Back to Journals » Vascular Health and Risk Management » Volume 17
Diagnostic Work-Up of Cardiac Amyloidosis Using Cardiovascular Imaging: Current Standards and Practical Algorithms
Authors Korosoglou G ![]() , Giusca S, André F, aus dem Siepen F, Nunninger P, Kristen AV, Frey N
, Giusca S, André F, aus dem Siepen F, Nunninger P, Kristen AV, Frey N
Received 6 August 2021
Accepted for publication 7 October 2021
Published 23 October 2021 Volume 2021:17 Pages 661—673
DOI https://doi.org/10.2147/VHRM.S295376
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Grigorios Korosoglou,1,2 Sorin Giusca,1,2 Florian André,3,4 Fabian aus dem Siepen,3,4 Peter Nunninger,5 Arnt V Kristen,3,6 Norbert Frey3,4
1GRN Hospital Weinheim, Department of Cardiology, Vascular Medicine and Pneumology, Weinheim, Germany; 2Cardiac Imaging Center Weinheim, Hector Foundation, Weinheim, Germany; 3Department of Cardiology, Pneumology and Angiology, University Hospital Heidelberg, Heidelberg, Germany; 4German Centre for Cardiovascular Research (DZHK), Partner Site Heidelberg, Heidelberg, Germany; 5Radiology and Nuclear Medicine Weinheim, Weinheim, Germany; 6Cardiovascular Center Darmstadt, Darmstadt, Germany
Correspondence: Grigorios Korosoglou
GRN Hospital Weinheim, Department of Cardiology & Vascular Medicine, Roentgenstrasse 1, Weinheim, D-69469, Germany
Tel +49 6201 89 2142
Fax +49 6201 89 2507
Email [email protected]
Abstract: Among non-ischemic cardiomyopathies, cardiac amyloidosis is one of the most common, being caused by extracellular depositions of amyloid fibrils in the myocardium. Two main forms of cardiac amyloidosis are known so far, including 1) light-chain (AL) amyloidosis caused by monoclonal production of light-chains, and 2) transthyretin (ATTR) amyloidosis, caused by dissociation of the transthyretin tetramer into monomers. Both AL and ATTR amyloidosis are progressive diseases with median survival from diagnosis of less than 6 months and 3 to 5 years, respectively, if untreated. In this regard, death occurs in most patients due to cardiac causes, mainly congestive heart failure, which can be prevented due to the presence of effective, life-saving treatment regimens. Therefore, early diagnosis of cardiac amyloidosis is crucial more than ever. However, diagnosis of cardiac amyloidosis may be challenging due to variable clinical manifestations and the perceived rarity of the disease. In this regard, clinical and laboratory reg flags are available, which may help clinicians to raise suspicion of cardiac amyloidosis. In addition, advances in cardiovascular imaging have already revealed a higher prevalence of cardiac amyloidosis in specific populations, so that the diagnosis especially of ATTR amyloidosis has experienced a > 30-fold increase during the past ten years. The goal of our review article is to summarize these findings and provide a practical approach for clinicians on how to use cardiovascular imaging techniques, such as echocardiography, cardiac magnetic resonance, bone scintigraphy and, if required, organ biopsy within predefined diagnostic algorithms for the diagnostic work-up of patients with suspected cardiac amyloidosis. In addition, two clinical cases and practical tips are provided in this context.
Keywords: cardiac amyloidosis, ATTR amyloidosis, AL amyloidosis, echocardiography, cardiac magnetic resonance, bone scintigraphy, myocardial biopsy, specific therapy
Introduction
Non-ischemic cardiomyopathies are a heterogeneous group of heart muscle diseases, which are frequently associated with genetic disorders.1 Among secondary non-ischemic cardiomyopathies, cardiac amyloidosis is one of the most common, being caused by extracellular depositions of amyloid fibrils of different precursor proteins in the myocardium.2 Two main forms of cardiac amyloidosis have been described so far, including (i) light-chain (AL) amyloidosis where plasma cell dyscrasia results in monoclonal production of light-chains, and (ii) transthyretin (ATTR) amyloidosis, caused by dissociation of the transthyretin tetramer into monomers. Transthyretin is a transport protein for thyroxine and retinol and is mainly produced in the liver.3 ATTR amyloidosis, encompasses 2 sub-forms, including (i) the familial or hereditary variant, due to pathologic variants in the transthyretin gene (ATTRv) and (ii) the sporadic, non-genetic, earlier referred to as the senile systemic form, caused by aggregation of wild-type transthyretin (ATTRwt).4 More than 130 pathologic mutations leading to instability of the tetramer have been reported for ATTR amyloidosis, resulting in amyloid deposition in the heart, the nerves, and other tissues, leading to variable phenotypes and clinical presentations.4,5
Both AL and ATTR amyloidosis are in part rapidly progressive disorders with median survival from diagnosis of less than 6 months and 3 to 5 years, respectively, if untreated.4,6 Amyloid deposition in the myocardium results in heart failure symptoms, such as dyspnea, fatigue, symptomatic hypotension, and syncope. In addition, deposition of amyloid in the conduction system can cause bundle-branch block, AV-blockage, or atrial fibrillation.4 The early diagnosis of cardiac amyloidosis is crucial since patients both with AL and ATTR amyloidosis nowadays profit from specific therapies. Especially with ATTR amyloidosis, tafamidis was recently shown to effectively reduce all-cause mortality and hospitalizations due to heart failure and decline of 6-minute walk distance and quality of life compared with placebo,7 whereas AL amyloidosis patients profit from specific chemotherapies. Tafamidis is a small molecule, which binds to transthyretin with high affinity and selectivity, thus inhibiting the dissociation of tetramers into monomers.8,9 In addition, tafamidis was also shown to inhibit the progression of neurologic impairments with mild (stage 1) hereditary ATTR polyneuropathy.10
Even wild-type transthyretin can misfold into the amyloid configuration, whereas the reason for loss of stability of the tetramer is unclear. ATTRwt amyloidosis is a late-onset disease, predominantly manifested in male patients over 60 years.4 The prevalence of ATTRwt amyloidosis is still a matter of debate but seems to be higher than earlier anticipated. Thus, studies aiming at the diagnosis of ATTRwt amyloidosis without systematically performing myocardial biopsies, have reported a prevalence of 13% in patients hospitalized for heart failure with preserved ejection fraction (HFpEF).11 A high prevalence of undiagnosed patients with ATTRwt amyloidosis may be related to the limited effectiveness of pharmacotherapy in earlier HFpEF trials.12 In addition, almost every sixth patient (16%) who is considered for transcatheter aortic valve replacement (TAVR) due to severe aortic stenosis has been shown to have concomitant ATTRwt amyloidosis.13 In surgically removed heart valves of elderly (more than 80 years) individuals, on the other hand, ATTRwt amyloid deposits were reported in every fourth (25%) patient.14 Furthermore, 5% of patients with increased left ventricular (LV) wall thickness and initial diagnosis of hypertrophic cardiomyopathy exhibit familial ATTR amyloidosis (ATTRv).15 In the same direction, in patients referred with presumed hypertrophic cardiomyopathy, cardiac amyloidosis was the underlying entity with a prevalence of (9%), the latter increasing with age.16 Thus, especially in patients over 80 years, ATTR amyloidosis was diagnosed in every fourth (26%) patient with presumed hypertrophic cardiomyopathy.
Due to the presumably high number of patients with ATTR amyloidosis, which are undiagnosed or initially classified as HFpEF, hypertrophic cardiomyopathy or aortic stenosis and the presence of specific therapies, which are more effective in the early stages of the disease (clinical heart failure NYHA class II), it becomes clear that early diagnosis of ATTR amyloidosis represents an unmet clinical need. However, diagnosis of ATTR amyloidosis may be challenging due to variable clinical manifestations and the perceived rarity of the disease. To date, advances in cardiovascular imaging have already revealed a higher prevalence of cardiac amyloid deposits in specific populations, so that the diagnosis especially of ATTR amyloidosis has experienced a >30-fold increase during the past ten years in specialized centers.17 The goal of our review is to summarize these findings and provide a practical approach for clinicians on how to use non-invasive imaging techniques, including echocardiography, cardiac magnetic resonance, bone scintigraphy and, if required, organ biopsy for the diagnostic work-up of patients with suspected cardiac amyloidosis.
Diagnosis of Cardiac Amyloidosis
Clinical Red Flags in AL and ATTR Amyloidosis
Renal impairment with albuminuria, gastrointestinal disorders such as weight loss and diarrhea, hepato- and splenomegaly, macroglossia, periorbital bleedings, and subcutaneous nodules, the latter easily assessed by clinical examination are typical red flags in individuals with AL amyloidosis.18
Special attention is also necessary when taking the clinical history in patients with suspected ATTR amyloidosis since extracardiac symptoms and findings may proceed cardiac symptoms for 1–2 decades. Thus, a history of bilateral carpal tunnel syndrome or non-traumatic distal biceps tendon rupture may be present in patients with ATTR amyloidosis, long time before heart failure symptoms.17,19 Lumbar spinal stenosis is also common especially in patients with ATTRwt amyloidosis, due to amyloid infiltration and consecutive thickening of the ligamentum flavum, causing lumbar spinal narrowing.20 In addition, peripheral and autonomic neuropathy can be present in AL, ATTRv, and less frequently in patients with ATTRwt amyloidosis.21 Amyloid neuropathy in ATTRv is characterized by a symmetric, axonal and distal polyneuropathy with beginning in the lower limbs.
ECG and Laboratory Markers
In contrast to myocyte hypertrophy in hypertrophic cardiomyopathy or hypertensive heart disease, in amyloidosis, increased LV-mass is largely attributed to the presence of electrical inactive amyloid, infiltrating the extracellular space of the myocardium.22 Therefore, low voltage on electrocardiogram (ECG) in the presence of moderate or severe hypertrophy is a typical hallmark for the presence of either ATTR or AL amyloidosis. In addition, cardiac troponin, and NTpro-BNP levels may be disproportionately increased with values above 50ng/L for hsTnT and 1500ng/L for NT-proBNP, respectively in the presence of mild or moderate clinical symptoms.
Echocardiography
Conventional Measures and Diastolic Function
Transthoracic echocardiography is usually the initial imaging technique for the evaluation of patients with suspected cardiac amyloidosis. Typical echocardiographic findings are increased left ventricular (LV) wall thickness over 12mm, accompanied by additional thickening of the right ventricle (RV), valves, and interatrial septum, and generally small pericardial effusion (reviewed in).23,24 Furthermore, bi-atrial dilatation is frequently observed in the presence of severe diastolic dysfunction.25 The characteristic speckled appearance of the myocardium, on the other hand, referred to as “granular sparkling”, has been described in patients with cardiac amyloidosis and previously deemed as a key factor in establishing the diagnosis.26 However, granular sparkling can also be present in other causes of LV hypertrophy, yielding low sensitivity for the identification of cardiac amyloidosis than previously anticipated.27,28 In addition, tissue harmonic imaging and image processing techniques may alter the myocardial echogenicity and, thus, impair the diagnostic value of this feature.
Generally, LV diastolic abnormalities occur already in the early stages of cardiac amyloidosis, whereas LV-ejection fraction, usually remains normal or mildly reduced until the late stages of the disease, despite diminished longitudinal deformation of the myocardium.29,30 Despite preserved ejection fraction, however, cardiac output is low due to decreased ventricular volumes.31,32 In this regard, myocardial contraction fraction, the ratio of (LV) stroke volume to myocardial volume measured by M-mode echocardiography was predictive of mortality in patients with ATTRv, ATTRwt, and AL amyloidosis.33,34
The Role of Echocardiographic Myocardial Deformation (Strain)
Changes of the myocardium in dimensions and shape during contraction and relaxation can be characterized by the assessment of myocardial “strain”. Strain is a measure of regional or global deformation, which originates from physics and can be applied to the heart to measure regional shortening, thickening, and lengthening of the myocardium. Strain can be quantitatively assessed by echocardiography using speckle tracking imaging and has demonstrated a characteristic pattern with markedly reduced strain in mid and basal and relatively preserved strain in apical segments. This pattern is referred to as “apical sparing” and is well suited for the differentiation of such patients from other causes of LV hypertrophy.35–37 The apical sparing pattern is easily recognizable on polar maps of myocardial strain and is observed both in patients with ATTR and AL cardiac amyloidosis. Even though absolute values of apical strain are lower in patients with ATTR compared to AL amyloidosis, this difference probably does not allow for the accurate differentiation of cardiac amyloidosis subtypes.35,38
In addition, myocardial strain was shown to bear prognostic implications in amyloidosis in several previous clinical studies, exhibiting incremental value over clinical and biochemical markers for the risk stratification of patients with AL amyloidosis.39–41 However, limited data are available so far for the prognostic value of echocardiographic derived strain in ATTR amyloidosis. Red flags in cardiac amyloidosis in terms of clinical data, ECG, and echocardiographic imaging markers can be depicted in Table 1.
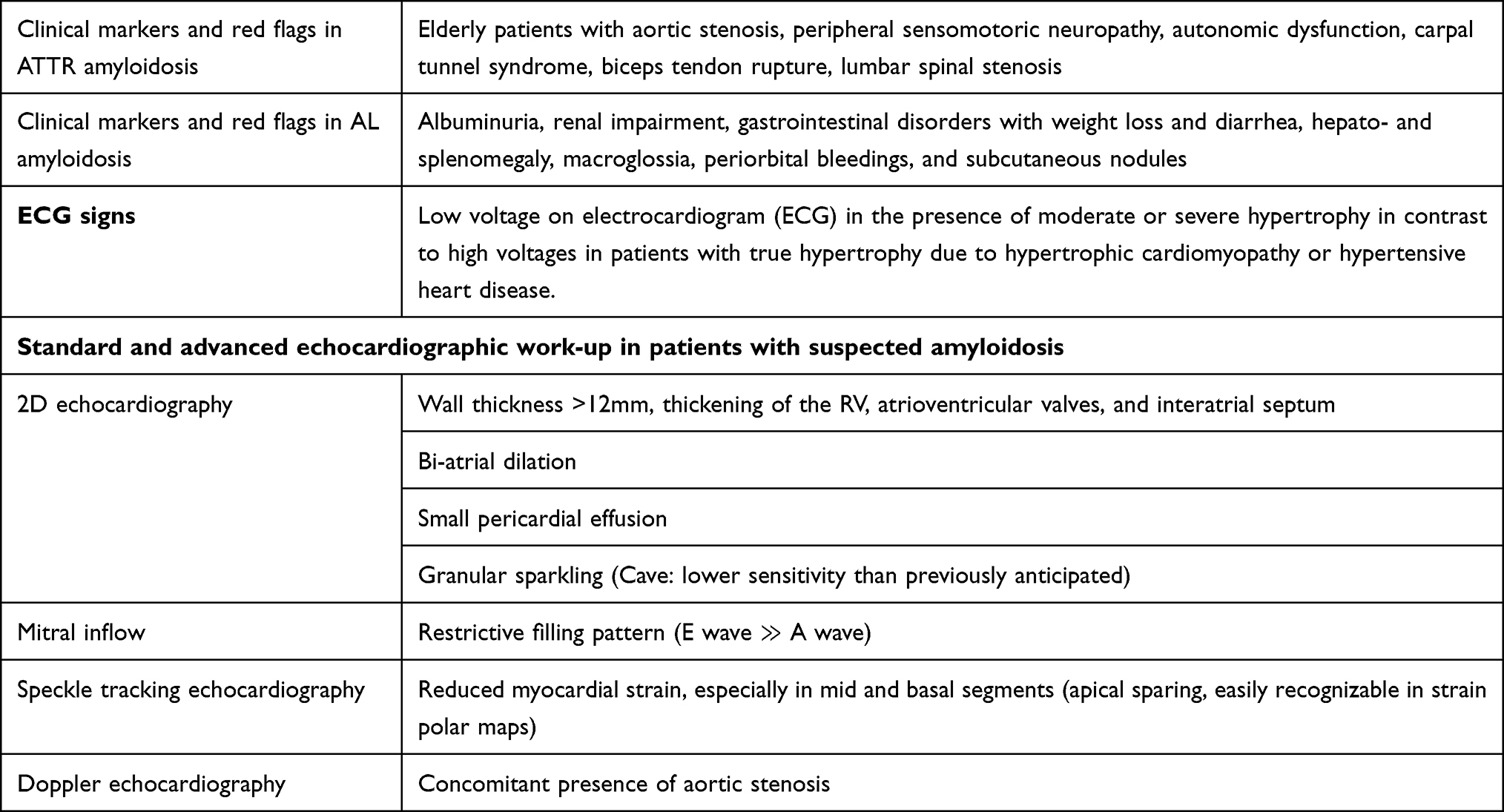 |
Table 1 Clinical, ECG and Echocardiographic Red Flags in Patients with Cardiac Amyloidosis |
Cardiac Magnetic Resonance (CMR)
Conventional Cine Measures by CMR
CMR allows for non-invasive imaging of cardiac structures with high spatial resolution and intrinsic blood-to-tissue contrast. The versatility of CMR can provide the assessment of myocardial function, perfusion, tissue characterization by late gadolinium enhancement (LGE) and mapping techniques and, if required, myocardial deformation using “strain” analyses within a single examination, independent of the patients’ acoustic windows and without radiation exposure.42 Morphological and functional alterations are comparable with echocardiography findings. In addition, CMR studies highlighted differences in terms of asymmetrical septal hypertrophy in ATTR amyloidosis, mimicking hypertrophic cardiomyopathy versus concentric hypertrophy seen in most patients with AL amyloidosis.43 Of note, tissue characterization techniques, which are highly sensitive for amyloidosis, like late Gadolinium Enhancement and T1 mapping can be implemented in a single CMR examination.
Late Gadolinium Enhancement
Gadolinium is an extracellular contrast agent. Therefore, gadolinium distributes preferentially in areas of increased extracellular volume (ECV), as in diseased myocardium with an expanded extracellular space in patients with cardiac amyloidosis, whereas the contrast agent rapidly washes out in healthy myocardium. LGE images are typically acquired approximately 10 min after contrast agent administration. LGE allows for visual and, if required, quantitative assessment of the myocardial amyloid burden, the latter albeit being challenging in cases of diffuse global LGE with progressed disease.
Several studies previously demonstrated the presence of LGE in patients with cardiac amyloidosis (44–50 reviewed in51) LGE patterns vary from patchy or diffuse to more specific patterns, such as global subendocardial or transmural, the latter associated with technical difficulties in terms of nulling, due to the absence of “healthy” myocardial tissue. Transmural LGE is associated with progressed diseased and adverse clinical outcomes in both, AL and ATTR amyloidosis.48 The sensitivity and specificity of LGE versus endomyocardial biopsy for the diagnosis of cardiac amyloidosis were reported to be 86% and 92%, respectively, based on a recent meta-analysis.51 Importantly, the technical approach to LGE imaging with amyloidosis needs to involve the use of phase-sensitive inversion recovery (PSIR) pulse sequences, which exhibit incremental value to conventional LGE acquisitions since they can address the nulling problems seen in cardiac amyloidosis especially in cases of global transmural affection of the myocardium.48 In addition, PSIR pulse sequences were shown to provide valuable prognostic information.48
A smaller number of clinical studies aimed at differentiating ATTR from AL amyloidosis using CMR and particularly LGE patterns. In this regard, transmural left ventricular LGE and LGE of the free RV wall and the atria was found to a larger extent in patients with ATTR versus AL amyloidosis (90% of ATTR patients demonstrated transmural LGE compared with 37% of AL patients).52 Based on these findings, an LGE scoring system was developed, which might help to differentiate ATTR from AL amyloidosis with a sensitivity and specificity of 87% and 96%, respectively. In the same direction, transmural LGE was present in more patients with ATTR versus AL amyloidosis (63% versus 27%),48 whereas the LGE contrast intensity, left atrial, RV free wall, and inter-atrial LGE was also more pronounced in ATTR versus AL amyloidosis in another study.53 In addition, consideration of light chain analysis and serum and urine protein immunofixation can further enhance the ability of CMR to differentiate between the 2 underlying pathologies ATTR and AL amyloidosis.54 Although these findings appear promising for the differentiation of the 2 main cardiac amyloidosis subtypes, larger prospective trials are warranted in this context.
Native T1 Mapping and Extracellular Volume (ECV)
Native T1
Considering the concomitant renal impairment especially in AL amyloidosis patients, a non-contrast CMR scan would be preferable in such patients, when the administration of gadolinium-based contrast agents is problematic. In this regard, measures of the myocardial T1 relaxation with non-contrast T1 mapping sequences have been used for the detection and quantification of interstitial expansion due to fibrosis.55 In cardiac amyloidosis, native T1 mapping was shown to exhibit significantly higher values in patients than in control subjects.56 In the same direction, T1 elevations were noted in AL and ATTR amyloidosis patients, allowing for differentiation between both subtypes of amyloidosis versus hypertrophic cardiomyopathy with high accuracy.57 In addition, native T1 values were shown to be related to functional echocardiographic markers of systolic and diastolic LV function and to be significantly increased even in asymptomatic amyloidosis patients, thus being potentially more sensitive than LGE.58 Furthermore, native T1 mapping easily enabled the differentiation between cardiac amyloidosis and the Anderson Fabry disease due to diametrically opposite T1 values, with elevated T1 in amyloidosis versus reduced T1 due to fat infiltration with the Fabry disease.59
Recently, 2 meta-analyses underscored the value of T1 mapping for the diagnostic classification and risk stratification of patients with cardiac amyloidosis.60,61 Based on the pooled values of native T1 in cardiac amyloidosis and control subjects, thresholds suggesting the absence, indeterminate, or presence of amyloidosis were <994 msec, 994–1073 msec, and >1073 msec, respectively, at 1.5T.60 Interestingly, native T1 showed similar diagnostic and prognostic value as LGE, without requiring contrast administration.61
Despite these promising data, some limitations of native T1 mapping need to be mentioned. Thus, heterogeneity of “normal” T1 values of the myocardium has been reported, which is attributed to differences in the sequences used for mapping, especially in terms of the applied flip angles, different field strengths (1.5 versus 3.0T, the latter exhibiting longer T values by 100–200ms) and difficulties with the precise identification between myocardial and fibrotic tissue.62 These discrepancies have been considered by recent guidelines in this field, which recommend each center to develop reference ranges with control subjects before performing clinical T1 scans.63
Extracellular Volume (ECV)
By measuring the T1 value of the myocardium before and after gadolinium administration, quantification of the myocardial ECV can be achieved. This parameter reflects the fraction of myocardium composed by the extracellular space, where the amyloid deposits accumulate in patients with cardiac amyloidosis. ECV requires the administration of gadolinium-based contrast agents but is considered less susceptible to technical issues, compared with the pulse sequences used for native T1 mapping.62,64 Like native T1, ECV has been extensively validated in cardiac amyloidosis,65 including studies where ECV was serially studied during treatment with novel pharmacologic agents both in AL and ATTR amyloidosis.66,67 Due to its high reproducibility and quantitative nature, the use of ECV was proposed for serial studies, monitoring the response to therapies in patients with cardiac amyloidosis. In addition, ECV has been validated against histology, exhibiting close correlation to histologic amyloid burden68,69 and was independently associated with patients’ outcomes.43 In ATTR amyloidosis, ECV was, in contrast to native T1 values, independently associated with mortality after adjustment for age, biochemical, functional markers, and LGE. This highlights the robustness of this marker,65 which was recently confirmed in a meta-analysis by a systematic comparison of ECV versus native T1 in AL and ATTR amyloidosis patient cohorts.61 Summarizing the relevance of native T1 and ECV, both markers bear strong diagnostic and prognostic implications and need to be considered together with clinical, laboratory, and other imaging parameters during the diagnostic work-up of patients with suspected cardiac amyloidosis.60 While T1 does not require the administration of contrast agents, ECV seems to be more robust in terms of both reproducibility and risk stratification for patients with cardiac amyloidosis. Besides CMR, cardiac computed tomography (CCT), which is routinely performed prior to TAVR, can also be used to the assessment of ECV.70 CCT based ECV measures can reliably detect interstitial expansion due to amyloid deposition, compared to bone scintigraphy, so that CCT may be an alternative to CMR in such patients who are frequently affected by ATTRwt amyloidosis and undergo contrast enhanced CCT imaging by clinical indication prior to TAVR.
The Role of CMR-Based Myocardial Deformation (Strain)
We and others have previously demonstrated the role of strain‐encoded magnetic resonance (MR) (SENC), an advanced tagging technique, which provides both, color-coded visual and quantitative assessment of myocardial strain, for the diagnostic classification and risk stratification of several cardiac disorders (71 reviewed in72). Recently, we demonstrated the value of myocardial strain measured by fast-SENC for the risk stratification of patients with heart failure due to non-ischemic cardiomyopathies.73 Likewise, fast-SENC aided the diagnostic classification of patients with different forms of LV hypertrophy, effectively differentiating patients with cardiac amyloidosis.74 Myocardial strain derived from the fast-SENC successfully differentiated between patients with hypertrophic cardiomyopathy and amyloidosis, the latter exhibiting lower global strain values. In addition, patients with amyloidosis showed a base-to-apex strain gradient, which was not the case in hypertrophic cardiomyopathy and hypertensive heart disease.74 This base-to-apex strain gradient, which was initially described by echocardiographic speckle tracking as “apical sparing”35–37 was also recently confirmed using strain analysis by CMR, being associated with less LGE burden in apical compared to mid and basal segments. Thus, relatively preserved strain in apical segments in patients with cardiac amyloidosis may be at least partially explained by the increased burden of amyloid deposition and fibrosis in mid and basal versus apical segments.75,76 Furthermore, circumferential strain by tagged CMR showed higher sensitivity than LGE for the early detection of cardiac amyloidosis.77
Overall, developments of CMR sequences and software analysis can aid the comprehensive evaluation of patients with suspected cardiac amyloidosis, using quantification of LV mass and volumes, symmetricity of LV-hypertrophy, LGE, native T1 mapping, ECV, and myocardial strain within a single examination. Red flags for cardiac amyloidosis for CMR imaging parameters are summarized in Table 2. Since the prevalence of amyloidosis may be higher than previously anticipated, varying between 5% and 15% in specific populations, CMR emerges as a first-line test, given its diagnostic versatility to detect cardiac amyloidosis, quantify the burden of amyloid deposition and identify other relevant myocardial disorders when amyloidosis is not present.
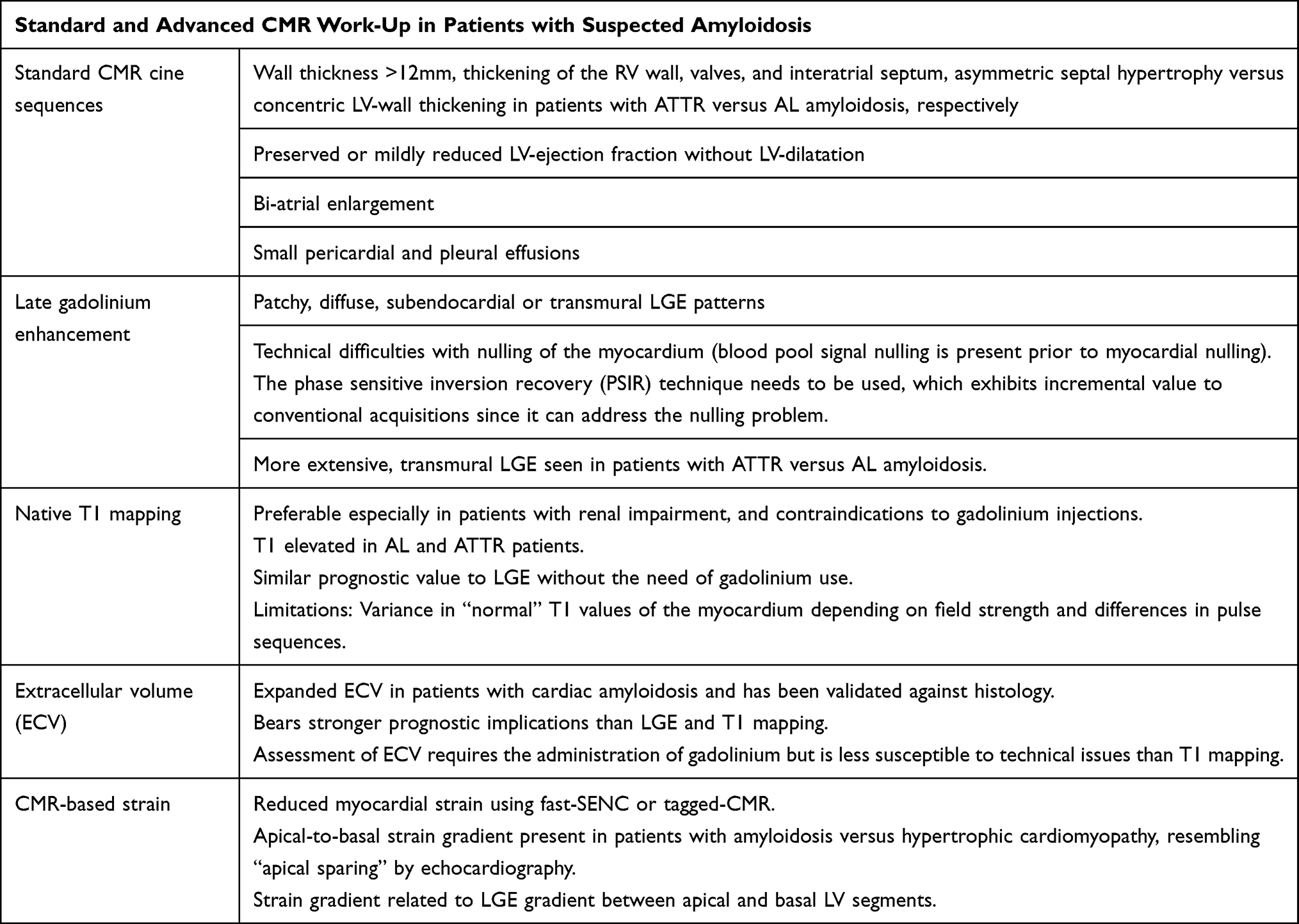 |
Table 2 CMR Red Flags in Patients with Cardiac Amyloidosis |
Bone Tracer Myocardial Scintigraphy
The ability of nuclear scintigraphy imaging to depict ATTR amyloid deposition in the heart with high sensitivity and specificity after exclusion of monoclonal gammopathy has revolutionized the diagnostic work-up of cardiac amyloidosis during the last decade, in many cases obviating the need for myocardial biopsy. Among radiotracers, 99mtechnetium-phosphate derivatives (99mTc-3,3- diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD), and Tc-pyrophosphate (Tc-PYP)) are the most widely used. Although the exact mechanism of tracer uptake into the heart is not completely understood, binding of the tracers seems to be related to a high calcium content with cardiac amyloid deposits. Calcium content is presumably higher in patients with ATTR compared to AL amyloidosis, which results in a much stronger uptake of pyrophosphate-based tracers in patients with ATTR amyloidosis, in contrast to the AL subtype.78,79 In hereditary TTR amyloidosis, amyloid fibrils are composed of either a mixture of full-length and TTR fragments (Type A) or only full length TTR (Type B). It should be noted that bone scintigraphy is false negative in all patients with Type B fibres80,81 and therefore ATTRv patients with some mutations (eg p.Val50Met late onset or p.Phe84Leu) cannot be diagnosed using scintigraphy. Furthermore, bone scintigraphy can provide false positive results in a very few cases, for example after intoxication with hydroxychloroquine.82 To avoid false positive results due to blood pooling, SPECT imaging should be used to confirm myocardial uptake.83 Limited data are available so far, comparing 99mTc-DPD versus 99mTc-PYP.
99mTc-DPD was the first radiotracer demonstrating the ability to distinguish AL from ATTR amyloidosis in previous studies.79,84 In these studies, 99mTc-DPD exhibited high accuracy for the differentiation between amyloidosis subtypes, whereas uptake was noticed even in patients without evident echocardiographic involvement, thus highlighting the high sensitivity of this technique and its ability to early diagnose the disease. In addition, the extent of 99mTc-DPD uptake was related to poorer clinical outcomes.84 In the subsequent study, performed by the same research group and including a larger number of AL amyloidosis patients, 99mTc-DPD uptake remained useful for the differentiation of amyloidosis subtypes, albeit exhibiting lower diagnostic yield due to mild tracer uptake in approximately one third of patients with AL amyloidosis. When tracer uptake was assessed using the semiquantitative Perugini visual score of 0, 1, 2, or 3 which represents no, mild (less than in bones), moderate (similarly high as in bones), or strong (higher than in bones) uptake, respectively,79 the positive and negative predictive values for a diagnosis of ATTR were 88 and 100%, respectively for a score of ≥2.85 These findings were confirmed within a multicenter study, where bone scintigraphy allowed for the non-invasive diagnosis of ATTR amyloidosis with a sensitivity of >99% but simultaneously relatively low specificity of 68% due to a low level of uptake in some AL amyloidosis patients. However, when only moderate or strong uptake (Perugini Score 2 or 3) was considered, the specificity increased to 97%. In addition, excellent positive predictive value, and specificity of 100% were achieved in patients with Perugini Score 2 or 3 and absence of a monoclonal protein in serum and urine.86 Like “apical sparing” in echocardiography and CMR, increased 99mTc-HMDP uptake was also noticed in mid and basal versus apical segments with ATTR amyloidosis patients.87
Another tracer used for bone scintigraphy, 99mTc-PYP, which in contrast to 99mTc-DPD is available in the United States, exhibits similar sensitivity and specificity to 99mTc-DPD for the diagnosis of ATTR amyloidosis. In this regard, quantitative analysis with 99mTc-PYP uptake, revealed high sensitivity and specificity of 97% and 100%, respectively for the identification of ATTR amyloidosis and was associated with all-cause mortality.88,89 In addition, apical sparing was also described with 99mTc-PYP, whereas pronounced apical sparing was associated with poorer outcomes in patients with ATTR amyloidosis.90
Beyond bone scintigraphy tracers, which are in the meanwhile clinically established for the diagnostic work-up of patients with suspected ATTR amyloidosis, increasing evidence is present that amyloid specific positron emission tomography (PET) tracers, including 18-F florbetapir and the 11-C Pittsburgh B compound, which may be able to specifically bind to AL amyloid.91,92
Comprehensive Algorithms Considering Clinical Data, Cardiovascular Imaging, and Myocardial Biopsy
In case of suspected cardiac amyloidosis based on clinical or echocardiographic findings, attention needs to be given to the so-called “red flags”, including systemic, multi-organ manifestations in patients with AL amyloidosis and carpal tunnel syndrome, lumbar spinal stenosis, peripheral sensomotoric and/or autonomic neuropathy, and non-traumatic distal biceps tendon rupture, which may proceed heart failure symptoms more than 10 years in patients with ATTR amyloidosis. In addition, ECG may show paradoxically low voltage in the presence of marked LV hypertrophy. Furthermore, care needs to be taken with standard echocardiography, giving special attention to (i) the presence of septal, asymmetric versus concentric hypertrophy in patients with ATTR versus AL amyloidosis, (ii) impaired diastolic function exhibiting restrictive filling patterns, (iii) thickening not only of the LV but also the RV and atrial wall including the interatrial septum, and atrioventricular valves and (iv) regional differences in myocardial strain reduction, resulting in “apical sparing”. In case of clinical suspicion and echocardiographic indices of amyloidosis, laboratory investigation including biomarkers like troponin and NT-proBNP, as well as CMR need to be the next steps in the diagnostic algorithm. With CMR, attention needs to be given to morphologic and functional parameters, as mentioned above for echocardiography. The assessment of LGE extent and patterns allows for the detection of amyloid deposits and new values as T1 mapping, ECV, and myocardial strain may provide additional valuable information. Furthermore, patients need to undergo screening for monoclonal gammopathy in blood and urine samples including measurement of free light chains and immunofixation. If monoclonal gammopathy is detected, subcutaneous abdominal tissue biopsy provides high diagnostic sensitivity for AL amyloidosis.93 In cases where monoclonal gammopathy is not detected, bone scintigraphy, using 99mTc-DPD or 99mTc -PYP can confirm the diagnosis of ATTR amyloidosis. In case of moderate or strong tracer uptake (Perugini Score 2 or 3), an endomyocardial biopsy is not required due to the high specificity and positive predictive value, allowing for the initiation of specific pharmacologic treatment. In case of equivocal findings, such as absent monoclonal gammopathy with absent or mild tracer uptake by bone scintigraphy, an endomyocardial biopsy may be however necessary. If ATTR amyloidosis is confirmed, further genetic testing is needed to confirm or exclude a variant in the TTR gene. The widespread adoption of CMR and bone scintigraphy has many-fold increased the diagnosis of ATTR amyloidosis, consequently enabling early treatment with specific and potentially life-saving therapies.7 A diagnostic algorithm based on the above considerations is proposed in Figure 1. Based on this algorithm, a typical patient case is described below.
 |
Figure 1 With clinical suspicion and echocardiographic indices of cardiac amyloidosis, laboratory markers and CMR are usually the next diagnostic steps. In addition, patients need to undergo screening for elevated free light chains and monoclonal gammopathy. If monoclonal gammopathy and/or elevated free light chains with abnormal kappa/lambda ratio are detected, subcutaneous abdominal tissue biopsy can provide a high diagnostic sensitivity for AL amyloidosis. In case of a negative biopsy of abdominal fat, a biopsy of another organ (salivary glands, rectal tissue, or endomyocardial biopsy) can be useful to confirm the diagnosis, increasing the sensitivity. If monoclonal gammopathy is not detected and free light chains are not elevated, AL amyloidosis can be excluded and bone scintigraphy with strong tracer uptake can confirm the diagnosis of ATTR amyloidosis. In case of equivocal findings however, endomyocardial biopsy may be however necessary. |
Patient Case and Practical Issues
An 81-year-old male patient presented with atypical angina and exertional dyspnea NYHA class II. He had a moderate atherogenic risk profile, including arterial hypertension and hyperlipidemia and had a prior coronary intervention with stent implantation of the LAD 3 years ago. External echocardiographic work-up had been performed, with echocardiographic images considering the presence of hypertrophic cardiomyopathy due to asymmetric LV-hypertrophy. In addition, carpal tunnel syndrome was diagnosed 7 years ago, and the patient had undergone surgical repair at that time. In domo echocardiography confirmed the presence of septal asymmetric hypertrophy (diastolic and systolic echocardiographic images of the long axis and 4-chamber view shown in Figure 2A–D) with preserved LV-function and ejection fraction of 55%. In addition, strain polar maps showed reduced strain values in mid- and basal LV segments with apical sparing (Figure 2K). Laboratory markers showed increased high-sensitivity troponin T of 53ng/L, creatinine of 1.4mg/dl, GFR of 38 mL/min/1.73m2 and NT-pro BNP of 3500ng/L. Further diagnostic work-up using CMR was deemed appropriate, where asymmetric LV hypertrophy was confirmed in the 4- and 3-chamber view (diastolic and systolic CMR images shown in Figure 2E–H. In addition, with LGE images transmural enhancement of the LV, RV, the atria, and the interatrial septum was present (Figure 2I and J). Subsequently, serum and urine immunofixation showed the absence of monoclonal free light chains, and suspicion of ATTR amyloidosis was confirmed using bone scintigraphy, which indeed showed strong tracer uptake in transversal and coronal images (Figure 2L). Molecular genetic testing excluded a variant of the TTR gene. Thus, ATTRwt amyloidosis was diagnosed and pharmacologic treatment with 61 mg tafamidis per day was initiated. Clinical improvement was observed already after 3 months of specific pharmacologic therapy regarding the patients’ heart failure symptoms.
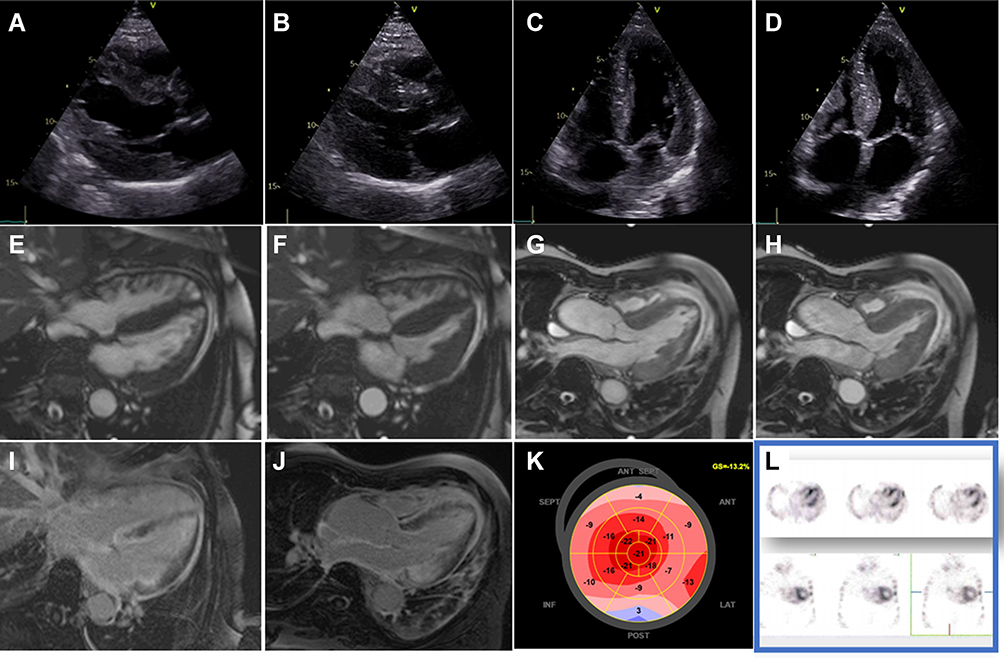 |
Figure 2 Echocardiography shows the presence of septal asymmetric hypertrophy (A–D) with preserved ejection fraction. In addition, strain polar maps show apical sparing (K). Further diagnostic work-up using CMR confirmed asymmetric LV hypertrophy (E–H) and showed transmural LGE of the LV, RV, the atria, and the interatrial septum (I and J). In addition, serum, and urine immunofixation showed no monoclonal free light chains, and suspicion of ATTR amyloidosis was confirmed using bone scintigraphy, which exhibited strong tracer uptake (L). |
Conclusions
Cardiac amyloidosis is a rapidly progressive disease with a median survival of 3–5 years and approximately 6 months in untreated patients with ATTR and AL amyloidosis, respectively.4,6,17,94 Death occurs in most patients due to cardiac causes, including sudden cardiac death and heart failure,94 which can be prevented to date due to the presence of effective, potentially life-saving treatment regimens for both subtypes of cardiac amyloidosis. Therefore, precise diagnosis of cardiac amyloidosis at the early stages of the disease is nowadays crucial more than ever. In this regard, combined cardiovascular imaging by echocardiography, CMR, bone scintigraphy and, if required, organ biopsy need to be applied within predefined diagnostic algorithms, facilitating (i) early and in most cases non-invasive diagnosis, (ii) quantification of disease burden and (iii) monitoring the effectiveness of pharmacologic treatment.
Abbreviations
AL, light-chain amyloid; BMI, body mass index; CCT, cardiac computed tomography; CMR, cardiovascular magnetic resonance; Fast-SENC, fast strain encoded sequence; GFR, glomerular filtration rate; HCM, hypertrophic cardiomyopathy; LAD, left anterior descending; LGE, late gadolinium enhancement; LV, left ventricle; RV, right ventricle; TAVR, transcatheter aortic valve replacement; TTR, transthyretin; ATTRv, variant transthyretin amyloid; ATTRwt, wild-type transthyretin amyloid.
Ethics and Consent
Written informed consent was provided for publishing the case details and all accompanying images. No institutional approval was required to publish the case details.
Disclosure
The authors report no conflicts of interest for this work.
References
1. McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res. 2017;121(7):722–730. doi:10.1161/CIRCRESAHA.117.309711
2. Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–1816.
3. Glenner GG, Terry W, Harada M, Isersky C, Page D. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172(3988):1150–1151. doi:10.1126/science.172.3988.1150
4. Ruberg FL, Berk JL. Transthyretin (TTR) Cardiac Amyloidosis. Circulation. 2012;126(10):1286–1300. doi:10.1161/CIRCULATIONAHA.111.078915
5. Maurer MS, Hanna M, Grogan M, et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161–172. doi:10.1016/j.jacc.2016.03.596
6. Merlini G, Palladini G. Light chain amyloidosis: the heart of the problem. Haematologica. 2013;98(10):1492–1495. doi:10.3324/haematol.2013.094482
7. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
8. Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163–178. doi:10.1007/s10741-014-9462-7
9. Hammarström P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293(5539):2459–2462. doi:10.1126/science.1062245
10. Coelho T, Merlini G, Bulawa CE, et al. Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther. 2016;5(1):1–25. doi:10.1007/s40120-016-0040-x
11. González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–2594. doi:10.1093/eurheartj/ehv338
12. Oghina S, Bougouin W, Bézard M, et al. The Impact of Patients With Cardiac Amyloidosis in HFpEF Trials. JACC Heart Fail. 2021;9(3):169–178. doi:10.1016/j.jchf.2020.12.005
13. Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38(38):2879–2887. doi:10.1093/eurheartj/ehx350
14. Kristen AV, Schnabel PA, Winter B, et al. High prevalence of amyloid in 150 surgically removed heart valves–a comparison of histological and clinical data reveals a correlation to atheroinflammatory conditions. Cardiovasc Pathol off J Soc Cardiovasc Pathol. 2010;19(4):228–235. doi:10.1016/j.carpath.2009.04.005
15. Damy T, Costes B, Hagège AA, et al. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J. 2016;37(23):1826–1834. doi:10.1093/eurheartj/ehv583
16. Maurizi N, Rella V, Fumagalli C, et al. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int J Cardiol. 2020;300:191–195. doi:10.1016/j.ijcard.2019.07.051
17. Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799–2806. doi:10.1093/eurheartj/ehx589
18. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol. 2005;79(4):319–328. doi:10.1002/ajh.20381
19. Nijst P, Tang WW. Recent advances in the diagnosis and management of amyloid cardiomyopathy. Fac Rev. 2021;10:31. doi:10.12703/r/10-31
20. Yanagisawa A, Ueda M, Sueyoshi T, et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol off J U S Can Acad Pathol Inc. 2015;28(2):201–207.
21. Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28(1):10–21. doi:10.1016/j.tcm.2017.07.004
22. Sperry BW, Vranian MN, Hachamovitch R, et al. Are classic predictors of voltage valid in cardiac amyloidosis? A contemporary analysis of electrocardiographic findings. Int J Cardiol. 2016;214:477–481. doi:10.1016/j.ijcard.2016.04.030
23. Cacciapuoti F. The role of echocardiography in the non-invasive diagnosis of cardiac amyloidosis. J Echocardiogr. 2015;13(3):84–89. doi:10.1007/s12574-015-0249-1
24. Mohty D, Damy T, Cosnay P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528–540. doi:10.1016/j.acvd.2013.06.051
25. Rubinshtein R, Glockner JF, Feng D, et al. Comparison of magnetic resonance imaging versus Doppler echocardiography for the evaluation of left ventricular diastolic function in patients with cardiac amyloidosis. Am J Cardiol. 2009;103(5):718–723. doi:10.1016/j.amjcard.2008.10.039
26. Siqueira-Filho AG, Cunha CL, Tajik AJ, Seward JB, Schattenberg TT, Giuliani ER. M-mode and two-dimensional echocardiographic features in cardiac amyloidosis. Circulation. 1981;63(1):188–196. doi:10.1161/01.CIR.63.1.188
27. Picano E, Pinamonti B, Ferdeghini EM, et al. Two-dimensional echocardiography in myocardial amyloidosis. Echocardiogr Mt Kisco N. 1991;8(2):253–259. doi:10.1111/j.1540-8175.1991.tb01395.x
28. Selvanayagam JB, Hawkins PN, Paul B, Myerson SG, Neubauer S. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol. 2007;50(22):2101–2110. doi:10.1016/j.jacc.2007.08.028
29. Klein AL, Hatle LK, Taliercio CP, et al. Serial Doppler echocardiographic follow-up of left ventricular diastolic function in cardiac amyloidosis. J Am Coll Cardiol. 1990;16(5):1135–1141. doi:10.1016/0735-1097(90)90545-Z
30. Swanton RH, Brooksby IA, Davies MJ, Coltart DJ, Jenkins BS, Webb-Peploe MM. Systolic and diastolic ventricular function in cardiac amyloidosis. Studies in six cases diagnosed with endomyocardial biopsy. Am J Cardiol. 1977;39(5):658–664. doi:10.1016/S0002-9149(77)80125-2
31. Mesquita ET, Jorge AJL, Souza CV, Andrade TR. Cardiac Amyloidosis and its New Clinical Phenotype: heart Failure with Preserved Ejection Fraction. Arq Bras Cardiol. 2017;109(1):71–80.
32. Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–1212. doi:10.1161/CIRCULATIONAHA.108.843334
33. Rubin J, Steidley DE, Carlsson M, Ong M-L, Maurer* MS. Myocardial Contraction Fraction by M-Mode Echocardiography is Superior to Ejection Fraction in Predicting Mortality in Transthyretin Amyloidosis. J Card Fail. 2018;24(8):504–511. doi:10.1016/j.cardfail.2018.07.001
34. Tendler A, Helmke S, Teruya S, Alvarez J, Maurer MS. The myocardial contraction fraction is superior to ejection fraction in predicting survival in patients with AL cardiac amyloidosis. Amyloid Int J Exp Clin Investig off J Int Soc Amyloidosis. 2015;22(1):61–66.
35. Phelan D, Collier P, Thavendiranathan P, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart Br Card Soc. 2012;98(19):1442–1448.
36. Liu D, Hu K, Niemann M, et al. Impact of Regional Left Ventricular Function on Outcome for Patients with AL Amyloidosis. PLoS One. 2013;8(3):e56923. doi:10.1371/journal.pone.0056923
37. Liu D, Hu K, Niemann M, et al. Effect of combined systolic and diastolic functional parameter assessment for differentiation of cardiac amyloidosis from other causes of concentric left ventricular hypertrophy. Circ Cardiovasc Imaging. 2013;6(6):1066–1072. doi:10.1161/CIRCIMAGING.113.000683
38. Tuzovic M, Yang EH, Baas AS, et al. Cardiac Amyloidosis: diagnosis and Treatment Strategies. Curr Oncol Rep. 2017;19(7):46. doi:10.1007/s11912-017-0607-4
39. Barros-Gomes S, Williams B, Nhola LF, et al. Prognosis of Light Chain Amyloidosis With Preserved LVEF: added Value of 2D Speckle-Tracking Echocardiography to the Current Prognostic Staging System. JACC Cardiovasc Imaging. 2017;10(4):398–407. doi:10.1016/j.jcmg.2016.04.008
40. Salinaro F, Meier-Ewert HK, Miller EJ, et al. Longitudinal systolic strain, cardiac function improvement, and survival following treatment of light-chain (AL) cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2017;18(9):1057–1064. doi:10.1093/ehjci/jew298
41. Buss SJ, Emami M, Mereles D, et al. Longitudinal left ventricular function for prediction of survival in systemic light-chain amyloidosis: incremental value compared with clinical and biochemical markers. J Am Coll Cardiol. 2012;60(12):1067–1076. doi:10.1016/j.jacc.2012.04.043
42. Korosoglou G, Giusca S, Gitsioudis G, Erbel C, Katus HA. Cardiac magnetic resonance and computed tomography angiography for clinical imaging of stable coronary artery disease. Diagnostic classification and risk stratification. Front Physiol. 2014;5:291. doi:10.3389/fphys.2014.00291
43. Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol. 2017;70(4):466–477. doi:10.1016/j.jacc.2017.05.053
44. Bhatti S, Watts E, Syed F, et al. Clinical and prognostic utility of cardiovascular magnetic resonance imaging in myeloma patients with suspected cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2016;17(9):970–977. doi:10.1093/ehjci/jew101
45. Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186–193. doi:10.1161/01.CIR.0000152819.97857.9D
46. Boynton SJ, Geske JB, Dispenzieri A, et al. LGE Provides Incremental Prognostic Information Over Serum Biomarkers in AL Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2016;9(6):680–686. doi:10.1016/j.jcmg.2015.10.027
47. Minutoli F, Di Bella G, Mazzeo A, et al. Comparison between (99m)Tc-diphosphonate imaging and MRI with late gadolinium enhancement in evaluating cardiac involvement in patients with transthyretin familial amyloid polyneuropathy. AJR Am J Roentgenol. 2013;200(3):W256–265. doi:10.2214/AJR.12.8737
48. Fontana M, Pica S, Reant P, et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation. 2015;132(16):1570–1579. doi:10.1161/CIRCULATIONAHA.115.016567
49. Austin BA, Tang WHW, Rodriguez ER, et al. Delayed hyper-enhancement magnetic resonance imaging provides incremental diagnostic and prognostic utility in suspected cardiac amyloidosis. JACC Cardiovasc Imaging. 2009;2(12):1369–1377. doi:10.1016/j.jcmg.2009.08.008
50. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155–164. doi:10.1016/j.jcmg.2009.09.023
51. Brownrigg J, Lorenzini M, Lumley M, Elliott P. Diagnostic performance of imaging investigations in detecting and differentiating cardiac amyloidosis: a systematic review and meta-analysis. ESC Heart Fail. 2019;6(5):1041–1051. doi:10.1002/ehf2.12511
52. Dungu JN, Valencia O, Pinney JH, et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):133–142. doi:10.1016/j.jcmg.2013.08.015
53. Kristen AV, Aus Dem Siepen F, Scherer K, et al. Comparison of different types of cardiac amyloidosis by cardiac magnetic resonance imaging. Amyloid Int J Exp Clin Investig off J Int Soc Amyloidosis. 2015;22(2):132–141.
54. Chatzantonis G, Bietenbeck M, Elsanhoury A, et al. Diagnostic value of cardiovascular magnetic resonance in comparison to endomyocardial biopsy in cardiac amyloidosis: a multi-centre study. Clin Res Cardiol off J Ger Card Soc. 2021;110(4):555–568. doi:10.1007/s00392-020-01771-1
55. Dass S, Suttie JJ, Piechnik SK, et al. Myocardial tissue characterization using magnetic resonance noncontrast t1 mapping in hypertrophic and dilated cardiomyopathy. Circ Cardiovasc Imaging. 2012;5(6):726–733. doi:10.1161/CIRCIMAGING.112.976738
56. Hosch W, Bock M, Libicher M, et al. MR-relaxometry of myocardial tissue: significant elevation of T1 and T2 relaxation times in cardiac amyloidosis. Invest Radiol. 2007;42(9):636–642. doi:10.1097/RLI.0b013e318059e021
57. Fontana M, Banypersad SM, Treibel TA, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):157–165. doi:10.1016/j.jcmg.2013.10.008
58. Karamitsos TD, Piechnik SK, Banypersad SM, et al. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging. 2013;6(4):488–497. doi:10.1016/j.jcmg.2012.11.013
59. Sado DM, White SK, Piechnik SK, et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging. 2013;6(3):392–398. doi:10.1161/CIRCIMAGING.112.000070
60. Wang TKM, Brizneda MV, Kwon DH, et al. Reference Ranges, Diagnostic and Prognostic Utility of Native T1 Mapping and Extracellular Volume for Cardiac Amyloidosis: a Meta-Analysis. J Magn Reson Imaging JMRI. 2020;2:548.
61. Pan JA, Kerwin MJ, Salerno M. Native T1 Mapping, Extracellular Volume Mapping, and Late Gadolinium Enhancement in Cardiac Amyloidosis: a Meta-Analysis. JACC Cardiovasc Imaging. 2020;13(6):1299–1310. doi:10.1016/j.jcmg.2020.03.010
62. Vo HQ, Marwick TH, Negishi K. Pooled summary of native T1 value and extracellular volume with MOLLI variant sequences in normal subjects and patients with cardiovascular disease. Int J Cardiovasc Imaging. 2020;36(2):325–336. doi:10.1007/s10554-019-01717-3
63. Messroghli DR, Moon JC, Ferreira VM, et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: a consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson off J Soc Cardiovasc Magn Reson. 2017;19(1):75.
64. Mongeon F-P, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovasc Imaging. 2012;5(9):897–907. doi:10.1016/j.jcmg.2012.04.006
65. Martinez-Naharro A, Kotecha T, Norrington K, et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovasc Imaging. 2019;12(5):810–819. doi:10.1016/j.jcmg.2018.02.006
66. Brahmanandam V, McGraw S, Mirza O, Desai AA, Farzaneh-Far A. Regression of cardiac amyloidosis after stem cell transplantation assessed by cardiovascular magnetic resonance imaging. Circulation. 2014;129(22):2326–2328. doi:10.1161/CIRCULATIONAHA.114.009135
67. Fontana M, Martinez-Naharro A, Chacko L, et al. Reduction in CMR Derived Extracellular Volume With Patisiran Indicates Cardiac Amyloid Regression. JACC Cardiovasc Imaging. 2021;14(1):189–199. doi:10.1016/j.jcmg.2020.07.043
68. Banypersad SM, Sado DM, Flett AS, et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging. 2013;6(1):34–39. doi:10.1161/CIRCIMAGING.112.978627
69. Duca F, Kammerlander AA, Panzenböck A, et al. Cardiac Magnetic Resonance T1 Mapping in Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2018;11(12):1924–1926. doi:10.1016/j.jcmg.2018.06.010
70. Scully PR, Patel KP, Saberwal B, et al. Identifying Cardiac Amyloid in Aortic Stenosis: ECV Quantification by CT in TAVR Patients. JACC Cardiovasc Imaging. 2020;13(10):2177–2189. doi:10.1016/j.jcmg.2020.05.029
71. Korosoglou G, Elhmidi Y, Steen H, et al. Prognostic value of high-dose dobutamine stress magnetic resonance imaging in 1493 consecutive patients: assessment of myocardial wall motion and perfusion. J Am Coll Cardiol. 2010;56(15):1225–1234. doi:10.1016/j.jacc.2010.06.020
72. Korosoglou G, Giusca S, Hofmann NP, et al. Strain-encoded magnetic resonance: a method for the assessment of myocardial deformation. ESC Heart Fail. 2019;6(4):584–602. doi:10.1002/ehf2.12442
73. Korosoglou G, Giusca S, Montenbruck M, et al. Fast Strain-Encoded Cardiac Magnetic Resonance for Diagnostic Classification and Risk Stratification of Heart Failure Patients. JACC Cardiovasc Imaging. 2021;14(6):1177–1188. doi:10.1016/j.jcmg.2020.10.024
74. Giusca S, Steen H, Montenbruck M, et al. Multi-parametric assessment of left ventricular hypertrophy using late gadolinium enhancement, T1 mapping and strain-encoded cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2021;23(1):92. doi:10.1186/s12968-021-00775-8
75. Bravo PE, Fujikura K, Kijewski MF, et al. Relative Apical Sparing of Myocardial Longitudinal Strain Is Explained by Regional Differences in Total Amyloid Mass Rather Than the Proportion of Amyloid Deposits. JACC Cardiovasc Imaging. 2019;12(7Part 1):1165–1173. doi:10.1016/j.jcmg.2018.06.016
76. Williams LK, Forero JF, Popovic ZB, et al. Patterns of CMR measured longitudinal strain and its association with late gadolinium enhancement in patients with cardiac amyloidosis and its mimics. J Cardiovasc Magn Reson off J Soc Cardiovasc Magn Reson. 2017;19(1):61.
77. Oda S, Utsunomiya D, Nakaura T, et al. Identification and Assessment of Cardiac Amyloidosis by Myocardial Strain Analysis of Cardiac Magnetic Resonance Imaging. Circ J Off J Jpn Circ Soc. 2017;81(7):1014–1021.
78. Aljaroudi WA, Desai MY, Tang WHW, Phelan D, Cerqueira MD, Jaber WA. Role of imaging in the diagnosis and management of patients with cardiac amyloidosis: state of the art review and focus on emerging nuclear techniques. J Nucl Cardiol off Publ Am Soc Nucl Cardiol. 2014;21(2):271–283.
79. Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076–1084. doi:10.1016/j.jacc.2005.05.073
80. Pilebro B, Suhr OB, Näslund U, Westermark P, Lindqvist P, Sundström T. 99mTc-DPD uptake reflects amyloid fibril composition in hereditary transthyretin amyloidosis. Ups J Med Sci. 2016;121(1):17–24. doi:10.3109/03009734.2015.1122687
81. Musumeci MB, Cappelli F, Russo D, et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020;13(6):1314–1321. doi:10.1016/j.jcmg.2019.10.015
82. Chang ICY, Bois JP, Bois MC, Maleszewski JJ, Johnson GB, Grogan M. Hydroxychloroquine-Mediated Cardiotoxicity With a False-Positive 99mTechnetium-Labeled Pyrophosphate Scan for Transthyretin-Related Cardiac Amyloidosis. Circ Cardiovasc Imaging. 2018;11(1):e007059. doi:10.1161/CIRCIMAGING.117.007059
83. Poterucha TJ, Elias P, Ruberg FL, et al. False Positive 99mTc-Pyrophosphate Scanning Leading to Inappropriate Tafamidis Prescriptions. JACC Cardiovasc Imaging. 2021;14:2042.
84. Rapezzi C, Quarta CC, Guidalotti PL, et al. Role of (99m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011;4(6):659–670. doi:10.1016/j.jcmg.2011.03.016
85. Rapezzi C, Quarta CC, Guidalotti PL, et al. Usefulness and limitations of 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. Eur J Nucl Med Mol Imaging. 2011;38(3):470–478. doi:10.1007/s00259-010-1642-7
86. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133(24):2404–2412. doi:10.1161/CIRCULATIONAHA.116.021612
87. Van Der Gucht A, Cottereau A-S, Abulizi M, et al. Apical sparing pattern of left ventricular myocardial 99mTc-HMDP uptake in patients with transthyretin cardiac amyloidosis. J Nucl Cardiol off Publ Am Soc Nucl Cardiol. 2018;25(6):2072–2079.
88. Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6(2):195–201. doi:10.1161/CIRCIMAGING.112.000132
89. Castano A, Haq M, Narotsky DL, et al. Multicenter Study of Planar Technetium 99m Pyrophosphate Cardiac Imaging: predicting Survival for Patients With ATTR Cardiac Amyloidosis. JAMA Cardiol. 2016;1(8):880–889. doi:10.1001/jamacardio.2016.2839
90. Sperry BW, Vranian MN, Tower-Rader A, et al. Regional Variation in Technetium Pyrophosphate Uptake in Transthyretin Cardiac Amyloidosis and Impact on Mortality. JACC Cardiovasc Imaging. 2018;11(2 Pt 1):234–242. doi:10.1016/j.jcmg.2017.06.020
91. Dorbala S, Vangala D, Semer J, et al. Imaging cardiac amyloidosis: a pilot study using 18F-florbetapir positron emission tomography. Eur J Nucl Med Mol Imaging. 2014;41(9):1652–1662. doi:10.1007/s00259-014-2787-6
92. Lee S-P, Lee ES, Choi H, et al. 11C-Pittsburgh B PET imaging in cardiac amyloidosis. JACC Cardiovasc Imaging. 2015;8(1):50–59. doi:10.1016/j.jcmg.2014.09.018
93. Quarta CC, Gonzalez-Lopez E, Gilbertson JA, et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J. 2017;38(24):1905–1908. doi:10.1093/eurheartj/ehx047
94. Grogan M, Scott CG, Kyle RA, et al. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J Am Coll Cardiol. 2016;68(10):1014–1020. doi:10.1016/j.jacc.2016.06.033
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.