Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Differential anti-inflammatory effects of budesonide and a p38 MAPK inhibitor AZD7624 on COPD pulmonary cells
Authors Higham A, Karur P, Jackson N, Cunoosamy DM, Jansson P, Singh D
Received 15 December 2017
Accepted for publication 18 March 2018
Published 19 April 2018 Volume 2018:13 Pages 1279—1288
DOI https://doi.org/10.2147/COPD.S159936
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Andrew Higham,1,2 Pradeep Karur,1,2 Natalie Jackson,2 Danen M Cunoosamy,3 Paul Jansson,3 Dave Singh1,2
1Division of Infection, Immunity and Respiratory Medicine, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, The University of Manchester and University Hospital of South Manchester, NHS Foundation Trust, Manchester, UK; 2Medicines Evaluation Unit, The University Hospital of South Manchester, Manchester, UK; 3RIA IMED Biotech Unit, AstraZeneca, Gothenburg, Sweden
Background: The effects of anti-inflammatory drugs in COPD patients may vary between different cell types. The aim of the current study was to assess the anti-inflammatory effects of the corticosteroid budesonide and a p38 MAPK inhibitor (AZD7624) on different cell types obtained from COPD patients and healthy controls.
Methods: Eight healthy smokers, 16 COPD infrequent exacerbators, and 16 frequent COPD exacerbators (≥2 exacerbations in the last year) were recruited for bronchoscopy and blood sampling. The anti-inflammatory effects of budesonide and AZD7624 were assessed on cytokine release from lipopolysaccharide-stimulated alveolar macrophages and peripheral blood mononuclear cells and polyinosinic:polycytidylic acid-stimulated bronchial epithelial cells.
Results: The anti-inflammatory effects of budesonide varied greatly within a patient according to the cell type studied. Bronchial epithelial cells showed the lowest sensitivity to budesonide, while peripheral blood mononuclear cells showed the greatest sensitivity. AZD7624 had a greater effect than budesonide on cytokine production from bronchial epithelial cells. Exacerbation frequency did not influence corticosteroid sensitivity.
Conclusion: We observed variable corticosteroid and p38 MAPK inhibitor anti-inflammatory responses within the same individual depending on the cell type studied. These findings support the use of multiple anti-inflammatory strategies in COPD patients due to differences between cell types.
Keywords: PBMC, corticosteroid, inflammation, exacerbation, macrophage, epithelial cell
Introduction
COPD is characterized by an abnormal innate immune response in the lungs, with increased numbers of macrophages and neutrophils.1 Bronchial epithelial cells (BECs) are involved in the pathophysiology of COPD through the release of cytokines and chemokines in response to cigarette smoke.2 COPD exacerbations are an acute worsening of symptoms, often caused by viral or bacterial infection and resulting in further amplification of airway inflammation.3
Inhaled corticosteroids (ICS) are anti-inflammatory drugs that prevent exacerbations and improve quality of life in COPD patients.4,5 The clinical effects of ICS vary between patients, and are greater in individuals with higher blood eosinophil counts.6 We have previously observed a wide variation between COPD patients in the effect of corticosteroids on the inflammatory response of alveolar macrophages, although no overall group mean difference was observed compared to controls.7 COPD peripheral blood mononuclear cells (PBMCs) have been used as surrogates for alveolar macrophages,8 as the latter are practically difficult to obtain, but the relationship between the corticosteroid sensitivity of PBMCs and alveolar macrophages from the same individual is not clearly understood.
p38 mitogen activated protein kinases (MAPK) are members of the serine/threonine protein kinase family which are activated by a plethora of environmental stimuli and are described as stress-activated protein kinases.9 The p38 MAPK pathway promotes inflammation in response to cytokines, toll-like receptor agonists, and components of cigarette smoke by enhancing gene transcription, stabilizing mRNAs, and increasing protein translation.10,11 p38 MAPK shows increased activation in the lungs of COPD patients,12 suggesting a role for this intracellular signaling pathway in the pathophysiology of airway inflammation in COPD. Indeed, p38 MAPK inhibitors reduce cytokine production from COPD PBMCs and lung cells such as alveolar macrophages.8,13–15 AZD7624 is a novel inhaled p38 MAPK inhibitor that has been developed for the treatment of COPD exacerbations.
The effects of anti-inflammatory drugs, such as corticosteroids and p38 MAPK inhibitors, may vary according to cell type.7,16 We have studied the anti-inflammatory effects of the corticosteroid budesonide and the p38 MAPK inhibitor AZD7624 in different cell types obtained from COPD patients and controls. PBMCs, alveolar macrophages, and BECs were obtained from the same individuals, allowing comparison of drug effects between different cell types. Furthermore, we carefully characterized the clinical features of the COPD patients, in order to evaluate whether drug sensitivity was related to clinical characteristics such as exacerbation frequency.
Methods
Study subjects
Eight healthy smokers (HS), sixteen COPD infrequent exacerbators (COPD IFE), and sixteen COPD frequent exacerbators (COPD FE) were recruited for sputum induction, phlebotomy, and bronchoscopy. HS were current smokers with a ≥10 pack-years smoking history, normal spirometry, and no airway colonization. COPD was diagnosed based on ≥10 pack-years smoking history, typical symptoms, and airflow obstruction. COPD FE were defined as having ≥2 exacerbations/yr for the last year, whereas COPD IFE were defined as having 0 exacerbations/yr for the last year. All subjects were nonatopic, with no history of respiratory illness or antibiotic use within 6 weeks of the study.
Demographics and clinical characteristics are presented in Table 1, and sputum, bronchoalveolar lavage (BAL), and blood cell counts are presented in Table S1. The study was approved by National Research Ethics Service (NRES) Committee North West – Greater Manchester Central (ref code 06/Q1403/156), and participants provided written and informed consent.
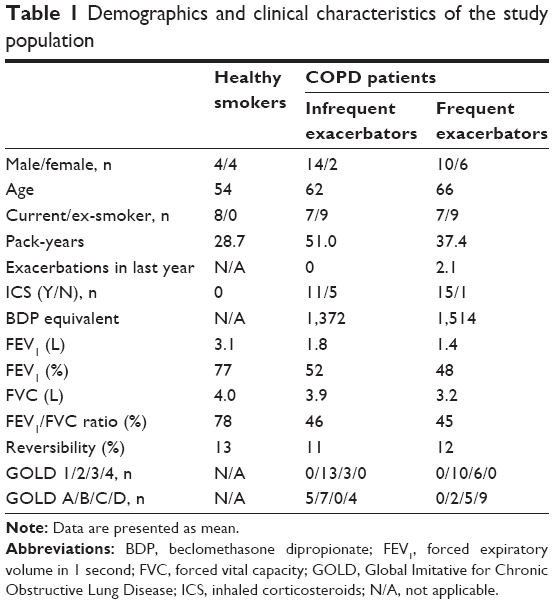 | Table 1 Demographics and clinical characteristics of the study population |
Sputum processing
Differential cell count
Sputum was induced using 3%, 4%, and 5% saline, inhaled in sequence for 5 min, for a maximum of 15 min via an ultrasonic nebulizer (EASYneb II, Flaemnuova, San Martino della Battaglia, Italy). To minimize contamination of saliva, all subjects were instructed to thoroughly rinse their mouth with distilled water and perform coughing prior to sputum expectoration. Sputum plugs were isolated from the saliva component, combined proportionately with phosphate-buffered saline (PBS) and vortexed for 10 sec, rocked for 15 min, and centrifuged (790× g for 10 min at 4°C). PBS supernatants were removed and 0.2% DTT was added and the suspension was vortexed for 10 sec, rocked for 15 min, and filtered using a 48 μm filter (Sefar Ltd, Bury, UK). The suspension was centrifuged (790× g for 10 min at 4°C) and DTT supernatants were removed, the cell pellet was resuspended in PBS and cytospins were prepared. Slides were air dried for 30 min and then fixed in methanol for 10 min before staining with RapiDiff (Triangle, Skelmersdale, UK) for differential cell counting.
PBMC processing
Heparinized blood was layered over Ficoll-paque (GE Healthcare, Buckinghamshire, UK) and centrifuged (400× g for 30 min at room temperature). PBMCs were harvested from the interface and cells were resuspended in Roswell Park Memorial Institute-1640 (Sigma-Aldrich, Gillingham, UK) supplemented with 10% v/v fetal calf serum, 2 mM L-glutamate (both Invitrogen, Paisley, UK), and 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich), and viable cell counts were determined by trypan blue exclusion. PBMCs were seeded onto flat bottomed 96-well plates at 1×105 PBMCs/well and used the same day.
Bronchoscopy sampling
Bronchoscopy was performed after the subjects had been sedated. BAL was collected from the right and/or left upper lobe. The bronchoscope was wedged in the bronchus and a maximum of 4×60 mL aliquots of prewarmed sterile 0.9% NaCl solution was instilled per lobe.
The aspirated fluid was stored on ice before filtration (100 μm filter, Becton Dickinson, Oxford, UK). The filtrate was centrifuged (400× g for 10 min at 4°C) and the BAL fluid removed. The cell pellet was treated with 3 mL ACK buffer for 3 min to lyse red blood cells, and the cell lysis quenched with PBS. BAL cells were resuspended in Roswell Park Memorial Institute media supplemented with 10% fetal calf serum, L-glutamine and Penicilin/Streoptomycin, and viable cell counts were determined by trypan blue exclusion. Macrophages were seeded onto flat bottomed 96-well plates at 1×105 macrophages/well for 1 h before nonadherent cells were removed, fresh media was added, and left overnight. Cytospins were prepared and slides were left to air dry for 30 min and then fixed in methanol for 10 min before staining with RapiDiff for differential cell counting.
Bronchial brushes were collected in bronchial epithelial basal medium (Lonza, Slough, UK) and stored on ice before centrifugation (400× g for 10 min at 4°C). The cell pellet was resuspended in bronchial epithelial growth medium (basal medium plus bullet kit; Lonza) and cells were seeded onto the base of bovine collagen (100 μg/mL; Stemcell, Cambridge, UK)-coated T25 flasks positioned vertically and incubated at 37°C and 5% CO2 for 10 days with medium changed every 2–3 days. Cells were then passaged onto the base of bovine collagen-coated T25 flasks positioned horizontally and grown to 80% confluence with medium changed every 2–3 days. When confluent, cells were passaged onto bovine collagen-coated flat bottomed 96-well plates at 30,000 cells/well and grown to 80% confluence.
Cell culture
PBMCs and lung macrophages were pretreated with AZD7624 or budesonide (both provided by AstraZeneca) for 1 h prior to stimulation with lipopolysaccharide (LPS) (1 μg/mL, Escherichia coli B6-026; Sigma-Aldrich) for 24 h. Supernatants were collected and analyzed for tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and IL-10 by enzyme linked immunosorbent assay according to manufacturer’s instructions (TNF-α and IL-6 R & D Systems, Abingdon, UK; IL-10 eBioscience). BECs were pretreated with AZD7624 or budesonide for 1 h prior to stimulation with poly I:C (100 μg/mL; Invivogen, Toulouse, France) for 24 h. Supernatants were collected and analyzed for IL-6 and C-X-C motif ligand 10 (CXCL10) by ELISA according to manufacturer’s instructions (R & D Systems). All compound-treated cells and unstimulated and stimulated controls contained 0.02% DMSO (Sigma-Aldrich).
Statistical analysis
All statistical analysis was performed using GraphPad InStat software (GraphPad Software Inc., La Jolla, CA, USA). Data distribution was determined by the Kolmogorov–Smirnov test. Clinical characteristics were normally distributed and comparisons between groups were made by a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc analysis, an unpaired t-test, or a χ2 test. Basal and stimulated cytokine production were analyzed by a one-way ANOVA followed by Tukey’s post hoc analysis (parametric) or a Kruskal–Wallis test followed by Dunn’s post hoc analysis (nonparametric). Comparison of compound effects between study groups and within the same study group was made by a two-way ANOVA followed by a Tukey’s post hoc test. IC50 values for budesonide (the concentration required to cause 50% inhibition) were calculated using sigmoidal curve fitting of group mean data. IC30 was also calculated as, often, it was not possible to calculate the IC50 due to inhibition not reaching above 50%.
Results
Study subjects
COPD patients had typically impaired lung function compared to HS, such as significantly lower forced expiratory volume in 1 second and forced expiratory volume in 1 second/forced vital capacity ratio (p<0.001); the demographics are shown in Table 1. The mean exacerbation rate in the previous year for the COPD FE group was 2.1. The majority of COPD FE and COPD IFE were ICS users. Apart from exacerbation history, COPD FE and COPD IFE groups had similar clinical characteristics. Induced sputum, BAL, and blood cell counts are shown in Table S1. There were few significant differences between groups.
PBMCs
PBMCs were collected from 8 HS, 16 COPD IFE, and 16 COPD FE. There were no differences between groups in the basal production or the LPS-induced production of TNF-α, IL-6, or IL-10 (Figure S1). There was no difference between groups in the inhibitory effect of budesonide (0.1–1,000 nM) on LPS-induced cytokine production (Figure 1). The IC50 and IC30 values were similar between groups (Table 2). Maximal inhibition for budesonide reached approximately 70% for TNF-α and IL-6, while IL-10 inhibition was lower at approximately 45%.
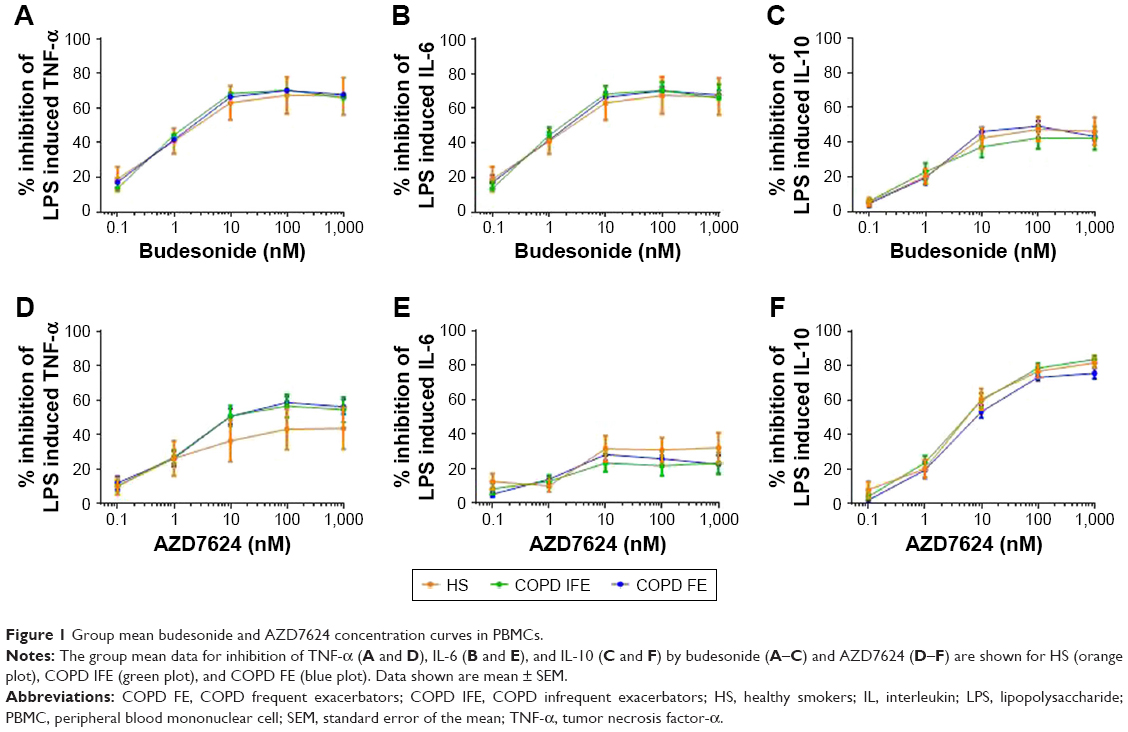 | Figure 1 Group mean budesonide and AZD7624 concentration curves in PBMCs. |
 | Table 2 IC50, IC30, and EC50 values calculated from sigmoidal curve fitting of budesonide inhibition in PBMCs |
There was no difference in the inhibitory effect of AZD7624 between groups (Figure 1). Maximal inhibition for AZD7624 reached approximately 50% for TNF-α, 30% for IL-6, and 80% for IL-10. In all patient groups, the maximal inhibition of TNF-α and IL-6 by budesonide was greater than AZD7624, while inhibition of IL-10 was lower for budesonide compared to AZD7624 (Table S2).
COPD patients were further analyzed according to current smoking and blood eosinophil counts (using a threshold of 300 cells/μL, which identifies patients with higher corticosteroid response clinically).17 The majority of patients were ICS users (n=26 out of 32), so subanalysis based on ICS use could not be performed. For smoking status (14 current smokers versus 18 ex-smokers) and eosinophil counts (n=12 >300 cells/μL versus n=20 <300 cells/μL), there were no significant differences between groups for drug effects (Figures S2 and S3).
Alveolar macrophages
Alveolar macrophages were collected from all 8 HS, 8 COPD IFE, and 9 COPD FE out of a possible 16 from each COPD group. It was not possible to use alveolar macrophages from all COPD patients as BAL fluid return or cell yield were too low. The basal production of IL-6 was significantly higher from COPD FE compared to HS and COPD IFE (p<0.05; Figure S4). LPS-induced production of TNF-α and IL-6 was significantly higher from HS compared to COPD IFE and COPD FE (p<0.01; Figure S4). There were no other differences between groups in basal or LPS-stimulated cytokine production.
Group mean inhibition of cytokine production by budesonide is shown in Figure 2. TNF-α inhibition was significantly greater in HS compared to COPD IFE and COPD FE. Greater IL-10 inhibition in HS was also observed compared to COPD IFE (at 100 nM only) and COPD FE (at 10–100 nM). There was no difference between groups in the degree of IL-6 inhibition. Budesonide had similar effects in COPD FE compared to COPD IFE.
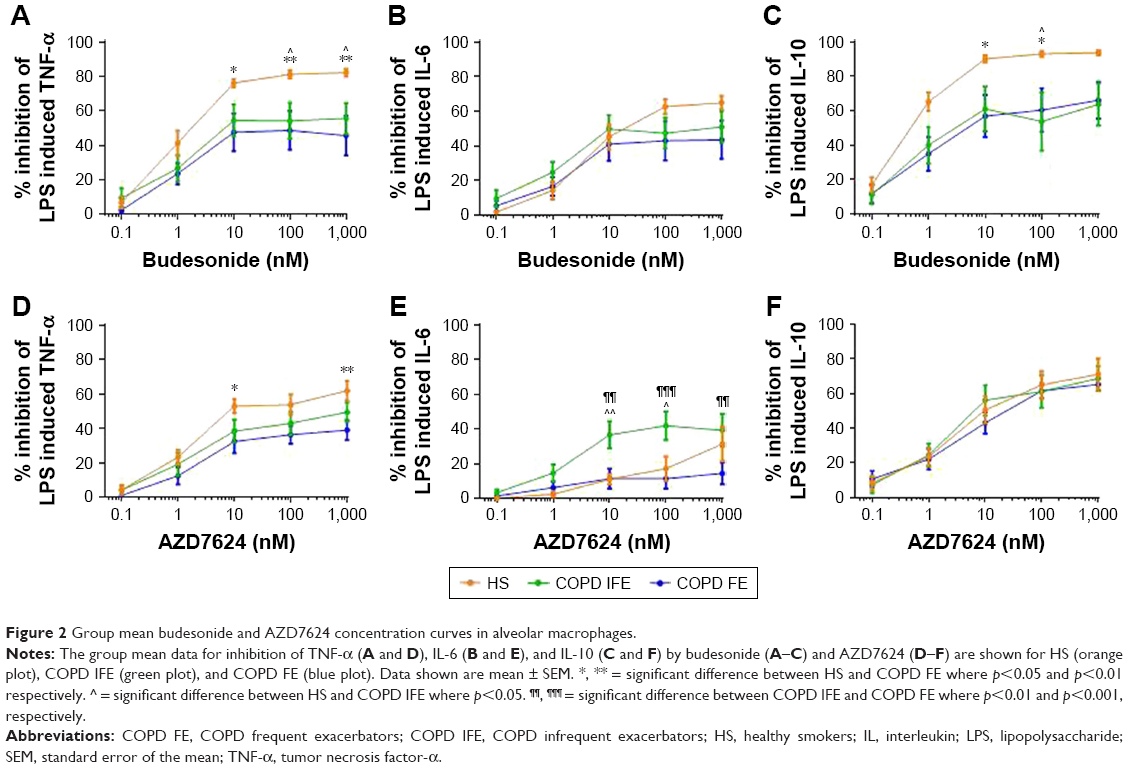 | Figure 2 Group mean budesonide and AZD7624 concentration curves in alveolar macrophages. |
The IC50 and IC30 values for all cytokines are shown in Table 3. In some cases in COPD patients, the IC50 values could not be calculated as the maximal effect was <50%. In general, IC50 and IC30 values showed a trend to be lower in HS compared to COPD patients.
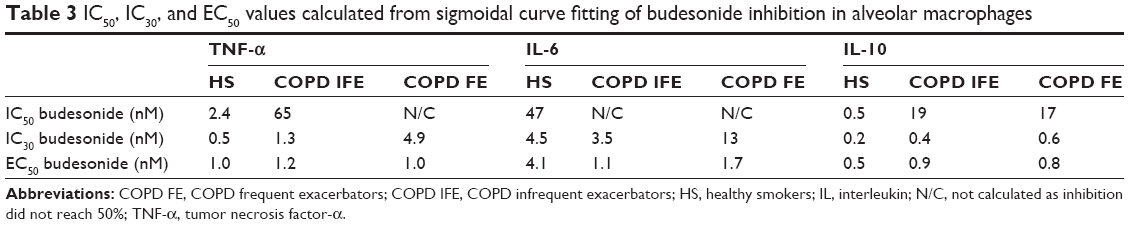 | Table 3 IC50, IC30, and EC50 values calculated from sigmoidal curve fitting of budesonide inhibition in alveolar macrophages |
The maximal effect of budesonide was reached at 100–1,000 nM for the majority of subjects (Figure 3). In the HS group there was little variation between subjects for the degree of maximal inhibition. In contrast, the individual responses for COPD IFE and COPD FE patients were more heterogeneous. In all subject groups, variation in the individual responses was evident at lower budesonide concentrations (1–10 nM). This variation became less evident in HS as the budesonide concentration increased, but was still evident in COPD patients.
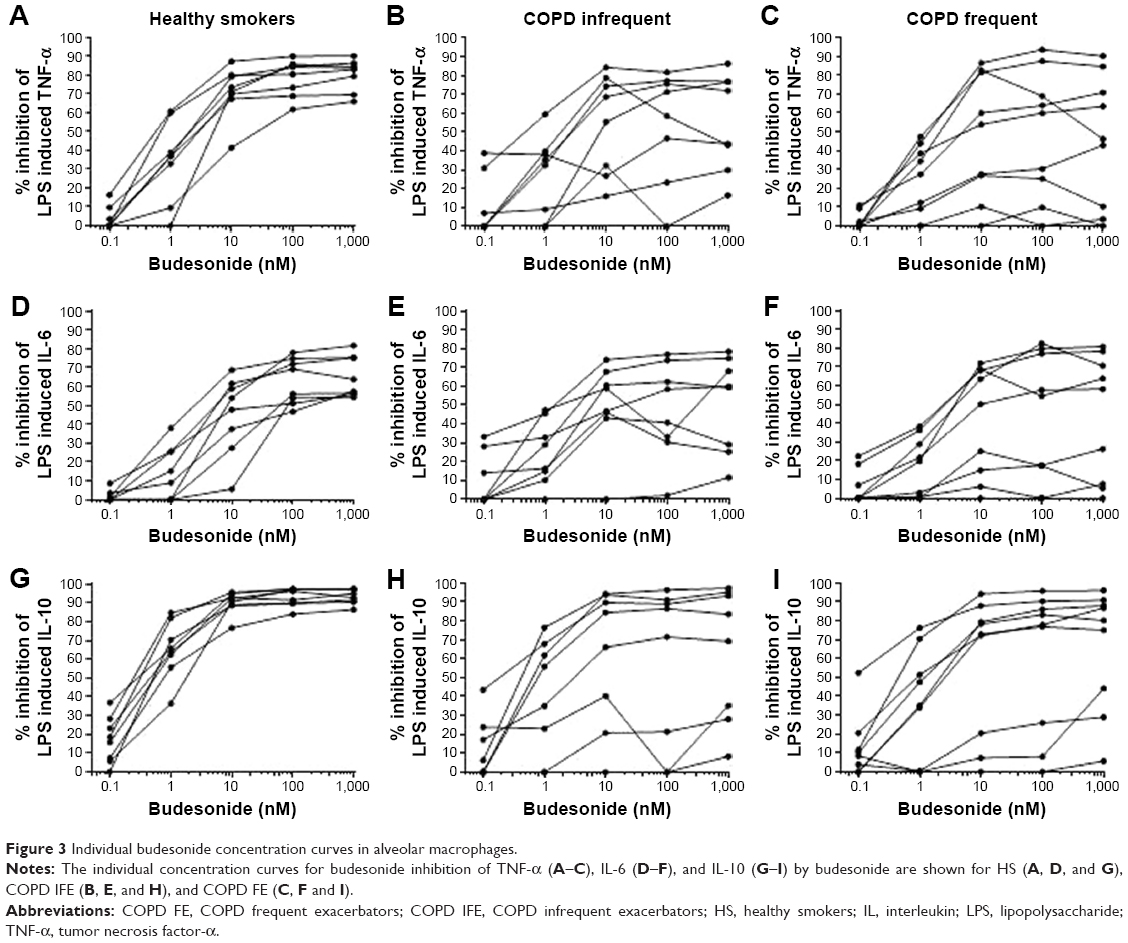 | Figure 3 Individual budesonide concentration curves in alveolar macrophages. |
Inhibition of TNF-α by AZD7624 was significantly higher in HS compared to COPD FE (Figure 2; 10 and 1,000 nM) while inhibition of IL-6 was significantly higher in COPD IFE compared to HS (10 and 100 nM) and COPD FE (10–1,000 nM). There was no difference between groups for IL-10 inhibition. In HS, maximal inhibition of all cytokines by budesonide was significantly higher than AZD7624 (Table S3). Conversely, the only difference in the COPD groups was for maximal inhibition of IL-6 in the COPD FE group, which reached 43% for budesonide and 11% for AZD7624.
When analyzing the COPD data based on smoking status and blood eosinophil counts, there were no differences between subgroups (Figures S5 and S6).
Relationship between PBMCs and alveolar macrophages
We investigated the relationship between PBMC and alveolar macrophage steroid responses by comparing maximal budesonide inhibition from paired samples (Figure 4; HS =8, COPD IFE =8, and COPD FE =9). There were no significant correlations between maximal inhibition of IL-6, TNF, or IL-10 release from paired PBMCs and alveolar macrophages when the groups were analyzed combined (Figure 4) or separately (data not shown).
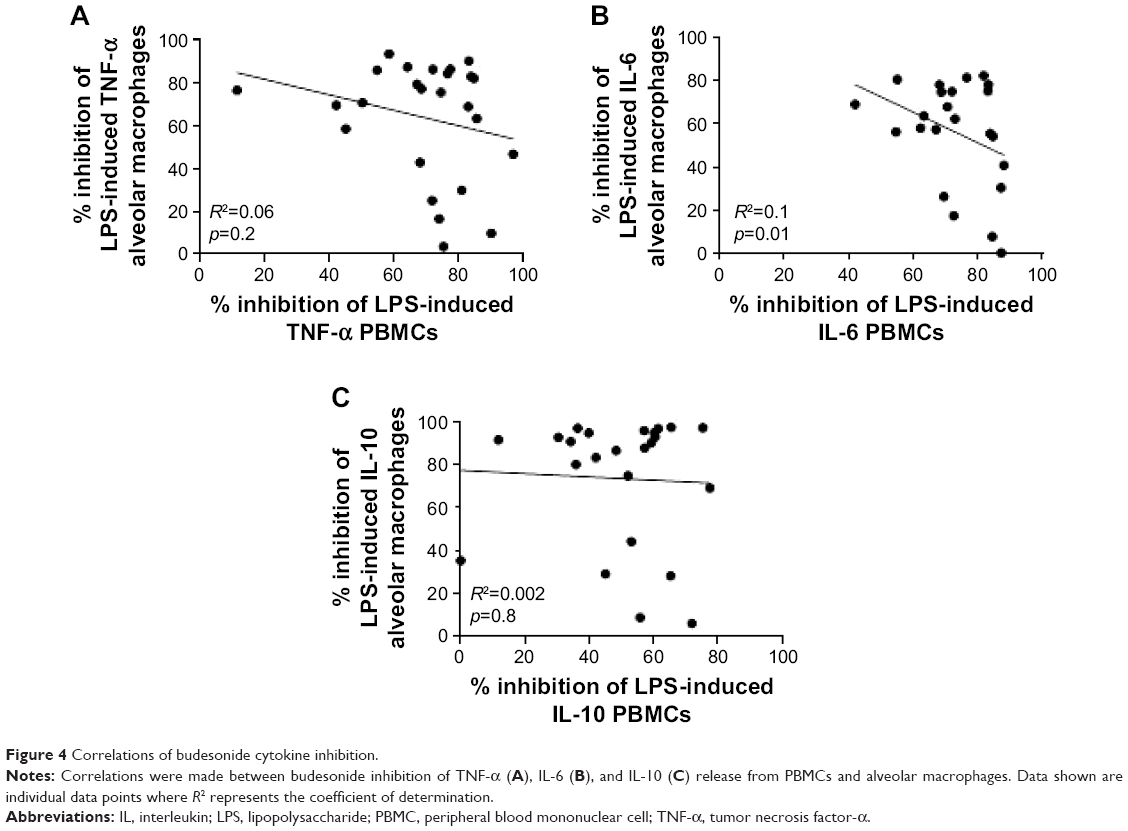 | Figure 4 Correlations of budesonide cytokine inhibition. |
BEC study
BECs were collected from six COPD IFE and five COPD FE. There was no difference in the basal production of IL-6 or poly I:C-induced production of IL-6 and CXCL10 (Figure S7). The basal production of IL-10 was undetectable. Budesonide had a small inhibitory effect on poly I:C-induced cytokine production, with no difference between groups; the maximal inhibition was approximately 20% for IL-6 and CXCL10 (Figure 5).
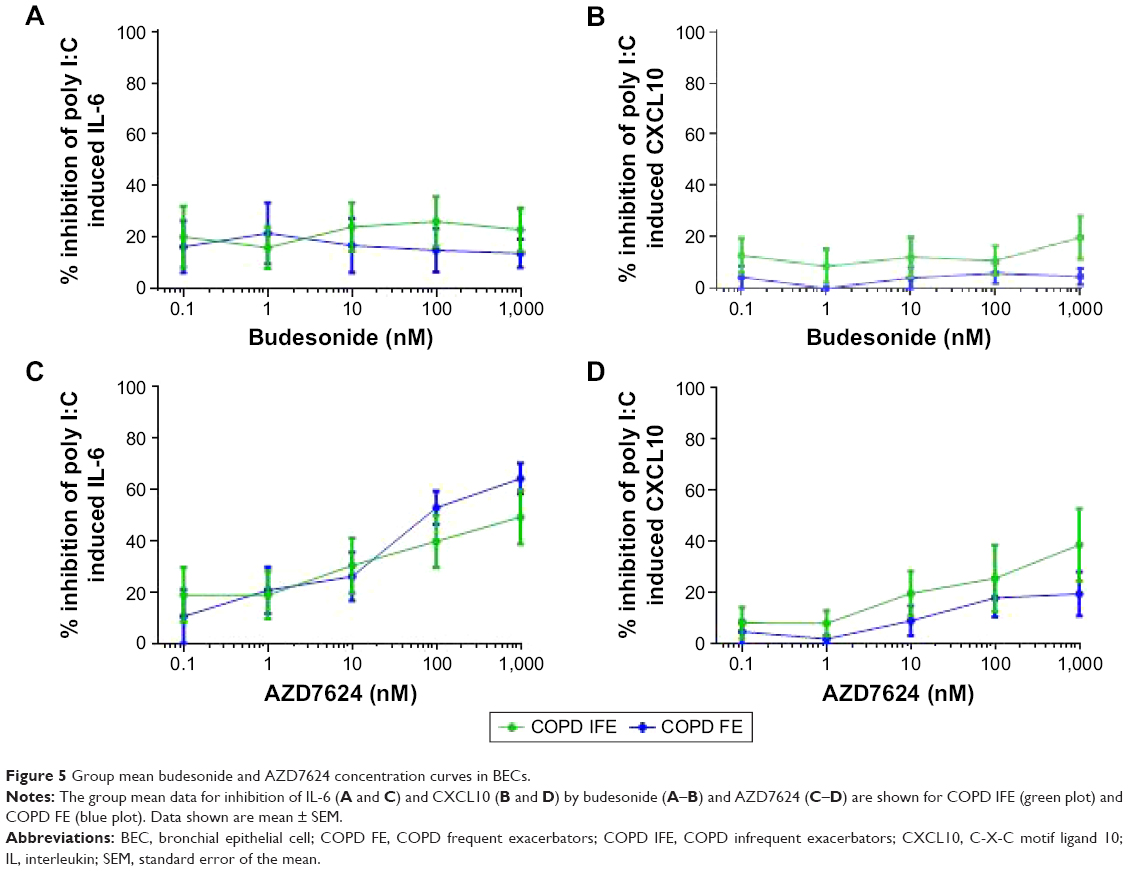 | Figure 5 Group mean budesonide and AZD7624 concentration curves in BECs. |
Maximal inhibition by AZD7624 was approximately 40%–60% for IL-6 and 20%–40% for CXCL10. For both COPD IFE and COPD FE groups, maximal inhibition of IL-6 by AZD7624 was significantly greater than budesonide (Table S4). Inhibition of CXCL10 was numerically greater with AZD7624 compared to budesonide, but this did not reach statistical significance.
Discussion
We show that the anti-inflammatory effects of the corticosteroid budesonide in COPD and controls varied greatly within a patient according to the cell type studied. BECs showed the greatest insensitivity to budesonide, while PBMCs showed the greatest sensitivity. There was no correlation between the results from PBMCs and alveolar macrophages, highlighting that PBMCs do not predict the corticosteroid sensitivity of lung cells. Clinical characteristics, including exacerbation frequency, did not influence corticosteroid sensitivity. The p38 MAPK inhibitor AZD7624 had a greater effect than budesonide on cytokine production from BECs.
An important aspect of this study was the comparison of drug effects on different cell types obtained from the same individual. Cytokine production from BECs showed little inhibition by corticosteroids. Poly I:C was used as a stimulus in these experiments, as this TLR3 agonist produces robust proinflammatory responses in BECs, greater than the TLR4 agonist LPS.18 LPS was used in PBMC and macrophage experiments, and perhaps some difference in drug sensitivity between cell types could be attributed to the stimuli used. Regardless, we show that budesonide had little effect on the response to the viral mimetic poly I:C; this is likely to be an important observation in the context of viral-induced COPD exacerbations. Indeed, it has been reported that a short-term course of systemic steroids did not reduce viral load or shedding in adults hospitalized with respiratory syncytial virus,19 further supporting the concept that corticosteroids have limited utility in treating viral-induced COPD exacerbations. AZD7624 had a greater effect on BEC cytokine production, suggesting that p38 MAPK inhibitors may be able to address the excessive inflammation that can occur after viral infection in COPD patients. Moreover, p38 MAPK inhibition in the bronchial epithelial BEAS2B cell line has been shown to reduce poly I:C-induced TNF-α and IL-1β release,20 further supporting a role for p38 inhibition in viral-induced bronchial inflammation.
We observed up to approximately 40% inhibition of CXCL10 by AZD7624. Although this may reduce the airway inflammatory burden, CXCL10 is an important component of the antiviral response.21,22 Reducing the levels of CXCL10 may have detrimental effects on the antiviral response in COPD patients. This must be considered when examining the utility of p38 inhibition as a potential COPD treatment.
Corticosteroids are poor inhibitors of cytokine release from COPD BECs. It has previously been shown that poly I:C-induced CXCL8 release from COPD BECs was inhibited by 22% by dexamethasone.23 In another study, budesonide inhibited TNF-α-induced granulocyte macrophage-colony stimulating factor and CXCL8 release from COPD BECs by approximately 40%.16 Here, we report similar findings in both COPD FE and IFE, with budesonide inhibition failing to reach above 25%.
PBMCs are often used as surrogates for alveolar macrophages due to the practical difficulties in obtaining the latter.8 We are unaware of any publications which have compared the corticosteroid response of paired PBMCs and alveolar macrophages from the same individual. We demonstrate a lack of correlation between the results from these different cell types. This questions the relevance of PBMC experiments when studying the effects of ICS that are used principally to address lung inflammation.
In contrast to our previous reports, this is the first time that we have observed a reduced effect of corticosteroids in HS compared to COPD alveolar macrophages. We have previously pooled the results from a number of studies (n=20 never smokers, n=27 smokers, and n=45 COPD patients) using dexamethasone to study the effects of corticosteroids on alveolar macrophages, and found no difference between COPD patients and controls.7 However, we reported considerable between-individual variation in corticosteroid sensitivity and now report the same phenomenon, albeit using budesonide. We suggest that the different corticosteroids are unlikely to be the reason for the different observations regarding COPD patients versus HS. More likely, the high degree of between-individual variation in COPD macrophage corticosteroid sensitivity means that the modest sample sizes used here contributed to observable differences between groups. It is interesting that clinical characteristics, including exacerbation frequency, had no influence on corticosteroid sensitivity in alveolar macrophages (or PBMCs).
In general, the effects of AZD7624 were lower than budesonide in PBMCs and alveolar macrophages, in contrast to the BEC findings. This highlights variation between the effects of different classes of anti-inflammatory drugs according to cell type and suggests that multiple anti-inflammatory drugs are needed in COPD to address different cellular components of inflammation. Differences between groups for the effects of AZD7624 were inconsistent, and we suspect are false-positive results due to small sample sizes.
IL-10 is an important mediator in the resolution of pulmonary inflammation. In IL-10-knockout mice infected with Streptococcus pneumoniae, there was increased mortality, higher expression of proinflammatory cytokines, and increased recruitment of neutrophils into the lungs compared to wild-type littermates.24 Furthermore, IL-10-knockout mice exposed to cigarette smoke demonstrated significantly higher levels of neutrophils and TNF-α in the BAL fluid compared to wild-type littermates.25 There is a reduction in the levels of IL-10 in COPD blood, and this negatively correlates with severity of disease.26 It is possible that the inhibition of IL-10 production in COPD patients by corticosteroids or p38 MAPK inhibitors may hinder the resolution of inflammation.
The dose of LPS used to stimulate an inflammatory response from alveolar macrophages and PBMCs in this study was 1 μg/mL. This is a concentration widely used in the literature,27–30 and we have previously shown that 1 μg/mL is a submaximal dose in alveolar macrophages.28 Although this may not accurately reflect in vivo levels of endotoxin,31 this is a well-recognized method for studying the anti-inflammatory effects of drugs in vitro. In conclusion, we have observed highly variable corticosteroid responses within the same individual depending on the cell type studied; PBMCs showed the greatest sensitivity and BECs showed the greatest insensitivity. In addition, the p38 MAPK inhibitor AZD7624 had a greater effect on poly I:C-induced cytokine release from BECs than budesonide. These findings demonstrate that a single class of drug cannot successfully suppress the complex inflammatory network that exists in COPD. A one-size-fits-all approach is therefore not only redundant in treating different individuals with COPD32 but also in treating inflammation within the same COPD patient.
Acknowledgment
This work was funded by AstraZeneca.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
DMC and PJ are employees of AstraZeneca, which funded the study. DS has received sponsorship to attend international meetings, honoraria for lecturing or attending advisory boards, and research grants from various pharmaceutical companies including Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, Genentech, GlaxoSmithKline, Glenmark, Johnson and Johnson, Merck, NAPP, Novartis, Pfizer, Skypharma, Takeda, Teva, Therevance, and Verona. The authors report no other conflicts of interest in this work.
References
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Murray LA, Dunmore R, Camelo A, et al. Acute cigarette smoke exposure activates apoptotic and inflammatory programs but a second stimulus is required to induce epithelial to mesenchymal transition in COPD epithelium. Respir Res. 2017;18(1):82. | ||
Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173(10):1114–1121. | ||
Calverley P, Pauwels R, Vestbo J, et al. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2003;361(9356):449–456. | ||
Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356(8):775–789. | ||
Siddiqui SH, Guasconi A, Vestbo J, et al. Blood eosinophils: a biomarker of response to extrafine beclomethasone/formoterol in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192(4):523–525. | ||
Higham A, Booth G, Lea S, Southworth T, Plumb J, Singh D. The effects of corticosteroids on COPD lung macrophages: a pooled analysis. Respir Res. 2015;16:98. | ||
Khorasani N, Baker J, Johnson M, Chung KF, Bhavsar PK. Reversal of corticosteroid insensitivity by p38 MAPK inhibition in peripheral blood mononuclear cells from COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:283–291. | ||
Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320–344. | ||
Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 2004;4(4):372–377. | ||
Moretto N, Bertolini S, Iadicicco C, et al. Cigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cells. Am J Physiol Lung Cell Mol Physiol. 2012;303(10):L929–L938. | ||
Gaffey K, Reynolds S, Plumb J, Kaur M, Singh D. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur Respir J. 2013;42(1):28–41. | ||
Armstrong J, Harbron C, Lea S, et al. Synergistic effects of p38 mitogen-activated protein kinase inhibition with a corticosteroid in alveolar macrophages from patients with chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2011;338(3):732–740. | ||
Higham A, Lea S, Ray D, Singh D. Corticosteroid effects on COPD alveolar macrophages: dependency on cell culture methodology. J Immunol Methods. 2014;405:144–153. | ||
Khalaf RM, Lea SR, Metcalfe HJ, Singh D. Mechanisms of corticosteroid insensitivity in COPD alveolar macrophages exposed to NTHi. Respir Res. 2017;18(1):61. | ||
Heijink I, van Oosterhout A, Kliphuis N, et al. Oxidant-induced corticosteroid unresponsiveness in human bronchial epithelial cells. Thorax. 2014;69(1):5–13. | ||
Kolsum U, Damera G, Pham TH, et al. Pulmonary inflammation in patients with chronic obstructive pulmonary disease with higher blood eosinophil counts. J Allergy Clin Immunol. 2017;140(4):1181.e7–1184.e7. | ||
Ritter M, Mennerich D, Weith A, Seither P. Characterization of Toll-like receptors in primary lung epithelial cells: strong impact of the TLR3 ligand poly(I:C) on the regulation of Toll-like receptors, adaptor proteins and inflammatory response. J Inflamm. 2005;2:16. | ||
Lee FE, Walsh EE, Falsey AR. The effect of steroid use in hospitalized adults with respiratory syncytial virus-related illness. Chest. 2011;140(5):1155–1161. | ||
Meusel TR, Imani F. Viral induction of inflammatory cytokines in human epithelial cells follows a p38 mitogen-activated protein kinase-dependent but NF-κ B-independent pathway. J Immunol. 2003;171(7):3768–3774. | ||
Yuan J, Liu Z, Lim T, et al. CXCL10 inhibits viral replication through recruitment of natural killer cells in coxsackievirus B3-induced myocarditis. Circ Res. 2009;104(5):628–638. | ||
Lokensgard JR, Hu S, Sheng W, et al. Robust expression of TNF-α, IL-1β, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J Neurovirol. 2001;7(3):208–219. | ||
Milara J, Morell A, Ballester B, et al. Roflumilast improves corticosteroid resistance COPD bronchial epithelial cells stimulated with toll like receptor 3 agonist. Respir Res. 2015;16:12. | ||
Penaloza HF, Nieto PA, Munoz-Durango N, et al. Interleukin-10 plays a key role in the modulation of neutrophils recruitment and lung inflammation during infection by Streptococcus pneumoniae. Immunology. 2015;146(1):100–112. | ||
Higaki M, Wada H, Mikura S, et al. Interleukin-10 modulates pulmonary neutrophilic inflammation induced by cigarette smoke exposure. Exp Lung Res. 2015;41(10):525–534. | ||
Wang H, Ying H, Wang S, et al. Imbalance of peripheral blood Th17 and Treg responses in patients with chronic obstructive pulmonary disease. Clin Respir J. 2015;9(3):330–341. | ||
Higham A, Lea S, Plumb J, et al. The role of the liver X receptor in chronic obstructive pulmonary disease. Respir Res. 2013;14:106. | ||
Armstrong J, Sargent C, Singh D. Glucocorticoid sensitivity of lipopolysaccharide-stimulated chronic obstructive pulmonary disease alveolar macrophages. Clin Exp Immunol. 2009;158(1):74–83. | ||
Ding X, Jin S, Tong Y, et al. TLR4 signaling induces TLR3 up-regulation in alveolar macrophages during acute lung injury. Sci Rep. 2017;7:34278. | ||
Isler P, de Rochemonteix BG, Songeon F, Boehringer N, Nicod LP. Interleukin-12 production by human alveolar macrophages is controlled by the autocrine production of interleukin-10. Am J Respir Cell Mol Biol. 1999;20(2):270–278. | ||
McSharry C, Spears M, Chaudhuri R, Cameron EJ, Husi H, Thomson NC. Increased sputum endotoxin levels are associated with an impaired lung function response to oral steroids in asthmatic patients. J Allergy Clin Immunol. 2014;134(5):1068–1075. | ||
Singh D, Roche N, Halpin D, Agusti A, Wedzicha JA, Martinez FJ. Current controversies in the pharmacological treatment of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;194(5):541–549. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.