Back to Journals » International Journal of General Medicine » Volume 14
Diagnostic Usefulness of MLPA Techniques for Recurrent Copy Number Variants Detection in Global Developmental Delay/Intellectual Disability
Authors Miclea D, Szucs A, Mirea AM ![]() , Stefan DM, Nazarie F, Bucerzan S, Lazea C
, Stefan DM, Nazarie F, Bucerzan S, Lazea C ![]() , Grama A, Pop TL
, Grama A, Pop TL ![]() , Farcas M
, Farcas M ![]() , Zaharie G
, Zaharie G ![]() , Matyas M, Mager Snr M, Vintan M
, Matyas M, Mager Snr M, Vintan M ![]() , Popp R, Alkhzouz C
, Popp R, Alkhzouz C
Received 21 May 2021
Accepted for publication 15 July 2021
Published 16 August 2021 Volume 2021:14 Pages 4511—4515
DOI https://doi.org/10.2147/IJGM.S320033
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Diana Miclea,1,2 Adriana Szucs,1,2 Andreea Mirea,1,2 Delia-Maria Stefan,1,2 Florina Nazarie,1,2 Simona Bucerzan,2,3 Cecilia Lazea,2,3 Alina Grama,2,3 Tudor Lucian Pop,2,3 Marius Farcas,4 Gabriela Zaharie,3,4 Melinda Matyas,3,4 Monica Mager Snr,2,3 Mihaela Vintan,2,3 Radu Popp,1 Camelia Alkhzouz2,3
1Department of Molecular Sciences, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania; 2Emergency Clinical Hospital for Children, Cluj-Napoca, Romania; 3Department of Mother and Child, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania; 4County Emergency Clinical Hospital, Cluj-Napoca, Romania
Correspondence: Diana Miclea
Department of Molecular Sciences, “Iuliu Hatieganu” University of Medicine and Pharmacy, Pasteur Street, No. 6, Cluj-Napoca, 400349, Romania
Email [email protected]
Background: Genetic testing has become a standardized practice in the diagnosis of patients with global developmental delay/intellectual disability (GDD/ID). The aim of this study is to observe the frequency of recurrent copy number variations (CNVs) in patients diagnosed with GDD/ID, using MLPA technique.
Methods: A total of 501 paediatric patients with GDD/ID were analysed using SALSA MLPA probemix P245 Microdeletion Syndromes-1A, and the technical steps were performed according to the MRC Holland MLPA general protocol.
Results: Twenty-five of 501 patients (5%) were diagnosed with a microdeletion/microduplication syndrome. Amongst them, 7 of 25 (30%) with clinical suggestion have a confirmed diagnosis, for the other cases the clinical features were not evocative for a specific syndrome.
Conclusion: This study showed that in cases with a specific clinical diagnosis the MLPA technique could be a useful alternative, less expensive and more efficient to indicate as first intention of a targeted diagnostic test, as it is the case of Williams syndrome, Prader–Willi syndrome or DiGeorge syndrome.
Keywords: MLPA, global developmental delay, intellectual disability, diagnostic
Background
Genetic testing has become a standardized practice in the diagnosis of patients with global developmental delay/intellectual disability (GDD/ID). Based on its definition from the Diagnostic and Statistical Manual of Mental Disorders 5 (DSM5) ID is characterized by significant intellectual disability and deficits in conceptual, social and practical adaptative functions, occurring in the growth period.1 GDD is confirmed if the values from the standardized tests are lower than two standard deviations below average, in a minimum of two areas of the following: global or fine motility, language and speech, cognition, personal or social field, daily activities.2 Children under the age of five are diagnosed with GDD, and after this age threshold with ID. GDD/ID is regarded as a pathology that belongs to the neurodevelopmental disorders.3
Genetic causes are responsible for over 50% of GDD/ID cases.4–6 Amongst them, the most frequent are numerical chromosomal anomalies, such as Down Syndrome, which is responsible for up to 10% of cases6,7 and structural chromosomal anomalies, frequently microdeletion/microduplication syndromes, known as recurrent copy number variations (CNVs). Anomalies given by microdeletion/microduplication syndromes are small chromosomal alterations, lower than 5 Mb, which cannot be detected using standard karyotyping due to its low-resolution capacity. The frequency of microdeletions/microduplications was found to be around 20% when using high-resolution genomic techniques, such as chromosomal microarray analysis (CMA).5
Karyotyping has been the golden standard in the diagnosis of chromosomal anomalies for a long time. However, in recent years, CMA has become a very useful technique and is used as the first intention diagnostic test when either numerical or structural anomalies are suspected.5 At a genomic level, certain CNVs are more frequent than others and these can be identified as recurrent CNVs. These recurrent CNVs can be observed in around 10% of GDD/ID cases8 and usually occur through non-allelic homologue recombination (NAHR) between region-specific low copy repeats (LCRs) in meiosis.9 These anomalies can be identified using multiplex ligation-dependent probe analysis (MLPA) or quantitative PCR techniques, which imply relatively low costs. It is known that MLPA evaluates fewer target regions than CMA, but these regions are very well chosen, generally according to their frequent involvement in human pathology. Thus, the less frequent regions associated with disorders could be omitted in MLPA but seen in CMA. However, the advantages of using MLPA instead of other high-resolution genomic techniques consist of the omission regarding incidental modifications such as consanguinity, malignancy predisposition, or other changes not related to the disorder.
The aim of this study is to observe the frequency of the recurrent CNVs for patients diagnosed with GDD/ID, using MLPA technique.
Materials and Methods
A total of 501 paediatric patients with GDD/ID were analysed. They were diagnosed at the Emergency Clinical Hospital for Children from Cluj-Napoca, Romania, from 1t October 2017 to 1 April 2019. Each patient was investigated by anamnesis, clinical exam, basic biochemical investigation in GDD/ID (blood count, serum iron and ferritin, alanine transaminase – ALT, aspartate aminotransferase – AST, serum creatinine, blood urea nitrogen – BUN, creatine phosphokinase, uric acid, thyroid stimulating hormone – TSH, free thyroxine – free T4, free triiodothyronine – free T3, blood glucose level), ammonia and lactic acid. Depending on the clinical context, neurological, ophthalmological, ENT consult or others were indicated. Also, for the investigation of any internal malformations, an ultrasound examination was performed when needed.
Ethical Issues
For each patient, an informed consent regarding their participation in the study, was obtained from their parents, as a signed consent form. The study was approved by the Ethics Committee of Emergency Clinical Hospital for Children, Cluj-Napoca. The study was conducted in accordance with the Declaration of Helsinki.
MLPA Genetic Testing
Three milliltres of blood in a vacutainer containing EDTA was collected from each of the patients. DNA extraction was performed using a DNA extraction kit (Wizard Genomic DNA Purification Kit, Promega, Madison, WI, USA). SALSA MLPA probemix P245 Microdeletion Syndromes-1A was used, and the technical steps were performed according to the MRC Holland MLPA general protocol.10 The probes detect sequences involved in a distinct subset of the most common microdeletion and microduplication disorders.
The specific chromosomal regions evaluated by the MLPA kit are described in Table 1. Statistical analysis was done using IBM SPSS Statistics software (IBM Corp., Armonk, NY, USA).
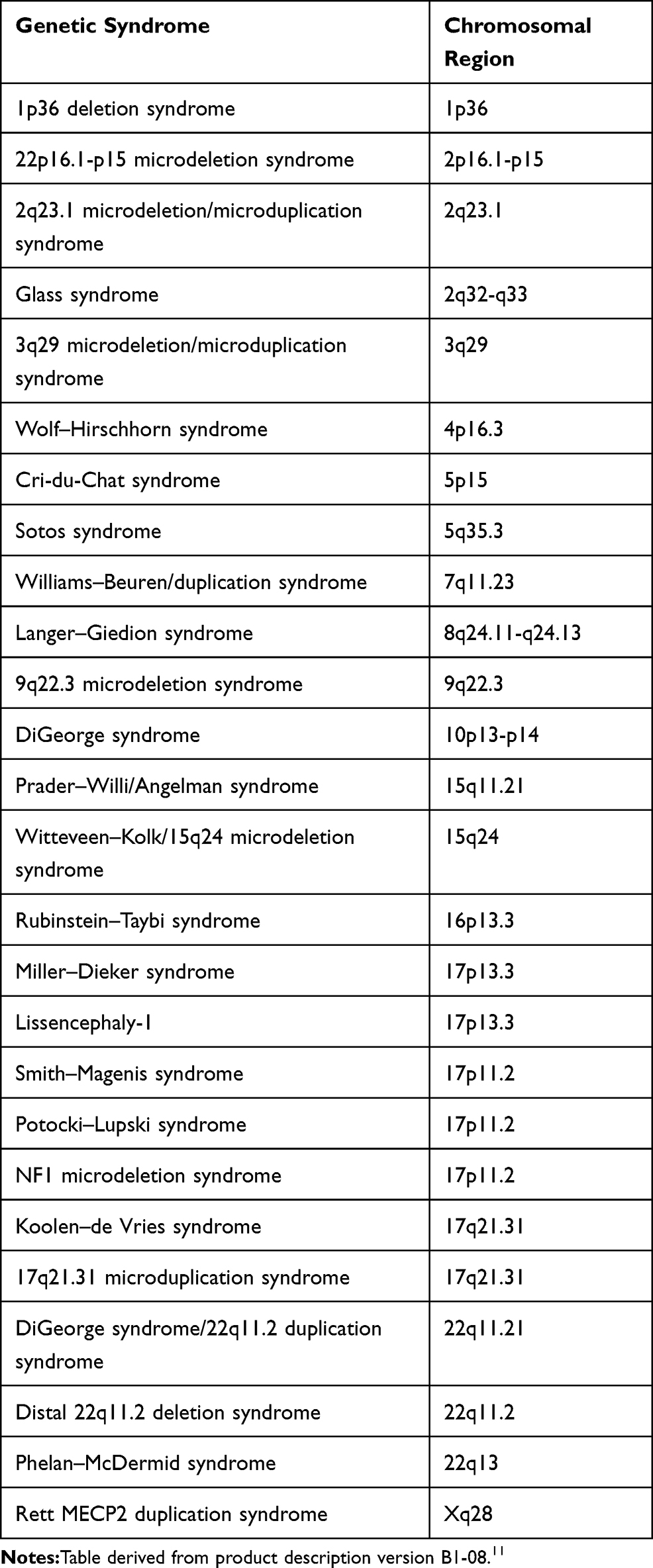 |
Table 1 Chromosomal Region Assessed by SALSA MLPA P245-B1 Microdeletion Syndromes-1A |
Results
The average age for diagnosis was 10.6 years (SD = 6.05 years); 160/501 (32%) of the patients were aged 2 years or younger, 144/501 (29%) of the patients were older than 2 years but younger or equal to 10 years, and 197/501 (39%) were older than 10 years. Concerning sex distribution, 223/501 patients (44.5%) were females and 278/501 (55.5%) were males. 315/501 (63%) had associated varying signs of cranio-facial dysmorphism. 128/501 (25%) of the patients presented associated malformation of internal organs.
25/501 patients (5%) were diagnosed with a microdeletion/microduplication syndrome. Amongst them, 7/25 (30%) had been clinically diagnosed, for the other cases the clinical features were not evocative for a specific syndrome. The aetiology observed in these cases is described in Table 2.
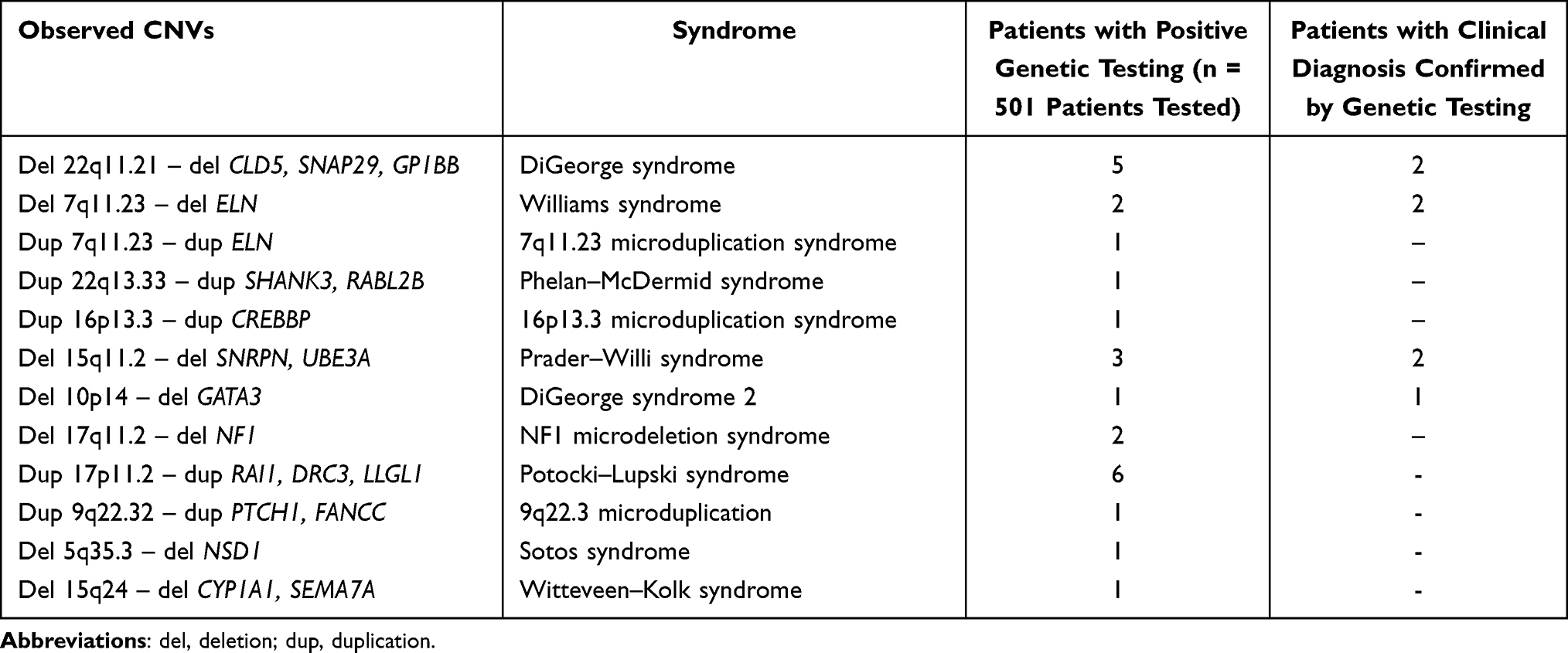 |
Table 2 CNVs Observed in Studied Patients with GDD/ID |
Considering a clinical approach, 7 out of 50 patients (14%) who had a different clinical diagnosis, possible to be evaluated in our panel, were also genetically confirmed. The other patients, who did not have a specific clinical diagnosis, were referred only with isolated or syndromic GDD/ID. Two of 11 patients (18%) who presented a clinical picture of DiGeorge syndrome were confirmed by genetic testing. For Prader–Willi syndrome, the clinical suggestion was confirmed in 2/15 patients (13%). For Williams syndrome, the clinical picture was confirmed in 2/5 patients (40%). GATA3 deletion was confirmed for the only patient who was clinically suspected, and whose clinical phenotype included hypoparathyroidism, deafness and renal dysplasia, a specific picture for this abnormality.
Discussions
The main result of this research is the 5% diagnostic rate in the investigation of microdeletion/microduplication syndromes using the MLPA technique, in patients with isolated/syndromic GDD/ID. This percentage was observed performing a “genotype first” approach. 14% of patients with suggestive clinical diagnosis, “phenotype first approach”, were confirmed after genetic testing. Patients with quite specific clinical diagnosis, such as DiGeorge syndrome (3 patients) or Prader–Willi syndrome (1 patient) were detected by MLPA without clinical suggestion, recommending MLPA as a possible first low cost investigation to rule out recurrent CNVs. However, although the MLPA test is useful for recurrent CNVs detection, rare CNVs, non-recurrent could not be seen by this technique, requiring further investigations (CMA or exome/genome sequencing with CNVs analysis), in the case of an undiagnosed patients with GDD/ID after this first test.
Regarding the different types of genetic testing used to establish an aetiological diagnosis in GDD/ID, an extensive review of the literature regarding the evaluation possibilities of the children affected by GDD determined the overall karyotype detection rate to be 3.7%, and the most frequent encountered anomalies were Down syndrome, sex chromosome aneuploidies and unbalanced translocations/deletion syndromes.2
A meta-analysis aiming to highlight the importance of the CMA in learning disability and congenital anomalies determined an overall diagnostic rate of 10% from 19 studies and nearly 14,000 cases.7 A study investigating GDD/ID with CMA and conventional karyotyping, reported a 32.2% diagnostic rate for CMA and 18.1% diagnostic rate for karyotyping.12 This comparison between the two tests regarding developmental delay diagnosis (among others), had already been stated in a prior study, in which the chromosomal microarray detection rate was rather modest – around 9%, but this rate was still twice as good as the karyotype diagnostic rate.13 This CMA and karyotype connection in this pathology was also approached by Siggberg et al.14 in a study that described CMA diagnostic rates of 10% with low-resolution and 15.8% with high-resolution in cases in which the karyotype had been negative.14
Trying to assess the diagnostic efficiency using karyotyping and MLPA, a study evaluating recurrent microdeletions/microduplications and karyotype indicated a diagnostic rate of 19%.15 Another study evaluating by MLPA the microdeletions and subtelomeric regions in children with GDD/ID with normal karyotype, reported a 9% detection rate.16 Another investigation on intellectual disability assessing only subtelomeric regions revealed a 4.2% prevalence of subtelomeric rearrangements.17 A cohort of 150 patients tested with MLPA subtelomere kits and MLPA kit for microdeletions, revealed 14% diagnostic rate, 7.3% subtelomeric rearrangements and 6.6% microdeletions, the most frequent, as in our study, being DiGeorge, Prader–Willi, Angelman, Langer–Giedion syndromes and 17q21.31, 15q24 microdeletions.18 Other MLPA assessment was applied in intellectual disability using the MLPA telomere kit. The diagnostic rate was 6.7%, but it was nearly doubled – 12.4% when a clinical selection was performed pre-test.19
A 10-year retrospective analysis on 36,325 cases with GDD/ID targeted the diagnostic outcomes from CMA, karyotyping and FISH. While the diagnostic yield of array-based tests was estimated to be a minimum of 19%, the karyotype detection rate was 4.5%, and 3.5% for FISH.20 The most frequent CNVs detected by CMA platforms were associated with microdeletion/microduplication syndromes: 15q11.2-q13.1 – Prader–Willi/Angelman; GRIA3 gene on X chromosome; 22q.13.3 deletion; 17p11.2 deletion.21 Similarly, a CMA and MLPA investigation indicated a 15.6% diagnostic rate for CMA and a 2.1% for MLPA.22
The syndromes which were clinically suggested from the start, were those presenting strongly defined clinical features, such as Prader–Willi syndrome, Williams syndrome and velo-cardio-facial syndrome. Regarding the other cases presenting non-specific clinical features, a certain aetiological diagnosis was not established.
This study regarding the frequency of the main microdeletion/microduplication syndromes in a group of 501 patients with isolated or syndromic GDD/ID is important and indicates that an attentive clinical assessment could elevate the diagnostic rate. Genomic evaluation of CNVs (using chromosomal microarray or next-generation sequencing) has become more and more useful for a better diagnosis, with a diagnostic efficiency of more than 50%. However, in cases with a specific clinical diagnosis, it could be less expensive and more efficient to indicate as first intention a low cost diagnostic test, as in the case of Williams syndrome, Prader–Willi syndrome or DiGeorge syndrome.
Ethical Issues
For each patient, an informed consent regarding their participation in the study, was obtained from their parents, as a signed consent form. The study was approved by the Ethics Committee of Emergency Clinical Hospital for Children, Cluj-Napoca.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest.
References
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association;2013:271–279. Available from: https://psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596.
2. Shevell M, Ashwal S, Donley D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the quality standards subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology. 2003;60(3):367–380. doi:10.1212/01.wnl.0000031431.81555.16
3. Moeschler JB, Shevell M, Saul RA, et al. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014;134(3):e903–18. doi:10.1542/peds.2014-1839
4. Lavillaureix A, Nava C, Mignot C, et al. New generation sequencing for the diagnosis of intellectual disabilities: exome sequencing or large panel? Eur J Paediatr Neurol. 2017;21:e155. doi:10.1016/j.ejpn.2017.04.1148
5. Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal Microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi:10.1016/j.ajhg.2010.04.006
6. Karam SM, Riegel M, Segal SL, et al. Genetic causes of intellectual disability in a birth cohort: a population-based study. Am J Med Genet A. 2015;167(6):1204–1214. doi:10.1002/ajmg.a.37011
7. Sagoo GS, Butterworth AS, Sanderson S, Shaw-Smith C, Higgins JP, Burton H. Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects. Genet Med. 2009;11(3):139–146. doi:10.1097/GIM.0b013e318194ee8f
8. Wolfe K, Strydom A, Morrogh D, et al. Chromosomal microarray testing in adults with intellectual disability presenting with comorbid psychiatric disorders. Eur J Hum Genet. 2017;25(1):66–72. doi:10.1038/ejhg.2016.107
9. Lee JA, Lupski JR. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52(1):103–121. doi:10.1016/j.neuron.2006.09.027
10. MLPA®, General Protocol Instructions For Use MLPA (Multiplex Ligation-dependent Probe Amplification) General Protocol for the detection and quantification of DNA sequences; 2019. Available from: www.mrcholland.com,www.mlpa.com.
11. MLPA®. MRC-Holland Product Description SALSA ® MLPA ® Probemix P245-B1 Microdeletion Syndromes-1A; 2020. Available from: https://www.mrcholland.com/products/30024/Product%20Description%20P245-B1%20Microdeletion%20Syndromes-1A-v08.pdf.
12. Lee CL, Lee CH, Chuang CK, et al. Array-CGH increased the diagnostic rate of developmental delay or intellectual disability in Taiwan. Pediatr Neonatol. 2019;60(4):453–460. doi:10.1016/j.pedneo.2018.11.006
13. Bejjani BA, Shaffer LG. Application of array-based comparative genomic hybridization to clinical diagnostics. J Mol Diagn. 2006;8(5):528–533. doi:10.2353/jmoldx.2006.060029
14. Siggberg L, Ala-Mello S, Jaakkola E, et al. Array CGH in molecular diagnosis of mental retardation - a study of 150 finnish patients. Am J Med Genet A. 2010;152A(6):1398–1410. doi:10.1002/ajmg.a.33402
15. David M, Dieterich K, Billette de Villemeur A, et al. Prevalence and characteristics of children with mild intellectual disability in a French county. J Intellect Disabil Res. 2014;58(7):591–602. doi:10.1111/jir.12057
16. John N, Rajasekhar M, Girisha K, Sharma PSN, Gopinath P. Multiplex ligation-dependant probe amplification study of children with idiopathic mental retardation in South India. Indian J Hum Genet. 2013;19(2):165–170. doi:10.4103/0971-6866.116115
17. Medina A, Piñeros L, Arteaga C, et al. Multiplex ligation-dependent probe amplification to subtelomeric rearrangements in idiopathic intellectual disability in Colombia. Pediatr Neurol. 2014;50(3):250–254. doi:10.1016/j.pediatrneurol.2013.10.017
18. Pohovski LM, Dumic KK, Odak L, Barisic I. Multiplex ligation-dependent probe amplification workflow for the detection of submicroscopic chromosomal abnormalities in patients with developmental delay/intellectual disability. Mol Cytogenet. 2013;6(1):7. doi:10.1186/1755-8166-6-7
19. Koolen DA, Nillesen WM, Versteeg MH, et al. Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA). J Med Genet. 2004;41(12):892–899. doi:10.1136/jmg.2004.023671
20. Hochstenbach R, van Binsbergen E, Engelen J, et al. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur J Med Genet. 2009;52(4):161–169. doi:10.1016/j.ejmg.2009.03.015
21. Rodríguez-Revenga L, Vallespín E, Madrigal I, et al. A parallel study of different array-CGH platforms in a set of Spanish patients with developmental delay and intellectual disability. Gene. 2013;521(1):82–86. doi:10.1016/j.gene.2013.02.043
22. Shin S, Yu N, Choi JR, Jeong S, Lee KA. Routine chromosomal microarray analysis is necessary in Korean patients with unexplained developmental delay/mental retardation/autism spectrum disorder. Ann Lab Med. 2015;35(5):510–518. doi:10.3343/alm.2015.35.5.510
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.