")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Development of Obesity: The Driver and the Passenger
Authors Kopp W
Received 2 September 2020
Accepted for publication 3 November 2020
Published 27 November 2020 Volume 2020:13 Pages 4631—4642
DOI https://doi.org/10.2147/DMSO.S280146
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Wolfgang Kopp
Diagnostikzentrum Graz, Graz, 8043, Austria
Correspondence: Wolfgang Kopp
Former Head of Diagnostikzentrum (retired), Mariatrosterstraße 41, Graz 8043, Austria
Email [email protected]
Abstract: Obesity has reached epidemic proportions and is one of the greatest challenges for public health in the twenty-first century. The macronutrient composition of diets, in particular the amount and ratio of carbohydrates, fat and protein, have received considerable attention in recent decades due to its potential relevance to the development of obesity and weight loss. The effects of various macronutrients on body weight regulation are still under debate. High-carbohydrate diets, and particularly high-fat diets, have been blamed for the increase in the prevalence of obesity. This paper shows that neither fat nor carbohydrates are fattening per se. Mixed diets with substantial amounts of fat and high-glycemic carbohydrates, like current WDs, are required to promote weight gain and obesity. High-glycemic carbohydrates are the active partner (the “driver”), which promotes fat storage through its insulinogenic effect, while fat is the passive partner (the “passenger”) on the way to obesity. Elevated insulin levels (postprandial, but more importantly due to hypersecretion and hyperinsulinemia) promote fat storage and play a key role in obesogenesis and the obesity epidemic. Furthermore, mixed diets high in high-glycemic carbohydrates and fat promote fetal programming, with long-term adverse impacts on the offspring, including insulin hypersecretion, (childhood) obesity and metabolic diseases. Maternal obesity and high weight gain during pregnancy have also been linked to deleterious effects on fetal programming. As the global obesity epidemic increasingly affects women of reproductive age, a significant percentage of fetuses will experience fetal programming with a tendency towards obesity – a self-reinforcing process that further fuels the epidemic. A change in lifestyle and diet composition is needed to prevent or limit the development of obesity and related diseases.
Keywords: hypersecretion, hyperinsulinemia, fetal programming, weight gain, insulinogenic Western diet
Introduction
Obesity has reached epidemic proportions and is one of the greatest challenges for public health in the twenty-first century. According to the World Health Organization, overweight and obesity remain the leading causes for premature death worldwide.1 The obesity epidemic, first noted in the US, has spread to all parts of the world during the last decades and continues to rise at an alarming rate in both developed and developing countries. Meanwhile, a dramatic increase in overweight and obesity has also been recognized in children and adolescents. Despite intensive research, the causes of the obesity epidemic remain incompletely understood.2,3
The traditional concept of energy balance implies that long-term weight maintenance requires a sustainable macronutrient balance so that the fuel mix consumed is equal to the fuel mix oxidized. Obesity results from a chronic surplus of energy intake compared to energy expenditure. Several factors are thought to interact to produce a state of positive energy balance, including diet, lifestyle, lack of physical activity, genetic, epigenetic, metabolic, behavioral and environmental influences.2–4
The macronutrient composition of diets, in particular the amount and ratio of carbohydrates, fat and protein, have received considerable attention in recent decades due to its potential relevance to the development of obesity and weight loss. The effects of various macronutrients on body weight regulation are still under debate. High-fat as well as high-carbohydrate diets have been blamed for the increase in the prevalence of obesity.5–8
Diet Composition and Metabolic Consequences
The macronutrients fat, carbohydrates and protein are broken down into glucose, fatty acids (FA) and amino acids. Adenosine triphosphate is the main source of energy in the human body. Most of it is produced in the mitochondria through oxidative phosphorylation of glucose and FAs. Glucose and FAs serve as main fuels, amino acids serve as building blocks or can be converted into glucose or fat for storage or oxidation.9
Macronutrient composition has significant metabolic consequences regarding substrate use and storage. Depending on diet composition, an “adipocentric” or a “glucocentric” metabolism develops.10 Low-carbohydrate diets (for definition see Westman et al10), like the traditional diets of some hunter-gatherer societies,11 are associated with an adipocentric metabolism. Low-carbohydrate diets are metabolically “simple”. FAs and ketone bodies, produced by the liver from FAs, serve as the main fuel sources. Fat is easily stored and released into circulation on demand, while glucose is primarily provided for glucose-dependent tissues. This clear separation of cell fuels is maintained during feeding and fasting. Glucose-dependent tissues (ie red blood cells, retina, lens, and renal medulla) receive glucose through gluconeogenesis and glycogenolysis. Proteins serve both as building blocks and as sources of energy. In contrast, mixed diets with substantial amounts of carbohydrates and fat, like current Western diets (WD), are associated with a glucocentric metabolism.10,12 Here, the situation is more complex, since glucose and FAs both serve as main fuels, competing for mitochondrial oxidization. Basically, mitochondria are able to select fuel in response to nutritional circumstances and switch freely between alternative fuels in order to adjust fuel oxidation to fuel availability (termed as “metabolic flexibility”), but function smoothly best when acetyl-CoA is produced from one fuel at a time. Therefore, a glucocentric metabolism requires nutrient partitioning in order to avoid mitochondrial congestion and metabolic “gridlock”.13
Nutrient partitioning is primarily mediated by the counter-regulatory hormones insulin and glucagon. During the fed state, a high insulin/glucagon ratio promotes lipid storage, while glucose serves as the main fuel. In the fasted state, a high glucagon/insulin ratio stimulates lipolysis, and FAs serve as the main fuel, while glucose supply to glucose-dependent tissues is provided through gluconeogenesis and glycogenolysis. Therefore, during the fed state, glucose serves as the main fuel and fat is stored, while during the fasted state, FAs serve as the main fuel and glucose is preserved for glucose-dependent tissues.10,12
Dietary Fat or Carbohydrates: Who is the Culprit?
Fat
It is widely held that diets high in fat are mainly responsible for the epidemic of obesity.5,6 However, if fat per se is fattening, diets containing very high amounts of fat should cause weight gain and obesity. Numerous studies have shown that ad libitum high-fat, low-carbohydrate ketogenic diets (<50 g carbohydrates/day, preferably of low-glycemic type) do not promote weight gain, but weight maintenance or weight loss.10 A number of ad libitum feeding studies showed that caloric intake decreases spontaneously and weight loss is induced in obese14,15 and non-obese individuals16,17 when carbohydrate intake is restricted. The fact that ketogenic diets are very high in fat contradicts or at least does not support the view that the energy density of diets is the main driver behind the development of obesity.18 Although the hunger-reducing effects of ketogenic diets are well documented, the mechanisms are still under debate. Evidence suggests that ketone bodies, mainly beta-hydroxybutyrate, may directly suppress appetite via changes in plasma levels of the “hunger hormone” ghrelin.19 Furthermore, a decrease in insulin levels may play a role. On the one hand, suppression of insulin, for instance with octreotide or diazoxide, leads to weight loss.20 On the other hand, several studies have found that insulin increases food intake and that foods with a high insulin response are less satiating.10
Carbohydrates
Carbohydrates currently play an extremely important role in human nutrition. WDs contain carbohydrate staples that are mostly high-glycemic in effect, like refined cereals (currently, 85% of the cereals consumed in the US diet are highly processed refined grains), corn, potatoes and sugars.11 Sugar (sucrose and fructose) in particular is consumed in large quantities, not only in beverages but also as sweets and as an additive in bakeries and ready meals. Carbohydrate is the macronutrient with the greatest impact on postprandial blood glucose and insulin response. The glycemic index (GI) and glycemic load (GL) are major determinants of postprandial glucose excursion. The GI ranks carbohydrates according to their effect on blood glucose levels. The higher the GI, the faster and higher the rise in postprandial blood glucose levels (and vice versa). GL is another method for evaluating the glycemic effect of carbohydrate foods, considering the serving size (GL = the product of GI and amount of carbohydrate of a meal).21,22 A high GI, a high GL and a high postprandial insulin response are characteristic features of current WDs.
The plasma concentration of glucose is strictly regulated, and excess glucose must be removed from the circulation immediately. Glucose homeostasis therefore requires a continuous balance between glucose transport, storage and metabolism. The fate of the glucose ingested depends on the nutritional and metabolic context. Glucose can be oxidized (for energy generation), dissipated (via thermogenesis), stored as glycogen (mainly in liver and skeletal muscles) or converted to fatty acids in the liver (without a significant contribution to the total fat balance).23 According to animal and human studies, protein and carbohydrate intake promotes its own oxidation.24 Therefore, the human body is extremely good at adapting carbohydrate oxidation to carbohydrate intake.
It has been suggested that high-carbohydrate diets represent a key factor in the development of obesity.7,8 However, if carbohydrates per se are fattening, diets containing very high amounts of carbohydrate should cause weight gain and obesity. A number of ad libitum feeding studies (in rodents and humans) with high-carbohydrate/low-fat diets do not support this view. These diets consumed ad libitum (and not designed as low-glycemic carbohydrate diets) did not cause weight gain, but rather (moderate) weight loss.25–28
It Takes Two to Tango
As shown above, neither carbohydrates nor fat are fattening per se. In fact, it requires mixed diets with substantial amounts of fat and high-insulinogenic carbohydrates, like current WDs, for weight gain, as feeding studies on rodents and humans have shown. So-called “high-fat diets” are not only high in fat, but usually also contain substantial amounts of high-glycemic carbohydrate. For example, while low-carbohydrate/high-fat ketogenic diets promote weight loss, mixed high-fat/high-glycemic carbohydrate research diets, like D12492 (60% fat, 20% carbohydrates [sucrose and maltodextrin], 20% protein) or D12451 (45% fat, 35% carbohydrates [sucrose, maltodextrin, cornstarch], 20% protein) are used to cause obesity in animal studies.29 An ad libitum feeding study in mice, comparing fat mass gain over 8 weeks of free access to unrefined low-fat, refined low-fat, and refined high-fat diets showed that significant weight gain only occurred in the refined high-fat group.30 In a prospective rodent feeding study comparing a low-fat/low-sugar standard diet and a high-carbohydrate/high-fat WD, mice on the WD quickly became hyperphagic, gained weight and became obese.31 Ad libitum feeding studies in humans comparing mixed carbohydrate/low-fat and carbohydrate/high-fat diets provided similar results.32 Weight gain was proportional to the fat content of a mixed diet in another feeding study. Female C57BL/6J mice were fed different levels of a safflower oil (10, 20, 30, 40, 50, and 60% of total energy) diet ad libitum for 15 weeks. Graded increments of body weight and fat mass were observed, and significant weight increases were manifested in diets with 30% fat and more.33
As mentioned before, mixed (high-fat, high-carbohydrate) diets have a glucocentric metabolism, in which glucose is oxidized and fat stored during feeding, while fatty acids are released and oxidized in the fasted state. Under normal physiological conditions, the fat stored during feeding is oxidized during the fasting interval between meals and especially during the night, which leads to a sustainable macronutrient balance and a stable body weight.
However, mobilization and oxidation of FFAs in the fasted state depend on the normalization of insulin levels, since high insulin levels prevent lipolysis while maintaining FFA re-esterification.34 Depending on diet and lifestyle, for instance, frequent snacking and consumption of sucrose-containing beverages,35 fasting intervals may be too short to oxidize all of the fat stored during the fed state. As a result, part of the (daily) stored fat can remain stored and cause temporary weight fluctuations or contribute to a gradual increase in fat mass. Even small differences in substrate oxidation may cause long-term gradual weight gain. High-glycemic carbohydrates produce a high insulin response that promotes postprandial oxidation of carbohydrates at the expense of fat oxidation.7,8 Therefore, the main regulator of fat oxidation in the human body is the amount and, in particular, the quality of carbohydrates consumed and the related insulin response. Postprandial insulin levels control fat oxidation and therefore, body weight. Lowering insulinemia through the use of low-GI foods leads to better access to fatty acids as a fuel source, which promotes greater fat oxidation.36 As an example, despite a relatively high proportion of fat, an ad libitum low-glycemic load diet (40% carbohydrate and 35% fat) promoted body fat loss over 6 months and completely prevented weight gain in a subgroup of obese young adults over the following 12 months.37
A Key Role of Insulin Hypersecretion/Hyperinsulinemia
Genetic/epigenetic or diet-induced insulin hypersecretion and (fasting) hyperinsulinemia may be more important for significant weight gain than the physiological postprandial insulin response. While insulin resistance (IR) and compensatory hyperinsulinemia are traditionally thought to be caused by visceral obesity, significant evidence suggests that insulin hypersecretion and hyperinsulinemia precede (and are involved in) the development of IR and obesity.7,38–44 Insulin hypersecretion is a key characteristic of obesity45 and is significantly more prevalent in obesity than IR.46 Hyperinsulinemia is the result of both compensatory (to IR) and primary hypersecretion of insulin.46 The increase in insulin secretion appears to be due to excessive insulin output from an unusually large functional ß-cell mass.45 A body of literature places hyperinsulinemia mechanistically upstream of diet-induced obesity. Insulin hypersecretion and fasting hyperinsulinemia significantly accelerate weight gain,38,40,46–51 especially in the presence of high-fat diets. The importance of abnormally increased insulin levels for weight gain is supported by a large number of studies in humans40,52,53 and animals.39,50,54–58 In a prospective study by Sigal et al52 on insulin-sensitive adults, weight gain over a 16-year period correlated with the magnitude of the acute insulin response to glucose. High rates of weight gain occurred in individuals who presented with an acute glucose-stimulated insulin hypersecretion, and this effect was particularly evident in insulin-sensitive individuals. In French children, an augmented early postprandial insulin response, likely due to ß-cell dysfunction and dysregulation, was the earliest metabolic change in adolescent obesity development.40 Also, in a cohort of 83 children who were followed for 6 years, an increase in fasting insulin levels was significantly associated with an increase in body fat.59 Similarly, fasting plasma insulin concentrations were found to correlate with the rate of weight gain in Pima Indian children.60 The Da Qing Children Cohort Study also showed that fasting plasma insulin levels in early childhood are an independent predictor of subsequent weight gain over a 5-year follow-up period,53 and a study of insulin dynamics among obese schoolchildren showed that insulin hypersecretion preceded the development of IR by several years.61 The CARDIA study examined 3095 young adults over a 7-year period and found that a 5 µU/ml increase in fasting insulin predicted a 5 kg/m2 increase in body mass index (BMI) after race and gender adjustment.62 Experimental hyperinsulinemia imposed on normal rats resulted in hyperphagia, weight gain and onset of IR within a few days.50 Also, randomized clinical studies show that treatment with insulin or an insulin secretagogue leads to increased fat deposition and weight gain.63
In an obesity rat model, ventromedial hypothalamic lesions caused excessive insulin secretion, hyperphagia and persistent weight gain that can be blocked by pancreatic vagotomy.57,58 Similarly, children with hypothalamic obesity after CNS insult exhibited insulin hypersecretion and weight gain which responded to octreotide-mediated insulin suppression with weight loss.64 Likewise, suppression of acute insulin secretion in a subset of obese adults manifesting insulin hypersecretion without IR promoted weight loss.65
WDs may play an important role in the development of insulin hypersecretion and hyperinsulinemia since these diets induce a high postprandial insulin response38 which may be further increased by the insulinotropic effect of high saturated fat intake.66 The high need for insulin in response to WDs and lifestyle (such as frequent snacking and consumption of sucrose-containing soft drinks) puts significant stress on β-cells, which may lead to hypertrophy and dysfunction and ultimately hypersecretion in response to normal meals.38,39,41–44 Indeed, hyperinsulinemia was found to be associated with ß-cell hyperplasia and enhanced secretory capacity.41,44 Animal feeding studies55,67–70 support a causal role of high-insulinemic/high-fat diets in the development of hyperinsulinemia and obesity. Female mice fed a high-fat research diet (D12492) for 12 weeks became hyperinsulinemic, obese and insulin-resistant. Additionally, they had enhanced insulin gene expression, increased β-cell mass and augmented glucose-induced insulin secretion.67 Prolonged feeding of Wistar rats with a diet rich in high-glycemic index starch and fat, similar to WDs, resulted in insulin hypersecretion and weight gain.68 Rats fed ad libitum with a sucrose-rich diet developed hyperinsulinemia and weight gain.55,69 In a prospective feeding study, nonhuman primates were randomized to either a Western or Mediterranean diet for 2.5 years. Compared to the Mediterranean group, the Western group developed an increased insulin response to glucose challenge, increased intake and significant weight gain.70 Further, in a rat model of obesity, four-day-old rats reared on a high-carbohydrate milk formula (56% of calories from carbohydrates compared to 8% in rat milk) responded immediately to the milk formula with the onset of hyperinsulinemia. A greater number of small-sized islets and an increase in the number of islets per unit area were observed in the pancreas. Chronic hyperinsulinemia was accompanied by development of obesity in the post-weaning period. Female rats spontaneously transmitted their metabolic characteristics to their progeny. The second generation spontaneously became chronically hyperinsulinemic and obese.71 On the other hand, attenuation of diet-induced insulin hypersecretion in young growing mice provided protection against obesity throughout adult life.72
Development of IR has been reported to limit insulin-mediated weight gain.73 Since insulin controls metabolic flux, storage and disposal of glucose, FAs and amino acids, insulin sensitivity is a prerequisite for functioning fuel partitioning. With development of IR, FAs are no longer adequately stored in adipose tissue and even released into circulation during the absorptive state.13,74 Increases in circulating FFAs produce or are associated with (immediate or delayed) increases in FA versus glucose oxidation in skeletal muscle, with effects on weight gain.73 Consistent with this, IR was associated with a lower rate of weight gain in a population of nondiabetic Pima Indians.75 Also, in the Rancho Bernardo cohort study, development of IR preceded weight loss among older, community-dwelling adults without diabetes.76
In addition to refined carbohydrates, fructose may play a particularly important role in the development of hyperinsulinemia, despite a low GI.21 Feeding studies in rats have shown that diets rich in fructose cause hypertriglyceridemia and significant hyperinsulinemia. It should be borne in mind that WDs are high in fructose (as part of the disaccharide sucrose and in the form of fructose corn syrup) as a sweetener for soft drinks and as an additive for ready meals.77 In fact, fructose corn syrup consumption increased by >1000% in the United States between 1970 and 1990.78
Increased ROS production and oxidative stress (OS) also have been implicated in the development of insulin hypersecretion/hyperinsulinemia.38,79,80 Consistent with this, WDs,81 diets high in sucrose82 and diets with a high GL83 produce high ROS levels and OS. Postprandial glucose excursions correlate directly with the ensuing increase in free radicals.84 In contrast, low-glycemic diets, like Paleolithic and Mediterranean diets85 and ketogenic diets86 are associated with low OS.
In summary, mixed diets with high-insulinogenic carbohydrates and fat are required to promote weight gain and obesity. Carbohydrates hardly contribute to fat mass, but they have control over insulin and thus over fat storage. Diet-related long-term elevated insulin levels (postprandial, but more importantly due to hypersecretion/hyperinsulinemia) can promote significant weight gain (Figure 1). Therefore, high-glycemic carbohydrates are the active component, that promotes fat storage via its insulinogenic effect, while fat is the passive component. In a simple analogy, high-insulinogenic carbohydrates are the “driver” while fat is the “passenger” on the way to obesity.
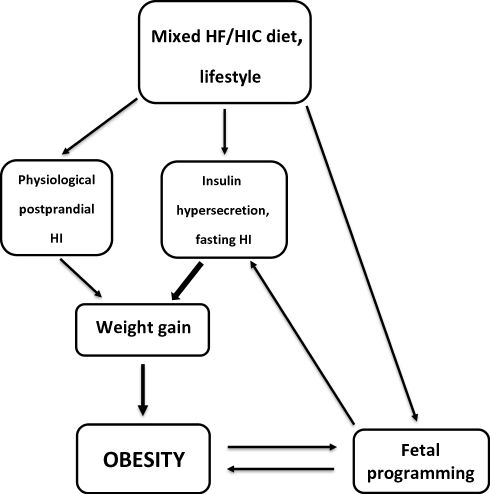 |
Figure 1 Proposed driver/passenger model of obesity. Abbreviations: HF/HIC, high-fat/high-insulinogenic carbohydrate; FOAD, fetal origin of adult disease; HI, hyperinsulinemia. |
Genetic/Epigenetic Aspects
ß-Cells
Genetic and epigenetic factors may be responsible for differences in sensitivity and reactivity of β-cells to high-glycemic carbohydrates as a cause of insulin hypersecretion. In a bidirectional Mendelian randomization study, increased genetically determined insulinemia was strongly associated with higher BMI, while higher genetically determined BMI was not associated with insulinemia.87 A study by LeStunff et al88 suggests that young obese patients with variants in the insulin promoter gene (variable number of tandem repeat locus) secrete more insulin than those with other genotypes. Increased susceptibility and insulin hypersecretion may – at least in part – be due to fetal programming.
Fetal Programming
Fetal programming is also involved in obesity development. In particular, the dramatic increase in overweight and obesity in children and adolescents2 can at least partially be attributed to this metabolic alteration. High-glycemic diets play an important role in this.
The Fetal Origins of Adult Disease (FOAD) Hypothesis
Prenatal development is a critical stage in the etiology of human diseases. While the fetus has a considerable ability to adapt metabolically to suboptimal environments, this adaptation often causes an increased susceptibility of the offspring to chronic diseases later in life, as both animal models and studies in humans have shown. The FOAD hypothesis has been the subject of much study in the last decades. Today, compelling evidence exists in support of the hypothesis. It implies that many maternal socio-demographic and lifestyle factors, such as diet, smoking, physical inactivity and psychosocial stress during sensitive development phases of pregnancy lead to changes in structure, physiology and metabolism of the fetus (“fetal programming”), that these effects are persistent and make people vulnerable to the development of obesity and related diseases.89–91
In humans, birth size serves as a surrogate marker of the intrauterine environment and fetal development. A number of epidemiological studies have confirmed a relationship between birth weight and increased risk of metabolic diseases.92 A large body of experimental evidence indicates that the primary maternal environmental factor which regulates fetoplacental growth is substrate delivery to the placenta.91,93–95 The nutritional milieu during pregnancy and proper nutrition of the fetus therefore play key roles. Both the maternal diet and the metabolism during pregnancy have a significant impact on fetal development and long-term health.96,97 Epigenetic dysregulation has been reported to mediate the effects of early nutrition on adult disease susceptibility.89,90,93,98 In an overfeeding-based rat model, exposure to maternal obesity resulted in changes in DNA methylation of pro-adipogenic genes in adipose tissue in the offspring, preceding the development of obesity.98
Early studies focused on the relationship between maternal malnutrition (protein and/or calorie restriction) and low birth weight of the offspring. Children with low birth weight have an increased risk of developing metabolic disorders like diabetes, hypertension, obesity and more later in life.91,99
Further studies showed that not only malnutrition, but also overeating can adversely affect the development of the fetus which manifests itself in high birth weight. Macrosomia, or high infant birth weight, is associated with an increased risk of complications for both mother and infant during delivery and in the perinatal period. Adverse health outcomes and birth weight were found to have a U-shaped relationship, with offspring born at low birth weights from under-nourished mothers and high birth weights from over-nourished mothers having the greatest risk for disease later in life.100 In the Western world, obesogenic WDs are commonly consumed during pregnancy. Both in human and animal models, overfeeding with WDs can lead to an adverse metabolic profile and a tendency towards obesity, IR and related diseases in adulthood.93,96,97,101 In addition, Western-style diets can increase the ß-cell mass in the offspring and cause insulin hypersecretion.102,103
Maternal hyperglycemia in particular is a major risk factor for fetal programming.91,93,95,99 Glucose is essential for normal fetal metabolism and fetal growth, and there is a direct correlation between maternal blood sugar levels and the size at birth, as a longitudinal study in non-diabetic, non-obese pregnant women showed. Maternal postprandial glycemia, even within the normal range, was strongly associated with birth weight in the third trimester.104 Another prospective randomized study confirmed that high-glycemic diets increase growth rate and size at birth while low-glycemic diets decrease it. In this study, 12 healthy pregnant women were randomized to a low-glycemic diet or an isoenergetic diet that mainly contained high-glycemic carbohydrates. The women in the high-glycemic group experienced excessive weight gain and were delivered of symmetrically larger infants (averaging 800g more than in the low-glycemic group) and larger placentas.94
A mismatch between an (evolutionary) genetic program and an inappropriate diet during pregnancy may play a key role in this regard.
Insulin Action During Pregnancy
Human pregnancy is accompanied by a physiologic transient IR, beginning around the 10th week. The second and third trimesters are characterized by IR with a nearly 50% decrease in insulin-mediated glucose disposal. After delivery, IR abates.105 The main purpose of this metabolic program is to provide the fetus with vital nutrients, especially glucose, the main energy substrate for the placenta and fetus and an important building block. Due to the IR of pregnancy, maternal metabolism uses more fat than glucose to generate energy, saving glucose for the metabolic needs of the developing fetus.105 It must be borne in mind that this metabolic program has evolved in connection with the low-glycemic diets that our ancestors lived on over a very long period of human evolution; the uncultivated high-fiber plant-based foods such as roots, tubers, wild herbs, berries, nuts, vegetables and fruits provided only relatively small amounts of glucose, compared to today’s cultivated vegetables, fruits and refined cereals.11,38 In a “low-glucose environment”, this metabolic program of pregnancy makes perfect sense. However, WDs interfere with this hereditary adaptation. Diets with a high content of highly processed, quickly digestible and absorbed carbohydrates provide large amounts of glucose. While glucose uptake in insulin-resistant maternal tissues is reduced, glucose can cross the placenta unhindered. Since insulin is not able to cross the placenta, the fetal pancreas is forced to produce large amounts of insulin for blood sugar control.12 According to Pedersen’s hyperglycemia-hyperinsulinism hypothesis,106 excessive maternal glucose crossing the placenta causes fetal hyperglycemia and hypertrophy of fetal islet tissue with insulin hypersecretion. Fetal hyperinsulinemia, plus a large supply of glucose substrate greatly enhance protein, lipid and glycogen synthesis, and thus promote fetal macrosomia95,106,107 and development of intrauterine IR.108
Birth weight is also influenced by the degree of maternal IR, which determines the nutrient flux to the fetus. In a retrospective observational study on non-obese, non-diabetic pregnant women to assess the relationship between maternal IR and birth weight in uncomplicated pregnancies, postprandial insulin levels and HOMA-IR were positively correlated with birth weight and the risk of giving birth to a large-for-gestational-age (LGA) infant.109 Overweight women begin their pregnancy with a higher IR (compared to their normal weight counterpart) which increases by an additional 50–60% in the course of pregnancy.110 Maternal obesity at conception therefore can have significant adverse effects on fetal development and increases the risk of both fetal macrosomia and growth-restricted infants, childhood obesity and related diseases.99,111,112 In addition to pre-existing obesity, high weight gain during pregnancy is also associated with increased fetal growth and later childhood obesity (Figure 1).113 Consistent with this, feeding studies showed that a high-GI diet is associated with greater maternal weight gain at birth,95 larger placentas, and increased birth weight compared to a nutritionally balanced, low-GI diet.95,114 Low-GI diets reduce the risk of LGA infants without increasing the number of small-gestational age infants.36,95,114 Cohort studies indicate that (low-glycemic) dietary patterns which are mainly based on fruits and vegetables, whole grain bread, milk, fish, chicken and lean meat have a positive effect on the formation of a normal placenta and the developing fetus and are associated with a reduced risk of pregnancy complications.96,101,115,116
Developmental programming is not limited to the in utero environment. Studies by Bayol et al117 have also identified the lactation period as a critical time window. Diet composition during this period can have a significant impact on the child’s development. “Junk food” as well as nutrient-enriched formula feed (containing extra protein and energy from carbohydrates or fat and other nutrients), commonly used as an alternative or supplement to breastfeeding, have been implicated in neonatal and infant growth acceleration and increased risk of childhood obesity.93,117,118 As noted already above, puppies fed a high-carbohydrate milk formula immediately after birth developed hyperinsulinemia and adult obesity, which was passed on to the next generation.71
OS seems to play a central role in fetal programming.119 As mentioned before, WDs,81 diets high in sucrose82 and diets with a high GL83 produce high ROS levels and OS, while low-glycemic diets are associated with low OS.120
Final Remark
WDs also play a key role in the development of obesity-related diseases (“civilization diseases”), including diabetes, heart disease, stroke, cancer and other more. The relationship between WDs and the development of civilization diseases has been described elsewhere.38
Summary and Conclusions
The evidence presented shows that diet composition plays a key role in the development of obesity. Neither fat nor carbohydrates are fattening per se. Mixed diets with high-glycemic carbohydrates and fat, like current WDs, are required to promote weight gain and obesity. Animal studies indicate that weight gain is proportional to the fat content (with significant weight increases at 30% fat and more) and that already 20% of high-insulinogenic carbohydrates in a high-fat diet are sufficient to cause significant weight gain.29,33 High-glycemic carbohydrates are the “driver” and promote fat storage through their insulinogenic effect, while fat is the “passenger” on the way to obesity. Elevated insulin levels (postprandial, but more importantly due to hypersecretion and hyperinsulinemia) promote significant fat storage and play a key role in obesogenesis and the obesity epidemic. The importance of high-insulinogenic carbohydrates in the development of obesity is illustrated best by direct comparison of two papers cited before: a Western ad libitum diet (40% fat, 43% high-glycemic carbohydrates, mostly sucrose) caused obesity31 while an ad libitum diet with 40% carbohydrates with a low glycemic load promoted fat loss despite a relatively high fat content of 35%.37
In addition, a maternal diet high in high-glycemic carbohydrates and fat promotes fetal programming, with long-term adverse impacts on the offspring, including insulin hypersecretion, (childhood) obesity and metabolic diseases. Maternal obesity and high weight gain during pregnancy have also been linked to deleterious effects on fetal programming. As the global obesity epidemic increasingly affects women of reproductive age, a significant percentage of fetuses will experience fetal programming with a tendency towards obesity - a self-reinforcing process that further fuels the epidemic.
Changing our lifestyle towards healthy eating and more physical activity may help reduce or prevent the development of obesity. A number of dietary patterns, both macronutrient- and food-based, have been suggested for weight loss and/or preventing the development of obesity, such as a Mediterranean eating pattern, a Paleolithic eating pattern, low-fat/high-carbohydrate diets, low-carbohydrate diets, ketogenic diets, low-glycemic diets and more.121 There is probably no one-size-fits-all dietary strategy for weight loss and obesity prevention. However, significantly reducing the intake of high-glycemic foods is certainly a prudent strategy, not only for weight management (especially in individuals with insulin hypersecretion) but also for the prevention of fetal programming and for health reasons.7,38,122 Consistent with this, a body of evidence, including prospective cohort observational studies, randomized controlled trials and mechanistic experiments in animal models, supports the effectiveness of low-GI and low-GL diets in preventing obesity.36
Abbreviations
FFA, free fatty acid; FOAD, fetal origins of adult disease; GI, glycemic index; GL, glycemic load; BMI, body mass index; IR, insulin resistance; WD, Western diet.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Disclosure
The author reports no conflicts of interest in this work.
References
1. GBD 2015 Obesity Collaborators, Afshin A, Forouzanfar MH, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377(1):13–27. doi:10.1056/NEJMoa1614362.
2. Seidell JC, Halberstadt J. The global burden of obesity and the challenges of prevention. Ann Nutr Metab. 2015;66(suppl 2):7–12. doi:10.1159/000375143
3. Abarca-Gómez L, Abdeen ZA, Hamid ZA, et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet. 2017;390(10113):2627–2642. doi:10.1016/S0140-6736(17)32129-3
4. Singh RK, Kumar P, Mahalingam K. Molecular genetics of human obesity: a comprehensive review. C R Biol. 2017;340(2):87–108. doi:10.1016/j.crvi.2016.11.007
5. Hall KD. Did the food environment cause the obesity epidemic? Obesity (Silver Spring). 2018;26(1):11–13. doi:10.1002/oby.22073
6. Wolfram G, Bechthold A, Boeing H, et al. Evidence-based guideline of the German Nutrition Society: fat intake and prevention of selected nutrition-related diseases. Ann Nutr Metab. 2015;67(3):141–204. doi:10.1159/000437243
7. Kopp W. High-insulinogenic nutrition - an etiologic factor for obesity and the metabolic syndrome? Metabolism. 2003;52(7):840–844. doi:10.1016/s0026-0495(02)05294-0
8. Ludwig DS, Ebbeling CB. The carbohydrate-insulin model of obesity: beyond “calories in, calories out”. JAMA Intern Med. 2018;178(8):1098–1103. doi:10.1001/jamainternmed.2018.2933
9. Alberts B, Bray D, Johnson A, et al. How cells obtain energy from food. In: Alberts B, Bray D, Johnson A, et al. editors. Essential Cell Biology. An Introduction to the Molecular Biology of the Cell. Vol. 4, New York: Garland Publishing Inc; 2014:419–466.
10. Westman EC, Feinman RD, Mavropoulos JC, et al. Low-carbohydrate nutrition and metabolism. Am J Clin Nutr. 2007;86(2):276–284. doi:10.1093/ajcn/86.2.276
11. Cordain L, Eaton SB, Sebastian A, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81(2):341–354. doi:10.1093/ajcn.81.2.341
12. Kopp W. Significant dietary changes during human evolution and the development of cancer: from cells in trouble to cells causing trouble. J Carcinog Mutagen. 2017;8(04):303. doi:10.4172/2157-2518
13. Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159(6):1253–1262. doi:10.1016/j.cell.2014.09.052
14. Nordmann AJ, Nordmann A, Briel M, et al. Effects of low-carbohydrate vs low-fat diets on weight loss and cardiovascular risk factors: a meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166(3):285–293. doi:10.1001/archinte.166.3.285
15. Brehm BJ, Seeley RJ, Daniels SR, D’Alessio DA. A randomized trial comparing a very low carbohydrate diet and a calorie-restricted low-fat diet on body weight and cardiovascular risk factors in healthy women. J Clin Endocrinol Metab. 2003;88(4):1617–1623. doi:10.1210/jc.2002-021480
16. McSwiney FT, Wardrop B, Hyde PN, Lafountain RA, Volek JS, Doyle L. Keto-adaptation enhances exercise performance and body composition responses to training in endurance athletes. Metabolism. 2018;81:25–34. doi:10.1016/j.metabol.2017.10.010
17. Greene DA, Varley BJ, Hartwig TB, Chapman P, Rigney M. A low-carbohydrate ketogenic diet reduces body mass without compromising performance in powerlifting and olympic weightlifting athletes. J Strength Cond Res. 2018;32(12):3373–3382. doi:10.1519/JSC.0000000000002904
18. Rolls BJ. Dietary energy density: applying behavioural science to weight management. Nutr Bull. 2017;42(3):246–253. doi:10.1111/nbu.12280
19. Stubbs BJ, Cox PJ, Evans RD, Cyranka M, Clarke K, de Wet H. A ketone ester drink lowers human ghrelin and appetite. Obesity (Silver Spring). 2018;26(2):269–273. doi:10.1002/oby.22051
20. Page MM, Johnson JD. Mild suppression of hyperinsulinemia to treat obesity and insulin resistance. Trends Endocrinol Metab. 2018;29(6):389–399. doi:10.1016/j.tem.2018.03.018
21. Björk I, Liljeberg H, Östan E. Low-glycemic index foods. Br J Nutr. 2000;83(Suppl 1):S149–S155. doi:10.1017/s0007114500001094
22. Wolever TM, Bolognesi C. Prediction of glucose and insulin responses of normal subjects after consuming mixed meals varying in energy, protein, fat, carbohydrate and glycemic index. J Nutr. 1996;126(11):2807–2812. doi:10.1093/jn/126.11.280
23. McDevitt RM, Bott SJ, Harding M, Coward WA, Bluck LJ, Prentice AM. De novo lipogenesis during controlled overfeeding with sucrose or glucose in lean and obese women. Am J Clin Nutr. 2001;74(6):737–746. doi:10.1093/ajcn/74.6.737
24. Proserpi C, Sparti A, Schutz Y, Di Vetta V, Milon H, Jéquier E. Ad libitum intake of a high-carbohydrate or high-fat diet in young men: effects on nutrient balances. Am J Clin Nutr. 1997;66(3):539–545. doi:10.1093/ajcn/66.3.539
25. Poppitt SD, Keogh GF, Prentice AM, et al. Long-term effects of ad libitum low-fat, high-carbohydrate diets on body weight and serum lipids in overweight subjects with metabolic syndrome. Am J Clin Nutr. 2002;75(1):11–20. doi:10.1093/ajcn/75.1.11
26. Hays NP, Starling RD, Liu X, et al. Effects of an ad libitum low-fat, high-carbohydrate diet on body weight, body composition, and fat distribution in older men and women: a randomized controlled trial. Arch Intern Med. 2004;164(2):210–217. doi:10.1001/archinte.164.2.210
27. Mueller-Cunningham WM, Quintana R, Kasim-Karakas SE. An ad libitum, very low-fat diet results in weight loss and changes in nutrient intakes in postmenopausal women. J Am Diet Assoc. 2003;103(12):1600–1606. doi:10.1016/j.jada.2003.09.017
28. Astrup A, Astrup A, Buemann B, Flint A, Raben A. Low-fat diets and energy balance: how does the evidence stand in 2002? Proc Nutr Soc. 2002;61(2):299–309. doi:10.1079/PNS2002149
29. Gajda AM. High fat diets for diet-induced obesity models. Research Diets, Inc.; 2008. Available from: http://www.researchdiets.com.
30. Dalby MJ, Ross AW, Walker AW, Morgan PJ. Dietary uncoupling of gut microbiota and energy harvesting from obesity and glucose tolerance in mice. Cell Rep. 2017;21(6):1521–1533. doi:10.1016/j.celrep.2017.10.056
31. Argueta DA, DiPatrizio NV. Peripheral endocannabinoid signaling controls hyperphagia in western diet-induced obesity. Physiol Behav. 2017;171:32–39. doi:10.1016/j.physbeh.2016.12.044
32. Lissner L, Levitsky DA, Strupp BJ, Kalkwarf HJ, Roe DA. Dietary fat and the regulation of energy intake in human subjects. Am J Clin Nutr. 1987;46(6):886–892. doi:10.1093/ajcn/46.6.886
33. Takahashi M, Ikemoto S, Ezaki O. Effect of the fat/carbohydrate ratio in the diet on obesity and oral glucose tolerance in C57BL/6J mice. J Nutr Sci Vitaminol (Tokyo). 1999;45(5):583–593. doi:10.3177/jnsv.45.583
34. Campbell PJ, Carlson MG, Hill JO, Nurjhan N. Regulation of free fatty acid metabolism by insulin in humans: role of lipolysis and reesterification. Am J Physiol. 1992;263(6 Pt 1):E1063–E1069. doi:10.1152/ajpendo.2006.263.6.E1063
35. Esmaillzadeh A, Kimiagar M, Mehrabi Y, Azadbakht L, Hu FB, Willett WC. Dietary patterns, insulin resistance, and prevalence of the metabolic syndrome in women. Am J Clin Nutr. 2007;85(3):910–918. doi:10.1093/ajcn/85.3.910
36. Brand-Miller J, McMillan-Price J, Steinbeck K, Caterson I. Carbohydrates–the good, the bad and the whole grain. Asia Pac J Clin Nutr. 2008;17(Suppl 1):16–19.
37. Ebbeling CB, Leidig MM, Feldman HA, Lovesky MM, Ludwig DS. Effects of a low–glycemic load vs low-fat diet in obese young adults: a randomized trial. JAMA. 2007;297(19):2092–2102. doi:10.1001/jama.297.19.2092
38. Kopp W. How Western diet and lifestyle drive the pandemic of obesity and civilization diseases. Diabetes Metab Syndr Obes. 2019;12:2221–2236. doi:10.2147/DMSO.S216791
39. Mehran AE, Templeman NM, Brigidi GS, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16(6):723–737. doi:10.1016/j.cmet.2012.10.019
40. Le Stunff C, Bougneres P. Early changes in postprandial insulin secretion, not in insulin sensitivity, characterize juvenile obesity. Diabetes. 1994;43(5):696–702. doi:10.2337/diab.43.5.696
41. Schofield CJ, Sutherland C. Disordered insulin secretion in the development of insulin resistance and type 2 diabetes. Diabet Med. 2012;29(8):972–979. doi:10.1111/j.1464-5491.2012.03655.x
42. Nolan CJ, Prentki M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: time for a conceptual framework shift. Diab Vasc Dis Res. 2019;16(2):118–127. doi:10.1177/1479164119827611
43. Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A. Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology. 2003;144(6):2717–2727. doi:10.1210/en.2002-221112
44. Mahler RJ. The relationship between the hyperplastic pancreatic islet and insulin insensitivity in obesity. Acta Diabetol Lat. 1981;18(1):1–17. doi:10.1007/BF02056101
45. Polonsky KS, Given BD, Van Cauter E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest. 1988;81(2):442–448. doi:10.1172/JCI113339
46. Ferrannini E, Natali A, Bell P, Cavallo-Perin P, Lalic N, Mingrone G. Insulin resistance and hypersecretion in obesity. J Clin Invest. 1997;100:1166–1173. doi:10.1172/JCI119628
47. Ferrannini E, Camastra S, Gastaldelli A, et al. Beta-cell function in obesity: effects of weight loss. Diabetes. 2004;53(Suppl 3):S26–S33. doi:10.2337/diabetes.53.suppl_3.s26
48. Erion KA, Corkey BE. Hyperinsulinemia: a cause of obesity? Curr Obes Rep. 2017;6(2):178–186. doi:10.1007/s13679-017-0261-z
49. Templeman NM, Skovsø S, Page MM, Lim GE, Johnson JD. A causal role for hyperinsulinemia in obesity. J Endocrinol. 2017;232(3):R173–183. doi:10.1530/JOE-16-0449
50. Cusin I, Rohner-Jeanrenaud F, Terrettaz J, Jeanrenaud B. Hyperinsulinemia and its impact on obesity and insulin resistance. Int J Obes. 1992;16(Suppl 4):S1–11.
51. Lustig RH. Autonomic dysfunction of the b-cell and the pathogenesis of obesity. Rev Endocr Metab Dis. 2003;4(1):23–32. doi:10.1023/A:1021819318484
52. Sigal RJ, El-Hashimy M, Martin BC, Soeldner JS, Krolewski AS, Warram JH. Acute post-challenge hyperinsulinemia predicts weight gain. Diabetes. 1997;46(6):1025–1029. doi:10.2337/diab.46.6.1025
53. Chen YY, Wang JP, Jiang YY, et al. Fasting plasma insulin at 5 years of age predicted subsequent weight increase in early childhood over a 5-year period: the Da Qing children cohort study. PLoS One. 2015;10(6):e0127389. doi:10.1371/journal.pone.0127389
54. Pénicaud L, Kinebanyan MF, Ferré P, et al. Development of VMH obesity: in vivo insulin secretion and tissue insulin sensitivity. Am J Physiol. 1989;257:E255–E260. doi:10.1152/ajpendo.1989.257.2.E255
55. Barnard RJ, Roberts CK, Varon SM, Berger JJ. Diet-induced insulin resistance precedes other aspects of the metabolic syndrome. J Appl Physiol. 1998;84(4):1311–1315. doi:10.1152/jappl.1998.84.6.1967
56. Grimditch GK, Barnard RJ, Hendricks L, Weitzman D. Peripheral insulin sensitivity as modified by diet and exercise training. Am J Clin Nutr. 1988;48(1):38–43. doi:10.1093/ajcn/48.1.38
57. Tokunaga K, Fukushima M, Kemnitz JW, Bray GA. Effect of vagotomy on serum insulin in rats with paraventricular or ventromedial hypothalamic lesions. Endocrinology. 1986;119:1708–1711.
58. Inoue S, Bray GA. The effects of subdiaphragmatic vagotomy in rats with ventromedial hypothalamic obesity. Endocrinology. 1977;100(1):108–114. doi:10.1210/endo-100-1-108
59. Johnson MS, Figueroa-Colon R, Huang TT, Dwyer JH, Goran MI. Longitudinal changes in body fat in African American and Caucasian children: influence of fasting insulin and insulin sensitivity. J Clin Endocrinol Metab. 2001;86(7):3182–3187. doi:10.1210/jcem.86.7.7665
60. Odeleye OE, de Courten M, Pettitt DJ, Ravussin E. Fasting hyperinsulinemia is a predictor of increased body weight gain and obesity in Pima Indian children. Diabetes. 1997;46(8):1341–1345. doi:10.2337/diab.46.8.1341
61. Preeyasombat C, Bacchetti P, Lazar AA, Lustig RH. Racial and etiopathologic dichotomies in insulin hypersecretion and resistance in obese children. J Pediatr. 2005;146(4):474–481. doi:10.1016/j.jpeds.2004.12.014
62. Folsom AR, Jacobs DR
63. Sinha A, Formica C, Tsalamandris C, et al. Effects of insulin on body composition in patients with insulin-dependent and non-insulin-dependent diabetes. Diabet Med. 1996;13(1):40–46. doi:10.1002/(SICI)1096-9136(199601)13:1<40::AID-DIA991>3.0.CO;2-U
64. Lustig RH, Rose SR, Burghen GA, et al. Hypothalamic obesity in children caused by cranial insult: altered glucose and insulin dynamics, and reversal by a somatostatin agonist. J Pediatr. 1999;135(2):162–168. doi:10.1016/s0022-3476(99)70017-x
65. Velasquez-Mieyer PA, Cowan PA, Buffington CK, et al. Suppression of insulin secretion promotes weight loss and alters macronutrient preference in a subset of obese adults. Int J Obes. 2003;27:219–226. doi:10.1038/sj.ijo.802227
66. Stein DT, Stevenson BE, Chester MW, et al. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J Clin Invest. 1997;100(2):398–403. doi:10.1172/JCI119546
67. Gonzalez A, Merino B, Marroquí L, et al. Insulin hypersecretion in islets from diet-induced hyperinsulinemic obese female mice is associated with several functional adaptations in individual β-cells. Endocrinology. 2013;154(10):3515–3524. doi:10.1210/en.2013-1424
68. Pawlak DB, Bryson JM, Denyer GS, Brand-Miller JC. High glycemic index starch promotes hypersecretion of insulin and higher body fat in rats without affecting insulin sensitivity. J Nutr. 2001;131(1):99–104. doi:10.1093/jn/131.1.99
69. Gonsolin D, Couturier K, Garait B, et al. High dietary sucrose triggers hyperinsulinemia, increases myocardial beta-oxidation, reduces glycolytic flux and delays post-ischemic contractile recovery. Mol Cell Biochem. 2007;295(1–2):217–228. doi:10.1007/s11010-006-9291-7
70. Shively CA, Appt SE, Vitolins MZ, et al. Mediterranean versus Western diet effects on caloric intake, obesity, metabolism, and hepatosteatosis in nonhuman primates [published correction appears in obesity (silver spring)]. Obesity (Silver Spring). 2019;27(5):777–784. doi:10.1002/oby.22436
71. Patel MS, Srinivasan M. Metabolic programming in the immediate postnatal life. Ann Nutr Metab. 2011;58(Suppl 2):18–28. doi:10.1159/000328040
72. Templeman NM, Clee SM, Johnson JD. Suppression of hyperinsulinaemia in growing female mice provides long-term protection against obesity. Diabetologia. 2015;58(10):2392–2402. doi:10.1007/s00125-015-3676-7
73. Eckel RA. Insulin resistance: an adaptation for weight maintenance. Lancet. 1992;340(8833):1452–1453. doi:10.1016/0140-6736(92)92633-q
74. Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60(10):2441–2449. doi:10.2337/db11-0425
75. Swinburn BA, Nyomba BL, Saad MF, et al. Insulin resistance associated with lower rates of weight gain in Pima Indians. J Clin Invest. 1991;88(1):168–173. doi:10.1172/JCI115274
76. Wedick NM, Mayer-Davis EJ, Wingard DL, Addy CL, Barrett-Connor E. Insulin resistance precedes weight loss in adults without diabetes: the Rancho Bernardo study. Am J Epidemiol. 2001;153(12):1199–1205. doi:10.1093/aje/153.12.1199
77. Sleder J, Chen YD, Cully MD, Reaven GM. Hyperinsulinemia in fructose-induced hypertriglyceridemia in the rat. Metabolism. 1980;29(4):303–305. doi:10.1016/0026-0495(80)90001-3
78. Galderisi A, Giannini C, Van Name M, Caprio S. Fructose consumption contributes to hyperinsulinemia in adolescents with obesity through a GLP-1-mediated mechanism. J Clin Endocrinol Metab. 2019;104(8):3481–3490. doi:10.1210/jc.2019-00161
79. Corkey BE. Diabetes: have we got it all wrong? Insulin hypersecretion and food additives: cause of obesity and diabetes? Diabetes Care. 2012;35(12):2432–2437. doi:10.2337/dc12-0825
80. Saadeh M, Ferrante TC, Kane A, et al. Reactive oxygen species stimulate insulin secretion in rat pancreatic islets: studies using mono-oleoyl-glycerol. PLoS One. 2012;7(1):e30200. doi:10.1371/journal.pone.0030200
81. Erdelyi I, Levenkova N, Lin EY, et al. Western-Style diets induce oxidative stress and dysregulate immune responses in the colon in a mouse model of sporadic colon cancer. J Nutr. 2009;139(11):2072–2078. doi:10.3945/jn.108.104125
82. Busserolles J, Rock E, Gueux E, Mazur A, Grolier P, Rayssiguier Y. Short-term consumption of a high-sucrose diet has a pro-oxidant effect in rats. Br J Nutr. 2002;87(4):337–342. doi:10.1079/BJNBJN2002524
83. Anderson C, Milne GL, Park YM, Sandler DP, Nichols HB. Dietary glycemic index and glycemic load are positively associated with oxidative stress among premenopausal women. J Nutr. 2018;148(1):125–130. doi:10.1093/jn/nxx022
84. Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295(14):1681–1687. doi:10.1001/jama.295.14
85. Whalen KA, McCullough ML, Flanders WD, Hartman TJ, Judd S, Bostick RM. Paleolithic and mediterranean diet pattern scores are inversely associated with biomarkers of inflammation and oxidative balance in adults. J Nutr. 2016;146(6):1217–1226. doi:10.3945/jn.115.224048
86. Greco T, Glenn TC, Hovda DA, Prins ML. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J Cereb Blood Flow Metab. 2016;36(9):1603–1613. doi:10.1177/0271678X15610584
87. Astley CM, Todd JN, Salem RM, et al. Genetic evidence that carbohydrate-stimulated insulin secretion leads to obesity. Clin Chem. 2018;64(1):192–200. doi:10.1373/clinchem.2017.280727
88. LeStunff C, Fallin D, Schork NJ, Bougneres P. The insulin gene VNTR is associated with fasting insulin levels and the development of juvenile obesity. Nat Genet. 2000;26:444–446.
89. Simeoni U, Armengaud JB, Siddeek B, Tolsa JF. Perinatal origins of adult disease. Neonatology. 2018;113(4):393–399. doi:10.1159/000487618
90. Moosavi A, Motevalizadeh Ardekani A. Role of epigenetics in biology and human diseases. Iran Biomed J. 2016;20(5):246–258. doi:10.22045/ibj.2016.01.185
91. Kwon EJ, Kim YJ. What is fetal programming?: a lifetime health is under the control of in utero health. Obstet Gynecol Sci. 2017;60(6):506–519. doi:10.5468/ogs.2017.60.6
92. Jones RH, Ozanne SE. Fetal programming of glucose-insulin metabolism. Mol Cell Endocrinol. 2009;297(1–2):4–9. doi:10.1016/j.mce.2008.06.020
93. Gali Ramamoorthy T, Allen TJ, Davies A, et al. Maternal overnutrition programs epigenetic changes in the regulatory regions of hypothalamic Pomc in the offspring of rats. Int J Obes (Lond). 2018;42(8):1431–1444. doi:10.1038/s41366-018-0094-1
94. Clapp JF. Diet, exercise and feto-placental growth. Arch Gynecol Obstet. 1997;261:101–107.
95. Clapp JF. Maternal carbohydrate intake and pregnancy outcome. Proc Nutr Soc. 2002;61(1):45–50. doi:10.1079/pns2001129
96. Paknahad Z, Fallah A, Moravejolahkami AR. Maternal dietary patterns and their association with pregnancy outcomes. Clin Nutr Res. 2019;8(1):64–73. doi:10.7762/cnr.2019.8.1.64
97. Damasceno DC, Dallaqua B, Lovizutto Iessi I, Volpato GT, Campos KE. Impact of maternal over-nutrition during pregnancy on maternal oxidative stress and fetal skeletal/visceral anomalies of the rats. J Nutr Disord Ther. 2016;6(01):1. doi:10.4172/2161-0509.1000185
98. Borengasser SJ, Zhong Y, Kang P, et al. Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology. 2013;154(11):4113–4125. doi:10.1210/en.2012-2255
99. Jornayvaz FR, Vollenweider P, Bochud M, Mooser V, Waeber G, Marques-Vidal P. Low birth weight leads to obesity, diabetes and increased leptin levels in adults: the CoLaus study. Cardiovasc Diabetol. 2016;15(1):73. doi:10.1186/s12933-016-0389-2
100. Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359(1):61–73. doi:10.1056/NEJMra0708473
101. Tzanetakou IP, Mikhailidis DP, Perrea DN. Nutrition during pregnancy and the effect of carbohydrates on the offspring’s metabolic profile: in search of the “perfect maternal diet”. Open Cardiovasc Med J. 2011;5(1):103–109. doi:10.2174/1874192401105010103
102. Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab. 2006;291(4):E792–799. doi:10.1152/ajpendo.00078.2006
103. Elsakr JM, Dunn JC, Tennant K, et al. Maternal Western-style diet affects offspring islet composition and function in a non-human primate model of maternal over-nutrition. Mol Metab. 2019;25:73–82. doi:10.1016/j.molmet.2019.03.010
104. Parretti E, Mecacci F, Papini M, et al. Third-trimester maternal glucose levels from diurnal profiles in nondiabetic pregnancies: correlation with sonographic parameters of fetal growth. Diabetes Care. 2001;24(8):1319–1323. doi:10.2337/diacare.24.8.1319
105. Sonagra AD, Biradar SM, Murthy KD. Normal pregnancy- a state of insulin resistance. J Clin Diagn Res. 2014;8(11):CC01–CC3. doi:10.7860/JCDR/2014/10068.5081
106. Pedersen J. Hyperglycaemia-hyperinsulinism theory and birthweight. In: The Pregnant Diabetic and Her Newborn: Problems and Management. Baltimore: Williams and Wilkins; 1977:211–220.
107. Susa JB, Schwarz R. Hyperinsulinemia in the primate fetus. Diabetes. 1985;34(Supplement_2):36–41. doi:10.2337/diab.34.2.S36
108. Simental-Mendía LE, Castañeda-Chacón A, Rodríguez-Morán M, Guerrero-Romero F. Birth-weight, insulin levels, and HOMA-IR in newborns at term. BMC Pediatr. 2012;12(1):94. doi:10.1186/1471-2431-12-94
109. Yamashita H, Yasuh I, Fukuda M, et al. The association between maternal insulin resistance in mid-pregnancy and neonatal birthweight in uncomplicated pregnancies. Endocr J. 2014;61(10):1019–1024. doi:10.1507/endocrj.EJ14-0163
110. Catalano PM, Hauguel-de Mouzon S. Is it time to revisit the Pedersen hypothesis in the face of the obesity epidemic? Am J Obstet Gynecol. 2011;204(6):479–487. doi:10.1016/j.ajog.2010.11.039
111. Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ, Badger TM. Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol. 2008;294(2):R528–R538. doi:10.1152/ajpregu.00316.2007
112. Shrestha N, Ezechukwu HC, Holland OJ, Hryciw DH. Developmental programming of peripheral diseases in offspring exposed to maternal obesity during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2020;319(5):R507–R516. doi:10.1152/ajpregu.00214.2020
113. McDowell M, Cain MA, Brumley J. Excessive gestational weight gain. J Midwifery Women's Health. 2019;64(1):46–54. doi:10.1111/jmwh.12927
114. Moses RG, Luebcke M, Davis WS, et al. Effect of a low-glycemic-index diet during pregnancy on obstetric outcomes. Am J Clin Nutr. 2006;84(4):807–812. doi:10.1093/ajcn/84.4.807
115. Hillesund ER, Bere E, Haugen M, Øverby NC. Development of a new nordic diet score and its association with gestational weight gain and fetal growth - a study performed in the Norwegian mother and child cohort study (MoBa). Public Health Nutr. 2014;17(9):1909–1918. doi:10.1017/S1368980014000421
116. Timmermans S, Steegers-Theunissen RP, Vujkovic M, et al. The Mediterranean diet and fetal size parameters: the Generation R Study. Br J Nutr. 2012;108(8):1399–1409. doi:10.1017/S000711451100691X
117. Bayol SA, Farrington SJ, Stickland NC. A maternal ‘junk food’ diet in pregnancy and lactation promotes an exacerbated taste for ‘junk food’ and a greater propensity for obesity in rat offspring. Br J Nutr. 2007;98(04):843–851. doi:10.1017/S0007114507812037
118. Ong KK, Emmett PM, Noble S, Ness A, Dunger DB. Dietary energy intake at the age of 4 months predicts postnatal weight gain and childhood body mass index. Pediatrics. 2006;117(3):e503–e508. doi:10.1542/peds.2005-1668
119. Rodríguez-Rodríguez P, Ramiro-Cortijo D, Reyes-Hernández CG. Implication of oxidative stress in fetal programming of cardiovascular disease. Front Physiol. 2018;9:602. doi:10.3389/fphys.2018.00602
120. Hu Y, Block G, Norkus EP, Morrow JD, Dietrich M, Hudes M. Relations of glycemic index and glycemic load with plasma oxidative stress markers. Am J Clin Nutr. 2006;84(1):70–76. doi:10.1093/ajcn/84.1.70
121. Smethers AD, Rolls BJ. Dietary management of obesity: cornerstones of healthy eating patterns. Med Clin N Am. 2018;102(1):107–124. doi:10.1016/j.mcna.2017.08.009
122. Reynolds A, Mann J, Cummings J, Winter N, Mete E, Te Morenga L. Carbohydrate quality and human health: a series of systematic reviews and meta-analyses. Lancet. 2019;393(10170):434–445. doi:10.1016/s0140-6736(18)31809-9
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.