")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Dexmedetomidine attenuates the neurotoxicity of propofol toward primary hippocampal neurons in vitro via Erk1/2/CREB/BDNF signaling pathways
Authors Tu Y, Liang Y, Xiao Y, Lv J, Guan R, Xiao F , Xie Y , Xiao Q
Received 22 September 2018
Accepted for publication 13 December 2018
Published 19 February 2019 Volume 2019:13 Pages 695—706
DOI https://doi.org/10.2147/DDDT.S188436
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Youbing Tu,1 Yubing Liang,2 Yong Xiao,1 Jing Lv,1 Ruicong Guan,1 Fei Xiao,1 Yubo Xie,1,* Qiang Xiao3,*
1Department of Anesthesiology, The First Affiliated Hospital of Guangxi Medical University, Nanning 530021, China; 2Department of Anesthesiology, The Affiliated Tumor Hospital of Guangxi Medical University, Nanning 530021, China; 3Department of Surgery, The First Affiliated Hospital of Guangxi Medical University, Nanning 530021, China
*These authors contributed equally to this work
Background: Propofol is a commonly used general anesthetic for the induction and maintenance of anesthesia and critical care sedation in children, which may add risk to poor neurodevelopmental outcome. We aimed to evaluate the effect of propofol toward primary hippocampal neurons in vitro and the possibly neuroprotective effect of dexmedetomidine pretreatment, as well as the underlying mechanism.
Materials and procedures: Primary hippocampal neurons were cultured for 8 days in vitro and pretreated with or without dexmedetomidine or phosphorylation inhibitors prior to propofol exposure. Cell viability was measured using cell counting kit-8 assays. Cell apoptosis was evaluated using a transmission electron microscope and flow cytometry analyses. Levels of mRNAs encoding signaling pathway intermediates were assessed using qRT-PCR. The expression of signaling pathway intermediates and apoptosis-related proteins was determined by Western blotting.
Results: Propofol significantly reduced cell viability, induced neuronal apoptosis, and downregulated the expression of the BDNF mRNA and the levels of the phospho-Erk1/2 (p-Erk1/2), phospho-CREB (p-CREB), and BDNF proteins. The dexmedetomidine pretreatment increased neuronal viability and alleviated propofol-induced neuronal apoptosis and rescued the propofol-induced downregulation of both the BDNF mRNA and the levels of the p-Erk1/2, p-CREB, and BDNF proteins. However, this neuroprotective effect was abolished by PD98059, H89, and KG501, further preventing the dexmedetomidine pretreatment from rescuing the propofol-induced downregulation of the BDNF mRNA and p-Erk1/2, p-CREB, and BDNF proteins.
Conclusion: Dexmedetomidine alleviates propofol-induced cytotoxicity toward primary hippocampal neurons in vitro, which correlated with the activation of Erk1/2/CREB/BDNF signaling pathways.
Keywords: hippocampus, propofol, dexmedetomidine, extracellular signal-regulated MAP kinases, cyclic AMP response element-binding protein, brain-derived neurotrophic factor
Introduction
Substantial preclinical data suggest that general anesthesia drugs affect brain development in young animals. More recently, increasing number of clinical research have reported the potential neurotoxicity of anesthetics in infants and young children, arousing widespread concern in the community.1–4 Propofol is a commonly used anesthetic for the induction and maintenance of anesthesia and critical care sedation in children. Based on accumulating evidence, propofol may induce developmental neurotoxicity, raising serious concerns regarding the use of propofol anesthesia in pediatric patients.5–7 Dexmedetomidine, a highly selective α2-adrenergic agonist, exerts a neuroprotective effect on propofol-induced neurotoxicity,8–10 but the associated mechanisms have not yet been fully clarified. The neuroprotective effects of dexmedetomidine are mediated by its binding to imidazoline I1 receptors and modulating histone acetylation via Erk1/2 pathways.11–13 In addition, dexmedetomidine has been shown to preserve neurological function by increasing the phosphorylation of protein kinase B and cAMP response element-binding protein (CREB) and subsequently upregulating the expression of the antiapoptotic factor Bcl-2 and BDNF.14 Primary rat hippocampal neurons were used in the present study to evaluate the effect of propofol toward primary hippocampal neurons in vitro and the possibly neuroprotective effect of dexmedetomidine pretreatment, as well as the possible involvement of Erk1/2/CREB/BDNF signaling pathways.
Materials and procedures
Hippocampal neuron culturing
The experimental procedure and protocols were approved by the Animal Use and Care Committee of Guangxi Medical University (No SCXK GUI 2004-0002) and were performed in accordance with the Guideline for ethical review of animal welfare (GB/T 35892-2018). Primary hippocampal neurons were cultured with fetal rat hippocampi from 16–18 days Sprague Dawley embryo according to a previously described protocol,15 with slight modifications. Briefly, Sprague Dawley rat pregnancy for 16–18 days, then anesthetized with 1% isoflurane, the uterus was quickly exposed and the fetal rats were removed to a sterile container. The fetal rats were anesthetized and decapitated and hippocampi were dissected in HBSS (Ca2+ and Mg2+-free) under a stereo microscope in a sterile environment. The harvested hippocampi were treated with a 0.25% trypsin-EDTA solution (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China, Cat#T1300) for 15 minutes at 37°C and dissociated by repeated trituration through a fire-polished glass pipette. The single cell suspension was seeded onto poly-lysine-coated plates (Sigma-Aldrich Chemical Co., St Louis, MO, USA, Cat# P4832) at a density of 1−2×106 cells/mL in plating medium containing 88% DMEM/F12 (Thermo Fisher Scientific Inc., Waltham, MA, USA, Cat# 11320082), 10% FBS (Gibco®; Thermo Fisher Scientific Inc., Cat#10099141), 1% glutamine (Sigma-Aldrich Chemical Co., Cat# G7513) and 1% penicillin/streptomycin (Beijing Solarbio Science & Technology Co., Cat# P1400) at 37°C in a humidified atmosphere of 5% CO2 and 95% room air. Four hours after plating, the plating medium was changed to serum-free maintenance medium, which is not conducive to the growth and survival of glial cells,15 and thus the surviving cell population was predominantly composed of neurons. The maintenance medium consisted of 96% neurobasal medium supplemented with 2% B27 (50×, Gibco®; Thermo Fisher Scientific Inc., Cat# 21103049), 1% 200 mM glutamine (100×, Sigma-Aldrich Chemical Co., Cat# G7513) and 1% penicillin/streptomycin (100×, Beijing Solarbio Science & Technology Co., Cat# P1400), and half of the maintenance medium was replaced twice every 7 days. All experiments were performed at 8 days in vitro (DIV).
Immunocytochemistry
Hippocampal neurons (8 DIV) were identified with rabbit monoclonal antibody against neuron-specific enolase (NSE) (Abcam, Cambridge, England, UK, Cat# ab79757). Cells were plated on poly-lysine-coated coverslips and fixed with 4% paraformaldehyde (Beijing Solarbio Science & Technology Co.) for 10 minutes at room temperature. After permeabilization with 0.1% Triton X-100 (Beijing Solarbio Science & Technology Co.), cells were blocked with 5% goat serum (Beijing Solarbio Science & Technology Co.) for 10 minutes. Then, cells were incubated with a primary antibody against NSE (dilution 1:200) overnight at 4°C, followed by an incubation with a horseradish peroxidase-labeled secondary antibody for 10 minutes at room temperature. The immune reaction was visualized by incubating cells with the chromogen 3,3′-diaminobenzidine. Cells were counterstained with Gill-2 hematoxylin and then imaged using a Leica (Wetzlar, Hessen, Germany) microscope.
Experiment groups and processing
The 8 DIV primary hippocampal neurons in different Petri dishes were serially numbered and were assigned to nine groups using a randomization table. Group C (control group), Group I (intralipid vehicle group), Group DMSO (dimethyl sulfoxide [DMSO] group), Group P (propofol group), Group D (dexmedetomidine group), Group PD (dexmedetomidine + propofol group), Group PDP (PD98059 + dexmedetomidine + propofol group), Group HDP (H89 + dexmedetomidine + propofol group), or Group KDP (KG501 + dexmedetomidine + propofol group) (Figure 1). In Group C, the culture medium was replaced with fresh maintenance medium. The cells in Group PDP, HDP, and KDP were preincubated with 25 μM PD98059 (inhibitor of Erk1/2 phosphorylation), 10 μM H89 (inhibitor of CREB phosphorylation), and 25 μM KG-501 (CREB inhibitor) for 30 minutes, respectively, and the cells in Group DMSO were preincubated with 0.25% DMSO, which served as a vehicle control. Then, cells in Group PDP, HDP, and KDP were treated with 10 μM dexmedetomidine and incubated for additional 30 minutes. Meanwhile, the cells in Group D and PD were pretreated with 10 μM dexmedetomidine for 30 minutes. Finally, the cells in Group P, PD, PDP, HDP, and KDP were incubated with 100 μM propofol for 3 hours. The neuronal cell in Group I was exposed to intralipid vehicle, which also served as a vehicle control (Figure 1).
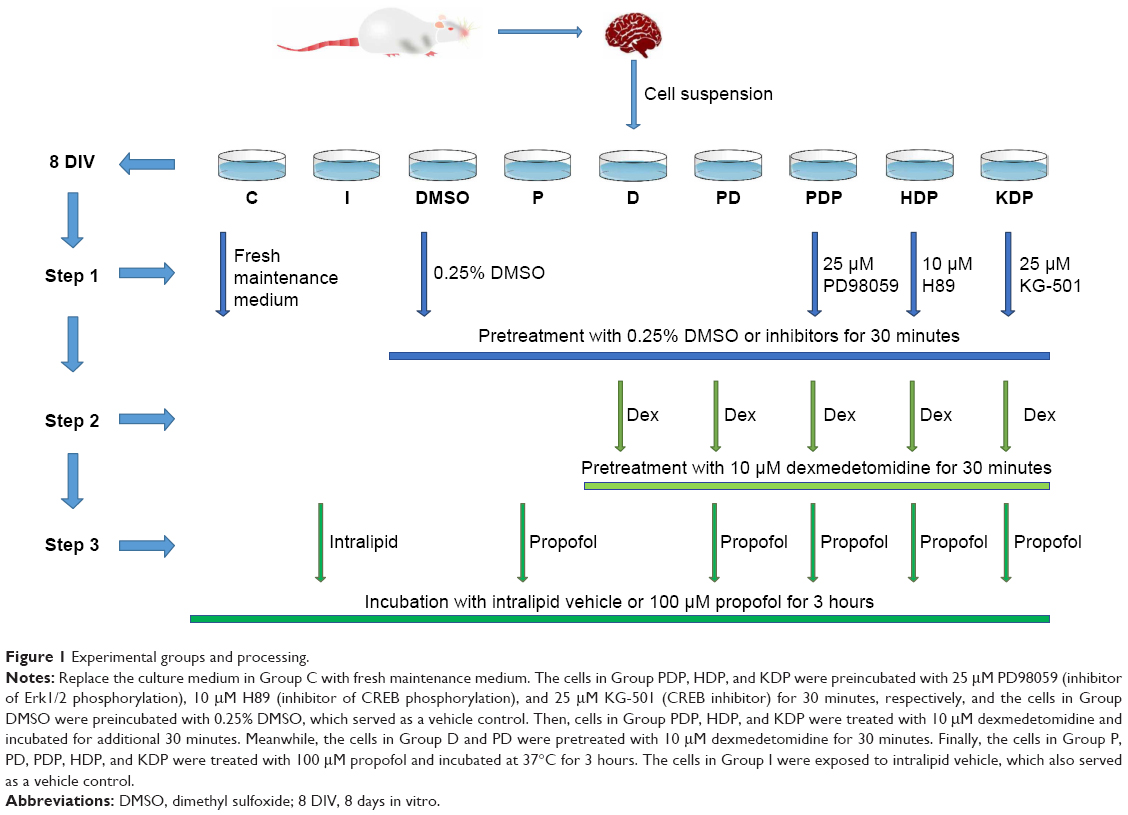 | Figure 1 Experimental groups and processing. |
Neuronal cell viability was measured by cell counting kit-8 (CCK-8), and neuronal cell death was analyzed by flow cytometry (Annexin V/propidium iodide). The expression of the Erk1/2, CREB, and BDNF mRNAs was determined using qRT-PCR. Additionally, we assessed the levels of total and phosphorylated Erk1/2 and CREB proteins, the BDNF protein, and the apoptosis-related proteins cleaved-caspase3, Bcl-2, and Bax. Samples for detection in each group at least repeated for three times, and each group has at least three independent batch samples.
Neuronal cell viability evaluations
Neuronal cell viability was determined by CCK-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan, Cat# CK04). One hundred microliters of suspended cells were plated in a 96-well plate at a density of 5×103 cells/well and preincubated for 7 days in a humidified incubator at 37°C with 5% CO2 atmosphere. After the experimental treatment, the entire volume of culture medium was replaced with 200 μL of fresh maintenance medium, and 10 μL of CCK-8 solution were added to each well of the plate. Absorbance at a 450 nm wavelength was detected by a microplate reader (Thermo Fisher Scientific Inc.) after the 96-well plate was incubated at 37°C for 2 hours.
Apoptosis evaluations
Transmission electron microscopy
Primary hippocampal neurons were treated as described above, trypsinized, and collected by centrifugation at 1,000 rpm for 5 minutes. The collected neurons were thoroughly fixed with 2.5% glutaraldehyde for 6 hours, rinsed with PBS (pH 7.4), dehydrated with increasing concentrations of ethanol, embedded, sliced, and double stained with uranyl acetate and lead citrate.16 Then, neuronal morphology and apoptosis were observed using a HITACHI H-7650 transmission electron microscope.
Flow cytometry analysis
Neuronal cell was harvested, stained with Annexin V/propidium iodide (BD Biosciences, Cat# 556547), and analyzed by the FACSCalibur flow cytometer (BD Biosciences). In present experiment, a nonstained control tube and a single color tube were used to control the gate according to the manufacturer’s constructions.
qRT-PCR
Total mRNA was extracted using RNAiso Plus (TaKaRa Bio Inc., Tokyo, Japan, Cat# 9108) and was reverse-transcribed into cDNA by PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa Bio Inc., Cat# RR047A). qPCR was performed with SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) (TaKaRa Bio Inc., Cat# RR820A). Primer sequences were as follows: MAPK3, forward 5′-CTACACGCAGCTGCAGTACATC-3′ and reverse 5′-GTGCGCTGACAGTAGGTTTGA-3′; MAPK1, forward 5′-GCGTTGGTACAGAGCTCCAGAA-3′ and reverse 5′-TGCAGCCCACAGACCAAATATC-3′; CREB, forward 5′-ACAGTTCAAGCCCAGCCACAG-3′ and reverse 5′-GCACTAAGGTTACAGTGGGAGCAGA-3′; BDNF, forward 5′-CAGCGCGAATGTGTTAGTGGTTA-3′ and reverse 5′-CAGTGGACAGCCACTTTGTTTCA-3′; and GAPDH, forward 5′-ACAGCAACAGGGTGGTGGAC-3′ and reverse 5′-TTTGAGGGTGCAGCGAACTT-3′. The qRT-PCR reaction conditions were predenaturation at 95°C for 30 seconds and PCR reaction at 95°C for 5 seconds and 60°C for 34 seconds (Applied Biosystems 7500 Real-Time PCR System).
Western blotting
Neuronal cell was scraped off the dish and homogenized with brief sonication in ice-cold lysis buffer (0.5 mL per 5×106 cells) (Beijing Solarbio Science & Technology Co.). After brief centrifugation at 12,000 rpm for 20 minutes to remove insoluble tissues, the protein concentration of each homogenate was determined using the BCA kit (Beijing Solarbio Science & Technology Co.). Each cell lysate was added with loading buffer (5×, Beijing Solarbio Science & Technology Co.) and then boiled at 100°C for 5 minutes. Equal amounts of neuronal cell lysate were separated on 12% SDS-PAGE gels (Beijing Solarbio Science & Technology Co.) and immunoblotted onto polyvinylidene fluoride membranes (0.22 μm; EMD Millipore, Billerica, MA, USA). Membranes were blocked with 5% BSA (Beijing Solarbio Science & Technology Co.) for 1 hour and incubated overnight at 4°C with antibodies against Erk1/2 (Cell Signaling Technology, Beverly, MA, USA, Cat#4695S), p-Erk1/2 (Cell Signaling Technology, Cat#4377S), CREB (Cell Signaling Technology, Cat#9197S), p-CREB (Cell Signaling Technology, Cat#9198S), BDNF (Abcam, Cat#ab108319), cleaved-caspase3 (Cell Signaling Technology, Cat#9661), Bcl-2 (Abcam, Cat#ab196495), Bax (Abcam, Cat#ab32503), or GAPDH (Proteintech, Chicago, IL, USA, Cat#10494-1-AP) overnight. Membranes were rinsed with tris buffered saline tween (TBST) (Beijing Solarbio Science & Technology Co.) and then incubated with a fluorescent dye-conjugated secondary antibody (LI-COR Biosciences, Lincoln, NE, USA) for 3 hours. Images were acquired by an Odyssey system (LI-COR Biosciences) after the membranes were rinsed with TBST three times for 5 minutes each.
Statistical analysis
SPSS 22.0 (IBM Corporation, Armonk, NY, USA) and GraphPad Prism 6.07 software (OriginLab, Northampton, MA, USA) were used for statistical analyses. Student’s t-test was used to compare two experimental groups and one-way ANOVA was used to compare several treatments groups. When appropriate, Bonferroni’s post hoc comparison test was used. P-values <0.05 were considered as statistically significant.
Results
Hippocampal neuron cultures
Changes in the morphology of primary hippocampal neurons were recorded from 4 hours after plating to 9 DIV (Figure 2). Additionally, primary hippocampal neurons cultured for 8 DIV were subjected to identification and purity analyses using immunocytochemistry with an NSE antibody. Under the light microscope, the hippocampal neuronal cell bodies, dendrites, and axons were stained brown, and the purity of hippocampal neurons was 95.2%±3.6% (Figure 3).
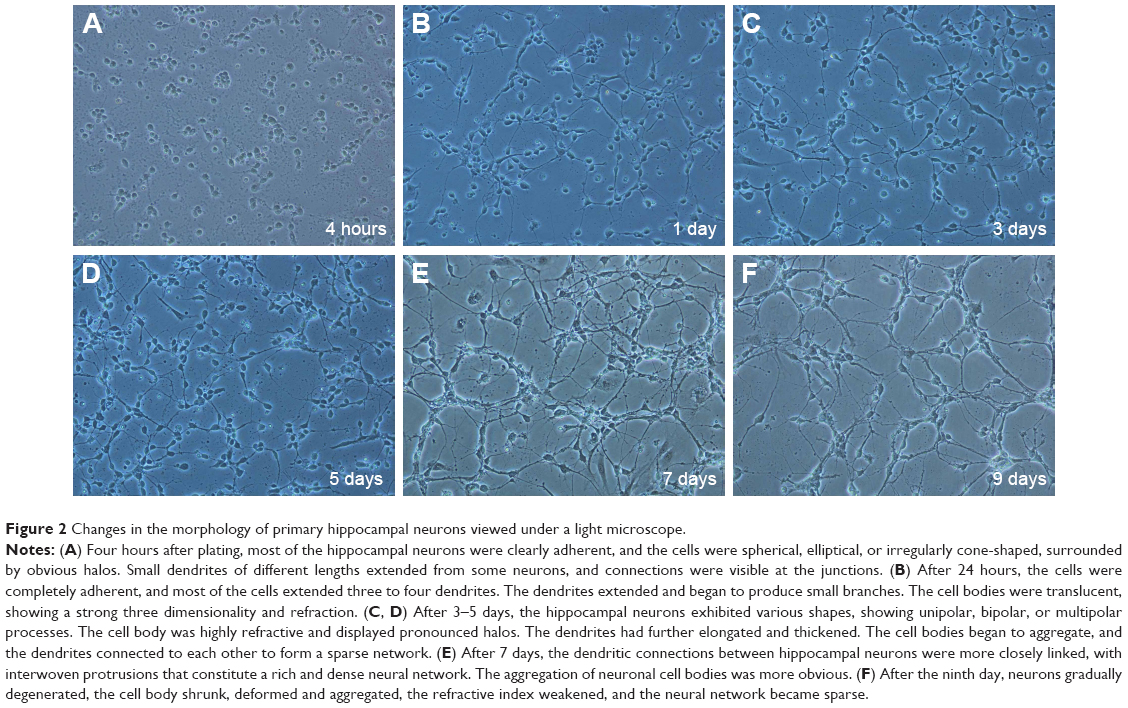 | Figure 2 Changes in the morphology of primary hippocampal neurons viewed under a light microscope. |
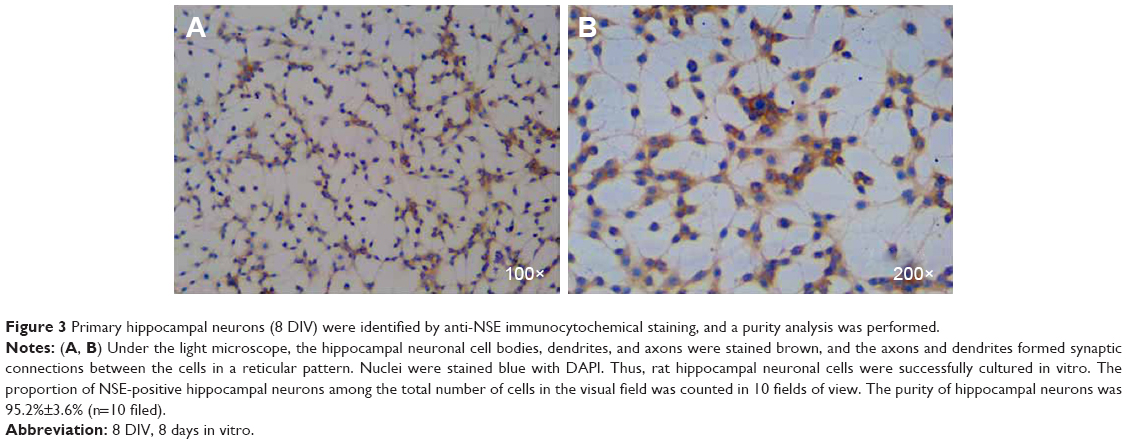 | Figure 3 Primary hippocampal neurons (8 DIV) were identified by anti-NSE immunocytochemical staining, and a purity analysis was performed. |
Dexmedetomidine alleviates propofol-induced neuroapoptosis and promotes neuronal survival
A significant increase in neuroapoptosis was observed under the transmission electron microscope after hippocampal neurons had been exposed to propofol (Figure 4A). CCK-8 assays revealed that the treatment with 100 μM propofol markedly reduced the neuronal viability by 32.71% compared with Group C (Figure 4B). Furthermore, propofol significantly increased the percentage of apoptotic cells (Figure 4C and D), increased the expression of the cleaved-caspase3 protein, and decreased the Bcl-2/Bax ratio in hippocampal neurons (Figure 4E–I). Whereas fewer neuroapoptosis was observed after dexmedetomidine pretreatment than in Group P (Figure 4A). Compared with neurons exposed to propofol, dexmedetomidine remarkably promoted neuronal viability (Figure 4B). Moreover, dexmedetomidine notably reduced the percentage of apoptotic cells (Figure 4C and D), increased the Bcl-2/Bax ratio, and decreased the level of the cleaved-caspase3 protein in hippocampal neurons (Figure 4E–I).
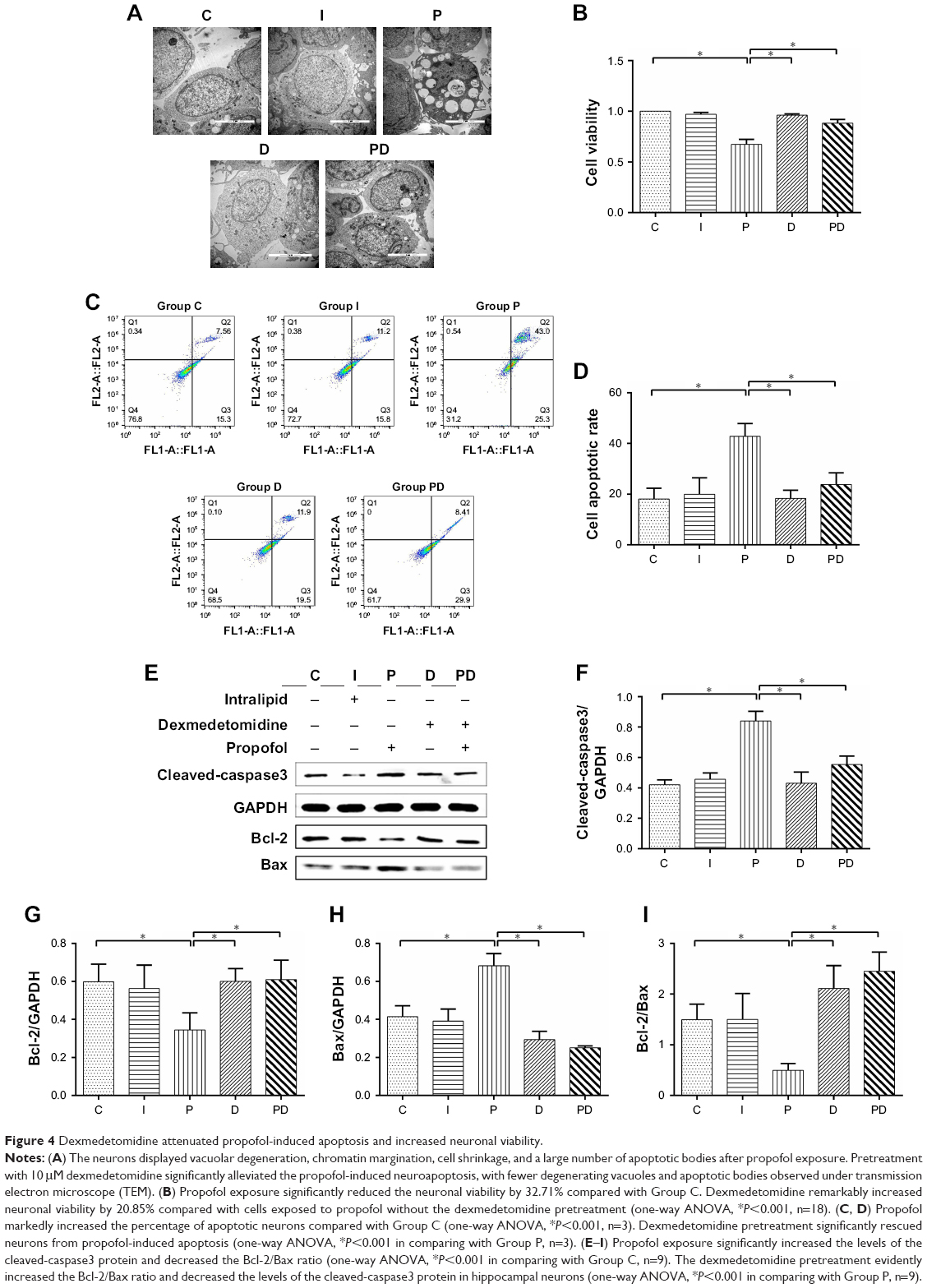 | Figure 4 Dexmedetomidine attenuated propofol-induced apoptosis and increased neuronal viability. |
Dexmedetomidine rescues the propofol-induced downregulation of p-Erk1/2, p-CREB, and BDNF
Propofol has been reported to induce significant alterations in the levels of neurotrophins and their receptors, as well as downstream effector kinases, which might occur through Akt/Erk signaling pathways,17 after 14-day-old Wistar rats were exposed to a single propofol dose of 25 mg/kg. In the present study, we proposed that Erk1/2 and its related downstream signaling pathways play a role in propofol-induced cytotoxicity. Therefore, we evaluated the effect of propofol on the expression of Erk1/2, p-Erk1/2, CREB, p-CREB, and BDNF in hippocampal neurons. The levels of the Erk1/2, p-Erk1/2, CREB, p-CREB, and BDNF proteins and the levels of Erk1/2, CREB, and BDNF mRNAs were determined by Western blotting and qRT-PCR, respectively. Compared with Group C, propofol markedly downregulated the expression of the BDNF mRNA (Figure 5A), but the expression of the Erk1/2 and CREB mRNAs and proteins was not significantly altered (Figures 5A and 6B–E). However, propofol reduced the levels of the p-Erk1/2, p-CREB, and BDNF proteins compared with the cells in Group C (Figure 6B–E).
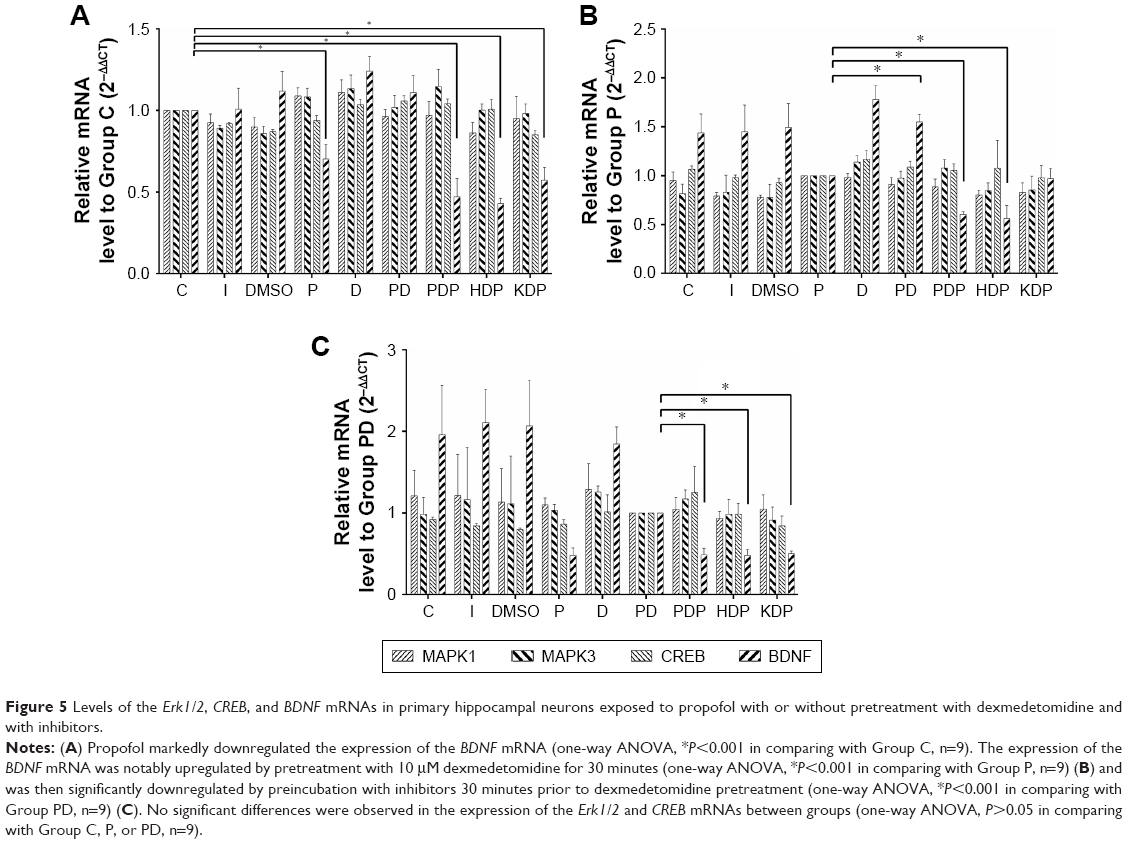 | Figure 5 Levels of the Erk1/2, CREB, and BDNF mRNAs in primary hippocampal neurons exposed to propofol with or without pretreatment with dexmedetomidine and with inhibitors. |
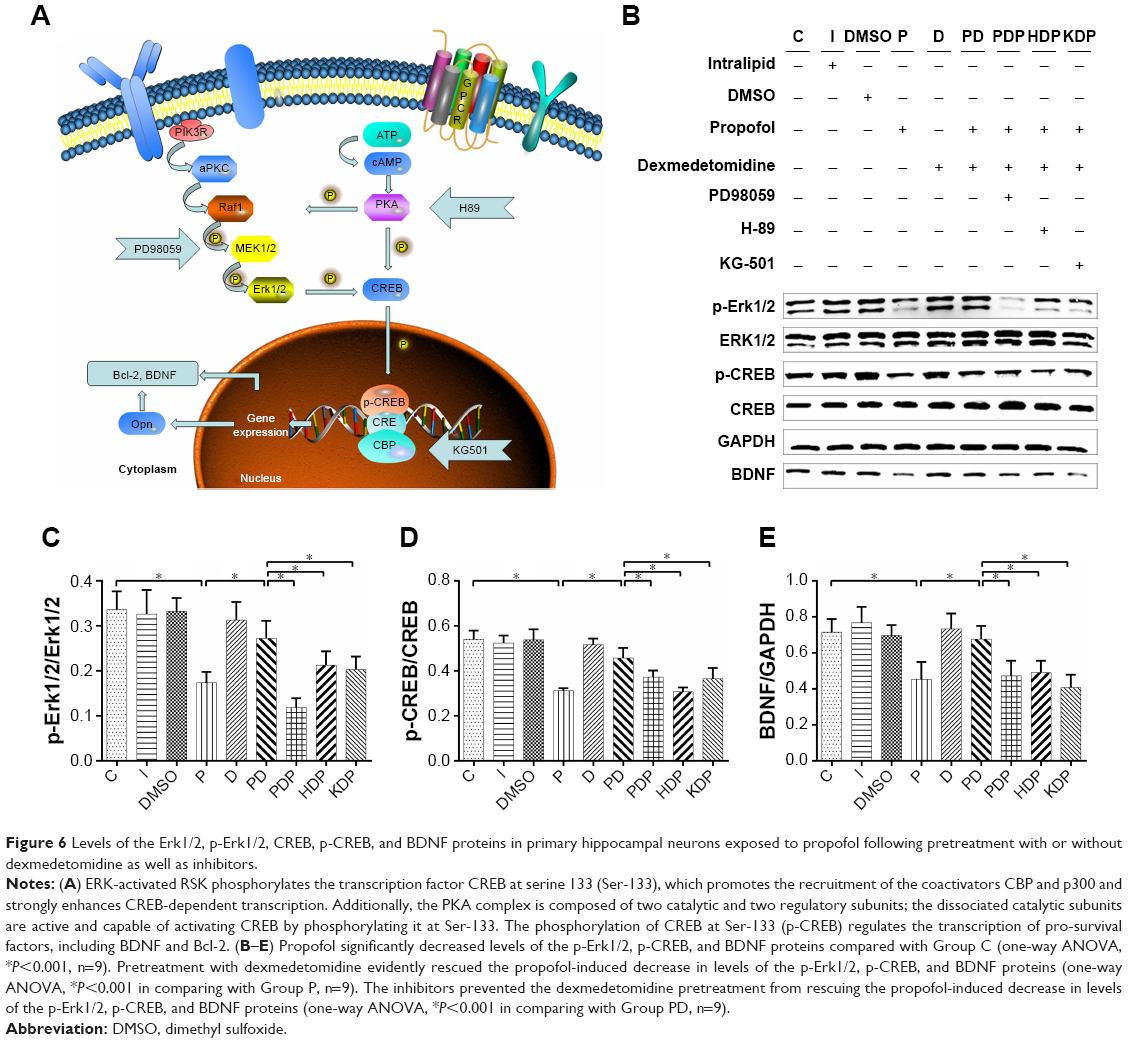 | Figure 6 Levels of the Erk1/2, p-Erk1/2, CREB, p-CREB, and BDNF proteins in primary hippocampal neurons exposed to propofol following pretreatment with or without dexmedetomidine as well as inhibitors. |
Dexmedetomidine exerts its neuroprotective effect by binding to imidazoline I1 receptors and modulating histone H3 acetylation in dopaminergic neurons in the striatum via the Erk1/2 signaling pathways.11–13 In the present study, pretreatment with dexmedetomidine evidently rescued the propofol-induced downregulation of both the BDNF mRNA (Figure 5B) and reduced levels of the p-Erk1/2, p-CREB, and BDNF proteins (Figure 6B–E). No notable difference was observed in the expression of the Erk1/2 and CREB mRNAs and proteins (Figures 5B and 6B–E) in cells pretreated with or without dexmedetomidine.
The protective effect of dexmedetomidine is partially abolished by inhibitors of p-Erk1/2, p-CREB, and CREB
Primary hippocampal neurons in Group PDP, HDP, and KDP were preincubated with PD98059, H89, and KG501, respectively, before pretreatment with dexmedetomidine. The inhibitors weakened the protective effect of dexmedetomidine on propofol-induced neurotoxicity and apoptosis (Figure 7C and D), leading to a significant increase in the number of apoptotic bodies (Figure 7A) and reduction in neuronal viability (Figure 7B), as well as the upregulation of the cleaved-caspase3 and Bax proteins (Figure 7E–I). Furthermore, phosphorylation inhibitors prevented the dexmedetomidine pretreatment from rescuing the propofol-induced downregulation of the BDNF mRNA (Figure 5C), decreased levels of the p-Erk1/2, p-CREB, and BDNF proteins (Figure 6B–E), and the reduced Bcl-2/Bax ratio (Figure 7E–I).
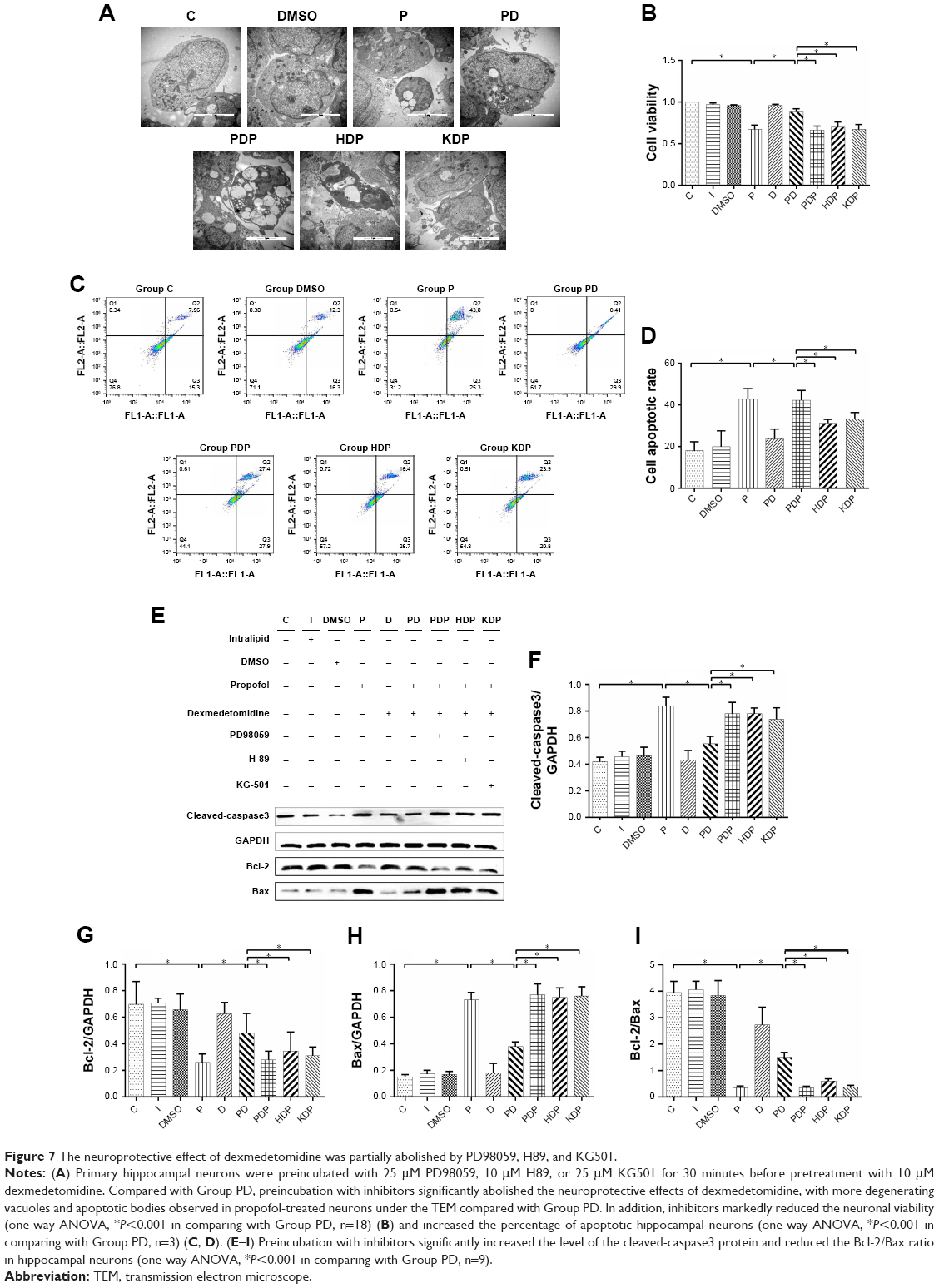 | Figure 7 The neuroprotective effect of dexmedetomidine was partially abolished by PD98059, H89, and KG501. |
Discussion
The present study used primary hippocampal neurons to evaluate the neuroprotective effect of dexmedetomidine on propofol-induced neuroapoptosis in vitro. The 100 μM propofol treatment markedly reduced neuronal viability, triggered neuroapoptosis, and downregulated the levels of p-Erk1/2, p-CREB, and BDNF. Preincubation of hippocampal neurons with 10 μM dexmedetomidine before propofol exposure significantly alleviated the propofol-induced neuroapoptosis and decrease in neuronal viability and upregulated the levels of p-Erk1/2, p-CREB, and BDNF. However, the protective effect of dexmedetomidine was eliminated by pretreatment with PD98059, H89, and KG501 prior to dexmedetomidine exposure. Thus, dexmedetomidine rescued propofol-induced neuroapoptosis in primary hippocampal neurons, which was related to upregulated levels of p-Erk1/2, p-CREB, and BDNF.
To date, compelling evidence from basic research, including studies of developing rodents,18 nonhuman primates,19 human stem cell-derived neurons,20 and preclinical studies,2–4 has raised serious concerns about the neurotoxicity of propofol and subsequent development of potential cognitive impairments. In the present study, primary hippocampal neurons at 7–8 DIV served as a model of developing brain and are extremely sensitive to general anesthetics. Incubated with 100 μM propofol for 3 hours is sufficient enough to induce significant neuronal apoptosis in the neonatal brain,21,22 thus establishing the developmental brain injury model. Propofol exposure remarkably reduced viability and increased the neuroapoptosis, as indicated by CCK-8 assays, transmission electron microscopy, flow cytometry, and Western blot analyses, indicating that the developmental brain injury model was successfully established. Meantime, these results further validated previous reports revealing the neurotoxicity of propofol in vivo9 and vitro.23,24
Dexmedetomidine has been reported to alleviate propofol-induced neuroapoptosis in the neonatal brain,22,25–27 but the underlying mechanism remains unclear. This study reassessed the protective effect of dexmedetomidine on the established propofol-induced developmental brain injury model in vitro. According to previous studies,26 0.1–100 μM dexmedetomidine attenuates cortical neuron apoptosis induced by either wortmannin or staurosporine in vitro in a dose-dependent manner. However, in the study by Laudenbach et al,28 dexmedetomidine concentrations greater than 10 μM provided less protection than lower concentrations. Therefore, we chose to pretreat primary hippocampal neurons with 10 μM dexmedetomidine prior to exposure to propofol based on the effective dose of dexmedetomidine and the possible neurotoxicity of high concentrations. An evident increase in neuronal cell viability and a reduction in neuronal cell apoptosis were observed in neurons that had been pretreated with 10 μM dexmedetomidine, as revealed by electron microscopy, CCK-8 assays, flow cytometry analysis, and Western blotting. Additionally, dexmedetomidine did not reduce neuronal viability and survival at this concentration, suggesting that 10 μM dexmedetomidine effectively attenuated the neurotoxic effect of propofol on primary hippocampal neurons without producing additional deleterious effects. As shown in other in vivo studies, high dexmedetomidine concentrations completely alleviate propofol-induced neurotoxicity,11,27 but these studies lacked an investigation of the potential neurotoxicity of high dexmedetomidine concentrations.26 Moreover, high dexmedetomidine concentrations failed to attenuate isoflurane-induced cortical neuron injury in one study.26 This difference may be due to differences in experimental methods and agent-specific neurotoxicity in these studies.
Although the neuroprotective effects of dexmedetomidine on different neural injury models have been elucidated,11,14 the underlying mechanism remains incompletely understood. Previous studies reported that the protective effects of dexmedetomidine pretreatment are associated with the activation of α2-adrenergic receptors,22 JNK, P38 MAPK ,29 PKC, and the phosphoinositide 3-kinase/Akt pathways.25,30 More recently, dexmedetomidine was reported to exert its neuroprotective effects partially by binding to imidazoline I1 receptors and modulating histone acetylation via Erk1/2 pathways.11,12 CREB is a transcription factor downstream of both Akt and Erk1/2 signaling pathways,31 and its transcriptional products are mainly pro-survival factors such as BDNF and Bcl-2. The cascade activation of CREB regulates survival, differentiation, synaptic plasticity, and memory maintenance.32,33 Pretreatment with dexmedetomidine was recently shown to preserve neurological function and attenuate neuronal injury following thoracic aortic occlusion in mice, which was associated with an increase in the phosphorylation of protein kinase B and CREB and the subsequent increases in the levels of the antiapoptotic protein Bcl-2 and BDNF.14,34 In addition, neurotrophic factors and BDNF are involved in isoflurane- and propofol-induced reductions in synapse density in the developing hippocampus.35 However, researchers have not yet completely elucidated whether Erk1/2/CREB/BDNF pathways are involved in the neuroprotective effects of dexmedetomidine on propofol-induced neurotoxicity.
Erk-activated RSK phosphorylates the transcription factor CREB at serine 133 (Ser-133), which allows recruitment of the coactivators, CREB-binding protein (CBP), and p300 and strongly enhances CREB-dependent transcription. Additionally, the PKA complex is composed of two catalytic and two regulatory subunits; the dissociated catalytic subunits are active and capable of activating CREB by phosphorylating it at Ser-133. Phosphorylation of CREB at Ser-133 (p-CREB) triggers the recruitment of the coactivator CBP and regulates the transcription of pro-survival factors such as BDNF and Bcl-2 (Figure 6A). In present study, we detected the levels of Erk1/2, p-Erk1/2, CREB, p-CREB, and BDNF following propofol administration in cells pretreated with or without dexmedetomidine. Additionally, PD98059, H89, and KG501 were administered to further determine the roles of Erk1/2/CREB/BDNF pathways in the protective effects of dexmedetomidine on propofol-induced neuroapoptosis. Notably, 100 μM propofol markedly downregulated the expression of the BDNF mRNA and levels of the p-Erk1/2, p-CREB, Bcl-2, and BDNF proteins. After pretreatment with 10 μM dexmedetomidine for 30 minutes, dexmedetomidine evidently rescued the propofol-induced downregulation of both the BDNF mRNA and the levels of the p-Erk1/2, p-CREB, Bcl-2, and BDNF proteins. However, the neuroprotective effects of dexmedetomidine were abolished by the administration of a p-Erk1/2 inhibitor (PD98059, 25 μM), p-CREB inhibitor (H89, 10 μM), and CREB inhibitor (KG501, 25 μM) 30 minutes prior to dexmedetomidine pretreatment, leading to significant increase in neuroapoptosis and a reduction in neuronal cell viability and the Bcl-2/Bax ratio, as well as increased levels of the cleaved-caspase3 protein. Furthermore, inhibitors prevented the dexmedetomidine pretreatment from rescuing the propofol-induced downregulation of the mRNA of BDNF, and the protein expression of p-Erk1/2, p-CREB, BDNF, and Bcl-2 revealed by qRT-PCR and Western blotting analyses, respectively. These findings indicated that propofol-induced neuroapoptosis was partially associated with the inhibition of Erk1/2/CREB/BDNF pathways, whereas dexmedetomidine could activate the Erk1/2/CREB/BDNF pathways, thus providing a neuroprotective effect on propofol-induced neurotoxicity.
Conclusion
Our study illustrated that dexmedetomidine alleviated propofol-induced cytotoxic effect on primary hippocampal neurons in vitro, which might be partially correlated with the activation of Erk1/2/CREB /BDNF signaling pathways.
Acknowledgments
This work was supported by National Natural Science Foundation of China (No 81373498 and 81060277) and Natural Science Foundation of Guangxi (2017GXNSFBA198108). We are grateful to the staff involved in this research, especially Ms. Zhou Sijia, for their concern, support, and understanding during our experiment and writing.
Author contributions
YX and QX made the same contributions to this paper. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Andropoulos DB, Greene MF. Anesthesia and developing brains – implications of the FDA warning. N Engl J Med. 2017;376(10):905–907. | ||
Davidson AJ, Disma N, de Graaff JC, et al. Neurodevelopmental outcome at 2 years of age after general anaesthesia and awake-regional anaesthesia in infancy (GAS): an international multicentre, randomised controlled trial. Lancet. 2016;387(10015):239–250. | ||
Gleich SJ, Flick R, Hu D, et al. Neurodevelopment of children exposed to anesthesia: design of the Mayo Anesthesia Safety in Kids (MASK) study. Contemp Clin Trials. 2015;41:45–54. | ||
Sun LS, Li G, Dimaggio CJ, et al. Feasibility and pilot study of the pediatric anesthesia neurodevelopment assessment (PANDA) project. J Neurosurg Anesthesiol. 2012;24(4):382–388. | ||
Twaroski DM, Yan Y, Zaja I, Clark E, Bosnjak ZJ, Bai X. Altered mitochondrial dynamics contributes to propofol-induced cell death in human stem cell-derived neurons. Anesthesiology. 2015;123(5):1067–1083. | ||
Creeley C, Dikranian K, Dissen G, Martin L, Olney J, Brambrink A. Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. Br J Anaesth. 2013;110(Suppl 1):i29–i38. | ||
Twaroski DM, Yan Y, Olson JM, Bosnjak ZJ, Bai X. Down-regulation of microRNA-21 is involved in the propofol-induced neurotoxicity observed in human stem cell-derived neurons. Anesthesiology. 2014;121(4):786–800. | ||
Lv J, Wei Y, Chen Y, et al. Dexmedetomidine attenuates propofol-induce neuroapoptosis partly via the activation of the PI3k/Akt/GSK3β pathway in the hippocampus of neonatal rats. Environ Toxicol Pharm. 2017;52:121–128. | ||
Wang Y, Wu C, Han B, et al. Dexmedetomidine attenuates repeated propofol exposure-induced hippocampal apoptosis, PI3K/Akt/Gsk-3β signaling disruption, and juvenile cognitive deficits in neonatal rats. Mol Med Rep. 2016;14(1):769–775. | ||
Xiao Y, Zhou L, Tu Y, et al. Dexmedetomidine attenuates the propofol-induced long-term neurotoxicity in the developing brain of rats by enhancing the PI3K/Akt signaling pathway. Neuropsychiatr Dis Treat. 2018;14:2191–2206. | ||
Hu SP, Zhao JJ, Wang WX, et al. Dexmedetomidine increases acetylation level of histone through ERK1/2 pathway in dopamine neuron. Hum Exp Toxicol. 2017;36(5):474–482. | ||
Schoeler M, Loetscher PD, Rossaint R, et al. Dexmedetomidine is neuroprotective in an in vitro model for traumatic brain injury. BMC Neurol. 2012;12:20. | ||
Wang Y, Han R, Zuo Z. Dexmedetomidine post-treatment induces neuroprotection via activation of extracellular signal-regulated kinase in rats with subarachnoid haemorrhage. Br J Anaesth. 2016;116(3):384–392. | ||
Bell MT, Puskas F, Bennett DT, et al. Dexmedetomidine, an α-2a adrenergic agonist, promotes ischemic tolerance in a murine model of spinal cord ischemia-reperfusion. J Thorac Cardiovasc Surg. 2014;147(1):500–507. | ||
Beaudoin GM, Lee SH, Singh D, et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7(9):1741–1754. | ||
Marsboom G, Toth PT, Ryan JJ, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110(11):1484–1497. | ||
Popic J, Pesic V, Milanovic D, et al. Propofol-induced changes in neurotrophic signaling in the developing nervous system in vivo. PLoS One. 2012;7(4):e34396. | ||
Koo E, Oshodi T, Meschter C, Ebrahimnejad A, Dong G. Neurotoxic effects of dexmedetomidine in fetal cynomolgus monkey brains. J Toxicol Sci. 2014;39(2):251–262. | ||
Li J, Xiong M, Nadavaluru PR, et al. Dexmedetomidine attenuates neurotoxicity induced by prenatal propofol exposure. J Neurosurg Anesthesiol. 2016;28(1):51–64. | ||
Gencer B, Karaca T, Tufan HA, et al. The protective effects of dexmedetomidine against apoptosis in retinal ischemia/reperfusion injury in rats. Cutan Ocul Toxicol. 2014;33(4):283–288. | ||
Cattano D, Young C, Straiko MM, Olney JW. Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth Analg. 2008;106(6):1712–1714. | ||
Sanders RD, Xu J, Shu Y, et al. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology. 2009;110(5):1077–1085. | ||
Spahr-Schopfer I, Vutskits L, Toni N, Buchs PA, Parisi L, Muller D. Differential neurotoxic effects of propofol on dissociated cortical cells and organotypic hippocampal cultures. Anesthesiology. 2000;92(5):1408–1417. | ||
Vutskits L, Gascon E, Tassonyi E, Kiss JZ. Clinically relevant concentrations of propofol but not midazolam alter in vitro dendritic development of isolated gamma-aminobutyric acid-positive interneurons. Anesthesiology. 2005;102(5):970–976. | ||
Li Y, Zeng M, Chen W, et al. Dexmedetomidine reduces isoflurane-induced neuroapoptosis partly by preserving PI3K/Akt pathway in the hippocampus of neonatal rats. PLoS One. 2014;9(4):e93639. | ||
Sanders RD, Sun P, Patel S, Li M, Maze M, Ma D. Dexmedetomidine provides cortical neuroprotection: impact on anaesthetic-induced neuroapoptosis in the rat developing brain. Acta Anaesthesiol Scand. 2010;54(6):710–716. | ||
Duan X, Li Y, Zhou C, Huang L, Dong Z. Dexmedetomidine provides neuroprotection: impact on ketamine-induced neuroapoptosis in the developing rat brain. Acta Anaesthesiol Scand. 2014;58(9):1121–1126. | ||
Laudenbach V, Mantz J, Lagercrantz H, Desmonts J-M, Evrard P, Gressens P. Effects of α2-adrenoceptor agonists on perinatal excitotoxic brain injury. Anesthesiology. 2002;96(1):134–141. | ||
Liao Z, Cao D, Han X, et al. Both JNK and P38 MAPK pathways participate in the protection by dexmedetomidine against isoflurane-induced neuroapoptosis in the hippocampus of neonatal rats. Brain Res Bull. 2014;107:69–78. | ||
do SH, Park SJ, Shin HJ, et al. Dexmedetomidine increases the activity of excitatory amino acid transporter type 3 expressed in Xenopus oocytes: the involvement of protein kinase C and phosphatidylinositol 3-kinase. Eur J Pharmacol. 2014;738:8–13. | ||
Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35(4):605–623. | ||
Impey S, McCorkle SR, Cha-Molstad H, et al. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119(7):1041–1054. | ||
Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294(5544):1030–1038. | ||
Huang W, Cao J, Liu X, et al. AMPK plays a dual role in regulation of CREB/BDNF pathway in mouse primary hippocampal cells. J Mol Neurosci. 2015;56(4):782–788. | ||
Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2009;110(4):813–825. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.