Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 13
Differential Diagnosis and Therapeutic Advances in Multiple Myeloma: A Review Article
Authors Hussain M, Yellapragada S, Al Hadidi S
Received 15 June 2023
Accepted for publication 7 September 2023
Published 15 September 2023 Volume 2023:13 Pages 33—57
DOI https://doi.org/10.2147/BLCTT.S272703
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wilson Gonsalves
Munawwar Hussain,1 Sarvari Yellapragada,2 Samer Al Hadidi1
1Myeloma Center, Winthrop P. Rockefeller Cancer Institute, University of Arkansas for Medical Sciences, Little Rock, AR, USA; 2Michael E. DeBakey VA Medical Center and Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX, USA
Correspondence: Samer Al Hadidi, Myeloma Center, Winthrop P. Rockefeller Cancer Institute, University of Arkansas for Medical Sciences, Little Rock, AR, USA, Tel +1 501-526-6990, Fax +1 501-320-7861, Email [email protected]
Abstract: Multiple myeloma (MM) is a hematologic malignancy characterized by the abnormal clonal proliferation of plasma cells that may result in focal bone lesions, renal failure, anemia, and/or hypercalcemia. Recently, the diagnosis and treatment of MM have evolved due to a better understanding of disease pathophysiology, improved risk stratification, and new treatments. The incorporation of new drugs, including proteasome inhibitors, immunomodulatory drugs, anti-CD38 antibodies and high-dose chemotherapy followed by hematopoietic stem cell transplantation, has resulted in a significant improvement in patient outcomes and QoL. In this review, we summarize differential diagnoses and therapeutic advances in MM.
Keywords: multiple myeloma, myeloma, smoldering myeloma, Waldenstrom macroglobulinemia, light-chain amyloidosis
Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by the abnormal clonal proliferation of plasma cells in the bone marrow with a subsequent increase in the production of immunoglobulins. Abnormal production of immunoglobulins leads to organ damage characterized by anemia, hypercalcemia, focal bone lesions, and/or renal impairment. Based on these outcomes, the differential diagnosis of MM is broad (Table 1). Differential diagnosis includes monoclonal gammopathy of undetermined significance (MGUS), smoldering multiple myeloma (SMM), Waldenström Macroglobulinemia (WM), Light-Chain (AL) Amyloidosis, and plasmacytoma. Thus, prompt diagnosis of MM is essential because timely treatment significantly impacts outcomes and patient quality of life (QoL).
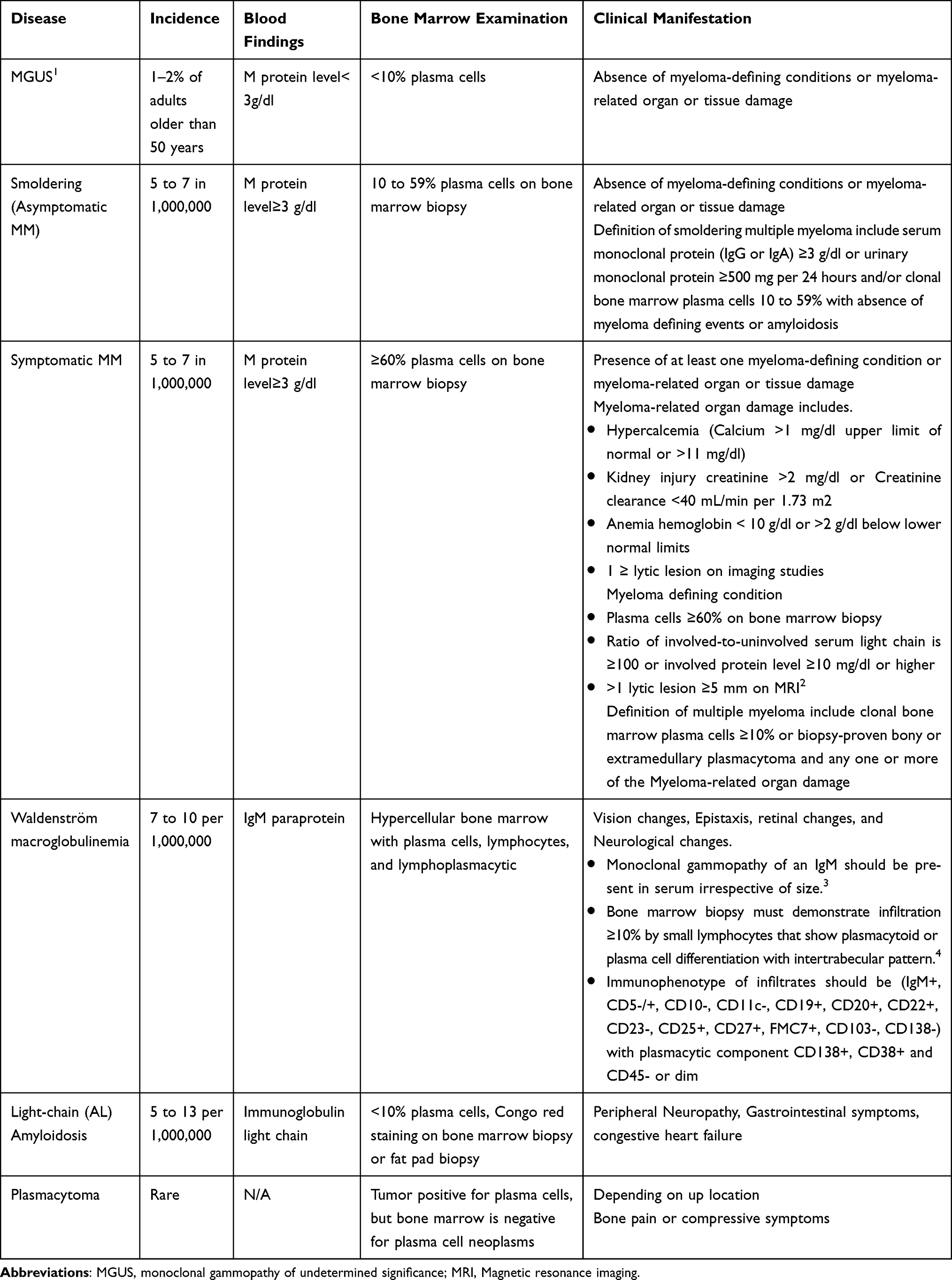 |
Table 1 Differential Diagnosis of MM |
MM is the second most common hematologic malignancy, with an estimated incidence of 34,470 adults (19,100 men and 15,370 women) in the US in 2022.5 MM is more common in Black compared to non-Hispanic White individuals and more common in men than women. The median age of initial diagnosis is 66 years.6,7
MM evolves from MGUS, a premalignant asymptomatic condition, which occurs in 3% of those over the age of 50.8,9 MGUS progresses to MM or related malignancies, including AL amyloidosis, lymphoma, or WM, at a rate of 1% per year.10 An intermediate, asymptomatic premalignant condition referred to as SMM carries a risk of progressing to MM of 10% per year in the first 5 years from initial diagnosis.10 A recent study of >75,000 individuals—the Iceland Screens, Treats, or Prevents Multiple Myeloma (iStopMM) study—showed that SMM had a prevalence of 0.5% in individuals over 40 years old, was more common in men (0.7%) than women (0.4%) and an incremental incidence with age.11 In most clinical cohorts of SMM, the median age of diagnosis is 65 years and is more common in Black individuals.
The introduction of newer therapies—immunomodulatory drugs, proteasome inhibitors (PIs), anti-CD38 antibodies, high-dose chemotherapy followed by hematopoietic stem cell transplantation, bispecific antibodies, and chimeric antigen T-cell therapy—has significantly improved progression-free survival (PFS) and/or overall survival (OS) in MM patients. In this review article, we will review the differential diagnosis of MM and treatment advances.
Monoclonal Gammopathy of Undetermined Significance
MGUS is a premalignant, asymptomatic plasma cell disorder and a precursor of MM, WM, and AL amyloidosis.12 In 1960, Jan Waldenström described MGUS as “essential hyperglobulinemia” or “benign monoclonal gammopathy.”13 In 1978, Robert Kyle introduced the current term “monoclonal gammopathy of undetermined significance”, based on a retrospective study of 241 patients with MGUS, of which a few progressed to MM, WM, or AL amyloidosis.14 MGUS is defined by <10% clonal plasma cells in the bone marrow, the presence of serum or urine M (monoclonal) protein, and the absence of diagnosis of MM or other plasma cell dyscrasias and no evidence of any organ dysfunction attributable to the monoclonal protein.2 Three different types of MGUS are recognized based on M-protein type: immunoglobulin M (IgM), non-IgM (IgG, IgA, or IgD), and light-chain MGUS. The risk of progression into a lymphoproliferative disorder differs for each subtype of MGUS.15
The prevalence of MGUS increases with age. It is observed in <0.3% of the population aged <40 years old, 3% of the population ≥50 years old, and 5% of the population ≥70 years old.10,16 The incidence and prevalence are higher in Black than White individuals and higher in men than women.17
According to the 2014 International Myeloma Working Group (IMWG), MGUS is diagnosed when the following 3 criteria are met: serum M protein <3 g/dL or presence of abnormal free light-chain (FLC) ratio, bone marrow plasma cells <10%, and absence of end-organ damage due to plasma cells.2
The risk of progression of MGUS is associated with several factors,18 the most important of which is an M protein spike of ≥15 g/L. Other risk factors include an IgA isotype, an abnormal free light-chain ratio of involved-to-uninvolved light chain ≥10, an increase in the M-protein level over time, bone marrow plasma cells >5%, a reduced level of uninvolved isotypes, and the presence of circulating plasma cells in the blood.
A model with the following biomarkers: M-protein ≥15 g/L, non-IgG MGUS, and abnormal FLC ratio of <0.125 or >8 was developed. Based on these factors, the 20-year risk of progression was 5% for those with no markers, 21% for those with 1, 37% for those with 2, and 58% for those exhibiting all of the markers (Table 2).19
 |
Table 2 Risk Stratification of MGUS-Based Mayo Clinic Risk Models |
Smoldering Multiple Myeloma
Smoldering Multiple Myeloma (SMM) is an intermediate condition between MGUS and MM. It is defined as an asymptomatic state where monoclonal protein ≥3 g/dL and/or 10–59% atypical plasma cells are present in the bone marrow, and there is no presence of end-organ damage (hypercalcemia, renal impairment, lytic lesions, or anemia) or other condition like amyloidosis or SLiM (60% or greater plasma cells in bone marrow, light-chain involvement with an involved-to-uninvolved ratio >100, and >1 focal lesion detected by magnetic resonance imaging [MRI]).20 The understanding of SMM is evolving. The risk of SMM progressing to MM is variable and is estimated to be 10% per year in the first 5 years following diagnosis, 3% in the next 5 years, and 1.5% thereafter.2 The median age of SMM onset is 67 years, with a high prevalence among Black individuals.21 Black individuals have a lower risk of progression to MM in both univariate (HR 0.57, cl 0.34–0.94) and multivariate models (HR 0.39, Cl 0.16–0.95) compared to White individuals.22 One-third of SMM patients never progress to MM.23 According to the iStopMM study, the prevalence of SMM in the general population is 0.5%. The iStopMM study collected blood samples from 75,422 patients, followed by bone marrow sampling in 1562. SMM prevalence increases with age and is more common in males than females.11
There are several risk stratification models that assess the risk of SMM progression to MM, including the Mayo Clinic 2018 (20/20/2) model, IMWG criteria, and PETHEMA (Programa de Estudio y Tratamiento de las Hemopatias Maligna) criteria, as shown (Table 3). It is important to note that there is variability of the results of those risk stratification models and none of them are perfect. The Mayo clinic 201824 model includes:
- Plasma cells >20%
- Free light-chain ratio >20
- Monoclonal protein M >2 g/dl
 |
Table 3 Various Risk Stratification Models of Smoldering Multiple Myeloma Progression |
Per the Mayo Clinic 2018 model, high-risk SMM includes 2–3 of the above factors with an estimated median time to progression of 29 months. The estimated risk of high-risk SMM progression to MM is 24% per year in the first 2 years, 11% per year within the next 3 years, and 5% in the next 5 years. The model defines intermediate-risk SMM as having one factor and estimates the time to progression to MM as 68 months. The estimated risk of intermediate-risk SMM progression to MM is 15% per year during the first 2 years, 7% per year during the next 3 years, and 4% per year for the next 5 years. Low-risk SMM is defined as having no factors present, and the estimated time to progression is 110 months. The estimated rate of low-risk SMM progression to MM is 5% per year during the first 10 years. IMWG validated Mayo Clinic’s risk stratification model in 1996 patients and found a 2-year risk of progression to MM or amyloidosis of 6% in low-, 18% in intermediate-, and 44% in high-risk SMM groups.25
The PETHEMA model includes immunoparesis and the percentage of plasma cells with aberrant immunophenotype, but the multiparameter flow cytometry requirement of the PETHEMA model makes it more difficult to implement clinically.26
Observation continues to be the most appropriate strategy for SMM management, regardless of risk stratification. This strategy is supported by the high incidence of SMM in large population studies and the lack of improvement in OS and/or QoL with earlier SMM treatment, and it is our recommended strategy at the University of Arkansas for Medical Sciences for all patients with SMM regardless of risk stratification.
Two treatment strategies were evaluated for high-risk SMM: control (low-intensity therapy aiming to delay time to organ damage) vs intensive chemotherapy to eradicate and potentially cure the disease.27
The QuiRedex Phase 3 multicenter trial studied a population of 119 patients with SMM28 and assigned 57 patients to a lenalidomide and dexamethasone treatment group and 62 to an observation-only group. The median follow-up was 75 months. The treatment arm showed a longer time of SMM progression to active MM compared with the observation group (median time to progression not reached [95% Cl 47 months-not reached] vs 23 months [16–31]; hazard ratio 0.24[95% CL 0.14–0.41]; p < 0.0001). In the observation group, 86% (53/62) of the patients progressed to MM compared to 39% (22/57) in the treatment group. The most important criticism of this study was that modern imaging techniques like MRI and whole-body positron emission tomography CT (PET-CT) were not used. It was also a possibility that a few MM patients were enrolled as SMM patients.
Another randomized phase 3 clinical trial29 in a population of 182 intermediate- or high-risk SMM patients assigned 92 patients to a lenalidomide treatment group and 90 to an observation-only group. Lenalidomide was administered on days 1 through 21 of a 28-day cycle. The primary endpoint was PFS, biochemical progression, and development of end-organ damage due to MM. The median follow-up was 35 months. PFS was longer in the lenalidomide treatment group compared to the observation-only group (hazard ratio 0.28; 95% Cl 0.12 to 0.62; p = 0.002). One-, two- and three-year progression-free survival was 98%,93%, and 91% for the lenalidomide arm vs 89%,76%, and 66% for the observation arm, respectively. Six deaths were reported, two in the lenalidomide arm vs four in the observation arm (hazard ratio for death, 0.46; 95% Cl, 0.08 to 2.53). This study had certain limitations. Initially, patients diagnosed in the previous year were enrolled in the study, but due to low recruitment, the protocol was altered to allow enrollment of patients diagnosed in the previous 5 years. It is possible that SMM patients that failed to progress to MM did not have high-risk SMM. Forty-seven percent of the patients had abnormal MRI at baseline, raising concerns for active MM as PET-CT was not a study requirement. In the observation arm, 24% of patients progressed to MM by 24 months, which was less than the anticipated 50%, suggesting a poor representation of high-risk SMM cases. According to the 2018 Mayo Clinic criteria, 58 patients were low risk. These criteria also identified 29 high-risk patients, of which 14 were assigned to the treatment arm. Due to an underpowered population, PFS statistics were not applied, and there were missing data on fluorescence in situ hybridization (FISH) genetics in 102/182 patients. This study included a QoL assessment with no significant difference in mean change score at 24 months. The low rate of progression and side effect concern makes it difficult to use this study in clinical practice.27
In the GEM-CESAR30 Phase 2, single-arm clinical trial of 90 patients with high-risk SMM or asymptomatic MM (based on 1 of 3 new biomarkers), 78 received KRD (carfilzomib, lenalidomide, dexamethasone) followed by autologous stem cell transplantation (ASCT) and KRD consolidation and maintenance for 2 years. At 30 months of follow-up, the overall response rate (ORR) was 100%, and complete response (CR) was 76%, with a minimal residual disease (MRD) rate of 63%. Three out of 90 patients died, four patients withdrew, and eight patients progressed from MRD negative to MRD positive. Thirty-one patients (34%) had at least one of the biomarkers considered myeloma-defining events that are currently classified as active MM. Grade 3–4 neutropenia and thrombocytopenia were reported in five (6%) and ten (18%) patients, respectively. Infection was observed in 16 patients (18%), and 8 (9%) patients had skin rash. Seven patients had to discontinue the maintenance regimen due to several complications, including cardiac arrest in one patient, four patients had hematological toxicity, and two patients had secondary malignancies.
In the ASCENT phase 2 clinical trial,31 46 SMM high-risk patients (70% male) with a median age of 63 years (range 47–76) were enrolled. Patients received 6 cycles of induction therapy with daratumumab (weekly for 8 weeks, then every other week for 16 weeks) and KRD (carfilzomib twice weekly, lenalidomide 25 mg daily for 3 weeks, dexamethasone 40 mg weekly) but in consolidation daratumumab every 4 weeks and dexamethasone 20 mg weekly. Patients received 12 cycles of maintenance with lenalidomide 10 mg daily for 3 weeks and daratumumab on day 1 every other cycle of a 4-week cycle. This study is still ongoing, but safety data showed concerning grade 3–4 adverse events, including cytopenia, infections, hypertension, diarrhea, and allergic reactions in less than 10% of patients, and 52% had at least one grade >2 adverse event. Given the asymptomatic nature of SMM and evolving definitions, high intensity treatments that do not improve OS and/or QoL while increase risk of serious adverse events should be discouraged unless planned in a clinical trial setting with an observational control arm.
Multiple Myeloma
MM is a hematological malignancy characterized by uncontrolled proliferation of plasma cells causing bone destruction, anemia, hypercalcemia, and/or acute kidney injury.32 There is no known etiology of MM, but risk factors are male sex, obesity, occupation (eg, firefighter), and dioxin and Agent Orange exposure.33 In newly diagnosed MM, typical findings include anemia (hemoglobin <12), one or more lytic lesions on a conventional radiograph in 79% of cases, elevated creatinine in 19% of cases, lymphadenopathy in 1% of cases, hypercalcemia in 13% of cases, and thrombocytopenia in 5% of cases.7 In newly diagnosed patients, 3.3% had extramedullary disease (presence of 1 or more extraosseous plasmacytomas on cross-sectional imaging), central nervous system involvement, and plasma cell leukemia. Extramedullary disease is aggressive both in newly diagnosed and relapsed refractory multiple myeloma (RRMM).34 About 10–15% of patients with MM are diagnosed with concurrent immunoglobulin light-chain amyloidosis during the course of the disease.35
Diagnostic evaluation of MM includes a complete blood count, imaging, urine studies, and bone marrow biopsy (Table 1). Blood tests included blood count with differential, serum creatinine, calcium level, albumin, lactate dehydrogenase, free serum light-chain level, beta-2 microglobulin levels, and serum protein electrophoresis with immunofixation. Serum protein electrophoresis shows monoclonal protein in 86% of patients.7 Urine tests include 24-hour urine collection to quantify Bence-Jones protein for baseline proteinuria, as secondary light-chain amyloidosis can have nephrotic range proteinuria. About 1–2% of all MM patients have nonsecretory MM, defined by no measurable serum or urine markers.36 In newly suspected MM, every patient should have a bone marrow biopsy, flow cytometry, cytogenetics, and FISH. Imaging includes MRI, PET-CT, whole-body low-dose CT, or bone survey in the absence of an advanced imaging modality.33 Revised IMWG diagnostic criteria for MM include 10% or greater plasma cells in the bone marrow and at least one type of end-organ damage (hypercalcemia, renal disease, anemia, and/or bone lytic lesion) or a myeloma defining SLiM criteria event (plasma cells >60% in bone marrow, free light-chain involved-to-uninvolved ratio >100, and more than one focal lesion on MRI) (Table 1).37
Waldenström Macroglobulinemia
Waldenström macroglobulinemia (WM) is a lymphoplasmacytic lymphoma characterized by an elevated level of immunoglobulin M (IgM).38 It is a rare disorder, with 1400 new cases in the US and an overall incidence of 3 per million persons per year.39,40 It is more prevalent in White men and has a median age of diagnosis of 70 years.41,42 Clinical features are due to infiltration of IgM and include anemia, peripheral neuropathy, lymphadenopathy, and hepatosplenomegaly.43 A study of 217 patients diagnosed with WM showed the following features.44
- Fundoscopic findings in 34% were characterized by dilated tortuous, segmented, sausage-shaped veins with hyperviscosity. Other findings include papilledema, hemorrhages, and papilledema, so patients with IgM levels >3000 mg/dL with hyperviscosity-related symptoms should have a fundoscopic examination.
- Bleeding in 23% of the patients, mainly due to hyperviscosity. Hyperviscosity causes platelet and clotting factor dysfunction.
- Constitutional “B” symptoms (fatigue, generalized weakness, weight loss, night sweats, and oronasal bleeding) in 23% of the patients.45
- Symptoms related to hyperviscosity in 31% of the patients included headache, vertigo, nystagmus, dizziness, blurring or loss of vision, deafness, or ataxia. Patients can have confusion, dementia, stroke, or coma in severe cases. Hyperviscosity can also precipitate or exacerbate congestive heart failure.44,46–48
- Neurological symptoms in 22% of the patients at the time of diagnosis, the most common being distal, symmetric, progressive sensorimotor neuropathy leading to generalized sensory loss and paresthesia.49,50
- Lymphadenopathy in 25% of patients and hepatomegaly in 24% of patients. Splenomegaly is often observed in newly diagnosed WM patients, and they can present with spontaneous spleen rupture.51
Diagnosis of WM is based on clinical presentation, bone marrow biopsy, and analysis of serum protein electrophoresis.4,52 For diagnosis of WM, criteria must be met, as shown in Table 1. Monoclonal gammopathy of an IgM should be present in serum, irrespective of level.3 Bone marrow biopsy must demonstrate small lymphocyte infiltration ≥10% that shows plasmacytoid or plasma cell differentiation with an intertrabecular pattern.4 Ninety percent of WM patients may have MYD88, L265P, and/or CXCR4 gene mutations, which are helpful in differentiating from other conditions for therapy selection and are prognostic.53,54
Light-Chain Amyloidosis
Light-chain (AL) amyloidosis is a multisystem disease caused by the deposition of fibrillary protein, producing dysfunction of affected organs.55 Because amyloidosis is nonspecific and has variable presentation, it is difficult to assess how many people are affected, and diagnosis is usually missed and delayed. About 4000 people are diagnosed with amyloid and AL amyloidosis every year in the US, with a median age between 50 and 65 years.56 The incidence of amyloidosis ranges from 9.4 to 14.0 cases per 1 million persons.57 Clinical features depend upon the type of protein and the extent and pattern of involvement.58 Systemic amyloidosis is due to the formation of insoluble amyloid fibrils that are due to the deposition of misfolded proteins. These proteins number over 30.59 AL amyloidosis is due to the deposition of a monoclonal light chain and can be associated with monoclonal gammopathy, MM, and B-cell lymphoma. AL amyloidosis has direct cardiotoxic, cytotoxic, and proapoptotic effects.60
ATTR amyloidosis is due to the deposition of transthyretin protein. Transthyretin is a protein produced by the liver, and its main function is the transportation of thyroid hormone and vitamin A.61 ATTR amyloidosis can be further differentiated into wild type (wtATTR) and hereditary subtypes (mutated vATTR).55 It is important to characterize the type of amyloid by mass spectrometry for all patients with amyloid deposition.
Amyloidosis predominantly involves the heart, kidney, liver, and gastrointestinal tract, but the lung, nervous system, muscles, and soft tissues can also be affected. These effects vary by subtype, with the vATTR subtype mainly affecting the heart and AL having more systemic involvement.62 Cardiac manifestation includes heart failure with preserved ejection fraction, exertional dyspnea, hypotension, angina, and cardiac arrhythmia. Renal involvement includes nonselective proteinuria or renal failure.63 Neuropathic effects involve both the somatic and autonomic systems and cause the loss of temperature and pain sensation, as well as numbness and weakness that lead to imbalance. Autonomic manifestations include altered bowel habits, orthostatic hypotension, urinary retention, and erectile dysfunction.64 Gastrointestinal tract symptoms include weight loss due to malabsorption, ulcers, perforation, and bowel dysmotility. Liver effects include hepatomegaly, hyposplenism, and liver failure.65 Patient can also present with muscle weakness, carpal tunnel syndrome, lumbar spinal stenosis, and alopecia.62
Therapeutic Advances in Multiple Myeloma
Due to the results of several clinical trials, MM treatment has been drastically changed both in newly diagnosed MM and RRMM settings. The introduction of novel treatments has changed OS, PFS, and QoL with less toxicity.66 The introduction of anti-CD38 monoclonal antibodies has revolutionized MM treatment across all settings and has now been followed by treatment with chimeric antigen receptor T (CAR-T) cells and bispecific antibodies both of which are showing early promising results. These drugs may become superior to conventional therapy due to their favorable toxicity profiles and high efficacy. New immune-based drugs with varying mechanisms of action, such as antibody-drug conjugates, immunomodulators, cereblon E3 ligase modulators, and fusion proteins are currently in development. Commonly used therapeutic agents are summarized in Table 4.
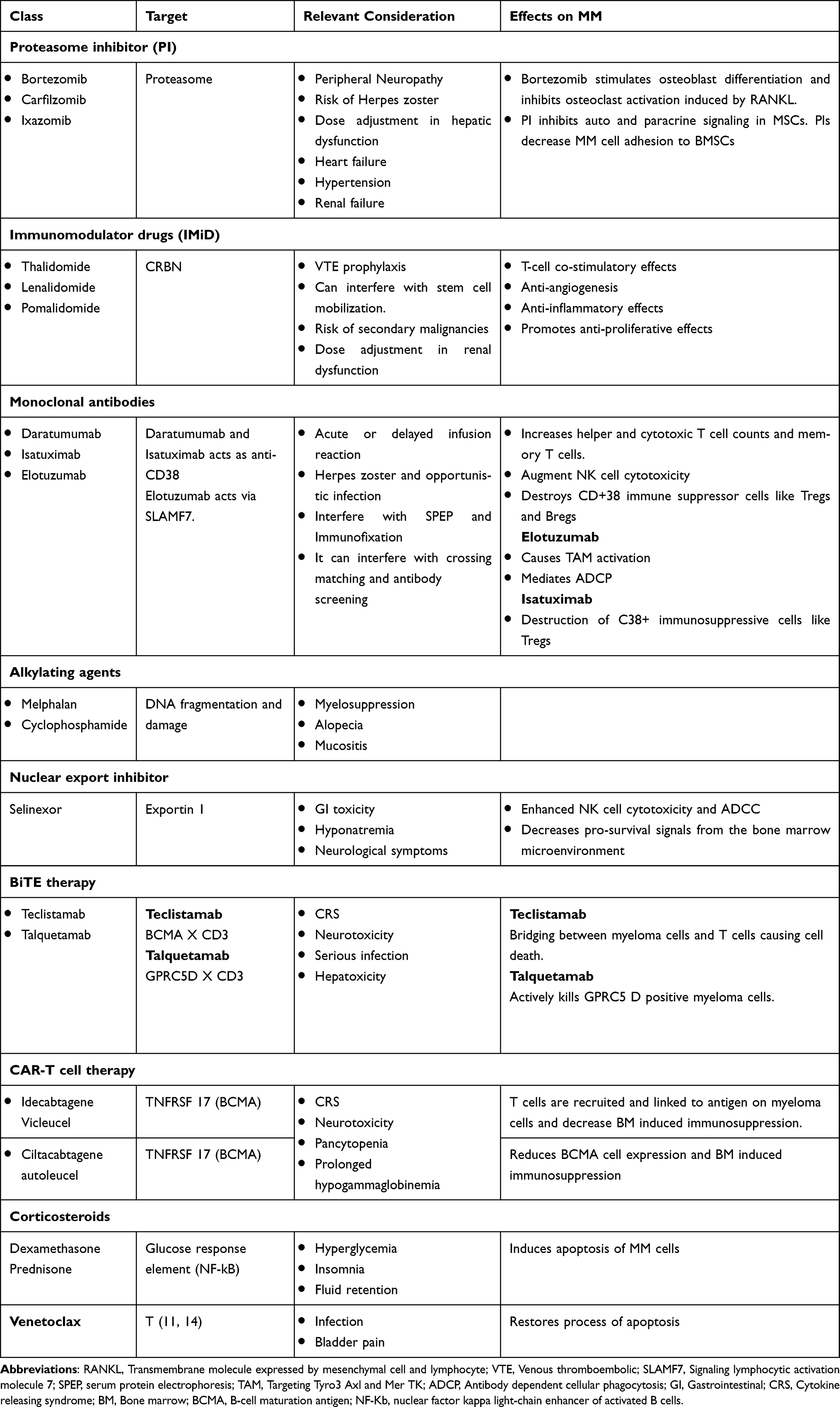 |
Table 4 Overview of Various Therapeutic Agents for Treatment of MM |
Newly diagnosed MM patients are treated with induction combination therapy. This treatment is usually at least a triple combination such as lenalidomide, dexamethasone, and bortezomib or different regimens depending upon the eligibility of patients for autologous stem cell transplant (ASCT).67 The main objective of the first phase of treatment is the reduction in tumor burden and better collection of stem cells.68 Melphalan-based regimens are no longer the standard of care in induction chemotherapy as they interfere with stem cell collection.69 After induction chemotherapy, patients are administered high-dose melphalan followed by ASCT.70 After ASCT, the patient will be treated with consolidation and/or maintenance/extended therapy based on various factors.71 For RRMM, there are several new treatment options, including novel combinations.71
Alkylating Agents
Alkylating agents have long been used in the treatment of MM. Those most commonly used are melphalan and cyclophosphamide. Alkylating agents work by breaking the double strand of DNA, leading to apoptosis.72 Melphalan was the first alkylating agent and the standard of care, in combination with prednisone, in 1961.72 In the last two decades, melphalan use has generally decreased with the development of novel agents; however, it is still used at a high dosage as a conditioning regimen before ASCT, and it is also used in combination with dexamethasone, thalidomide, cisplatin, Adriamycin, cyclophosphamide, and etoposide in aggressive disease.73
Cyclophosphamide, another commonly used alkylating agent, has strong immunomodulatory effects through the activation of natural killer cells, macrophages, and helper T cells and is currently used in combination with other standards of care.72 Another alkylating agent, melflufen, causes irreversible damage to DNA and produces apoptosis through a P53-independent mechanism. Melflufen is no longer used due to inferior outcomes compared to other drug therapies.74
Proteasome Inhibitors
Proteasome inhibitors (PIs) are one of the most important therapeutic agents in MM management. Proteasome is a large protease complex that causes the degradation of proteins in the nucleus and cytoplasm. In MM cells, several proteins are produced. Inhibiting proteasome causes these proteins to accumulate in the cytoplasm and endoplasmic reticulum, causing what is called endoplasmic reticulum stress and inducing apoptosis and the destruction of MM cells.75 In addition to apoptosis, PIs inhibit angiogenesis and cell cycle arrest, as shown in Figure 1.
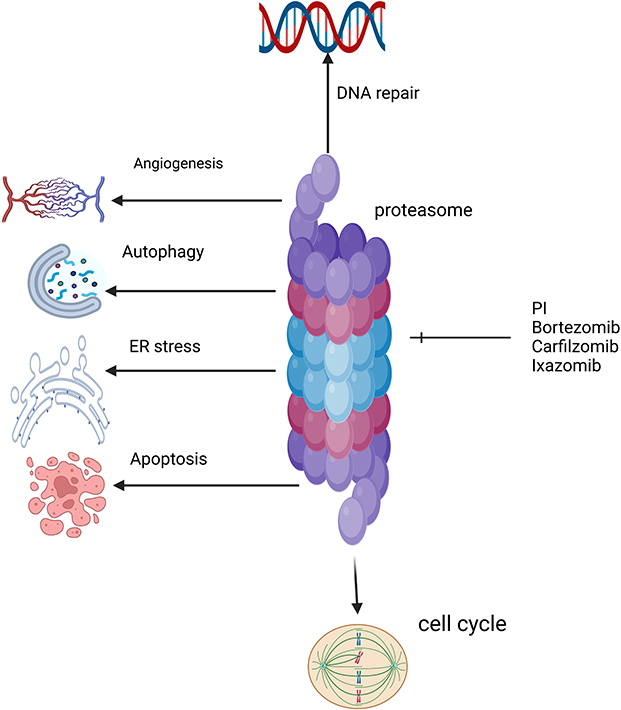 |
Figure 1 Mechanism of proteasome inhibitor action on multiple myeloma cells. |
Bortezomib was the first PI to gain FDA approval in 2003. Bortezomib is a boronic acid dipeptide that binds to the chymotrypsin and caspase and inhibits its activities, causing myeloma cell destruction.76
Carfilzomib and ixazomib are two next-generation PIs.75 Carfilzomib, approved in 2012, irreversibly inhibits proteasome by binding to the β5 subunit.77 It has different adverse effects compared to other PIs. The most concerning adverse effect is cardiotoxicity, which is due to the autophagy pathway and upregulation of protein phosphatase-2A activity and not due to proteasome inhibition.78 Ixazomib is an oral and reversible PI. It binds to the β5 subunit of the 20S proteasome and inhibits its chymotrypsin-like activity. Its half-life is short compared to other PIs,79 as shown in Figure 1. Ixazomib should not be used in maintenance setting in the context of modern therapy given inferior outcomes, and its use in general in treating MM is not recommended in our practice.
Corticosteroids
Steroids are the backbone of MM treatment, both for newly diagnosed MM and RRMM. In modern treatment regimens, steroids are combined with novel agents to increase the depth of clinical response. Glucocorticoids induce apoptosis in MM cells either by transactivation of glucocorticoid response elements, phosphorylation of RAFTK (Pyk2), or transrepression of NF-Kappa B, but its exact mechanism of action is still unknown.80
The Eastern Cooperative Oncology Group (ECOG) pilot study81 assessed the efficacy and safety of dexamethasone in 32 patients with advanced MM, of which 26 had received several lines of prior treatment. Patients received 40 mg of oral dexamethasone 4 days per week for 8 weeks. Those patients who responded to treatment were maintained on the same treatment administered at a 2-week interval. On analysis, 13/32 (40%) patients responded based on ECOG criteria. Moderate-to-severe side effects were observed in 9 patients (55%), including 9 with central nervous system effects, 3 with gastrointestinal bleeding, 2 with pulmonary emboli, and 1 with psychosis. Coadministration of corticosteroids with bortezomib has been shown to decrease the severity of peripheral neuropathy.82
Autologous Stem Cell Transplantation
ASCT (single and tandem) is a consolidation therapy in transplant-eligible, newly diagnosed MM patients with no age cut-off in the US.83 The first melphalan-based ASCT treatment of 9 patients was performed successfully in 1983.84 Barlogie et al showed that melphalan-induced myelosuppression could effectively be rescued with ASCT.85 Wildes et al86 showed prolonged median OS in older patients aged 65–77 years treated with ASCT compared to a non-ASCT group. Median OS was 56.0 months in the ASCT group (95% CI [49.1–65.4]) compared to 33.1 months in the non-ASCT group (24.3–43.1) (p = 0.004) with no increased mortality after adjusting for performance status, comorbidities, and disease status.
An open-label randomized phase 3 clinical trial87 compared melphalan-prednisone-lenalidomide (MPR) treatment to treatment with ASCT followed by lenalidomide and dexamethasone. Both PFS and OS were longer in the ASCT group when compared to the MPR group (4-year OS 81.6% vs 65.3%, p = 0.02; median PFS 43.0 months vs 22.4 months).
Cavo et al88 conducted a randomized phase 3 clinical trial that showed that bortezomib-based induction therapy followed by ASCT had improved PFS compared to VMP alone (Table 5).
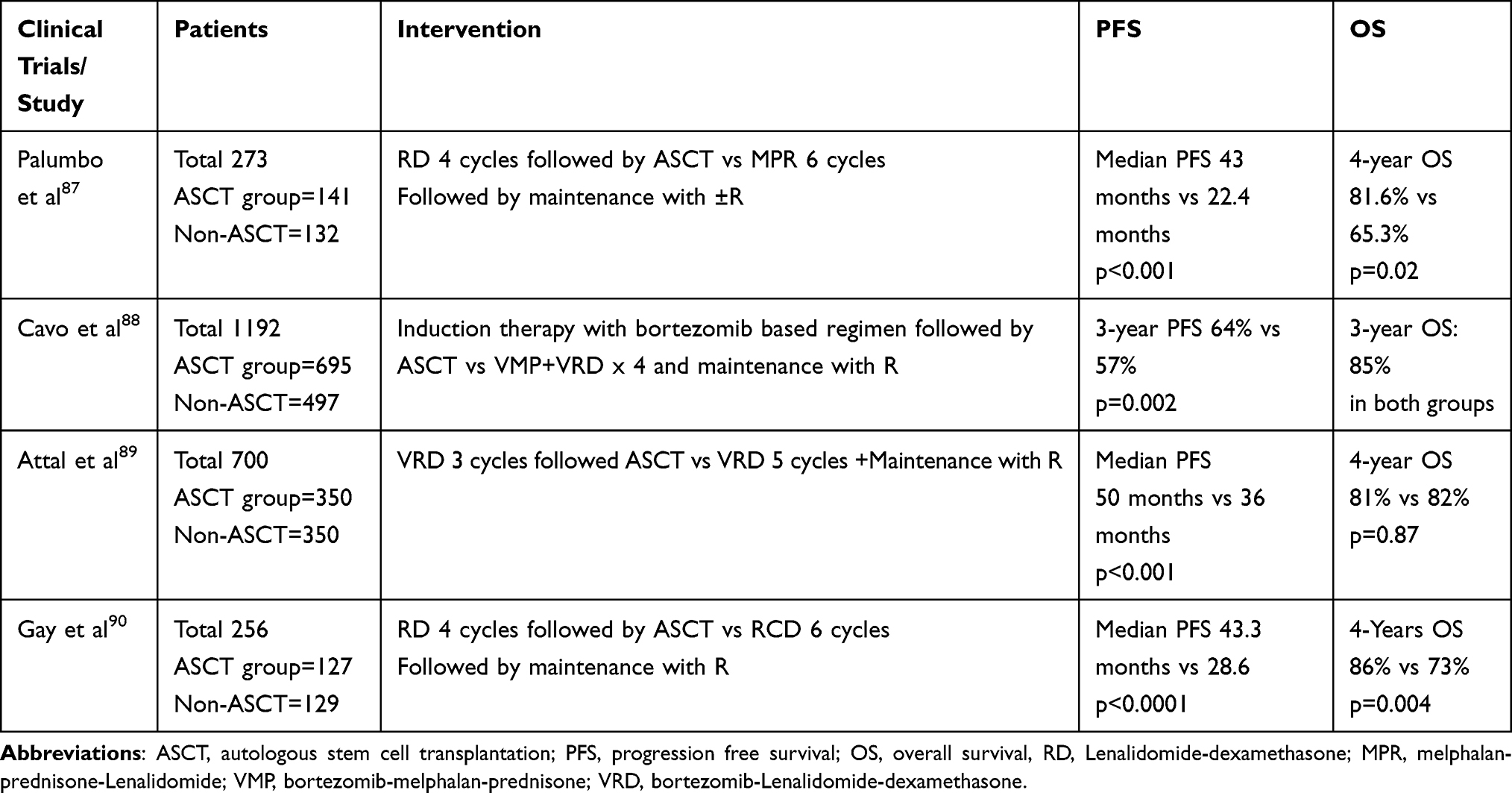 |
Table 5 Various Phase III Clinical Trials Showed Improved Outcomes with Autologous Stem Cell Transplantation |
An open-label, large phase 3 trial by Attal et al89 randomized patients into groups receiving either induction therapy with bortezomib, lenalidomide, and dexamethasone (VRD) followed by consolidation therapy with VRD or ASCT and followed by additional cycles of VRD. Both groups received lenalidomide maintenance therapy for up to 1 year. The median PFS and complete response were significantly longer in the ASCT group than in the non-transplant group (50 months vs 35 months; p < 0.001, 59% vs 0.48%, p = 0.03).
An open-label, phase 3 clinical trial by Gay et al randomized patients into groups receiving either chemotherapy plus lenalidomide or induction chemotherapy (Lenalidomide and dexamethasone) followed by ASCT. This trial showed improved PFS in the ASCT group compared to that in the other treatment group (median PFS 43.3 months vs 28.6 months, p < 0.0001),90 as shown in Table 5.
Whether to perform ASCT early or to delay it is sometimes debated. It is our recommendation to proceed with upfront ASCT in patients who are eligible for it. A retrospective study91 of 363 MM patients showed that ASCT performed <12 months after diagnosis improved PFS and demonstrated a higher response rate. The median age of patients was 52 years (range 20 to 72 years), and 233 (64.2%) were male. The median time from diagnosis to transplant was 11.5 months (range 4–67.5); 201 (55.4%) patients had ASCT within 12 months of diagnosis (early), and 162 (44.6%) patients had ASCT >12 months since diagnosis (delayed). Post ASCT analysis showed better CR (77.1% vs 64.8%, p < 0.025) and improved very good partial response (VGPR, 89% vs 81.5%, p < 0.03) in the early ASCT group compared to the delayed group. Transplant-related mortality at 100 days was similar among both groups. (3.5% vs 3.7%; p = 0.564) DETERMINATION trial92 compared RVd plus melphalan-based ASCT to RVd alone. Median PFS was better in the transplant group 67.5 vs 46.2 months (HR 1.53; 95% CI, 1.23–1.91; p < 0.0001) with no difference in 5-year OS between both groups.
Immunomodulatory Drugs
Immunomodulatory drugs (IMiDs) include thalidomide, lenalidomide, and pomalidomide. IMiDs have pleiotropic effects on MM with the ability to modulate host immune response and angiogenesis, impact cytokine secretion and inflammation, and produce direct cytotoxic effects on MM, including growth arrest and caspase-8-mediated apoptosis.93,94 These drugs represent a paradigm shift in the treatment of newly diagnosed MM and RRMM.95 Thalidomide is a synthetic derivative of glutamic acid with two active enantiomers, S and R. The S enantiomer is responsible for antitumor effects, and the R enantiomer has sedative effects.96 The combination of Bortezomib, Thalidomide, and dexamethasone (VTD) is a standard induction chemotherapy regimen in transplant-eligible, newly diagnosed MM patients. Several clinical trials have shown the superior efficacy of VTD over other drug combinations used in pre-ASCT induction treatments.97,98
A phase 3 study by Cavo et al97 compared VTD to the combination of thalidomide and dexamethasone as induction therapy before ASCT and showed CR or near complete response (nCR) of 33.1% vs 13.7% (p < 0.0001). Three-year PFS was longer in the VTD group (60% vs 48%, p = 0.042).
Another phase 3 trial99 compared VTD to bortezomib, cyclophosphamide, and dexamethasone (VCD) as pre-ASCT induction therapy and showed overall response rates (ORR) of 92.3% vs 83.4% (p = 0.01), respectively. In addition, VTD showed a VGPR of 66.3% compared to 56.2% for VCD (p = 0.05).
Phase 3 PETHEMA/GEM 12 study100 evaluated the efficacy and safety of VRD as an induction regimen. In this study 458 NDMM, patients aged <65 years received 6 cycles of VRD followed by ASCT with a conditioning regimen of busulfan and melphalan vs melphalan. The patient received consolidation with 2 cycles of VRD. In grouped response analysis of 6 induction cycles (n = 426), VGPR or better was achieved at 55.6% by cycle 3, 63.8% by cycle 4 and 68.3% by cycle 5, and 70.4% by cycle 6. About 33.4% had CR after induction in the intent-to-treat population (ITT) which was similar in the 92 patients with high-risk cytogenetics (34.8%). This response further deepened to 44.1% after ASCT and 50.2% after consolidation. In the ITT, the rate of undetected minimal residual disease (sensitivity 3 × 106) increased from induction (28.8%) to transplant (42.1%) and consolidation (45.2%). During induction, common adverse events grade 3 were neutropenia (12.9%) and infection (9.2%). Seventeen percent of patients had grade 2 peripheral neuropathy, 3.7% grade 3, and 0.2% grade 4 during induction. VRD is a well-tolerated and effective induction regimen in NDMM patients.
The CASSIOPEIA randomized phase 3 trial compared a quadruplet regimen of daratumumab and VTD to a triplet VTD regimen as induction therapy for ASCT eligible patients and showed a CR or better in 39% vs 26%, respectively (P < 0.0001).101
New IMiDs include iberdomide and mezigdomide (MEZI). Iberdomide is a cereblon E3 ligase modulator with stronger anti-tumor and enhanced immune stimulatory effects compared to other IMiDs. In Phase 1/2 trial,102 iberdomide was studied with oral dexamethasone in a dose-escalation cohort and a dose-expansion cohort. The dose-escalation cohort contained individuals who had been administered at least 2 previous lines of therapy, including lenalidomide or pomalidomide and PIs. Patients received escalating doses of Iberdomide (0.3–1.6 mg on days 1–21 of each 28-day cycle) and oral dexamethasone (40 mg or 20 mg [if age ≥75 years]) once a week. The dose-expansion cohort contained patients with RRMM who had received at least 3 previous lines of therapy and had triple-class refractory disease (refractory to IMiDs, PIs, and CD38 antibodies). Patients were treated with the recommended phase 2 dose, and treatment continued until their disease progressed or unacceptable toxicity was observed. ORR was 32% (95% CI 23–43) across all doses in the dose-escalation cohort and 26% (95% CI 18–36) in the dose-expansion cohort, respectively. Overall, the most common grade 3 or worse adverse effects were neutropenia in 45% of the patients, anemia in 25%, infection in 27%, and thrombocytopenia in 22%. There was 1% treatment-related mortality, and 5% of the patients discontinued treatment due to intolerable toxicity.
MEZI is an oral cereblon E3 ligase modulator with strong antimyeloma activity that has shown strong synergy with dexamethasone, PIs, and anti-CD38 monoclonal antibodies in the treatment of RRMM.103
Bcl (a B-Cell Lymphoma-2) Inhibitor
Venetoclax is BCL-2 inhibitor able to reinstate the apoptotic potential of cancer cells. Patients with MM cells with translocation t (11; 14) have higher BCL-2 expression and can benefit from venetoclax-based therapy.104
The BELLINI trial (phase 3, randomized, double-blind, multicenter) demonstrated that combining venetoclax with dexamethasone and bortezomib improved median PFS in patients with RRMM who had received one to three prior therapies. Patients were randomly assigned to receive either venetoclax (800 mg orally daily) or placebo, both with bortezomib (1.3 mg/m2 subcutaneously or intravenously) and dexamethasone (20 mg orally). Median follow-up was 18.7 months. The independent review committee found a median PFS of 22.4 months (venetoclax group) vs 11.5 months (placebo group) with a hazard ratio of 0.63 (p = 0.010). The most common grade 3 or worse adverse events in the venetoclax group were neutropenia (18%), pneumonia (16%), thrombocytopenia (15%), diarrhea (15%), and anemia (15%), with 8 fatal infections noted in the venetoclax group. Nevertheless, the venetoclax group exhibited higher mortality, primarily attributed to a heightened infection rate, underscoring the significance of carefully selecting patients suitable for this therapeutic approach.
Monoclonal Antibodies
CD38 is a glycoprotein expressed on MM cells. Daratumumab is an anti-CD38 monoclonal antibody. Daratumumab works through several mechanisms, including antibody-dependent cellular toxicity, antibody-dependent cellular phagocytosis, complement-mediated cytotoxicity, direct apoptosis, and immunomodulation by depleting CD38 positive immune suppressive cells with the expansion of T effector cells.105 Daratumumab is used in upfront in newly diagnosed MM as well as RRMM.
Isatuximab is another anti-CD38 monoclonal antibody with a different mechanism of action from daratumumab.106 Isatuximab binds to a specific epitope on the human CD38 receptor. Like daratumumab, isatuximab can induce direct apoptosis and has demonstrated antitumor activity in xenograft models of MM, acute lymphoid leukemia, and non-Hodgkin’s lymphoma.107
A phase 3 prospective randomized open-label trial (IKEMA)108 compared the combination of isatuximab with carfilzomib-dexamethasone (isatuximab group) to carfilzomib-dexamethasone alone (control group) in RRMM patients. Median PFS was not reached in the isatuximab group, in contrast to 19.15 months (95% CI 15.77 to not reached) in the control group, yielding a hazard ratio of 0.53 (99% CI 0.32 to 0.89; one-sided p = 0.0007). Grade 3 or worse treatment-related adverse events (AEs) occurred in 136/177 (77%) of the isatuximab group and 82 (67%) of 122 in the control group. AEs leading to treatment discontinuation were observed in 15 (8%) vs 17 (14%) in the isatuximab vs control groups, respectively. The addition of isatuximab to carfilzomib-dexamethasone led to improved PFS and response depth in RRMM.
In the ICARIA trial, a randomized open-label multicenter phase 3 study,109 isatuximab was compared to pomalidomide and dexamethasone (isatuximab group) in RRMM patients aged >18 years. Eligible patients had received at least two lines of therapy, including lenalidomide and PI. Patients refractory to anti-CD38 therapy or those who had received pomalidomide were excluded. Of the 307 patients, 154 were assigned to the isatuximab group and 153 to the control group. Median OS was 24.6 months (95% CI 20.3 to 31.3) in the isatuximab group and 17.7 months (14.4 to 26.2) in the control group, with a hazard ratio of 0.76 (95% CI 0.57 to 1.01). Grade 3 or worse treatment-related AEs included neutropenia (76 [50%] of 152 patients vs 52 [35%] of 149 patients) and other adverse events like pneumonia (35 [23%] vs 31 [21%]) and thrombocytopenia (20 [13%] vs 18 [12%]) in the isatuximab and control groups, respectively.
In the ALCYONE randomized trial,108 newly diagnosed transplant-ineligible MM patients received nine cycles of bortezomib, melphalan, and prednisone alone (control group) or in combination with daratumumab until disease progression. Analyses showed that PFS was 71.6% (95% Cl 65.5–76.8%) in the daratumumab group vs 50.2% (95% CI, 43.2–65.7%) in the control group (hazard ratio of disease progression or death, 0.50; 95% Cl 0.38–0.65; p < 0.001). The ORR was 90.9% in the daratumumab group vs 73.9% in the control group (p < 0.001). CR or better was achieved in 42.6% of the patients in the daratumumab group vs 24.4% in the control group. The main adverse event observed was cytopenia and infection in the daratumumab group.
The MAIA phase 3 clinical trial109 compared the use of daratumumab, Revlimid, and dexamethasone (DRd) to Revlimid and dexamethasone (RD) in previously untreated transplant-ineligible MM patients. DRd vs RD patients achieved better CR (47.6% vs 24.9%) and MRD (24.2% vs 7.3%) (p < 0.001). PFS at 30 months was 70.6% vs 55.6% (HR 0.56, 95% Cl 0.43–0.73, p < 0.001). The daratumumab group had a higher incidence of neutropenia (50.0% vs 35.3%) and pneumonia (13.7% vs 7.9%) than the RD group.
In the CASSIOPEIA study,110 newly diagnosed transplant-eligible MM patients received four pre-transplant inductions and two post-transplant consolidation cycles of bortezomib, thalidomide, and dexamethasone (VTd) alone (n = 542) or in combination with daratumumab (D-VTd) (n = 543). At day 100 post-transplantation, 29% of the D-VTd group and 20% of the VTd group achieved a stringent CR (OR 1.60, 95% Cl 1.21–2.12, p = 0.001). The D-VTd group exhibited improved median PFS compared to the VTd group (hazard ratio 0.47, 95% Cl 0.33–0.67, p < 0.0001). The most observed adverse effects included neutropenia (28% in the D-VTd group vs 15% in the Vtd group), stomatitis (13% vs 16%), and lymphopenia (17% vs 10%).
Elotuzumab is a humanized monoclonal antibody against a cell surface protein called CS1 (also known as SLAMF7). This surface protein is highly expressed on MM cells, NK cells, and a subset of CD+8 T cells. The use of Elotuzumab in combination with IMiDs and Pls has shown benefits in relapsed MM patients, however its use is limited given the availability of other active agents.111,112
Bispecific Antibodies
The use of novel agents and monoclonal antibody-based therapies has improved PFS of patients with MM. Bispecific antibodies include bispecific antibodies (BisAbs) and bispecific T-cell engagers (BiTEs). These molecules work by encouraging immune cells to lyse MM cells by binding antigens on MM and immune effector cells simultaneously. BisAbs are engineered antibodies, while BiTEs are recombinant proteins. Both cause T-cell activation, tumor cell lysis, and T-cell proliferation. Current bispecific antibodies target either B cell maturation antigen (BCMA), G protein-coupled receptor, class C group 5, member D (GPRC5D), or Fc Receptor-homolog 5 (FcRH5).113–117 Table 6 summarizes various bispecific antibodies.
 |
Table 6 Different Bispecific Antibodies Targets Used in MM |
Bispecific Antibodies early clinical trials showed promising results with roughly 2 out of 3 heavily pre-treated patients having a response. Follow-up duration for those studies are short. Ongoing trials include products: alnuctamab118, linvoseltamab119, Pacanalotamab,120 Pavurutamab,121,122 Teclistamab,123–125 Talquetamab,126–128 Cevostamab129 and Elranatamab.130 Most of those products are being used until disease progression and this carries increased risk of infections which needs to be monitored. Phase 3 clinical trials to compare those agents to standard of care are being done and will help in comparing their promising activity with other active agents. Table 7 summarizes data of the 2 FDA-approved BCMA bispecific antibodies in MM.
 |
Table 7 Summary of the Approved BCMA Bispecific Antibodies |
Talquetamab got approved by the FDA in August 2023. MonumenTAL-1 reported data on 232 patients who received talquetamab, with 102 receiving it intravenously and 130 subcutaneously. In this phase 2 study, two recommended subcutaneous doses were tested: 405 μg per kilogram weekly (30 patients) and 800 μg per kilogram every other week (44 patients). Noteworthy side effects at these doses included cytokine release syndrome (experienced by 77% and 80% of the patients respectively), skin-related issues (reported by 67% and 70%), and dysgeusia (seen in 63% and 57%). Most cases of cytokine release syndrome were of grade 1 or 2, except for one case of grade 3 rash linked to the 800-μg dose. Response rates were evaluated at median follow-ups of 11.7 months (for the 405-μg dose) and 4.2 months (for the 800-μg dose), with response percentages of 70% (95% confidence interval [CI], 51 to 85) and 64% (95% CI, 48 to 78), respectively. Duration of response was 10.2 months and 7.8 months for the respective doses.
Chimeric Antigen Receptor T-Cell Therapy
The emergence of chimeric antigen receptor T-cell therapy (CAR T-cell therapy) has changed the landscape of treatment of patients with RRMM. The first anti-BCMA CAR T-cell therapy for MM was studied in 2013.131 BCMA is a tumor necrosis factor receptor found on plasma cells, including MM cells. BCMA causes the proliferation and survival of MM cells through protein kinase B and nuclear factor signaling cascade.114 Two CAR T products, idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel), are approved by FDA for patients with RRMM, and several ongoing clinical trials are for new CAR T cell therapy as Table 8. Both are second-generation anti-BCMA therapies approved for patients that have previously received four or more lines of treatment, including PI, IMiD, and anti-CD38 therapy. Both are administered after lymphodepleting chemotherapy with cyclophosphamide 300 mg/m2 and fludarabine 30 mg/m2.132
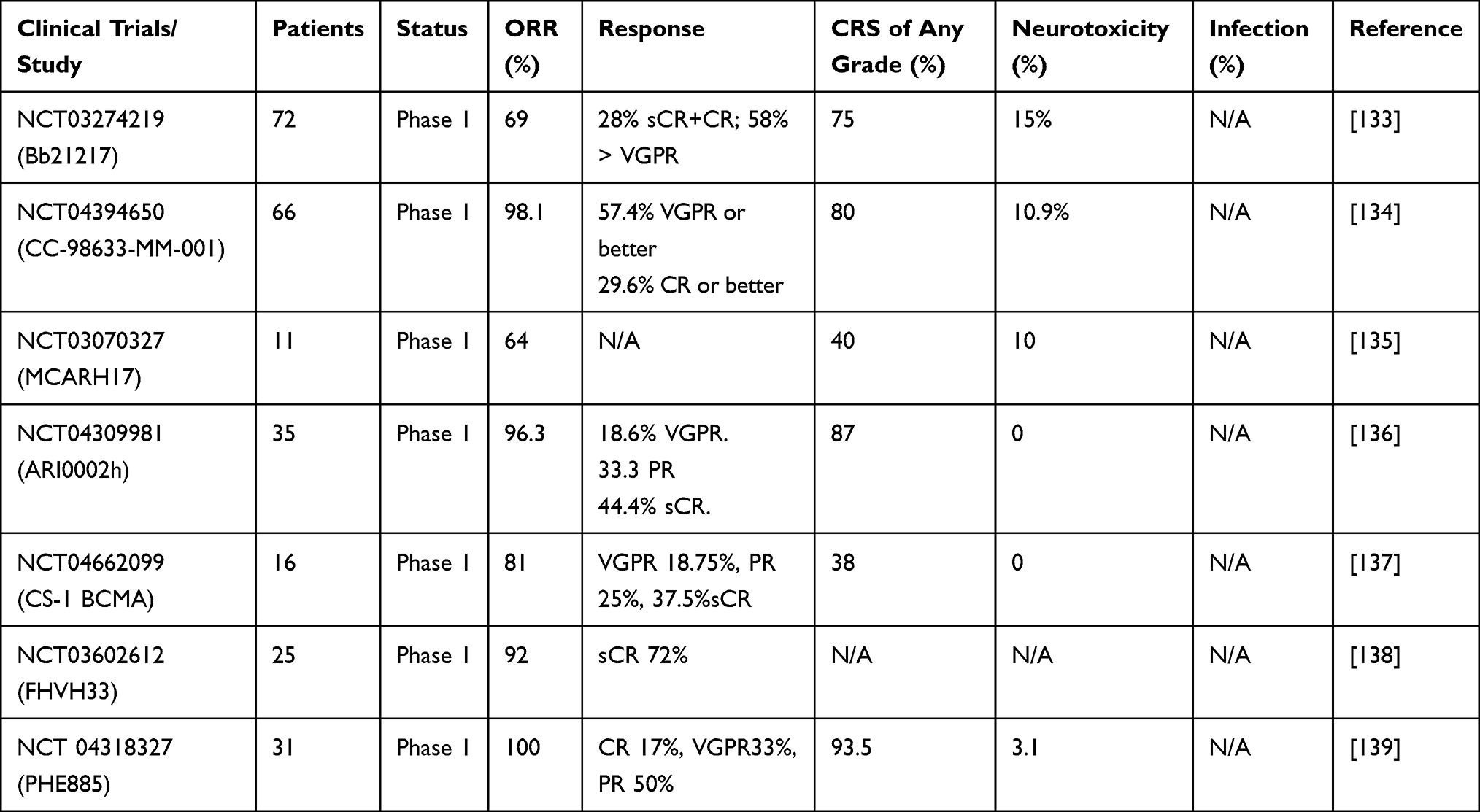 |
Table 8 Ongoing Clinical Trials for CAR-T Cell Therapy |
Ide-Cel
Ide-cel showed antimyeloma activity in the phase 1 CRB-401 clinical trial.140 In this trial, Ide-cel CAR T cells were administered to 30 patients in a dose escalation regimen—50 × 106, 150 × 106, 450 × 106, or 800 × 106 CAR T cells per kilogram. Patients administered 150 × 106 CAR T cells per kilogram or greater exhibited VGPR and CR, so doses of 150 × 106 CAR T cells per kilogram or greater were used in the subsequent phase. With the higher target dose, all patients responded, with a median PFS of 11.8 months, and CAR T cells persisted for up to 1 year after infusion.
In the phase 2 KarMMa study of ide-cel,141 140 patients with RRMM who had received 3 or more lines of therapy (including PI, IMiD, and anti-CD 38 directed therapy) were enrolled. Patients were 18 years or older with a median age of 61 (range 33–78), and 59% were male. Thirty-five percent of patients had high-risk cytogenetics, 94% had prior ASCT, 84% were triple refractory, and 26% were penta-refractory. Eighty-four percent of patients received bridging therapy (including dexamethasone, cyclophosphamide, daratumumab, carfilzomib, and pomalidomide) with a response in 4%. The primary endpoint was ORR, and the secondary endpoint was CR, including stringent CR. Of the 140 patients, 128 patients received ide-cel with a median follow-up of 13.3 months. ORR was 73%. Patients reaching CR were 33%, MRD negativity were 26%, and median PFS were 8.8 months. OS was 24.8 months (about 2 years). A dose–response relationship was demonstrated at the 450 × 106 target dose, with CR rates reaching 39%, ORRs reaching 81%, and PFS reaching 12.1 months. Depth of response and length of response were correlated in the 42 patients with a CR or stringent CR, and duration of response extended to 19, and median PFS extended to 20.2 months. The median time to first response was 1 month, ranging from 0.5 to 8.8 months, and the median time to CR or better was 2.8 months, ranging from 1.0 to 11.8 months. Data from the KarMMa trial were reanalyzed, revealing an OS of 24.8 months. Hematologic adverse events included grade 3 or 4 neutropenia in 89% of patients, anemia in 60% of patients, thrombocytopenia in 52% of patients, and leukopenia in 39% of patients. Another common adverse event, neurotoxicity, was observed in 18% of patients. Neurotoxicity reached no greater than grade 3, which only occurred in 3% of patients. CRS was observed in 84% of patients, but only 5% exhibited grade 3 or above. Within the population of treated patients (n = 128), 44 (34%) deaths were observed, 35 of which were due to progressive disease or complications resulting from progression. Treatment-related deaths were 3% and were due to gastrointestinal hemorrhage, cytomegalovirus pneumonia, pulmonary aspergillosis, and CRS.
Cilta-Cel
The CARTITUDE-2142 phase 2 multicohort study evaluated the efficacy and safety of cilta-cel in 20 MM patients. Patients in this study had received PIs, IMiDs, anti-CD38 antibodies, and non-cellular anti-BCMA therapy. A single cilta-cel infusion was given after lymphodepletion regimen. The primary endpoint was MRD negativity, and 7 out of 20 (35%) were MRD negative at a median follow-up of 11.3 months (range 0.6–16.0) ORR was 60% (CI 95% 36.1–80.9). The median duration of response was 11.5, and PFS was 9.1 months. Twelve patients (60%) had CRS (grade 1–2); four patients had immune effector cell-associated neurotoxicity (two had grades 3–4). Seven (35%) patients died, three due to progressive disease and four due to adverse events, one of which was treatment-related. Overall, patients with RRMM showed a favorable response to cilta-cel. Process of collection of CAR T cells, manufacturing and infusion are summarized in Figure 2.
Recently, both Idel-cel and Cilta-cel were studied in separate phase III clinical trials in KarMMa-3 and CARTITUDE-4 studies summarized in Table 9.
 |
Figure 2 Mechanism of CAR T cell therapy in MM. It includes collection of T cells, multiplication and infusion to patient. Notes: Adapted from Parikh RH, Lonial S. Chimeric antigen receptor T-cell therapy in multiple myeloma: A comprehensive review of current data and implications for clinical practice. CA. 2023;73(3):275–85. © 2023 The Authors. CA: A Cancer Journal for Clinicians published by Wiley Periodicals LLC on behalf of American Cancer Society.143 |
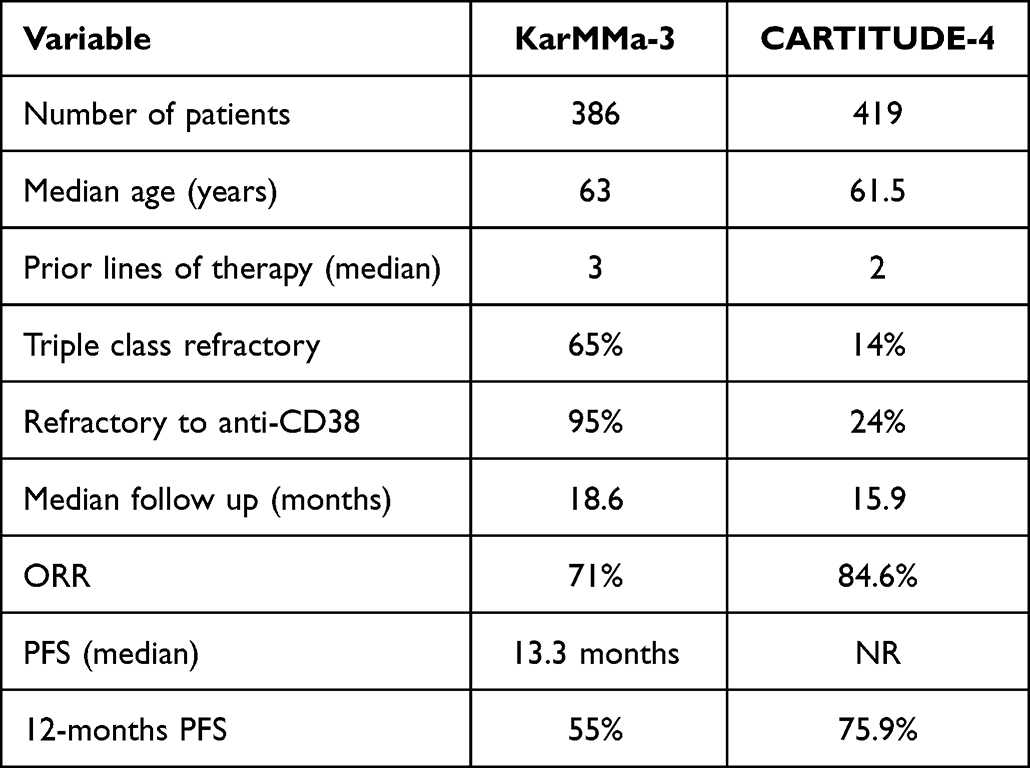 |
Table 9 Summary of Phase III Trials of CAR-T Cell Therapy in Multiple Myeloma |
New Developments
OS and PFS of MM have improved with new drugs, but 20–25% of newly diagnosed MM patients and 50% of patients with RRMM do not respond to PI.144 Thus, new drugs or combinations are needed.
Bcl-2 and Mcl-1 are therapeutic targets in the treatment of MM. Venetoclax is effective in patients with t(11; 14) and high Bcl-2 expression.145 Mcl-1 is reported in MM associated with 1q21 amplification. The overexpression of Mcl-1 is associated with poor prognosis.146 Several clinical trials of Mcl-1 inhibitors like MIK 665, PRT1419, and AMG 176 are currently in phase 1, as shown in Table 10. Maritoclax is a selective Mcl-1 antagonist that causes caspase-3 activation by direct binding to Mcl-1 and causes proteasomal degradation.
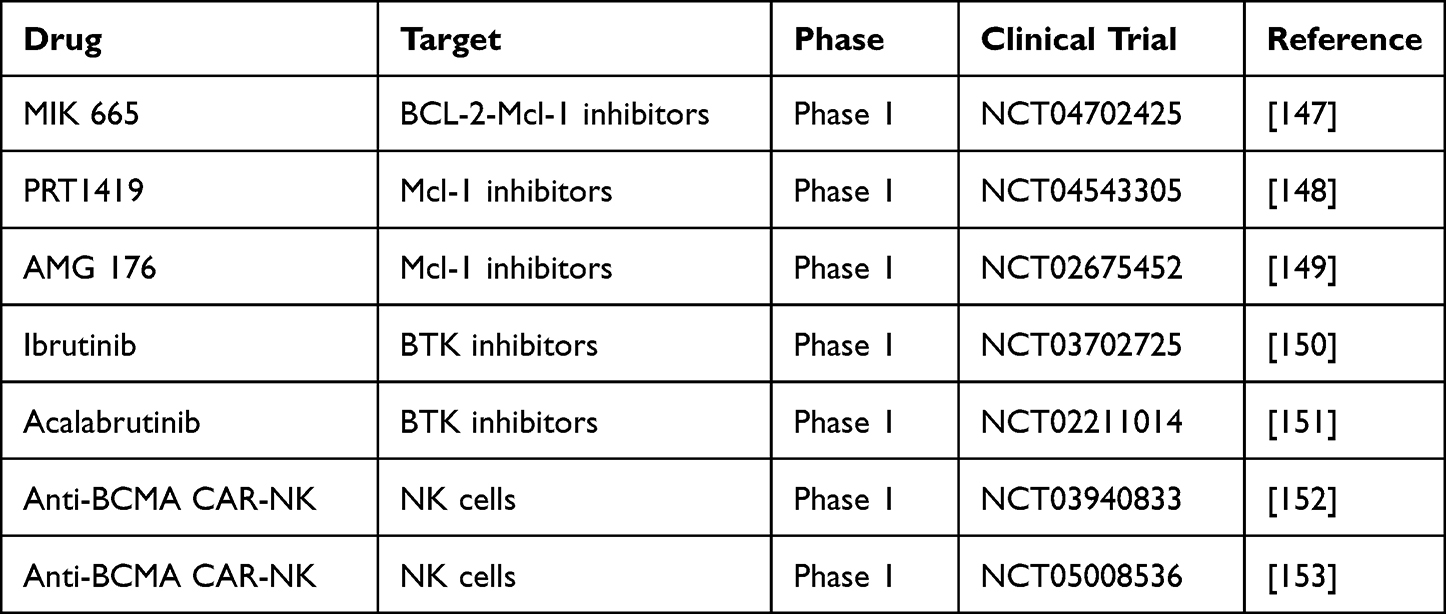 |
Table 10 Ongoing Trials of New Therapies for Multiple Myeloma |
Bruton’s tyrosine kinase (BTK) is overexpressed in MM cells and stem cells.154 Ibrutinib and acalabrutinib are two FDA-approved BTK inhibitors for the treatment of B-cell malignancies that are being investigated in clinical trials for MM, as shown in Table 10.155
Anti-BCMA CAR-T has promising results in MM, but there are concerns about toxicity, including CRS, neurologic toxicity, and bone marrow suppression.156 Therefore, NK cells can be an option.157 Trials of NK CAR T cells and several other clinical trials are currently ongoing, as shown in Table 8
Modakafusp is an immunocytokine that is aimed at delivering interferon alpha-2b to CD38+ cells. In a phase 1 clinical trial, 83 patients with RRMM (at least 3 previous lines of treatment) received 1- to 4-hour madakafusp infusions of 11 doses from 0.001 to 6 mg/kg following a 3+3 dose-escalation design. ORR was 42%. The median PFS was 5.7 months. Modakafusp showed promising antimyeloma activity in extensively pretreated MM patients, including those that were anti-CD38 refractory.158
Conclusions
It is important to differentiate between different forms of plasma cell dyscrasias and related disorders as delaying treatment or missing diagnosis has dire outcome. With wide availability and use of new imaging techniques including PET-CT and MRI, patients benefit from early treatment.
With the emergence of many new treatments, the future of MM therapy is bright. The introduction of immunological agents has revolutionized MM treatment with good efficacy and tolerable toxicity profile. Anti-CD38 drugs, like daratumumab, can be the backbone of MM treatment in both upfront newly diagnosed MM and relapsed refractory settings. Isatuximab is a new-generation anti-CD38 and an addition to this group.
New immunotherapeutics (CAR-T and bispecific antibodies) are promising treatment options in heavily treated RRMM settings. It is possible that armed immunotherapy can challenge the current standard of care and is a great option for high-risk MM. Other immunomodulators, such as antibody-drug conjugates and cereblon E3 ligase modulators, will continue to strengthen the field of various immune-based treatments. Non-immune-based treatment regimens must continue as patients on immunotherapy will relapse.
Despite significant development in the therapeutic modalities of MM, many patients relapse and develop drug resistance. Novel targeted therapies with great safety and efficacy are needed to combat MM, and a better understanding of the genetic and epigenetic basis of MM is the key. Next-generation sequencing coupled with genome editing will improve prognostication, risk stratification, and therapy response prediction. These methods will also help in the discovery of new strategies to prevent drug resistance as well as to improve the outcomes of MM.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kaseb H, Annamaraju P, Babiker HM. Monoclonal gammopathy of undetermined significance. In: StatPearls. Treasure Island (FL): StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC; 2022.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–48. doi:10.1016/S1470-2045(14)70442-5
3. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi:10.1182/blood-2016-01-643569
4. Dimopoulos MA, Kyle RA, Anagnostopoulos A, Treon SP. Diagnosis and management of Waldenstrom’s macroglobulinemia. J Clin Oncol. 2005;23(7):1564–1577. doi:10.1200/JCO.2005.03.144
5. ASCO. Cancer.net: ASCO; 2022.
6. Landgren O. Monoclonal gammopathy of undetermined significance and smoldering myeloma: new insights into pathophysiology and epidemiology. Hematology Am Soc Hematol Educ Program. 2010;2010:295–302. doi:10.1182/asheducation-2010.1.295
7. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21–33. doi:10.4065/78.1.21
8. Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418–5422. doi:10.1182/blood-2008-12-195008
9. Murray D, Kumar SK, Kyle RA, et al. Detection and prevalence of monoclonal gammopathy of undetermined significance: a study utilizing mass spectrometry-based monoclonal immunoglobulin rapid accurate mass measurement. Blood Cancer J. 2019;9(12):102. doi:10.1038/s41408-019-0263-z
10. Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378(3):241–249. doi:10.1056/NEJMoa1709974
11. Thorsteinsdottir S, Kristinsson SY. The consultant’s guide to smoldering multiple myeloma. Hematology Am Soc Hematol Educ Program. 2022;2022(1):551–559. doi:10.1182/hematology.2022000355
12. Go RS, Rajkumar SV. How I manage monoclonal gammopathy of undetermined significance. Blood. 2018;131(2):163–173. doi:10.1182/blood-2017-09-807560
13. Waldenstrom J. Studies on conditions associated with disturbed gamma globulin formation (gammopathies). Harvey Lect. 1960;56:211–231.
14. Kyle RA. Monoclonal gammopathy of undetermined significance. Natural history in 241 cases. Am J Med. 1978;64(5):814–826. doi:10.1016/0002-9343(78)90522-3
15. Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351(18):1860–1873. doi:10.1056/NEJMra041875
16. Pang L, Rajkumar SV, Kapoor P, et al. Prognosis of young patients with monoclonal gammopathy of undetermined significance (MGUS). Blood Cancer J. 2021;11(2):26. doi:10.1038/s41408-021-00406-6
17. Therneau TM, Kyle RA, Melton LJ, et al. Incidence of monoclonal gammopathy of undetermined significance and estimation of duration before first clinical recognition. Mayo Clin Proc. 2012;87(11):1071–1079. doi:10.1016/j.mayocp.2012.06.014
18. van de Donk NW, Palumbo A, Johnsen HE, et al. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica. 2014;99(6):984–996. doi:10.3324/haematol.2013.100552
19. Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106(3):812–817. doi:10.1182/blood-2005-03-1038
20. Kaseb H, Pavan A, Babiker HM. Monoclonal Gammopathy of Undetermined Significance. Treasure Island (FL): StatPearls; 2022.
21. Kyle RA, Remstein ED, Therneau TM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–2590. doi:10.1056/NEJMoa070389
22. Akhlaghi T, Maclachlan K, Korde N, et al. P886: African American patients with smoldering multiple myeloma may have a lower risk of progression compared to white patients. HemaSphere. 2022;6:778–779. doi:10.1097/01.HS9.0000846420.96485.de
23. Ahn IE, Mailankody S, Korde N, Landgren O. Dilemmas in treating smoldering multiple myeloma. J Clin Oncol. 2015;33(1):115–123. doi:10.1200/JCO.2014.56.4351
24. Tessier C, Allard T, Boudreault J-S, et al. Testing Mayo clinic’s new 20/20/20 risk model in another cohort of smoldering Myeloma patients: a retrospective study. Curr Oncol. 2021;28(3):2029–2039. doi:10.3390/curroncol28030188
25. Mateos MV, Kumar S, Dimopoulos MA, et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 2020;10(10):102. doi:10.1038/s41408-020-00366-3
26. Pérez-Persona E, Vidriales MB, Mateo G, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110(7):2586–2592. doi:10.1182/blood-2007-05-088443
27. Vaxman I, Gertz MA. How I approach smoldering multiple myeloma. Blood. 2022;140(8):828–838. doi:10.1182/blood.2021011670
28. Mateos MV, Hernández MT, Giraldo P, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med. 2013;369(5):438–447. doi:10.1056/NEJMoa1300439
29. Lonial S, Jacobus S, Fonseca R, et al. Randomized trial of lenalidomide versus observation in smoldering multiple Myeloma. J Clin Oncol. 2020;38(11):1126–1137. doi:10.1200/JCO.19.01740
30. Mateos M-V, Martinez Lopez J, Rodríguez-Otero P, et al. Curative strategy (GEM-CESAR) for high-risk Smoldering Myeloma (SMM): carfilzomib, lenalidomide and dexamethasone (KRd) as induction followed by HDT-ASCT, consolidation with Krd and maintenance with Rd. Blood. 2021;138(Supplement 1):1829. doi:10.1182/blood-2021-148423
31. Kumar SK, Abdallah A-O, Badros AZ, et al. Aggressive Smoldering Curative Approach Evaluating Novel Therapies (ASCENT): a phase 2 trial of induction, consolidation and maintenance in subjects with high risk Smoldering Multiple Myeloma (SMM): initial analysis of safety Data. Blood. 2020;136(Supplement 1):35–36. doi:10.1182/blood-2020-142584
32. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA. 2020;70(1):7–30. doi:10.3322/caac.21590
33. Cowan AJ, Green DJ, Kwok M, et al. Diagnosis and management of multiple Myeloma: a review. JAMA. 2022;327(5):464–477. doi:10.1001/jama.2022.0003
34. Jurczyszyn A, Grzasko N, Gozzetti A, et al. Central nervous system involvement by multiple myeloma: a multi-institutional retrospective study of 172 patients in daily clinical practice. Am J Hematol. 2016;91(6):575–580. doi:10.1002/ajh.24351
35. Bahlis NJ, Lazarus HM. Multiple myeloma-associated AL amyloidosis: is a distinctive therapeutic approach warranted? Bone Marrow Transplant. 2006;38(1):7–15. doi:10.1038/sj.bmt.1705395
36. Dupuis MM, Tuchman SA. Non-secretory multiple myeloma: from biology to clinical management. Onco Targets Ther. 2016;9:7583–7590. doi:10.2147/OTT.S122241
37. Ghobrial IM. Myeloma as a model for the process of metastasis: implications for therapy. Blood. 2012;120(1):20–30. doi:10.1182/blood-2012-01-379024
38. Gertz MA. Waldenström macroglobulinemia: 2021 update on diagnosis, risk stratification, and management. Am J Hematol. 2021;96(2):258–269. doi:10.1002/ajh.26082
39. Fonseca R, Hayman S. Waldenström macroglobulinaemia. Br J Haematol. 2007;138(6):700–720. doi:10.1111/j.1365-2141.2007.06724.x
40. Groves FD, Travis LB, Devesa SS, Ries LA, Fraumeni JF. Waldenström’s macroglobulinemia: incidence patterns in the United States, 1988–1994. Cancer. 1998;82(6):1078–1081. doi:10.1002/(SICI)1097-0142(19980315)82:6<1078::AID-CNCR10>3.0.CO;2-3
41. Castillo JJ, Olszewski AJ, Kanan S, Meid K, Hunter ZR, Treon SP. Overall survival and competing risks of death in patients with Waldenström macroglobulinaemia: an analysis of the surveillance, epidemiology and end results database. Br J Haematol. 2015;169(1):81–89. doi:10.1111/bjh.13264
42. Kyle RA, Larson DR, McPhail ED, et al. Fifty-year incidence of waldenström macroglobulinemia in Olmsted County, Minnesota, from 1961 through 2010: a population-based study with complete case capture and hematopathologic review. Mayo Clin Proc. 2018;93(6):739–746. doi:10.1016/j.mayocp.2018.02.011
43. Dimopoulos MA, Panayiotidis P, Moulopoulos LA, Sfikakis P, Dalakas M. Waldenström’s macroglobulinemia: clinical features, complications, and management. J Clin Oncol. 2000;18(1):214–226. doi:10.1200/JCO.2000.18.1.214
44. García-Sanz R, Montoto S, Torrequebrada A, et al. Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases. Br J Haematol. 2001;115(3):575–582. doi:10.1046/j.1365-2141.2001.03144.x
45. Kyle RA, Ansell SM, Kapoor P. Prognostic factors and indications for treatment of Waldenström’s Macroglobulinemia. Best Pract Res Clin Haematol. 2016;29(2):179–186. doi:10.1016/j.beha.2016.08.014
46. Pavy MD, Murphy PL, Virella G. Paraprotein-induced hyperviscosity. A reversible cause of stroke. Postgrad Med. 1980;68(3):109–112. doi:10.1080/00325481.1980.11715533
47. Mueller J, Hotson JR, Langston JW. Hyperviscosity-induced dementia. Neurology. 1983;33(1):101–103. doi:10.1212/WNL.33.1.101
48. Bloch KJ, Maki DG. Hyperviscosity syndromes associated with immunoglobulin abnormalities. Semin Hematol. 1973;10(2):113–124.
49. Nobile-Orazio E, Marmiroli P, Baldini L, et al. Peripheral neuropathy in macroglobulinemia: incidence and antigen-specificity of M proteins. Neurology. 1987;37(9):1506–1514. doi:10.1212/WNL.37.9.1506
50. Chad DA, Harris NL. Case records of the Massachusetts general hospital. Weekly clinicopathological exercises. Case 16-1999. A 71-year-old man with progressive weakness and a gammopathy. N Engl J Med. 1999;340(21):1661–1669. doi:10.1056/NEJM199905273402108
51. Charakidis M, Russell DJ. Spontaneous splenic rupture in Waldenstrom’s macroglobulinemia: a case report. J Med Case Rep. 2010;4:300. doi:10.1186/1752-1947-4-300
52. Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the second international workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30(2):110–115. doi:10.1053/sonc.2003.50082
53. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367(9):826–833. doi:10.1056/NEJMoa1200710
54. Zanwar S, Abeykoon JP. Treatment paradigm in Waldenström macroglobulinemia: frontline therapy and beyond. Ther Adv Hematol. 2022;13:20406207221093962. doi:10.1177/20406207221093962
55. Ihne S, Morbach C, Sommer C, Geier A, Knop S, Störk S. Amyloidosis-the diagnosis and treatment of an underdiagnosed disease. Dtsch Arztebl Int. 2020;117(10):159–166. doi:10.3238/arztebl.2020.0159
56. ASCO. Amyloidosis: statistics; 2022.
57. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046–1053. doi:10.1182/bloodadvances.2018016402
58. Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799–2806. doi:10.1093/eurheartj/ehx589
59. Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215–219. doi:10.1080/13506129.2018.1549825
60. Palladini G, Merlini G. What is new in diagnosis and management of light chain amyloidosis? Blood. 2016;128(2):159–168. doi:10.1182/blood-2016-01-629790
61. Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86(9):1036–1043. doi:10.1136/jnnp-2014-308724
62. Muchtar E, Dispenzieri A, Magen H, et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med. 2021;289(3):268–292. doi:10.1111/joim.13169
63. Ash S, Shorer E, Ramgobin D, et al. Cardiac amyloidosis-A review of current literature for the practicing physician. Clin Cardiol. 2021;44(3):322–331. doi:10.1002/clc.23572
64. Shin SC, Robinson-Papp J. Amyloid neuropathies. Mt Sinai J Med. 2012;79(6):733–748. doi:10.1002/msj.21352
65. Rowe K, Pankow J, Nehme F, Salyers W. Gastrointestinal Amyloidosis: review of the Literature. Cureus. 2017;9(5):e1228. doi:10.7759/cureus.1228
66. Pinto V, Bergantim R, Caires HR, Seca H, Guimarães JE, Vasconcelos MH. Multiple myeloma: available therapies and causes of drug resistance. Cancers. 2020;12(2):407. doi:10.3390/cancers12020407
67. Mikhael J, Ismaila N, Cheung MC, et al. Treatment of multiple Myeloma: ASCO and CCO joint clinical practice guideline. J Clin Oncol. 2019;37(14):1228–1263. doi:10.1200/JCO.18.02096
68. Harousseau JL. How to select among available options for the treatment of multiple myeloma. Ann Oncol. 2012;23(Suppl 10):x334–8. doi:10.1093/annonc/mds311
69. Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3:17046. doi:10.1038/nrdp.2017.46
70. Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk‐stratification, and management. Am J Hematol. 2018;93(8):981–1114. doi:10.1002/ajh.25117
71. Kumar SK. Recycling therapies for myeloma: the need for prospective trials. Cancer. 2019;125(17):2920–2922. doi:10.1002/cncr.32177
72. Poczta A, Rogalska A, Marczak A. Treatment of multiple Myeloma and the role of melphalan in the era of modern therapies-current research and clinical approaches. J Clin Med. 2021;10(9):1841. doi:10.3390/jcm10091841
73. Dima D, Jiang D, Singh DJ, et al. Multiple Myeloma therapy: emerging trends and challenges. Cancers. 2022;14(17):4082. doi:10.3390/cancers14174082
74. Mateos MV, Bladé J, Bringhen S, et al. Melflufen: a peptide-drug conjugate for the treatment of multiple myeloma. J Clin Med. 2020;9(10):3120. doi:10.3390/jcm9103120
75. Ito S. Proteasome inhibitors for the treatment of multiple Myeloma. Cancers. 2020;12(2):265. doi:10.3390/cancers12020265
76. Moreau P, Richardson PG, Cavo M, et al. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120(5):947–959. doi:10.1182/blood-2012-04-403733
77. Herndon TM, Deisseroth A, Kaminskas E, et al. U.S. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013;19(17):4559–4563. doi:10.1158/1078-0432.CCR-13-0755
78. Efentakis P, Kremastiotis G, Varela A, et al. Molecular mechanisms of carfilzomib-induced cardiotoxicity in mice and the emerging cardioprotective role of metformin. Blood. 2019;133(7):710–723. doi:10.1182/blood-2018-06-858415
79. Tzogani K, Florez B, Markey G, et al. European Medicines Agency review of ixazomib (Ninlaro) for the treatment of adult patients with multiple myeloma who have received at least one prior therapy. ESMO Open. 2019;4(5):e000570. doi:10.1136/esmoopen-2019-000570
80. Sharma S, Lichtenstein A. Dexamethasone-induced apoptotic mechanisms in myeloma cells investigated by analysis of mutant glucocorticoid receptors. Blood. 2008;112(4):1338–1345. doi:10.1182/blood-2007-11-124156
81. Friedenberg WR, Kyle RA, Knospe WH, Bennett JM, Tsiatis AA, Oken MM. High-dose dexamethasone for refractory or relapsing multiple myeloma. Am J Hematol. 1991;36(3):171–175. doi:10.1002/ajh.2830360303
82. Kumar SK, Laubach JP, Giove TJ, et al. Impact of concomitant dexamethasone dosing schedule on bortezomib-induced peripheral neuropathy in multiple myeloma. Br J Haematol. 2017;178(5):756–763. doi:10.1111/bjh.14754
83. Devarakonda S, Efebera Y, Sharma N. Role of stem cell transplantation in multiple Myeloma. Cancers. 2021;13(4):863. doi:10.3390/cancers13040863
84. McElwain TJ, Powles RL. High-dose intravenous melphalan for plasma-cell leukaemia and myeloma. Lancet. 1983;2(8354):822–824. doi:10.1016/S0140-6736(83)90739-0
85. Barlogie B, Alexanian R, Dicke KA, et al. High-dose chemoradiotherapy and autologous bone marrow transplantation for resistant multiple myeloma. Blood. 1987;70(3):869–872. doi:10.1182/blood.V70.3.869.869
86. Wildes TM, Finney JD, Fiala M, et al. High-dose therapy and autologous stem cell transplant in older adults with multiple myeloma. Bone Marrow Transplant. 2015;50(8):1075–1082. doi:10.1038/bmt.2015.106
87. Palumbo A, Cavallo F, Gay F, et al. Autologous transplantation and maintenance therapy in multiple myeloma. N Engl J Med. 2014;371(10):895–905. doi:10.1056/NEJMoa1402888
88. Cavo M, Gay F, Beksac M, et al. Autologous haematopoietic stem-cell transplantation versus bortezomib-melphalan-prednisone, with or without bortezomib-lenalidomide-dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): a multicentre, randomised, open-label, phase 3 study. Lancet Haematol. 2020;7(6):e456–e68. doi:10.1016/S2352-3026(20)30099-5
89. Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. 2017;376(14):1311–1320. doi:10.1056/NEJMoa1611750
90. Gay F, Oliva S, Petrucci MT, et al. Chemotherapy plus lenalidomide versus autologous transplantation, followed by lenalidomide plus prednisone versus lenalidomide maintenance, in patients with multiple myeloma: a randomised, multicentre, phase 3 trial. Lancet Oncol. 2015;16(16):1617–1629. doi:10.1016/S1470-2045(15)00389-7
91. Kumar L, Hussain MM, Chethan R, et al. Multiple Myeloma: impact of time to transplant on the outcome. Clin Lymphoma Myeloma Leuk. 2022;22(9):e826–e35. doi:10.1016/j.clml.2022.04.020
92. Richardson PG, Jacobus SJ, Weller E, et al. Lenalidomide, bortezomib, and dexamethasone (RVd) ± autologous stem cell transplantation (ASCT) and R maintenance to progression for newly diagnosed multiple myeloma (NDMM): the phase 3 DETERMINATION trial. J Clin Oncol. 2022;40(17_suppl):LBA4–LBA. doi:10.1200/JCO.2022.40.17_suppl.LBA4
93. Davies F, Baz R. Lenalidomide mode of action: linking bench and clinical findings. Blood Rev. 2010;24(Suppl 1):S13–9. doi:10.1016/S0268-960X(10)70004-7
94. Sedlarikova L, Kubiczkova L, Sevcikova S, Hajek R. Mechanism of immunomodulatory drugs in multiple myeloma. Leuk Res. 2012;36(10):1218–1224. doi:10.1016/j.leukres.2012.05.010
95. Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357(21):2123–2132. doi:10.1056/NEJMoa070594
96. Rajkumar SV. Thalidomide: tragic past and promising future. Mayo Clin Proc. 2004;79(7):899–903. doi:10.4065/79.7.899
97. Cavo M, Pantani L, Petrucci MT, et al. Bortezomib-thalidomide-dexamethasone is superior to thalidomide-dexamethasone as consolidation therapy after autologous hematopoietic stem cell transplantation in patients with newly diagnosed multiple myeloma. Blood. 2012;120(1):9–19. doi:10.1182/blood-2012-02-408898
98. Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127(21):2569–2574. doi:10.1182/blood-2016-01-693580
99. Hipp S, Tai YT, Blanset D, et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia. 2017;31(8):1743–1751. doi:10.1038/leu.2016.388
100. Rosiñol L, Oriol A, Rios R, et al. Bortezomib, lenalidomide, and dexamethasone as induction therapy prior to autologous transplant in multiple myeloma. Blood. 2019;134(16):1337–1345. doi:10.1182/blood.2019000241
101. Cho SF, Lin L, Xing L, et al. The immunomodulatory drugs lenalidomide and pomalidomide enhance the potency of AMG 701 in multiple myeloma preclinical models. Blood Adv. 2020;4(17):4195–4207. doi:10.1182/bloodadvances.2020002524
102. Lonial S, Popat R, Hulin C, et al. Iberdomide plus dexamethasone in heavily pretreated late-line relapsed or refractory multiple myeloma (CC-220-MM-001): a multicentre, multicohort, open-label, phase 1/2 trial. Lancet Haematol. 2022;9(11):e822–e32. doi:10.1016/S2352-3026(22)00290-3
103. Richardson PG, Trudel S, Quach H, et al. Mezigdomide (CC-92480), a potent, novel Cereblon E3 Ligase Modulator (CELMoD), combined with Dexamethasone (DEX) in patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM): preliminary results from the dose-expansion phase of the CC-92480-MM-001 trial. Blood. 2022;140(Supplement 1):1366–1368.
104. Lasica M, Anderson MA. Review of Venetoclax in CLL, AML and multiple Myeloma. J Pers Med. 2021;11(6):463. doi:10.3390/jpm11060463
105. Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128(3):384–394. doi:10.1182/blood-2015-12-687749
106. Bobin A, Leleu X. Recent advances in the treatment of multiple myeloma: a brief review. Fac Rev. 2022;11:28. doi:10.12703/r/11-28
107. Ghobrial I, Cruz CH, Garfall A, et al. Immunotherapy in multiple Myeloma: accelerating on the path to the patient. Clin Lymphoma Myeloma Leuk. 2019;19(6):332–344. doi:10.1016/j.clml.2019.02.004
108. Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518–528. doi:10.1056/NEJMoa1714678
109. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104–2115. doi:10.1056/NEJMoa1817249
110. Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019;394(10192):29–38. doi:10.1016/S0140-6736(19)31240-1
111. Collins SM, Bakan CE, Swartzel GD, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother. 2013;62(12):1841–1849. doi:10.1007/s00262-013-1493-8
112. Wudhikarn K, Wills B, Lesokhin AM. Monoclonal antibodies in multiple myeloma: current and emerging targets and mechanisms of action. Best Pract Res Clin Haematol. 2020;33(1):101143. doi:10.1016/j.beha.2020.101143
113. Seckinger A, Delgado JA, Moser S, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell. 2017;31(3):396–410. doi:10.1016/j.ccell.2017.02.002
114. Tai YT, Acharya C, An G, et al. April and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. 2016;127(25):3225–3236. doi:10.1182/blood-2016-01-691162
115. Ghermezi M, Li M, Vardanyan S, et al. Serum B-cell maturation antigen: a novel biomarker to predict outcomes for multiple myeloma patients. Haematologica. 2017;102(4):785–795. doi:10.3324/haematol.2016.150896
116. Martin T, Usmani SZ, Berdeja JG, et al. Ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J Clin Oncol. 2023;41(6):1265–1274. doi:10.1200/JCO.22.00842
117. Smith EL, Harrington K, Staehr M, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019;11(485). doi:10.1126/scitranslmed.aau7746
118. Wong SW, Bar N, Paris L, et al. Alnuctamab (ALNUC; BMS-986349; CC-93269), a B-Cell Maturation Antigen (BCMA) x CD3 T-Cell Engager (TCE), in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM): results from a phase 1 first-in-human clinical study. Blood. 2022;140(Supplement 1):400–402. doi:10.1182/blood-2022-159009
119. Bumma N, Richter JR, Dhodapkar MV, et al. LINKER-MM1 study: linvoseltamab (REGN5458) in patients with relapsed/refractory multiple myeloma. J Clin Oncol. 2023;41(16_suppl):8006. doi:10.1200/JCO.2023.41.16_suppl.8006
120. Topp MS, Duell J, Zugmaier G, et al. Anti-B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J Clin Oncol. 2020;38(8):775–783. doi:10.1200/JCO.19.02657
121. Cho SF, Yeh TJ, Anderson KC, Tai YT. Bispecific antibodies in multiple myeloma treatment: a journey in progress. Front Oncol. 2022;12:1032775. doi:10.3389/fonc.2022.1032775
122. Tan CR, Shah UA. Targeting BCMA in Multiple Myeloma. Curr Hematol Malig Rep. 2021;16(5):367–383. doi:10.1007/s11899-021-00639-z
123. Pillarisetti K, Powers G, Luistro L, et al. Teclistamab is an active T cell-redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020;4(18):4538–4549. doi:10.1182/bloodadvances.2020002393
124. Frerichs KA, Broekmans MEC, Marin Soto JA, et al. Preclinical activity of JNJ-7957, a novel BCMA×CD3 bispecific antibody for the treatment of multiple myeloma, is potentiated by daratumumab. Clin Cancer Res. 2020;26(9):2203–2215. doi:10.1158/1078-0432.CCR-19-2299
125. Janssen Research & Development, LLC. Janssen research & development; 2020.
126. Verkleij CPM, Broekmans MEC, van Duin M, et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv. 2021;5(8):2196–2215. doi:10.1182/bloodadvances.2020003805
127. Pillarisetti K, Edavettal S, Mendonça M, et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood. 2020;135(15):1232–1243. doi:10.1182/blood.2019003342
128. Chari A, Minnema MC, Berdeja JG, et al. Talquetamab, a T-cell-redirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med. 2022;387(24):2232–2244. doi:10.1056/NEJMoa2204591
129. Lesokhin AM, Richter J, Trudel S, et al. Enduring responses after 1-year, fixed-duration cevostamab therapy in patients with relapsed/refractory multiple myeloma: early experience from a phase I study. Blood. 2022;140(Supplement 1):4415–4417. doi:10.1182/blood-2022-157547
130. Raje N, Bahlis NJ, Costello C, et al. Elranatamab, a BCMA targeted T-cell engaging bispecific antibody, induces durable clinical and molecular responses for patients with relapsed or refractory multiple myeloma. Blood. 2022;140(Supplement 1):388–390. doi:10.1182/blood-2022-166494
131. Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–2060. doi:10.1158/1078-0432.CCR-12-2422
132. Rendo MJ, Joseph JJ, Phan LM, DeStefano CB. CAR T-cell therapy for patients with multiple myeloma: current evidence and challenges. Blood Lymphat Cancer. 2022;12:119–136. doi:10.2147/BLCTT.S327016
133. Study of bb21217 in multiple myeloma; 2021.
134. bluebird bio. A Phase 1 Study of bb21217, an Anti-BCMA CAR T Cell Drug Product, in Relapsed and/or Refractory Multiple Myeloma [Internet]. clinicaltrials.gov; 2021 Mar [cited 2023 Sep 11]. Report No.: NCT03274219. Available from: https://clinicaltrials.gov/study/NCT03274219.
135. Juno Therapeutics, a Subsidiary of Celgene. A Phase I, Multi Center, Open Label Study of CC-98633, BCMA Targeted NEX-T Chimeric Antigen Receptor (CAR) T Cells, in Subjects With Relapsed and/or Refractory Multiple Myeloma [Internet]. clinicaltrials.gov; 2023 Feb [cited 2023 Sep 11]. Report No.: NCT04394650. Available from: https://clinicaltrials.gov/study/NCT04394650.
136. clinicaltrial.gov. BCMA Targeted CAR T Cells With or Without Lenalidomide for the Treatment of Multiple Myeloma. NCT03070327; Available from: https://classic.clinicaltrials.gov/ct2/show/NCT03070327.
137. Latorre SV. Pilot Study of the Infusion of Differentiated Autologous T-cells From Peripheral Blood, Expanded and Transduced With a Lentivirus to Express a Chimeric Antigen Receptor With Anti-BCMA (TNFRSF17) Specificity Humanized Conjugated With the Co-stimulatory Region 4-1BB and Signal-transduction CD3z (ARI0002h) in Patients With Relapsed/Refractory Multiple Myeloma With Previous Treatment With Proteasome Inhibitor, Immunomodulatory Drug and Anti-CD38 Monoclonal Antibody [Internet]. clinicaltrials.gov; 2023 Aug [cited 2023 Sep 11]. Report No.: NCT04309981. Available from: https://clinicaltrials.gov/study/NCT04309981.
138. clinicaltrial.gov. T cells expressing a novel fully-human anti-BCMA CAR for treating multiple myeloma. NCT03602612; 2023.
139. clinicaltrial.gov. BCMA-directed CAR-T cell therapy in adult patients with multiple myeloma. NCT04318327; 2023.
140. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–1737. doi:10.1056/NEJMoa1817226
141. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–716. doi:10.1056/NEJMoa2024850
142. Cohen AD, Mateos MV, Cohen YC, et al. Efficacy and safety of cilta-cel in patients with progressive multiple myeloma after exposure to other BCMA-targeting agents. Blood. 2023;141(3):219–230. doi:10.1182/blood.2022015526
143. Parikh RH, Lonial S. Chimeric antigen receptor T-cell therapy in multiple myeloma: a comprehensive review of current data and implications for clinical practice. CA. 2023;73(3):275–285. doi:10.3322/caac.21771
144. Castella M, Fernández de Larrea C, Martín-Antonio B. Immunotherapy: a novel era of promising treatments for multiple myeloma. Int J Mol Sci. 2018;19(11):3613. doi:10.3390/ijms19113613
145. Shimony S, Stone RM, Stahl M. Venetoclax combination therapy in acute myeloid leukemia and myelodysplastic syndromes. Curr Opin Hematol. 2022;29(2):63–73. doi:10.1097/MOH.0000000000000698
146. Slomp A, Moesbergen LM, Gong J-N, et al. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 2019;3(24):4202–4214. doi:10.1182/bloodadvances.2019000702
147. ClinicalTrials.gov. Novartis Pharmaceuticals. NCT04702425; 2023.
148. ClinicalTrials.gov. A study of PRT1419 in patients with relapsed/refractory hematologic malignancies. NCT04543305; 2023.
149. Caenepeel S, Brown SP, Belmontes B, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8(12):1582–1597. doi:10.1158/2159-8290.CD-18-0387
150. Chari A, Cornell RF, Gasparetto C, et al. Final analysis of a phase 1/2b study of ibrutinib combined with carfilzomib/dexamethasone in patients with relapsed/refractory multiple myeloma. Hematol Oncol. 2020;38(3):353–362. doi:10.1002/hon.2723
151. Von Suskil M, Sultana KN, Elbezanti WO, et al. Bruton’s tyrosine kinase targeting in multiple myeloma. Int J Mol Sci. 2021;22(11):5707. doi:10.3390/ijms22115707
152. ClinicalTrials.gov. Clinical research of adoptive BCMA CAR-NK cells on relapse/refractory MM. NCT03940833; 2023.
153. ClinicalTrials.gov. Anti-BCMA CAR-NK cell therapy for the relapsed or refractory multiple myeloma. NCT05008536; 2023.
154. Yang Y, Shi J, Gu Z, et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res. 2015;75(3):594–604. doi:10.1158/0008-5472.CAN-14-2362
155. Elbezanti WO, Al-Odat OS, Chitren R, et al. Development of a novel Bruton’s tyrosine kinase inhibitor that exerts anti-cancer activities potentiates response of chemotherapeutic agents in multiple myeloma stem cell-like cells. Front Pharmacol. 2022;13:894535. doi:10.3389/fphar.2022.894535
156. Berdeja JG, Lin Y, Raje NS, et al. First-in-human multicenter study of bb2121 anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: updated results. J Clin Oncol. 2017;35(15_suppl):3010. doi:10.1200/JCO.2017.35.15_suppl.3010
157. Pittari G, Vago L, Festuccia M, et al. Restoring natural killer cell immunity against multiple myeloma in the era of new drugs. Front Immunol. 2017;8:1444. doi:10.3389/fimmu.2017.01444
158. Vogl DT, Kaufman JL, Holstein SA, et al. Modakafusp Alfa (TAK-573), an immunocytokine, shows clinical activity in patients with relapsed/refractory multiple myeloma; updated results from a first-in-human phase 1 study. Blood. 2021;138(Supplement 1):898. doi:10.1182/blood-2021-148463
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.