reasonably well understood. One such mechanism is mediated by chemical cascades that are initiated when NMDA receptors are activated by glutamate, which begins the phosphorylation of calcium/calmodulin-dependent protein kinase II (CaMKII).116 During a brief period of time (0.3–2 seconds)117 when CaMKII is partially phosphorylated, a different chemical cascade that is initiated when DA binds to D1 receptors can potentiate the LTP-inducing effects of CaMKII. The critical step in this DA-induced cascade may be the phosphorylation of DARPP-32 (dopamine and cAMP-regulated phosphoprotein) because the phosphorylated version of DARPP-32 deactivates proteins (eg, PP-1) that reduce the LTP effects of CaMKII. Thus, when DA binds to D1 receptors, an important LTP-inhibiting action is reduced. The effect of phosphorylating Ca2+-dependent enzymes, such as CaMKII is to signal the activation of transcription factors and genes that encode for the synthesis of membrane and cytoskeletal proteins to mediate actual structural changes in dendritic spines.118
In the machine-learning literature, a distinction is made between Hebbian learning and reinforcement learning.119,120 In Hebbian learning, any synapses that are (simultaneously) highly active are strengthened and any synapses that are weakly active are weakened. Reinforcement learning is similar to Hebbian learning with the additional requirement of a learning (or reinforcement) signal. If the signal is present, then the learning rules are the same as in Hebbian learning, but if the signal is absent, then all synapses are weakened, regardless of activity. Many researchers have argued that DA plays the role of the reinforcement signal in the basal ganglia.6,121
A necessary feature of any reinforcement signal is high temporal resolution. Consider a task in which the goal is to learn a number of stimulus–response associations. Following a correct response, DA must be released into the relevant synapses quickly, before the critical traces disappear. But after the correct synapses have been strengthened, it is also essential that excess DA be quickly cleared from the synapse. If it is not, and the response on the next trial is an error, then the residual DA will strengthen inappropriate synapses, namely, those responsible for producing the incorrect response. This would undo the beneficial learning that occurred following correct responses, and prevent discrimination learning. Within the striatum, DA is quickly cleared from synapses by DAT and, as a result, the temporal resolution of DA in the striatum is high enough for DA to serve as a trial-by-trial reinforcement signal. However, the low concentrations of DAT in the frontal cortex cause cortical DA levels to change slowly. Thus, the first rewarded behavior in a training session is likely to cause frontal cortical DA levels to rise, and the absence of DAT will cause DA levels in the frontal cortex to remain high throughout the training session. As a result, all synapses that are activated during the session are likely to be strengthened, regardless of whether the associated behavior is appropriate or not. Thus, although DA may facilitate LTP in the frontal cortex, it appears to operate too slowly to serve as a trial-by-trial reinforcement signal.4 Instead, it is thought that cortical LTP/LTD follows Hebbian learning rules,121,122 which means that cortical LTP can occur regardless of whether the resulting behavior was rewarded. Recent work suggests, however, that within the PFC at least, this Hebbian learning depends on baseline DA levels. If the background DA is too low, then activity that might normally facilitate LTP can actually induce LTD.123,124 Thus, disorders that reduce tonic cortical DA levels could have profound effects on the learning of PFC-mediated behaviors.
In summary, DA has two prominent neuromodulatory effects, ie, a fast effect in which the signal-to-noise ratio of the postsynaptic glutamate response is increased, and a slower effect in which synaptic plasticity is potentiated. The consequences of these two different effects are vastly different in the basal ganglia, where DAT concentrations are high, and in other targets of VTA and SNpc DA neurons where DAT concentrations are low (such as in the frontal cortex). For example, in the frontal cortex, a single phasic burst from DA neurons could increase signal-to-noise ratio for many minutes, whereas this increase would likely only last a few seconds in the basal ganglia. Furthermore, these regional differences in DAT concentration mean that synaptic plasticity follows reinforcement learning rules within the striatum and Hebbian rules in the frontal cortex and in other DAT-poor targets of VTA and SNpc DA neurons.
Effect of DA on behaviors mediated by declarative learning and memory systems
Declarative learning and memory systems receive a dense DA projection, mainly from the VTA. Furthermore, DAT concentrations are low in almost all brain regions that contribute to declarative systems. Thus, we should expect that increases in brain DA levels (at least up to some optimal level) should cause persistent increases in signal-to-noise ratio and enhanced Hebbian learning for any behavior mediated by declarative systems.
A wide range of research supports these general predictions, although as we will see, the evidence suggests that there is an optimal DA level for cognitive function, that is, performance is compromised if DA levels are either higher or lower than this optimal level.
Deficits due to DA reductions
PD is caused by the accelerated death of DA-producing neurons. Although the earliest damage is typically to the nigrostriatal system, there is also concomitant damage to the mesocorticolimbic system. In particular, production of DA in the VTA is substantially diminished as the disease progresses.125,126 Since DA neurons die in both the SNpc and the VTA, DA levels decrease in both the PFC and the striatum.2 Within the SNpc, cell loss is predominantly found in the ventral tier with less (but still extensive) damage in the dorsal tier.127,128 This is opposite to the dorsal-to-ventral pattern of the less severe cell loss occurring in normal aging. Parkinsonian motor symptoms appear after a 60%–70% loss of SNpc neurons and a 70%–80% loss of DA levels in the striatal nuclei.128,129 These motor symptoms include tremor, rigidity, bradykinesia, and akinesia.
In addition to motor deficits, non-demented PD patients present cognitive symptoms that resemble those observed in patients with PFC damage (especially those with dorsolateral PFC lesions).130 Numerous studies documenting the cognitive deficits of PD patients have revealed impairment in a wide variety of tasks mediated by declarative memory systems, including working memory, logical reasoning, recall from episodic memory, rule learning, and tasks that require executive attention.131–133
For example, PD patients are impaired in rule-based category learning, which requires hypothesis testing about what simple explicit rules can separate perceptual stimuli into categories.133 The most widely known example of a rule-based task is the Wisconsin Card Sorting Test (WCST),134 and as expected, PD patients are impaired on the WCST.135 Specifically, they show a tendency to perseverate on rules that are no longer successful.
Owen et al argued that perseverative errors can occur on the WCST for two reasons.130 One is a failure to select an appropriate alternative stimulus feature when feedback suggests that the feature that is the current focus of attention is incorrect, and the second is a failure to switch attention from the current unsuccessful feature to the newly selected feature. Using a clever design that isolated these two cognitive operations, Owen et al found that patients with frontal damage were impaired on attentional switching but not on feature selection, whereas (unmedicated) PD patients were impaired on both operations. Based on these and other results, Ashby et al proposed that feature selection is mediated by a circuit that includes the ACC and the PFC, whereas attentional switching is mediated by a circuit that includes the PFC and the head of the caudate nucleus.136
One line of evidence supporting this hypothesis is that lesioning the DA fibers that project from the VTA into the PFC actually improves the performance of monkeys on a simplified version of the WCST.137 If switching occurs entirely within the PFC, then such lesions should impair switching performance (as seen, for example, in patients with PD). If the basal ganglia play a key role, however, then lesioning DA fibers into the PFC should have no direct effect on switching. How then can one explain that lesioning DA fibers into the PFC actually improves performance on the WCST? An important clue to this apparent paradox comes from reports that such lesions tend to increase DA levels in the basal ganglia.137 If the basal ganglia are responsible for switching, if switching is enhanced by DA, and if lesioning the DA fibers that enter the PFC increases DA levels in the basal ganglia, then lesioning the DA fibers in the PFC should improve switching. Data such as these support the hypothesis that switching is mediated primarily by the basal ganglia whereas selection is mediated primarily by the ACC.
PD patients also suffer from proactive interference in rule application, which also has switching and selection elements. In testing PD patients in the Odd-Man-Out choice discrimination task (a task where subjects need to pick the odd-man-out from a grouping of three stimuli), Flowers and Robertson138 found that PD patients were relatively unimpaired on the first block of trials using one rule (ie, performance was quite close to controls), but were subsequently impaired in later blocks using either a different rule or the same original rule. In fact, their performance decrement was only slightly improved when told explicitly what rule to use when selecting the Odd-Man-Out stimulus: subjects never reacquired the same performance level as at the beginning of the test. In a similar task where subjects were required to alternate their response strategy on a trial-by-trial basis, PD patients produced more false alarms than controls, but only for long time intervals between targets.139 Taken together, these results show that PD patients are susceptible to proactive interference, which reflects deficits both in switching away from an early response strategy or rule, and selecting a new one.
Episodic memory can be tested by recall (eg, “What did you have for breakfast?”) or by recognition (“Did you have cereal?”). Compared with recognition, recall depends much more heavily on the ability to select the appropriate retrieval strategy, maintain it, and switch to a new strategy if the current strategy is ineffective.140 These processes are similar to those required in rule-based category learning. Not surprisingly, therefore, PD patients are more impaired in recall than in recognition, recalling fewer words than controls while still showing the usual serial position effects of recency and primacy.141 Evidence that this deficit is due to PFC dysfunction comes from studies showing that patients with frontal lobe damage use fewer strategies than healthy controls during episodic memory encoding and recall,142 and have difficulty remembering the sequence (temporal order) and the source (who said what) of items.143
Facilitation due to DA increases
Whereas many studies have shown that DA depletion impairs the performance of behaviors that depend on declarative memory systems, many fewer have directly investigated the effects of increasing DA levels in healthy subjects. Even so, some promising results have been reported. For example, several studies have reported improvements in the working memory of healthy humans who were given small doses of a DA agonist.144,145 Although high doses of DA-targeting drugs, such as psychostimulants, impair PFC function, low doses preferentially improve PFC cognitive function of both healthy subjects and patients with neuropsychiatric disorders such as schizophrenia and ADHD.146 Even so, most of the data on how DA increases might affect declarative memory-based tasks are indirect.
Ashby et al proposed that events that induce mild positive affect (ie, improved mood) in healthy adults are also often associated with increased cortical DA levels.147 For example, receiving an unexpected gift improves mood and it should also cause cortical DA levels to rise since DA neurons are well known to increase their firing following unexpected rewards.114 There is other evidence linking positive mood to DA. First, drugs that mimic the effects of DA (ie, DA agonists) or that enhance dopaminergic activity elevate feelings. These drugs include morphine and apomorphine (agonists), cocaine (which blocks DA reuptake), amphetamines (which increase DA release), and naturally produced endorphins.148 Finally, DA antagonists (ie, neuroleptics) are thought to flatten affect and render reward ineffective.149
If positive affect increases cortical DA levels, and if increased cortical DA improves executive function, then mild positive affect should be associated with improved executive function. Considerable evidence supports this prediction. In particular, many studies have shown that mild positive affect improves creative problem-solving, facilitates recall, and generally improves cognitive flexibility.147 Perhaps the strongest such evidence comes from studies of creative problem-solving using tasks such as the Remote Associates Test150 and the Duncker candle task.151 Success in both of these requires the selection of a nondominant response (eg, finding unusual compound words that relate usually unrelated words and using the match box as a platform for the candle instead of just storing matches). Thus, both tasks make heavy demands on the selection operation identified by Owen et al130 and hypothesized to depend on an ACC/PFC circuit.136 A number of studies have reported that positive affect improves performance in both tasks.152–154
More recently, Nadler et al showed that relative to neutral affect controls, positive affect subjects performed better in rule-based categorization, but not in information-integration categorization.155 These results are expected because the abundant DAT in the striatum should cause the DA released to the positive affect-inducing event to be cleared quickly out of the striatum, and therefore not influence the subsequent information-integration category learning. In contrast, the relative lack of cortical DAT means that the DA released to the positive affect-inducing event should elevate cortical DA levels above baseline for 20–30 minutes, ie, long enough to facilitate the selection and attentional switching processes needed for success in the rule-based task.
Positive affect has also been shown to facilitate the encoding of neutral and positive episodic memories, which is demonstrated by better later recall.156,157 One possibility is that the positive affect facilitates hippocampal-mediated memory consolidation. VTA DA projects heavily to the hippocampus, an important part of the medial temporal lobes thought to be necessary for the consolidation of episodic memories.158,159 Normal hippocampal function depends critically on the neurotransmitter acetylcholine (eg, reductions in hippocampal cholinergic activity produce spatial memory deficits in rats).160 DA has been shown to increase acetylcholine release in the hippocampus161 and to improve memory consolidation in a brightness discrimination task.162 Thus, it is plausible that positive affect-induced DA release improves episodic memory consolidation.
Optimal DA levels
Although declarative memory-mediated behaviors seem to improve when cortical DA levels rise modestly, evidence also strongly suggests that large increases impair performance. In other words, DA appears to have an “inverted U-shaped” influence on PFC function, where either decreases or increases from an optimal level of DA impair declarative memory-mediated behaviors.163 For example, blockade and excessive stimulation of D1 receptors both impair spatial working memory.164,165
Other supporting evidence comes from studies that induced negative affect. One might speculate that if positive affect is associated with increased cortical DA levels, then negative affect may be associated with decreased levels. However, stressful or anxiety-provoking events, which presumably would produce a negative affective state in humans, actually appear to increase DA levels in certain brain regions. In particular, animal studies indicate that stressful events (eg, foot shock, tail pinch) cause increased DA release from the VTA but have little or no effect on DA release from the SNpc.166,167 Working memory deficits during stress exposure are likely mediated by excessive D1 receptor stimulation, because they are prevented by D1 antagonist administration. At high levels of D1 receptor stimulation, the responsiveness and signal-to-noise ratio of PFC neurons are both reduced. Thus, these adverse effects of stress could be due to overstimulation of the DA system.
Changing environmental demands and internal states may necessitate shifts of control from one memory system to another, or a fine-tuned coordination between them. It appears that DA participates in influencing this interplay between memory systems. Under optimal levels of DA, PFC functions such as working memory and selective attention are supported, thereby enabling deliberate, flexible regulation of behavior within a changing environment. Sliding to either side of the “inverted U” function of DA compromises this state. Under chronic stress exposure (eg, over 2 weeks), a decrease in baseline PFC DA levels is observed.168 Such DA reductions are associated with anhedonia, social withdrawal (as in schizophrenia),169 and depression.170
On the other hand, acute uncontrollable stress can produce high levels of DA and norepinephrine, with the effect that control of behavior is switched to the amygdala and other more primitive brain circuits.171 Several lines of evidence suggest that DA may initiate this shift: DA levels in the amygdala are increased under stress;172,173 DA produces disinhibition in the basolateral amygdala;174 and release of DA enhances amygdala-related behavior,175 whereas inhibition of the DA system depresses learned fear responses.176 Shifting control of behavior from the PFC to the amygdala and other subcortical systems creates a bias toward sensory-driven emotional responses, fear conditioning, and stressor-associated procedural and spatial memory formation. The adaptive value of such switches may be that responding becomes more rapid and less effortful. This might save one’s life when there is danger and a need to react rapidly, but it could be detrimental in situations that require thoughtful analysis and inhibitory control.
Effect of DA on behaviors mediated by procedural learning and memory systems
Procedural learning and memory systems receive a dense DA projection, from both the SNpc and the VTA. In addition, DAT concentrations are high in the most critical procedural system brain regions, namely the basal ganglia. Thus, we should expect that increases in DA levels will cause transient increases in signal-to-noise ratio and enhanced reinforcement learning for any behavior mediated by procedural systems. Again, the available data support these general predictions.
First, the induction of positive affect improves learning on declarative memory-mediated rule-based categorization, but not on procedural memory-mediated information-integration categorization.155 This result follows from the assumption that the event that induced the positive affect increases cortical signal-to-noise ratios throughout the subsequent period of category learning, whereas the increased striatal signal-to-noise ratios caused by the same event are too brief to affect the category learning.
Second, many studies have shown that PD patients, who have reduced striatal DA levels, are impaired in procedural learning. The two most heavily studied procedural-learning tasks are information-integration category learning and the serial reaction time task,177 which requires subjects to press keys as quickly as possible in response to stimuli that appear in various locations on the screen. A large improvement in response time is observed when the stimulus sequence is repeated, even when subjects are unaware that a sequence exists. Several studies have reported that patients with PD show reduced implicit learning in the serial reaction time task.178,179 Other studies have reported that PD patients are impaired in information-integration learning, especially when the task is difficult.52,53,180 These results are consistent with the hypothesis that reductions in striatal DA levels decrease cortical-striatal synaptic plasticity.
Effect of DA on behaviors mediated by other learning and memory systems
Perceptual representation memory system
The perceptual representation memory system is mediated primarily within sensory association areas of the cortex. The DA systems do not project in any significant way to the visual or auditory cortex,9,10 so the effects of changes in brain DA levels should be minimal on behaviors mediated by perceptual representation memory.
This prediction is surprisingly difficult to test. On the one hand, some seemingly counter results have been reported. For example, transient stimulation of VTA DA neurons during presentation of an auditory tone increases the extent of the representation of this tone in the auditory cortex.181 Similarly, several functional magnetic resonance imaging studies have reported reward-based learning effects in the visual cortex that are consistent with DA-mediated synaptic plasticity.182,183 One complication, however, in interpreting these results is that the effects of reward in sensory association areas should be similar to the effects of attention, because attention is biased toward stimuli that predict reward.184 In fact, it has been proposed that within the visual cortex, plasticity is mediated by attention-gated reinforcement learning that uses acetylcholine as the reinforcement signal, rather than DA.185 In support of this prediction, stimulation of cholinergic neurons in the basal forebrain during presentation of an auditory tone also increases the extent of the representation of this tone in the auditory cortex.186 Other studies have shown that cortical learning is impaired following acetylcholine depletion.187 To complicate matters even more, there is abundant evidence that the DA and cholinergic systems interact, so altering DA levels is likely to have concomitant effects on acetylcholine levels.188,189 Clearly, more work is needed on this issue.
Memory of automatic behaviors
If automatic behaviors are stored in the cortex, as hypothesized by Ashby et al,66 then their production should be modestly affected by changes in brain DA levels. The motor and premotor cortex receive a relatively dense DA projection from the VTA, but the visual and auditory cortex do not. These predictions are largely untested, although, as noted earlier, PD patients, who have DA reductions and striatal dysfunction, are impaired in procedural learning but are relatively normal in producing automatic skills.65
Individual differences in DA function
Many factors are known to affect brain DA levels, including age, genetic predisposition, drug-taking history, and neuropsychological patient status.147 For example, brain DA levels are known to decrease by approximately 7% per decade of life during normal aging. However, adolescence is associated with sustained overexpression of DA receptors in the PFC that persists until adulthood.190 Interestingly, this inverted U-shaped peak and decline of DA receptor levels occur earlier in the striatum than in the PFC.191 The firing rate of midbrain DA neurons also peaks during this distinctive developmental period.192 Not surprisingly, adolescents differ from adults on cognitive tasks that are PFC-dependent and DA-dependent, such as decision-making, planning, working memory, and inhibitory control.193,194 These and other developmental shifts in DA function191 may contribute to the heightened vulnerability to drug abuse and stress during adolescence.195
Neurological diseases that impact the DA system may result from aberrant development at different stages. For example, schizophrenia typically develops during late adolescence and early adulthood,191 while ADHD develops in early childhood, but in 60%–70% of cases symptoms persist into adulthood.196,197 Similarly, single gene mutation diseases such as Fragile X syndrome and Rett syndrome are neurodevelopmental autism spectrum disorders marked by derailment of brain development (including synaptic plasticity) from infancy, and especially during the critical period from ages 1 to 3 years.198–200
Not surprisingly, different aspects of the DA system malfunction in these three DA-related disorders. A recent meta-analysis201 found that dopaminergic abnormality in schizophrenia is mainly presynaptic, which affects DA synthesis, baseline synaptic DA levels, and DA release. Current treatment mechanisms rely on D2 receptor blockade, which fail to target these abnormalities. ADHD stimulant medications target DAT levels, which are lower in ADHD patients.202 In Fragile X syndrome and Rett syndrome, the DA system is disrupted and hypofunctional,79,108 and dopaminergic treatment shows promise in ameliorating at least some of the devastating symptoms.203–205
It is important to note that DA medications do not have uniform effects throughout the brain. When PD patients are given DA-related medications to improve their motor function (eg, L-dopa), they show a modest improvement on many of the memory and executive tasks on which they are normally impaired.131 However, such drugs may improve some processes but have a minimal, or detrimental, effect on others. For example, Cools et al reported that DA replacement therapy improved the task-switching performance of PD patients, but made the same patients impulsive.206 They argued that by increasing levels of DA, normal function was restored to the system supporting task-switching but the system responsible for decision-making became “overdosed”.
Could DA agonists ameliorate memory problems in people with other kinds of frontal lobe dysfunction, such as due to aging or focal lesion? In cases of focal lesion, this may be only successful if enough tissue is left to respond to the DA drug.207 In one such successful attempt, McDowell et al208 gave either a DA agonist (bromocriptine) or placebo to traumatic brain injury patients (who showed impaired executive function, and probably had some frontal damage). The drug selectively improved performance on many of the executive measures, such as reducing perseverative errors on the WCST.
Another source of variability is genetic factors that affect DA neurotransmission.209 Thanks to recent progress in genomic research, individual differences in a variety of DA-relevant genotypes have been associated with a range of cognitive functions, such as working memory, attention, episodic memory, and reward processing, as well as with psychiatric disorders210,211 and addiction.212 At this point it is too soon to draw any strong conclusions because of possible complex interactions among genes and between genes and the environment. Even so, guided by the framework laid out in this article, some predictions are possible and even supported by recent genomic studies.
For example, several studies have shown that subjects with a COMT genotype (met/met) associated with higher PFC DA levels have better working memory and perform better on the WCST test than subjects with a COMT genotype (val/val) associated with lower levels of PFC DA.213,214 Another example is a recent study showing that carriers of a specific DRD2 genotype that is associated with higher striatal D2 receptor availability were better at information-integration category learning than participants with a genotype associated with lower D2 availability.215 Similarly, other studies have examined the physiological and behavioral effects of DARPP-32.97,216
Conclusion
The multiple roles via which DA affects behavior have been topics of scientific research for half a century. In contrast, the widespread acceptance of multiple learning and memory systems has occurred only recently. To date, research on DA function has proceeded mostly independently of research on memory systems. But to predict the effects of DA-related medications on a cognitive behavior, it is vital to know what memory systems are mediating that behavior. This knowledge will indicate which brain regions are relevant and whether those regions receive dopaminergic input. The next step in the prediction process is to determine the density of DAT in these relevant regions. Since DAT concentrations are high only in the basal ganglia, essentially this step reduces to determining whether the basal ganglia play a significant role in mediating the behavior. For behaviors that are mediated within brain regions that receive a rich DA projection but lack high concentrations of DAT, the general prediction should be that even a brief rise in brain DA levels could enhance performance for a period of 20–30 minutes. The major exception is if the person already has high DA levels, perhaps because he or she is a healthy adolescent or under the influence of some DA-related drug, in which case the opposite effect should be expected. For behaviors primarily mediated within the basal ganglia, the same brief rise in DA levels should have little if any effect because DAT will quickly clear the excess DA from basal ganglia synapses. However, a treatment that can raise DA levels for a more prolonged period of time (eg, L-dopa) should improve procedural learning, facilitate the initiation of novel movements, and improve attentional switching. Finally, behaviors mediated in brain regions that do not receive a prominent DA projection (eg, the visual cortex) should be relatively unaffected by DA-related treatments, although this prediction is complicated by the known interactions between DA and acetylcholine.
Acknowledgment
Preparation of this article was supported in part by a grant from the National Institute of Mental Health (2R01MH063760).
Disclosure
The authors report no conflicts of interest in this work.
References
Björklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. | ||
Cools R. Dopaminergic modulation of cognitive function – implications for L-dopa treatment in Parkinson’s disease. Neurosci Biobehav Rev. 2006;30:1–23. | ||
Seamans JK, Robbins TW. Dopamine modulation of the prefrontal cortex and cognitive function. In: Neve K, editor. The Dopamine Receptors. New York, NY, USA: Humana Press; 2010. | ||
Lapish CC, Kroener S, Durstewitz D, Lavin A, Seamans JK. The ability of the mesocortical dopamine system to operate in distinct temporal modes. Psychopharmacology (Berl). 2007;191:609–625. | ||
Cragg S, Rice M, Greenfield S. Heterogeneity of electrically evoked dopamine release and reuptake in substantia nigra, ventral tegmental area, and striatum. J Neurophysiol. 1997;77:863–873. | ||
Houk JC, Adams JL, Barto AG. A model of how the basal ganglia generate and use neural signals that predict reinforcement. In: Houk JC, Davis JL, Beiser DG, editors. Models of Information Processing in the Basal Ganglia. Cambridge, MA, USA: MIT Press; 1995. | ||
Eichenbaum H, Cohen NJ. From Conditioning to Conscious Recollection: Memory Systems of the Brain. New York, NY, USA: Oxford University Press; 2001. | ||
Squire LR. Declarative and nondeclarative memory: multiple brain systems supporting learning and memory. J Cogn Neurosci. 1992;4:232–243. | ||
Thierry AM, Blanc G, Sobel A, Stinus L, Glowinski J. Dopaminergic terminals in the rat cortex. Science. 1973;182:499–501. | ||
Goldman-Rakic PS, Leranth C, Williams SM, Mons N, Geffard M. Dopamine synaptic complex with pyramidal neurons in primate cerebral cortex. Proc Natl Acad Sci U S A. 1989;86:9015–9019. | ||
Cowan N. The magical mystery four: how is working memory capacity limited, and why? Curr Dir Psychol Sci. 2010;19:51–57. | ||
Baddeley A. Working memory: looking back and looking forward. Nat Rev Neurosci. 2003;4:829–839. | ||
Miller EK. The prefrontal cortex and cognitive control. Nat Rev Neurosci. 2000;1:59–65. | ||
Posner MI, DiGirolamo GJ. Conflict, target detection and cognitive control. In: Parasuraman R, editor. The Attentive Brain. Cambridge, MA, USA: MIT Press; 1998. | ||
Kane MJ, Engle RW. Working-memory capacity and the control of attention: the contributions of goal neglect, response competition, and task set to Stroop interference. J Exp Psychol Gen. 2003;132:47–70. | ||
Smith EE, Jonides J. Neuroimaging analyses of human working memory. Proc Natl Acad Sci U S A. 1998;95:2061–2068. | ||
Cabeza R, Nyberg L. Imaging cognition II: an empirical review of 275 PET and fMRI studies. J Cogn Neurosci. 2000;12:1–47. | ||
Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. | ||
Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014;157:163–186. | ||
Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. | ||
Fuster JM. Unit activity in prefrontal cortex during delayed-response performance: neuronal correlates of transient memory. J Neurophysiol. 1973;36:61–78. | ||
Constantinidis C, Steinmetz MA. Neuronal activity in posterior parietal area 7a during the delay periods of a spatial memory task. J Neurophysiol. 1996;76:1352–1355. | ||
Fuster JM, Alexander GE. Firing changes in cells of the nucleus medialis dorsalis associated with delayed response behavior. Brain Res. 1973;61:79–91. | ||
Hikosaka O, Sakamoto M, Usui S. Functional properties of monkey caudate neurons. III. Activities related to expectation of target and reward. J Neurophysiol. 1989;61:814–832. | ||
Schultz W, Romo R. Role of primate basal ganglia and frontal cortex in the internal generation of movements I. Preparatory activity in the anterior striatum. Exp Brain Res. 1992;91:363–384. | ||
Mushiake H, Strick PL. Pallidal neuron activity during sequential arm movements. J Neurophysiol. 1995;74:2754–2758. | ||
Ashby FG, Ell SW, Valentin VV, Casale MB. FROST: a distributed neurocomputational model of working memory maintenance. J Cogn Neurosci. 2005;17:1728–1743. | ||
Tulving E. Episodic and semantic memory. In: Tulving E, Donaldson W, editors. Organization of Memory. London, UK: Academic Press; 1972. | ||
Rovee-Collier C, Hayne H, Colombo M. The Development of Implicit and Explicit Memory. Volume 24. Philadelphia, PA, USA: John Benjamin Publishing; 2000. | ||
Damasio AR. Looking for Spinoza: Joy, Sorrow, and the Feeling Brain. New York, NY, USA: Random House; 2004. | ||
Duncan J, Owen AM. Common regions of the human frontal lobe recruited by diverse cognitive demands. Trends Neurosci. 2000;23:475–483. | ||
Eichenbaum H. Hippocampus: cognitive processes and neural representations that underlie declarative memory. Neuron. 2004;44:109–120. | ||
Yonelinas AP. The nature of recollection and familiarity: a review of 30 years of research. J Mem Lang. 2002;46:441–517. | ||
Vargha-Khadem F, Gadian DG, Watkins KE, et al. Differential effects of early hippocampal pathology on episodic and semantic memory. Science. 1997;277:376–380. | ||
Squire LR, Dede AJ. Conscious and unconscious memory systems. Cold Spring Harb Perspect Biol. 2015;7:a021667. | ||
Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. | ||
Willingham DB. A neuropsychological theory of motor skill learning. Psychol Rev. 1998;105:558–584. | ||
Maddox WT, Ashby FG, Bohil CJ. Delayed feedback effects on rule-based and information-integration category learning. J Exp Psychol Learn Mem Cogn. 2003;29:650–662. | ||
Ashby FG, Ell SW, Waldron EM. Procedural learning in perceptual categorization. Mem Cognit. 2003;31:1114–1125. | ||
Willingham DB, Wells LA, Farrell JM, Stemwedel ME. Implicit motor sequence learning is represented in response locations. Mem Cognit. 2000;28:366–375. | ||
Ashby FG, Gott RE. Decision rules in the perception and categorization of multidimensional stimuli. J Exp Psychol Learn Mem Cogn. 1988;14:33–53. | ||
Maddox WT, Bohil CJ, Ing AD. Evidence for a procedural-learning-based system in perceptual category learning. Psychol Bull Rev. 2004;11:945–952. | ||
Maddox WT, Ing AD. Delayed feedback disrupts the procedural-learning system but not the hypothesis-testing system in perceptual category learning. J Exp Psychol Learn Mem Cogn. 2005;31:100–107. | ||
Worthy DA, Markman AB, Maddox WT. Feedback and stimulus-offset timing effects in perceptual category learning. Brain Cogn. 2013;81:283–293. | ||
Carelli RM, Wolske M, West MO. Loss of lever press-related firing of rat striatal forelimb neurons after repeated sessions in a lever pressing task. J Neurosci. 1997;17:1804–1814. | ||
Merchant H, Zainos A, Hernadez A, Salinas E, Romo R. Functional properties of primate putamen neurons during the categorization of tactile stimuli. J Neurophysiol. 1997;77:1132–1154. | ||
Eacott MJ, Gaffan D. The role of monkey inferior parietal cortex in visual discrimination of identity and orientation of shapes. Behav Brain Res. 1991;46:95–98. | ||
Gaffan D, Eacott MJ. Uncinate fascicle section leaves delayed matching-to-sample intact, with both large and small stimulus sets. Exp Brain Res. 1995;105:175–180. | ||
McDonald RJ, White NM. Parallel information processing in the water maze: evidence for independent memory systems involving dorsal striatum and hippocampus. Behav Neural Biol. 1994;61:260–270. | ||
Packard MG, McGaugh JL. Double dissociation of fornix and caudate nucleus lesions on acquisition of two water maze tasks: further evidence for multiple memory systems. Behav Neurosci. 1992;106:439–446. | ||
Filoteo JV, Maddox WT, Davis JD. A possible role of the striatum in linear and nonlinear categorization rule learning: evidence from patients with Huntington’s disease. Behav Neurosci. 2001;115:786–798. | ||
Filoteo JV, Maddox WT, Salmon DP, Song DD. Information-integration category learning in patients with striatal dysfunction. Neuropsychology. 2005;19:212–222. | ||
Knowlton BJ, Mangels JA, Squire LR. A neostriatal habit learning system in humans. Science. 1996;273:1399–1402. | ||
Nomura EM, Maddox WT, Filoteo JV, et al. Neural correlates of rule-based and information-integration visual category learning. Cereb Cortex. 2007;17:37–43. | ||
Seger CA, Cincotta CM. The roles of the caudate nucleus in human classification learning. J Neurosci. 2005;25:2941–2951. | ||
Waldschmidt JG, Ashby FG. Cortical and striatal contributions to automaticity in information-integration categorization. Neuroimage. 2011;56:1791–1802. | ||
Gazzaniga MS, Ivry RB, Mangun GR. Cognitive Neuroscience: The Biology of the Mind. 4th ed, Cambridge, MA, USA: MIT Press; 2002. | ||
Casale MB, Ashby FG. A role for the perceptual representation memory system in category learning. Percept Psychophys. 2008;70:983–999. | ||
Schacter DL. Perceptual representation systems and implicit memory. Ann N Y Acad Sci. 1990;608:543–571. | ||
Wiggs CL, Martin A. Properties and mechanisms of perceptual priming. Curr Opin Neurobiol. 1998;8:227–233. | ||
Cave CB. Very long-lasting priming in picture naming. Psychol Sci. 1997;8:322–325. | ||
Schacter DL, Buckner RL. Priming and the brain. Neuron. 1998;20:185–195. | ||
Desimone R, Miller EK, Chelazzi L, Lueschow A. Multiple memory systems in the visual cortex. In: Gazzaniga MS, editor. The Cognitive Neurosciences. Cambridge, MA, USA: MIT Press; 1995. | ||
Ashby FG, Crossley MJ. Automaticity and multiple memory systems. Wiley Interdiscip Rev Cogn Sci. 2012;3:363–376. | ||
Asmus F, Huber H, Gasser T, Schöls L. Kick and rush paradoxical kinesia in Parkinson disease. Neurology. 2008;71:695. | ||
Ashby FG, Ennis JM, Spiering BJ. A neurobiological theory of automaticity in perceptual categorization. Psychol Rev. 2007;114:632–656. | ||
Sánchez-González MA, Garcia-Cabezas MA, Rico B, Cavada C. The primate thalamus is a key target for brain dopamine. J Neurosci. 2005;25:6076–6083. | ||
Hauber W. Dopamine release in the prefrontal cortex and striatum: temporal and behavioural aspects. Pharmacopsychiatry. 2010;43 Suppl 1:S32–S41. | ||
Williams SM, Goldman-Rakic PS. Widespread origin of the primate mesofrontal dopamine system. Cereb Cortex. 1998;8:321–345. | ||
Porrino LJ, Goldman-Rakic PS. Brainstem innervation of prefrontal and anterior cingulate cortex in the rhesus monkey revealed by retrograde transport of HRP. J Comp Neurol. 1982;205:63–76. | ||
Williams SM, Goldman-Rakic PS. Characterization of the dopaminergic innervation of the primate frontal cortex using a dopamine-specific antibody. Cereb Cortex. 1993;3:199–222. | ||
Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20:2369–2382. | ||
Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13:266–271. | ||
Cardinal RN, Parkinson JA, Hall J, et al. Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev. 2002;26:321–352. | ||
Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. | ||
Salamone JD, Correa M, Farrar A, et al. Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology (Berl). 2007;191:461–482. | ||
Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. | ||
Kaye WH, Frank GKW, McConaha C. Altered dopamine activity after recovery from restricting type anorexia nervosa. Neuropsychopharmacology. 1999;21:503–506. | ||
Paul K, Venkitaramani DV, Cox CL. Dampened dopamine-mediated neuromodulation in prefrontal cortex of fragile X mice. J Physiol. 2013;(591)4:1133–1143. | ||
Goto Y, Otani S, Grace AA. The Yin and Yang of dopamine release: a new perspective. Neuropharmacology. 2007;53:583–587. | ||
Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. | ||
Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. | ||
Giros B, Mestikawy S, Godinot N, et al. Cloning, pharmacological characterization and chromosome assignment of the human dopamine transporter. Mol Pharmacol. 1992;3:383–390. | ||
Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. | ||
Jaber M, Jones S, Giros B, Caron MG. The dopamine transporter: a crucial component regulating dopamine transmission. Mov Disord. 1997;12:629–633. | ||
Ito H, Takahashi H, Arakawa R, Takano H, Suhara T. Normal database of dopaminergic neurotransmission system in human brain measured by positron emission tomography. Neuroimage. 2008;39:555–565. | ||
Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–57. | ||
Tzschentke T. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol. 2001;63:241–320. | ||
Feenstra MG, Botterblom MH. Rapid sampling of extracellular dopamine in the rat prefrontal cortex during food consumption, handling and exposure to novelty. Brain Res. 1996;742:17–24. | ||
Vidgren J, Svensson LA, Liljas A. Crystal structure of catechol O-methyltransferase. Nature. 1994;368:354–358. | ||
Meador-Woodruff JH. Update on dopamine receptors. Ann Clin Psychiatry. 1994;6:79–90. | ||
Hall H, Sedvall G, Magnusson O, Kopp J, Halldin C, Farde L. Distribution of D1- and D2-dopamine receptors and its metabolites in the human brain. Neuropsychopharmacology. 1994;11:245–256. | ||
Lidow MS, Goldman-Rakic PS, Gallagher DW, Rakic P. Distribution of dopaminergic receptors in the primate cerebral cortex: quantitative autoradiographic analysis using [3H]Raclopride, [3H]Spiperone and [3H]Sch23390. Neuroscience. 1991;40:657–671. | ||
Kessler RM, Whetsell WO, Ansari MS, et al. Identification of extrastriatal dopamine D2 receptors in post moterm human brain with [125I]epidepride. Brain Res. 1993;609:237–243. | ||
Cortés R, Camps M, Gueye B, Probst A, Palacios JM. Dopamine receptors in human brain: Autoradiographic distribution of D1 and D2 sites in Parkinson syndrome of different etiology. Brain Res. 1989;483:30–38. | ||
Nemoda Z, Szekely A, Sasvari-Szekely M. Psychopathological aspects of dopaminergic gene polymorphisms in adolescence and young adulthood. Neurosci Biobehav Rev. 2011;35:1665–1686. | ||
Frank MJ, Doll BB, Oas-Terpstra J, Moreno F. Prefrontal and striatal dopaminergic genes predict individual differences in exploration and exploitation. Nat Neurosci. 2009;12:1062–1068. | ||
Cepeda C, Radisavljevic Z, Peacock W, Levine MS, Buchwald NA. Differential modulation by dopamine of responses evoked by excitatory amino acids in human cortex. Synapse. 1992;11:330–341. | ||
Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci U S A. 1994;91:5720–5724. | ||
Del Arco A, Mora F. NMDA and AMPA/kainate glutamatergic agonists increase the extracellular concentrations of GABA in the prefrontal cortex of freely moving rat: modulation by endogenous dopamine. Brain Res Bull. 2002;57:623–630. | ||
Urban NN, Gonzalez-Burgos G, Henze DA, Lewis DA, Barrionuevo G. Selective reduction by dopamine of excitatory synaptic inputs to pyramidal neurons in primate prefrontal cortex. J Physiol. 2001;539:707–712. | ||
Wang J, O’Donnell P. D1 dopamine receptors potentiate NMDA-mediated excitability increase in layer V prefrontal cortical pyramidal neurons. Cereb Cortex. 2001;11:452–462. | ||
Ronesi J, Lovinger DM. Induction of striatal long-term synaptic depression by moderate frequency activation of cortical afferents in rat. J Physiol. 2005;562:245–256. | ||
Arbuthnott GW, Ingham CA, Wickens JR. Dopamine and synaptic plasticity in the neostriatum. J Anat. 2000;196:587–596. | ||
Hemmings HC, Walaas SI, Ouimet CC, Greengard P. Dopaminergic regulation of protein phosphorylation in the striatum: DARRP-32. Trends Neurosci. 1987;10:377–383. | ||
Wickens JR. Striatal dopamine in motor activation and reward mediated learning: steps towards a unifying model. J Neural Transm Gen Sect. 1990;80:9–31. | ||
Ashby FG, Casale MB. A model of dopamine modulated cortical activation. Neural Netw. 2003;16:973–984. | ||
Servan-Schreiber D, Printz H, Cohen JD. A network model of catecholamine effects: gain, signal-to-noise ratio, and behavior. Science. 1990;249:892–895. | ||
Yang SN. Sustained enhancement of AMPA receptor-and NMDA receptor-mediated currents induced by dopamine d1/d5 receptor activation in the hippocampus: an essential role of postsynaptic Ca2+. Hippocampus. 2000;10:57–63. | ||
Levine MS, Li S, Cepeda C, Cromwell HC, Altemus KL. Neuromodulatory actions of dopamine on synaptically-evoked neostriatal responses in slices. Synapse. 1996;24:65–78. | ||
Umemiya M, Raymond LA. Dopaminergic modulation of excitatory postsynaptic currents in rat neostriatal neurons. J Neurophysiol. 1997;78:1248–1255. | ||
Chen L, Yang CR. Interaction of dopamine D1 and NMDA receptors mediates acute clozapine potentiation of glutamate EPSPs in rat prefrontal cortex. J Neurophysiol. 2002;87:2324–2336. | ||
Hollerman JR, Schultz W. Dopamine neurons report an error in the temporal prediction of reward during learning. Nat Neurosci. 1998;1:304–309. | ||
Mirenowicz J, Schultz W. Importance of unpredictability for reward responses in primate dopamine neurons. J Neurophysiol. 1994;2:1024–1027. | ||
Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. | ||
Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. | ||
Yagishita S, Hayashi-Takagi A, Ellis-Davies GC, Urakubo H, Ishii S, Kasai H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science. 2014;345:1616–1620. | ||
Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. | ||
Dayan P, Abbott LF. Theoretical Neuroscience. Cambridge, MA, USA: MIT Press; 2001. | ||
Sutton RS, Barto AG. Introduction to Reinforcement Learning. Cambridge, MA, USA: MIT Press; 1998. | ||
Doya K. Complementary roles of basal ganglia and cerebellum in learning and motor control. Curr Opin Neurobiol. 2000;10:732–739. | ||
Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. | ||
Matsuda Y, Marzo A, Otani S. The presence of background dopamine signal converts long-term synaptic depression to potentiation in rat prefrontal cortex. J Neurosci. 2006;26:4803–4810. | ||
Kolomiets B, Marzo A, Caboche J, Vanhoutte P, Otani S. Background dopamine concentration dependently facilitates long-term potentiation in rat prefrontal cortex through postsynaptic activation of extracellular signal-regulated kinases. Cereb Cortex. 2009;19:2708–2718. | ||
Javoy-Agid F, Agid Y. Is the mesocortical dopaminergic system involved in Parkinson disease? Neurology. 1980;30:1326–1330. | ||
Scatton B, Rouquier L, Javoy-Agid F, Agid Y. Dopamine deficiency in the cerebral cortex in Parkinson disease. Neurology. 1982;32:1039–1040. | ||
Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. | ||
Gibb WR, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1991;54:388–396. | ||
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington: clinical, morphological and neurochemical correlations. J Neurosci. 1973;20:415–455. | ||
Owen AM, Roberts AC, Hodges JR, Robbins TW. Contrasting mechanisms of impaired attentional set-shifting in patients with frontal lobe damage or Parkinson’s disease. Brain. 1993;116:1159–1175. | ||
Gotham AM, Brown RG, Marsden CD. ‘Frontal’ cognitive function in patients with Parkinson’s disease ‘on’ and ‘off’ levodopa. Brain. 1988;111:299–321. | ||
Levin BE, Labre MM, Weiner WJ. Cognitive impairments associated with early Parkinson’s disease. Neurology. 1989;39:557–561. | ||
Price A, Filoteo JV, Maddox WT. Rule-based category learning in patients with Parkinson’s disease. Neuropsychologia. 2009;47:1213–1226. | ||
Heaton RK, Chelune GJ, Talley JL, Kay GG, Curtiss G. Wisconsin Card Sorting Test Manual. Odessa, FL, USA: Psychological Assessment Resources; 1993. | ||
Lees AJ, Smith E. Cognitive deficits in the early stages of Parkinson’s disease. Brain. 1983;106:257–270. | ||
Ashby FG, Alfonso-Reese L, Turken AU, Waldron EM. A neuropsychological theory of multiple systems in category learning. Psychol Rev. 1998;105:442–481. | ||
Roberts AC, De Salvia MA, Wilkinson LS, et al. 6-Hydroxydopamine lesions of the prefrontal cortex in monkeys enhance performance on an analog of the Wisconsin Card Sort Test: possible interactions with subcortical dopamine. J Neurosci. 1994;14:2531–2544. | ||
Flowers K, Robertson C. The effect of Parkinson’s disease on the ability to maintain a mental set. J Neurol Neurosurg Psychiatry. 1985;48:517–529. | ||
Ravizza SM, Ivry RB. Comparison of the basal ganglia and cerebellum in shifting attention. J Cogn Neurosci. 2001;13:285–297. | ||
Burgess PW. Strategy application disorder: the role of the frontal lobes in human multitasking. Psychol Res. 2000;63:279–288. | ||
Breen EK. Recall and recognition memory in Parkinson’s disease. Cortex. 1993;29:91–102. | ||
Alexander MP, Stuss DT, Fansabedian N. California Verbal Learning Test: performance by patients with focal frontal and non-frontal lesions. Brain. 2003;126:1493–1503. | ||
Thaiss L, Petrides M. Source versus content memory in patients with a unilateral frontal cortex or a temporal lobe excision. Brain. 2003;126:1112–1126. | ||
Luciano M, Depue RA, Arbisi P, Leon A. Facilitation of working memory in humans by a D2 dopamine receptor agonist. J Cogn Neurosci. 1992;4:58–68. | ||
Müller U, von Cramon DY, Pollmann S. Dl-versus D2-receptor modulation of visuospatial working memory in humans. J Neurosci. 1998;18:2720–2728. | ||
Berridge CW, Arnsten AF. Catecholamine mechanisms in the prefrontal cortex: proven strategies for enhancing higher cognitive function. Curr Opin Behav Sci. 2015;4:33–40. | ||
Ashby FG, Isen AM, Turken AU. A neuropsychological theory of positive affect and its influence on cognition. Psychol Rev. 1999;106:529–550. | ||
Harte JL, Eifert GH, Smith R. The effects of running and meditation on beta-endorphin, corticotropin-releasing hormone and cortisol in plasma, and on mood. Biol Psychol. 1995;40:251–265. | ||
Wise RA. Neuroleptics and operant behavior: the anhedonia hypothesis. Behav Brain Sci. 1982;5:39–88. | ||
Mednick MT, Mednick SA, Mednick EV. Incubation of creative performance and specific associative priming. J Abnorm Psychol. 1964;69:84–88. | ||
Duncker K. On problem solving. Psychol Monogr. 1945;58:1–113. | ||
Estrada C, Young M, Isen AM. Positive affect influences creative problem solving and reported source of practice satisfaction in physicians. Motiv Emot. 1994;18:285–299. | ||
Greene TR, Noice H. Influence of positive affect upon creative thinking and problem solving in children. Psychol Rep. 1988;63:895–898. | ||
Isen AM, Daubman KA, Nowicki GP. Positive affect facilitates creative problem solving. J Pers Soc Psychol. 1987;52:1122–1131. | ||
Nadler RT, Rabi R, Minda JP. Better mood and better performance learning rule-described categories is enhanced by positive mood. Psychol Sci. 2010;21:1770–1776. | ||
Isen AM, Shalker TE, Clark M, Karp L. Affect, accessibility of material in memory, and behavior: a cognitive loop? J Pers Soc Psychol. 1978;36:1–12. | ||
Teasdale JD, Fogarty SJ. Differential effects of induced mood on retrieval of pleasant and unpleasant events from episodic memory. J Abnorm Psychol. 1979;88:248–257. | ||
McClelland JL, McNaughton BL, O’Reilly RC. Why there are complementary learning systems in the hippocampus and neocortex: insights from the successes and failures of connectionist models of learning and memory. Psychol Rev. 1995;102:419–457. | ||
Squire LR, Alvarez P. Retrograde amnesia and memory consolidation: a neurobiological perspective. Curr Opin Neurobiol. 1995;5:169–177. | ||
Kim JS, Levin ED. Nicotinic, muscarinic and dopaminergic actions in the ventral hippocampus and the nucleus accumbens: effects on spatial working memory in rats. Brain Res. 1996;725:231–240. | ||
Imperato A, Puglisi-Allegra S, Casolini P, Angelucci L. Changes in brain dopamine and acetylcholine release during and following stress are independent of the pituitary-adrenocortical axis. Brain Res. 1991;538:111–117. | ||
Grecksch G, Matthies H. The role of dopaminergic mechanisms in the rat hippocampus for the consolidation in a brightness discrimination. Psychopharmacology (Berl). 1981;75:165–168. | ||
Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10:376–384. | ||
Sawaguchi T, Goldman-Rakic PS. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science. 1991;251:947–950. | ||
Zahrt J, Taylor JR, Mathew RG, Arnsten AFT. Supranormal stimulation of dopamine D1 receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci. 1997;17:8528–8535. | ||
Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem. 1989;52:1655–1658. | ||
Sorg BA, Kalivas PW. Effects of cocaine and footshock stress on extracellular dopamine levels in medial prefrontal cortex. Neuroscience. 1993;53:695–703. | ||
Gresch PJ, Sved AF, Zigmond MJ, Finlay JM. Stress-induced sensitization of dopamine and norepinephrine efflux in medial prefrontal cortex of the rat. J Neurochem. 1994;63:575–583. | ||
Andreasen NC, Rezai K, Alliger R, et al. Hypofrontality in neuroleptic-naive patients and in patients with chronic schizophrenia: assessment with xenon 133 single-photon emission computed tomography and the Tower of London. Arch Gen Psychiatry. 1992;49:943–958. | ||
Galynker II, Cai J, Ongseng F, Finestone H, Dutta E, Serseni D. Hypofrontality and negative symptoms in major depressive disorder. J Nucl Med. 1998;39:608–612. | ||
Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. | ||
Inglis FM, Moghaddam B. Dopaminergic innervation of the amygdala is highly responsive to stress. J Neurochem. 1999;72:1088–1094. | ||
Yokoyama M, Suzuki E, Sato T, Maruta S, Watanabe S, Miyaoka H. Amygdalic levels of dopamine and serotonin rise upon exposure to conditioned fear stress without elevation of glutamate. Neurosci Lett. 2005;379:37–41. | ||
Grace AA, Rosenkranz JA. Regulation of conditioned responses of basolateral amygdala neurons. Physiol Behav. 2002;77:489–493. | ||
Borowski TB, Kokkinidis L. The effects of cocaine, amphetamine, and the dopamine D1 receptor agonist SKF 38393 on fear extinction as measured with potentiated startle: implications for psychomotor stimulant psychosis. Behav Neurosci. 1998;112:952–965. | ||
Nader K, LeDoux JE. Inhibition of the mesoamygdala dopaminergic pathway impairs the retrieval of conditioned fear associations. Behav Neurosci. 1999;113:891–901. | ||
Nissen MJ, Bullemer P. Attentional requirements of learning: evidence from performance measures. Cogn Psychol. 1987;19:1–32. | ||
Jackson GM, Jackson SR, Harrison J, Henderson L, Kennard C. Serial reaction time learning and Parkinson’s disease: evidence for a procedural learning deficit. Neuropsychologia. 1995;33:577–593. | ||
Siegert RJ, Taylor KD, Weatherall M, Abernethy DA. Is implicit sequence learning impaired in Parkinson’s disease? A meta-analysis. Neuropsychology. 2006;20:490–495. | ||
Sage JR, Anagnostaras SG, Mitchell S, et al. Analysis of probabilistic classification learning in patients with Parkinson’s disease before and after pallidolomy surgery. Learn Mem. 2003;10:226–236. | ||
Bao S, Chan VT, Merzenich MM. Cortical remodelling induced by activity of ventral tegmental dopamine neurons. Nature. 2001;412:79–83. | ||
Arsenault JT, Nelissen K, Jarraya B, Vanduffel W. Dopaminergic reward signals selectively decrease fMRI activity in primate visual cortex. Neuron. 2013;77:1174–1186. | ||
Weil RS, Furl N, Ruff CC, et al. Rewarding feedback after correct visual discriminations has both general and specific influences on visual cortex. J Neurophysiol. 2010;104:1746–1757. | ||
Anderson BA, Laurent PA, Yantis S. Value-driven attentional capture. Proc Natl Acad Sci U S A. 2011;108:10367–10371. | ||
Roelfsema PR, van Ooyen A, Watanabe T. Perceptual learning rules based on reinforcers and attention. Trends Cogn Sci. 2010;14:64–71. | ||
Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science. 1998;279:1714–1718. | ||
Winkler J, Suhr ST, Gage FH, Thal LJ, Fisher LJ. Essential role of neocortical acetylcholine in spatial memory. Nature. 1995;375:484–487. | ||
Calabresi P, Picconi B, Parnetti L, Di Filippo M. A convergent model for cognitive dysfunctions in Parkinson’s disease: the critical dopamine–acetylcholine synaptic balance. Lancet Neurol. 2006;5:974–983. | ||
Zmarowski A, Sarter M, Bruno JP. NMDA and dopamine interactions in the nucleus accumbens modulate cortical acetylcholine release. Eur J Neurosci. 2005;22:1731–1740. | ||
Naneix F, Marchand AR, Di Scala G, Pape JR, Coutureau E. Parallel maturation of goal-directed behavior and dopaminergic systems during adolescence. J Neurosci. 2012;32:16223–16232. | ||
McCutcheon JE, Marinelli M. Age matters. Eur J Neurosci. 2009;29:997–1014. | ||
Marinelli M, White FJ. Enhanced vulnerability to cocaine self-administration is associated with elevated impulse activity of midbrain dopamine neurons. J Neurosci. 2000;20:8876–8885. | ||
Crone EA, van der Molen MW. Developmental changes in real life decision making: performance on a gambling task previously shown to depend on the ventromedial prefrontal cortex. Dev Neuropsychol. 2004;25:251–279. | ||
Luciana M, Collins PF, Olson EA, Schissel AM. Tower of London performance in healthy adolescents: the development of planning skills and associations with self-reported inattention and impulsivity. Dev Neuropsychol. 2009;34:461–475. | ||
Crews F, He J, Hodge C. Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol Biochem Behav. 2007;86:189–199. | ||
Biederman J, Mick E, Faraone SV. Age-dependent decline of symptoms of attention deficit hyperactivity disorder: impact of remission definition and symptom type. Am J Psychiatry. 2000;157:816–818. | ||
Kessler RC, Chiu WT, Demler O, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. | ||
Naidu S, Murphy M, Moser HW, Rett A. Rett syndrome: natural history in 70 cases. Am J Med Genet. 1986;24:61–72. | ||
Armstrong VL. What is so critical? A commentary on the reexamination of critical periods. Dev Psychobiol. 2006;48:326–336. | ||
Rice D, Barone S Jr. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108 Suppl 3:511–533. | ||
Howes OD, Kambeitz J, Kim E, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment: meta-analysis of imaging studies. Arch Gen Psychiatry. 2012;69:776–786. | ||
Volkow ND, Wang GJ, Newcorn J, et al. Brain dopamine transporter levels in treatment and drug naive adults with ADHD. Neuroimage. 2007;34:1182–1190. | ||
Szczesna K, de la Caridad O, Petazzi P, et al. Improvement of the Rett syndrome phenotype in a Mecp2 mouse model upon treatment with levodopa and a dopa-decarboxylase inhibitor. Neuropsychopharmacology. 2014;39:2846–2856. | ||
Lim CS, Hoang ET, Viar KE, Stornetta RL, Scott MM, Zhu JJ. Pharmacological rescue of Ras signaling, GluA1-dependent synaptic plasticity, and learning deficits in a fragile X model. Genes Dev. 2014;28:273–289. | ||
Wang H, Wu LJ, Kim SS, et al. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron. 2008;59: 634–647. | ||
Cools R, Barker RA, Sahakian BJ, Robbins TW. L-Dopa medication remediates cognitive inflexibility, but increases impulsivity in patients with Parkinson’s disease. Neuropsychologia. 2003;41:1431–1441. | ||
Arnsten AF, Robbins TW. Neurochemical modulation of prefrontal cortical function in humans and animals. In: Stuss DT, Wills H, Knight RT, editors. Principles of Frontal Lobe Function. New York, NY, USA: Oxford Press; 2002. | ||
McDowell S, Whyte J, D’Esposito M. Differential effect of a dopaminergic agonist on prefrontal function in traumatic brain injury patients. Brain. 1998;121:1155–1164. | ||
Haile CN, Kosten TR, Kosten TA. Genetics of dopamine and its contribution to cocaine addiction. Behav Genet. 2007;37:119–145. | ||
Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: a meta-analytic review. Hum Genet. 2009;126:51–90. | ||
Meyer-Lindenberg A, Nichols T, Callicott JH, et al. Impact of complex genetic variation in COMT on human brain function. Mol Psychiatry. 2006;11:867–877. | ||
Le Foll B, Gallo A, Le Strat Y, Lu L, Gorwood P. Genetics of dopamine receptors and drug addiction: a comprehensive review. Behav Pharmacol. 2009;20:1–17. | ||
Bruder GE, Keilp JG, Xu H, et al. Catechol-O-methyltransferase (COMT) genotypes and working memory: associations with differing cognitive operations. Biol Psychiatry. 2005;58:901–907. | ||
Dickinson D, Elvevåg B. Genes, cognition and brain through a COMT lens. Neuroscience. 2009;164:72–87. | ||
Xie Z, Maddox WT, McGeary JE, Chandrasekaran B. The C957T polymorphism in the dopamine receptor D2 (DRD2) gene modulates domain-general category learning. J Neurophysiol. 2015;113:3281–3290. | ||
Meyer-Lindenberg A, Straub RE, Lipska BK, et al. Genetic evidence implicating DARPP-32 in human frontostriatal structure, function, and cognition. J Clin Invest. 2007;117:672–682. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.