Back to Journals » Infection and Drug Resistance » Volume 14
Diagnosing MonoMAC Syndrome in GATA2 Germline Mutated Myelodysplastic Syndrome via Next-Generation Sequencing in a Patient with Refractory and Complex Infection: Case Report and Literature Review
Authors Shen Y, Li Y, Li H ![]() , Liu Q, Dong H, Wang B, Ye B
, Liu Q, Dong H, Wang B, Ye B ![]() , Lin S, Shen Y, Wu D
, Lin S, Shen Y, Wu D ![]()
Received 8 February 2021
Accepted for publication 19 March 2021
Published 6 April 2021 Volume 2021:14 Pages 1311—1317
DOI https://doi.org/10.2147/IDR.S305825
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Yingying Shen,1 Yuzhu Li,2 Hangchao Li,2 Qi Liu,2 Huijie Dong,2 Bo Wang,1 Baodong Ye,1 Shenyun Lin,1 Yiping Shen,1 Dijiong Wu1
1Department of Hematology, First Affiliated Hospital of Zhejiang Chinese Medical University, Zhejiang Provincial Hospital of TCM, Hangzhou, Zhejiang Province, People’s Republic of China; 2First Clinical Medical College, Zhejiang Chinese Medical University, Zhejiang Provincial Hospital of TCM, Hangzhou, Zhejiang Province, People’s Republic of China
Correspondence: Dijiong Wu; Yiping Shen
Department of Hematology, First Affiliated Hospital of Zhejiang Chinese Medical University, No. 54 Youdian Road, Hangzhou, Zhejiang Province, People’s Republic of China
Email [email protected]; [email protected]
Abstract: Monocytopenia and mycobacterial infection (MonoMAC) syndrome is a rare disease. Herein, we reported a 65-year-old Asian woman, previously diagnosed with myelodysplastic syndrome (MDS), suffering from recurrent pneumonia, intermittent fever, fatigue, and chest tightness lasting for five months. She was ultimately diagnosed with MonoMAC syndrome with Mycobacterium kansasii (M. kansasii) infection and GATA2 mutation through metagenomic generation sequencing (mNGS) of peripheral blood specimen, for which she was given anti-NTM therapy. Her situation significantly improved within 2 weeks of therapy. We discussed the clinical features, genetic characteristic, and prognosis of this disorder, aiming to further elucidate this rare syndrome. For MDS/AML patient with recurrent mixed infection and pancytopenia (especially with monocyte absence), MonoMAC syndrome should be highly suspected, and germline mutation and pathogen sequencing should be performed.
Keywords: monocytopenia and mycobacterial infection syndrome, MonoMAC, M. kansasii, GATA-2 germline mutation, nontuberculous mycobacteria, next-generation sequencing
Introduction
Monocytopenia and mycobacterial infection (MonoMAC) syndrome is a rare autosomal dominant syndrome associated with monocytopenia, deficiency of B and natural killer (NK) cells, as well as mycobacterial, viral, fungal, and bacterial opportunistic infections. More than 50% of the patients with MonoMAC are diagnosed with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML).1,2 Patients with MonoMAC syndrome typically present with severe or recurrent infection of nontuberculous mycobacterial (NTM) or opportunistic fungal infection;1 the mortality rate in those patients is 28%.1
The allogeneic hematopoietic stem cell transplantation (HSCT) has been shown to be an effective treatment to reconstitute the depleted hematopoietic systems and reverse the clinical phenotype seen in affected patients.2 However, if the diagnosis is performed too late, the treatment may not be effective. Thus, early diagnosis is crucial. Herein, we diagnosed a MonoMAC syndrome in a MDS patient with GATA2 mutation by next-generation sequencing of the blood specimen, and the mutation was further confirmed as germline mutation with oral mucosal specimen from her son. To better understand the sensitivity and specificity of a blood specimen by NTM detection, we further reviewed the available reported MonoMAC cases and analyzed the disease’s diagnosing process, which may hopefully help clinicians perform earlier diagnoses of this kind of rare disease.
Case Report
A 65-year-old Asian woman was admitted to our clinic in August 2019 due to fever (38.3–39 °C), chest tightness, cough producing sputum, and shortness of breath for one day. Her clinical history could be defined by long-term leukopenia. She was diagnosed with MDS in 2014, after which she received supportive treatments. In 2019, she was admitted to the hospital with increased bone marrow blasts (10% vs 4%) and decreased megakaryocytes with small round separated nuclei (Figure 1A–E, Table 1). She received four courses of amifostine, but her symptoms did not improve. During hospitalization, she was infected with recurrent pneumonia, which could be relieved by multiple antibiotic treatments (Figure 2A and B). She received voriconazole after the last course in July 2019. Two months later, the symptoms of fever and pneumonia re-appeared again (Figure 2C).
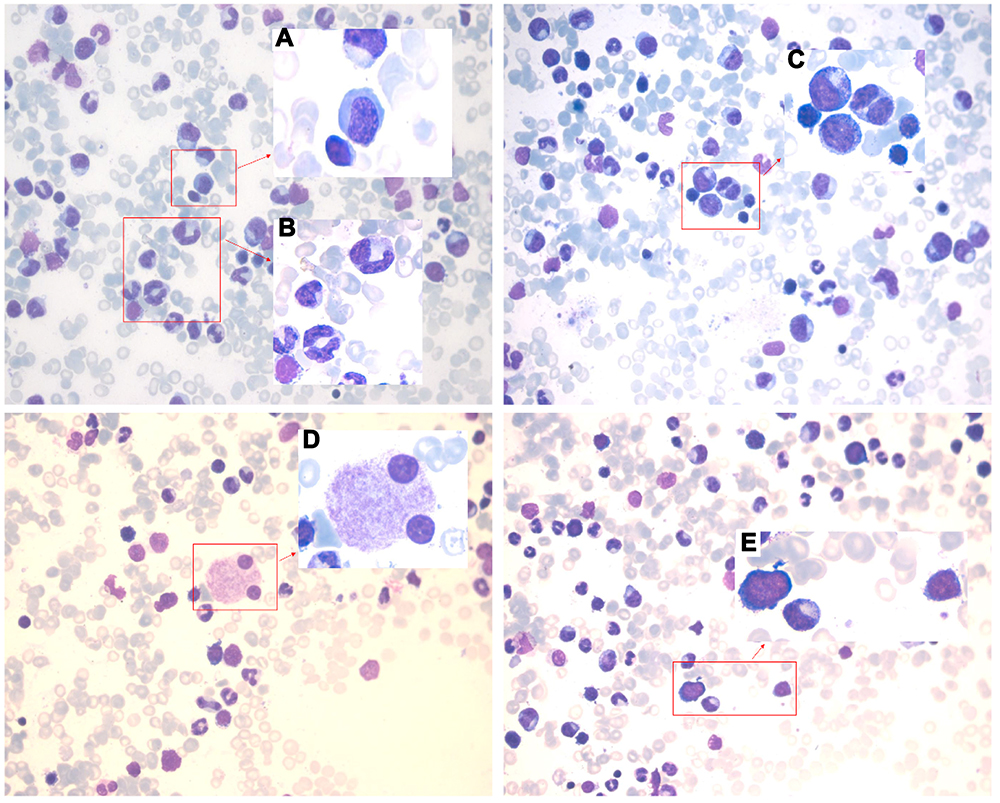 |
Figure 1 Bone marrow images at the first admission. Pathological change with multi-lineage myelodysplasia, including erythropathy (megaloblastic changes, (A)), granulocytopathy (rod thickening of neutrophils, (B); binuclear, (C)), megakaryocytopathy (binuclear, (D)) and blasts (E). |
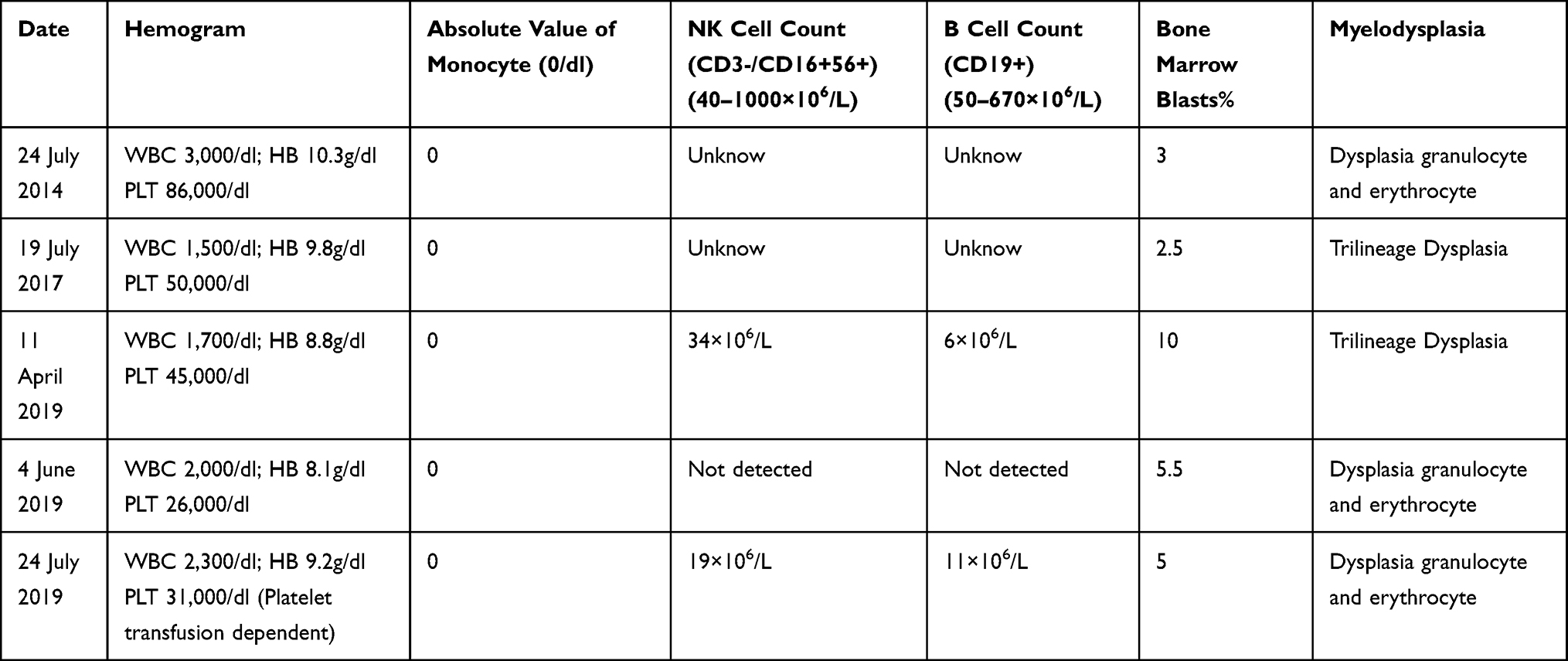 |
Table 1 Changes in Peripheral Blood and Bone Marrow During the Disease Process |
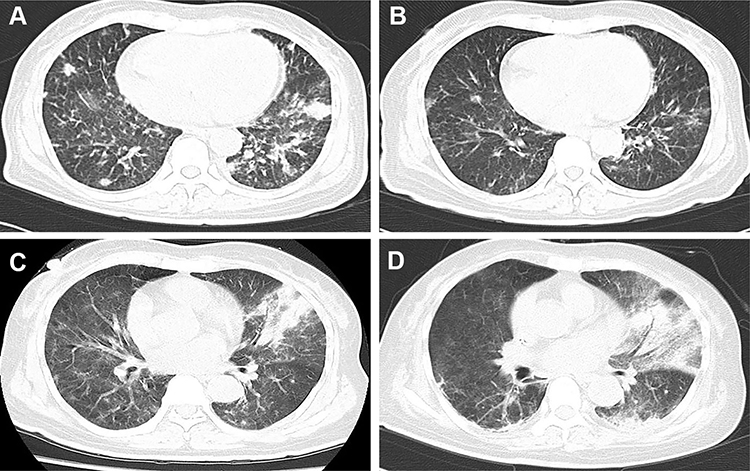 |
Figure 2 Computed tomography (CT) manifestation of recurrent pulmonary infection. The recurrent pneumonia was observed during the hospitalization (A), and could be relieved by multiple antibiotic treatments (B). (C) showed exacerbation of infection before the detection of mycobacterium kansasii (with air bronchogram and pulmonary consolidation), and the situation did not get improved after multiple treatments (D). |
On admission, she presented with fever and chest pain, and blood cell count at that time revealed lymphocytopenia and monocytopenia. The blood routine test showed a white blood cell (WBC) count of 1800 cells/mm3, a low monocyte count of 0–0.1 cells/mm3; a hemoglobin level of 7.6 g/dL; and a platelet count of 17 × 109/L. The absolute count of B cell (CD19+) was 6×106/L (50–670×106), NK cell (CD3-/CD16+56+) count was 34×106/L(40–1000×106/L). The CD4+ T-cell count was 141 cells/mm3 and the CD4/CD8 ratio was 0.99. She also had splenomegaly (4.1 cm in size) and iron overload (ferritin: 2361.4ng/mL). The results of Epstein-Barr virus (EBV)-DNA and cytomegalovirus (CMV)-DNA were negative. Next, the patient was treated with tigecycline, ceftazidime, and voriconazole according to the manifestation of lung computed tomography (CT) scan (Figure 2D). Furthermore, Klebsiella pneumoniae infection was confirmed by sputum culture analysis, after which she received therapy with caspofungin, posaconazole, cefoperazone, linezolid, ganciclovir, sulfamethoxazole, amphotericin B, and polymyxin subsequently. However, no significant improvement was observed with reference to fever and pneumonia. During this time, she developed chest tightness and dyspnea which required continuous high-flow oxygen inhalation. She was not able to undergo a bronchoscopy. Complicated with severe infection, the patient also developed acute heart failure. Eventually, we sequenced the patient’s peripheral blood specimens despite her repeated negative sputum culture, after which the M. kansasii infection was confirmed. Consequently, she was given antibiotic therapy, consisting of ethambutol, rifabutin, and clarithromycin as recommended.3 Her situation significantly improved within 2 weeks of therapy (without fever). C-reactive protein (CRP) level remarkably decreased from 250.87 mg/L to an almost normal level. According to the above-mentioned signs and symptoms, the patient was suspected of having MonoMAC syndrome. The next-generation sequencing (NGS, peripheral blood) found a heterozygous mutation in GATA2 (exon6:c.1126_1128del:p.K376del) along with U2AF1 (exon2:c.C101T:p.S34F), SETBP1 (exon4, cG2602A:pD868N, exon4:cG2608A:pG870S), ASXL1 (exon12:c.G2548T:p.E850X) as well as KMT2D, BRAF, EPPK1, and ETV6. Her son was further confirmed with the same GATA2 mutation with oral mucosal specimen. Unfortunately, the patient died of carbapenem-resistant Klebsiella pneumoniae bloodstream infection on October 2019, 41 days after the MonoMAC diagnosing.
Discussion
MonoMAC syndrome was firstly reported in 2010. Approximately 50% of MonoMAC patients reported so far presented with MDS or AML. Studies have found that MonoMAC syndrome is closely related to GATA2 germline mutation. Heterozygous mutation in GATA2 causes the loss of gene function that regulates many aspects of development from hematopoiesis to lymphatic, leading to immunodeficiency and bone marrow failure.4 Among individuals with GATA2 deficiency progressing to MDS/AML, acquired secondary mutations in the ASXL1 that encode chromatin-binding protein ASXL1 have been detected in approximately 30% of cases;5 this was also a case of our patient. Up to date, at least 30 cases of MonoMAC in MDS patients have been reported, some of whom were confirmed based on family GATA2 mutation. Various GATA mutation site have been reported, mostly in exon 6 and exon 7, and some in exon 4 and56–15 (Figure 3). In our case, a GATA2 mutation (p.K376 del), located in exon6 was found. The results showed that there might not be a close relationship between mutation site and the prognosis. Although HSCT has been shown to be an effective treatment for MonoMAC, the overall survival rate of 57% at 36 months.16
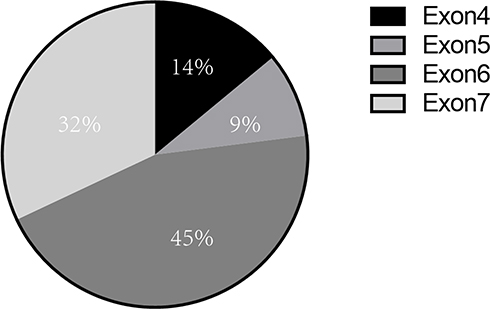 |
Figure 3 Distribution of GATA2 germline mutation sites in MonoMAC syndrome. Various site of GATA germline mutation had been reported, mostly in exon 6 (45%) and exon7 (32%), and some in exon 4 (14%) and exon 5 (9%). |
Early diagnosis improves prognosis and treatment outcomes. Unlike other microbial infection s, culturing nontuberculous mycobacterial (NTM) is challenging ad requires a long time for analysis (7days to several weeks),17 while the rate of positive sputum test is mostly under 35%.18 In order to increase the specificity and sensitivity of detection, NGS assays are very commonly used for gene screening in those with predisposition syndrome. Usually, bronchoalveolar lavage fluid (BALF) is an ideal specimen for mNGS detection. However, patients with hematological disorders, especially those with severe thrombocytopenia, cannot tolerant bronchoscopy.19 In our case, we preferred the peripheral blood specimen; other reported specimen including secretion and tissue fluid, were optional samples for mNGS (Table 2).
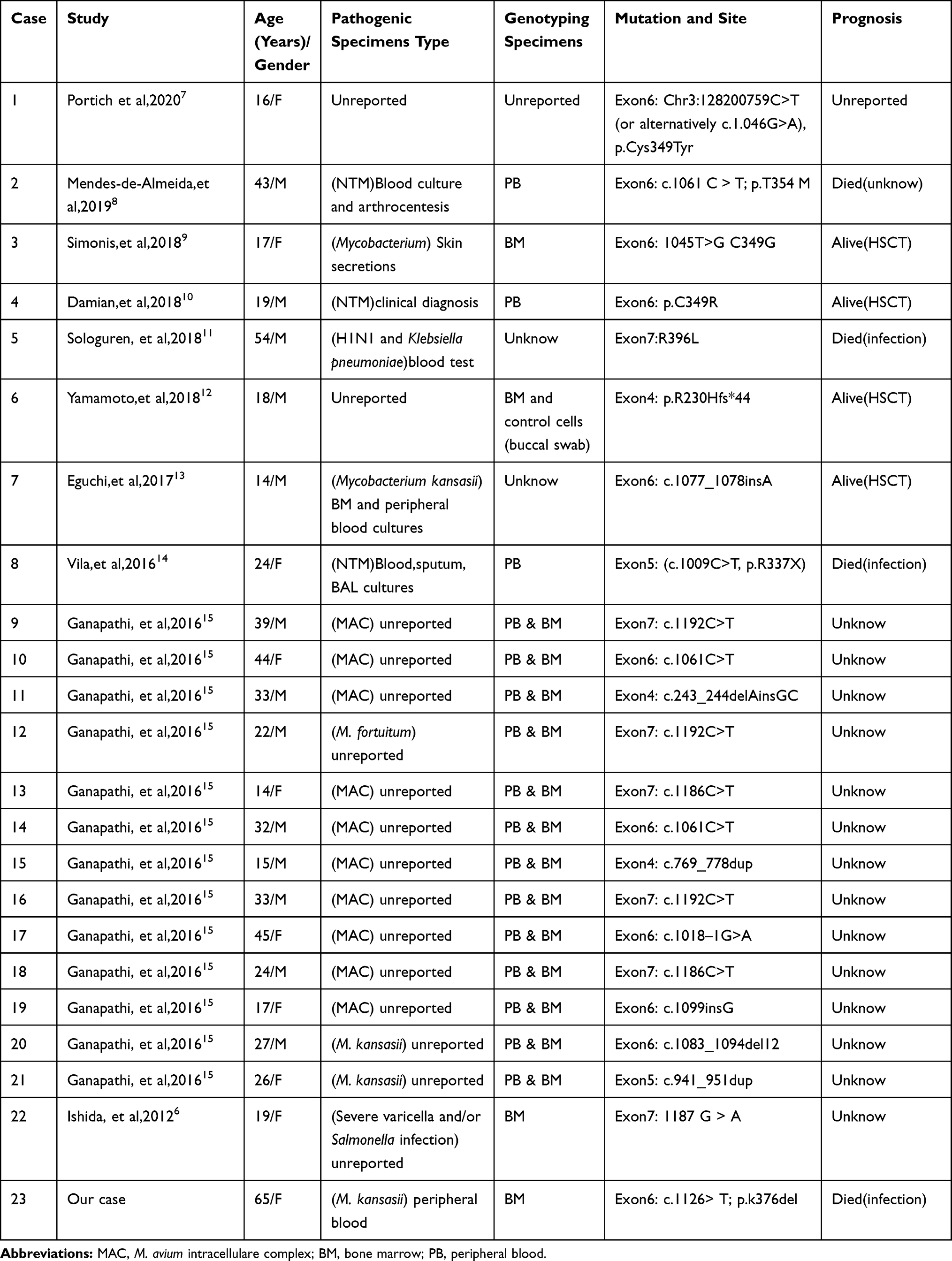 |
Table 2 Mutation Sites of the Related Literature in MDS Patients with MonoMAC Syndrome |
In our case, the NTM was finally confirmed as M. kansasii, which was firstly isolated in 1953.19 M. kansasii is ubiquitous in the environment as a group I non-tuberculous mycobacterium (NTM).20 The M. kansasii extrapulmonary disease is rare.21 In patients with poor immunity, M. kansasii infection may lead to underlying systemic disease or abscess formation,22 with a 1-year mortality rate of 43% and median survival of 71 days.23 The current therapies for M. kansasii infection are similar but not the same. The British Thoracic Society (BTS) recommends daily two-drug therapy with rifampicin (450–600mg/day) and ethambutol (15mg/kg) for 9 months.24 The American Thoracic Society (ATS) and the Infectious Diseases Society of America (IDSA) guidelines recommend daily therapy with isoniazid (5mg/kg/day), rifampicin (10mg/kg/day), ethambutol (15mg/kg/day) and pyridoxine (50mg/d)) until sputum culture is negative for at least 12 months.25 The standard treatment regimen is usually effective, but is always time-consuming and not suitable for all the patients.26 Alternative therapies include clarithromycin (500–1000mg/day) plus rifampicin (10mg/kg/day) and ethambutol (15–25mg/kg/day).27 Rifampicin is the main drug of the therapy; ethambutol can prevent acute resistance to rifampicin. If rifampicin resistance occurs, varonones, macrolides, nitroimidazoles and clofazimine can be used as therapeutic options. Physicians should make the therapeutic schedule individually considering the hepatorenal function and bone marrow hematopoiesis during the practice. In our case, Rifampicin, ethambutol and clarithromycin were selected, and the fever, respiratory, constitutional symptoms were improved within 2 weeks.
In conclusion, we reported a case of a rare MDS patient with refractory infection and immune deficiency ascribe to MonoMAC syndrome. The sequencing of NTM and germline GATA2 mutation should be performed as early as possible.
Data Sharing Statement
The data used and/or analyzed during the current study are available from the corresponding author upon a reasonable request.
Ethics Approval and Consent to Participate
This study was approved by the ethical committee of First Affiliated Hospital of Zhejiang Chinese Medical University.
Patient Consent for Publication
Written informed consent was obtained from the patient’s son for the publication of clinical results.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was supported by National Natural Science Foundation of China (NO. 81603573), Zhejiang Traditional Chinese Medicine Scientific Research Foundation (NO. 2020ZB085) and Science and Technology Department of Zhejiang Research Foundation (NO.2019C03047).
Disclosure
The authors declare that they have no competing interests.
References
1. Vinh DC, Patel SY, Uzel G, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115(8):1519–1529. doi:10.1182/blood-2009-03-208629
2. Bigley V, Haniffa M, Doulatov S, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med. 2011;208(2):227–234. doi:10.1084/jem.20101459
3. Destefano MS, Shoen CM, Cynamon MH. Therapy for Mycobacterium kansasii infection: beyond 2018. Front Microbiol. 2018;9:2271. doi:10.3389/fmicb.2018.02271
4. Kazenwadel J, Secker GA, Liu YJ, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119(5):1283–1291. doi:10.1182/blood-2011-08-374363
5. Bödör C, Renneville A, Smith M, et al. Germ-line GATA2 p.THR354MET mutation in familial myelodysplastic syndrome with acquired monosomy 7 and ASXL1 mutation demonstrating rapid onset and poor survival. Haematologica. 2012;97(6):890–894. doi:10.3324/haematol.2011.054361
6. Ishida H, Imai K, Honma K, et al. GATA-2 anomaly and clinical phenotype of a sporadic case of lymphedema, dendritic cell, monocyte, B- and NK-cell (DCML) deficiency, and myelodysplasia. Eur J Pediatr. 2012;171(8):1273–1276. doi:10.1007/s00431-012-1715-7
7. Portich JP, CONDINO NETO A, Faulhaber GAM. Humoral deficiency in a novel GATA2 mutation: a new clinical presentation successfully treated with hematopoietic stem cell transplantation. Pediatr Blood Cancer. 2020;67(9):e28374. doi:10.1002/pbc.28374
8. Mendes-de-almeida DP, Andrade FG, Borges G, et al. GATA2 mutation in long stand Mycobacterium kansasii infection, myelodysplasia and MonoMAC syndrome: a case-report. BMC Med Genet. 2019;20(1):64. doi:10.1186/s12881-019-0799-6
9. Simonis A, Fux M, Nair G, et al. Allogeneic hematopoietic cell transplantation in patients with GATA2 deficiency-a case report and comprehensive review of the literature. Ann Hematol. 2018;97(10):1961–1973. doi:10.1007/s00277-018-3388-4
10. Damian L, Sauvetre G, Marguet F, et al. Pseudo-sarcoidosis revealing MonoMAC syndrome. J Clin Immunol. 2018;38(7):739–741. doi:10.1007/s10875-018-0551-6
11. Sologuren I, Martinez-saavedra MT, Sole-violan J, et al. Lethal influenza in two related adults with inherited GATA2 deficiency. J Clin Immunol. 2018;38(4):513–526. doi:10.1007/s10875-018-0512-0
12. Yamamoto H, Hattori H, Takagi E, et al. [MonoMAC syndrome patient developing myelodysplastic syndrome following persistent EBV infection]. Rinsho Ketsueki. 2018;59(3):315–322. doi:10.11406/rinketsu.59.315.Japanese.
13. Eguchi K, Ishimura M, Sonoda M, et al. Nontuberculous mycobacteria-associated hemophagocytic lymphohistiocytosis in MonoMAC syndrome. Pediatr Blood Cancer. 2018;65(7):e27017. doi:10.1002/pbc.27017
14. Vila A, Dapas JI, Rivero CV, et al. Multiple opportunistic infections in a woman with GATA2 mutation. Int J Infect Dis. 2017;54:89–91. doi:10.1016/j.ijid.2016.11.408
15. Ganapathi KA, Townsley DM, Hsu AP, et al. GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood. 2015;125(1):56–70. doi:10.1182/blood-2014-06-580340
16. Chu VH, Curry JL, Elghetany MT, et al. MonoMAC versus idiopathic CD4+ lymphocytopenia. Comment to Haematologica. 2011;96(8):1221–5. Haematologica. 2012;97(4):
17. Sharma SK, Upadhyay V. Epidemiology, diagnosis & treatment of non-tuberculous mycobacterial diseases. Indian J Med Res. 2020;152(3):185–226. doi:10.4103/ijmr.IJMR_902_20
18. Hatta M, Sultan AR, Tandirogang N, et al. Detection and identification of mycobacteria in sputum from suspected tuberculosis patients. BMC Res Notes. 2010;3:1–6. doi:10.1186/1756-0500-3-72
19. Sugihara E, Hirota N, Niizeki T, et al. Usefulness of bronchial lavage for the diagnosis of pulmonary disease caused by Mycobacterium avium-intracellulare complex (MAC) infection. J Infect Chemother. 2003;9(4):328–332. doi:10.1007/s10156-003-0267-1
20. Buhler VB, Pollak A. Human infection with atypical acid-fast organisms; report of two cases with pathologic findings. Am J Clin Pathol. 1953;23(4):363–374. doi:10.1093/ajcp/23.4.363
21. Kaustova J, Chmelik M, Ettlova D, et al. Disease due to Mycobacterium kansasii in the Czech Republic: 1984–89. Tuber Lung Dis. 1995;76(3):205–209. doi:10.1016/S0962-8479(05)80006-1
22. Smith MB, Molina CP, Schnadig VJ, et al. Pathologic features of Mycobacterium kansasii infection in patients with acquired immunodeficiency syndrome. Arch Pathol Lab Med. 2003;127(5):554–560. doi:10.5858/2003-127-0554-PFOMKI
23. Liu CJ, Huang HL, Cheng MH, et al. Outcome of patients with and poor prognostic factors for Mycobacterium kansasii-pulmonary disease. Respir Med. 2019;151:19–26. doi:10.1016/j.rmed.2019.03.015
24. Subcommittee of the Joint Tuberculosis Committee of the British Thoracic Society. Management of opportunist mycobacterial infections: Joint Tuberculosis Committee Guidelines 1999. Thorax. 2000;55(3):210–218. doi:10.1136/thorax.55.3.210
25. Griffith DE, Aksamit T, Brown-elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367–416.
26. Santin M, Dorca J, Alcaide F, et al. Long-term relapses after 12-month treatment for Mycobacterium kansasii lung disease. Eur Respir J. 2009;33(1):148–152. doi:10.1183/09031936.00024008
27. Shitrit D, Priess R, Peled N, et al. Differentiation of Mycobacterium kansasii infection from Mycobacterium tuberculosis infection: comparison of clinical features, radiological appearance, and outcome. Eur J Clin Microbiol Infect Dis. 2007;26(10):679–684. doi:10.1007/s10096-007-0331-3
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.