Back to Journals » Drug Design, Development and Therapy » Volume 11
Development of new promising antimetabolite, DFP-11207 with self-controlled toxicity in rodents
Authors Fukushima M, Iizuka K, Jin C, Zhang C, Hong M, Eshima K ![]()
Received 22 November 2016
Accepted for publication 4 May 2017
Published 7 June 2017 Volume 2017:11 Pages 1693—1705
DOI https://doi.org/10.2147/DDDT.S128420
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Anastasios Lymperopoulos
Masakazu Fukushima, Kenzo Iizuka, Cheng Jin, Chun Zhang, Mei Hong, Kiyoshi Eshima
Division of Oncology Research and Development, Delta-Fly Pharma Inc., Kawauchi-cho, Tokushima, Japan
Abstract: To reduce 5-fluorouracil (5-FU)-induced serious toxicities without loss of antitumor activity, we have developed DFP-11207, a novel fluoropyrimidine, which consists of 1-ethoxymethyl-5-fluorouracil (EM-FU; a precursor form of 5-FU), 5-chloro-2,4-dihydroxypyridine (CDHP; an inhibitor of 5-FU degradation), and citrazinic acid (CTA; an inhibitor of 5-FU phosphorylation). In vitro studies of DFP-11207 indicated that it strongly inhibited the degradation of 5-FU by dihydropyrimidine dehydrogenase (DPD) in homogenates of the rat liver, and also inhibited the phosphorylation of 5-FU by orotate phosphoribosyltransferase (OPRT) in tumor tissues in a similar magnitude of potency by CDHP and CTA, respectively. Especially, DFP-11207 inhibited the intracellular phosphorylation of 5-FU in tumor cells in a dose-dependent manner whereas CTA alone did not protect intracellular 5-FU phosphorylation. These results postulate that DFP-11207 rapidly entered into the cell and the free CTA produced from DFP-11207 inhibited the phosphorylation of 5-FU in the cell. Furthermore, following oral administration of DFP-11207, CTA was found to be highly retained in the gastrointestinal (GI) tract compared to other tissues in rats. Interestingly, EM-FU, the prodrug of 5-FU was found to specifically produce 5-FU by various species of liver microsomes. When DFP-11207 was administered to rats, the plasma level of 5-FU was persisted for a long-time with lower Cmax and longer half-life than that from other 5-FU prodrugs. The antitumor activity of DFP-11207 was evaluated in human tumor xenografts in nude rats and found that DFP-11207 showed an antitumor activity in a dose-dependent fashion and its efficacy is equivalent to reference 5-FU drugs. In striking contrast, DFP-11207 manifested no or less 5-FU-related toxicities, such as a decrease in body weights, GI injury, and myelosuppression, especially thrombocytopenia. Taken together, the preclinical evaluation of DFP-11207 strongly indicates that DFP-11207 be a potential new version of the oral fluoropyrimidine prodrug for further clinical development.
Keywords: antimetabolite, 5-FU, citrazinic acid, 5-chloro-2,4-dihydroxypyridine, antitumor activity, myelo-toxicity
Introduction
Recent advance in new anticancer drugs has focused on low molecular weight tyrosine kinase inhibitors and monoclonal antibodies for various growth factor receptors to specifically that have been shown abnormal in tumors to minimize toxicities by traditional chemotherapies in various advanced and/or metastatic cancers. In parallel, the surrogate biomarker discovery and development have been capable for detecting various gene amplifications and mutations such as EGFR, HER-2 neu, and RAS in tumors and support molecular-targeted drugs development or precision medicine for tumor patients. However, only one-third of patients respond to such new medicines with a high refractory matter and much higher health care burden being created. However, the traditional cytotoxic agents including platinum drugs, taxanes, and antimetabolites are still the main stream of anticancer therapy that has been used to improve a clinical response and quality of life (QOL) of cancer patients.
5-Fluorouracil (5-FU), one of antimetabolites, discovered and synthesized since late 1950s, introduced clinically,1 has been widely used as a single agent or in combination with other cytotoxic drugs called as fluorouracil/leucovorin/oxaliplatin (FOLFOX),2 fluorouracil/leucovorin/irinotecan (FOLFIRI),3 fluorouracil/cisplatin (FP),4 and so on for cancer patients with mainly advanced and metastatic gastrointestinal (GI) or GI cancers. Clinical response and toxicity of 5-FU have been remarkably influenced by its dosing schedule and a continuous infusion (CI) of 5-FU has been found to increase the response rates of patients with GI cancers.5–8 Furthermore, Lokich et al9 showed that a long-term CI of 5-FU resulted in a higher response with mild to severe GI toxicity and a rapid bolus injection of 5-FU showed in a lower response with severe myelosuppression in their randomized Phase III study. Also, 5-FU has been shown to be rapidly hydrolyzed to its inactive form inducing cardio- and neurotoxicity and/or hand-foot-syndrome (HFS), while, a phosphorylated form of 5-FU has been indicated to cause GI- and myelotoxicity in addition to its antitumor activity.
To adopt the advantage of a long-time CI of 5-FU and patient compliance from a viewpoint of QOL in patients, several oral 5-FU derivatives such as capecitabine (Hoffman-La Roche Ltd., Basel, Switzerland),10 tegafur-uracil (UFT; Taiho Pharmaceutical Ltd, Tokyo, Japan),11 and tegafur-gimeracil-oteracil (S-1; Taiho Pharmaceutical Ltd)12 have been developed and widely applied to the treatment of cancer patients. However, these oral fluoropyrimidines do not completely separate the efficacy from 5-FU-induced toxicities, and therefore, dose-reduction or pause of the drug treatment are required for cancer patients. Incidentally, the primary dose-limiting toxicity of capecitabine, UFT, and S-1 has been shown to be HFS,13 GI toxicity,14 and hematological toxicity,15 respectively. Therefore, it is an unmet medical need for developing a new version of oral 5-FU drug that minimizes such adverse events without compromising the antitumor activity of oral fluoropyrimidines.
Recently, we have developed a new promising oral fluoropyrimidine prodrug, DFP-11207 (5-chloro-2-(3-(3-(ethoxymethyl)-5-fluoro-2,6-dioxo-1,2,3,6-tetrahydopyrimidine-1-carbonyl)benzoyloxy)pyridine-4-yl-2,6-bis(propionyloxy)isonicotinate) shown in Figure 1. This single molecule is intelligently designed and consists of three important components, 1-ethoxymethyl-5-fluorouracil (EM-FU), 5-chloro-2,4-dihydroxypyridine (CDHP), and citrazinic acid (CTA) to exhibit augmented antitumor activity in various human cancers without the toxicities described above by cooperating the three distinctive functional components. This study reports pharmacokinetics (PKs) and pharmacodynamics profiles in vitro and in vivo, and then the antitumor activity of DFP-11207 in human GI tumor xenografts in nude rats.
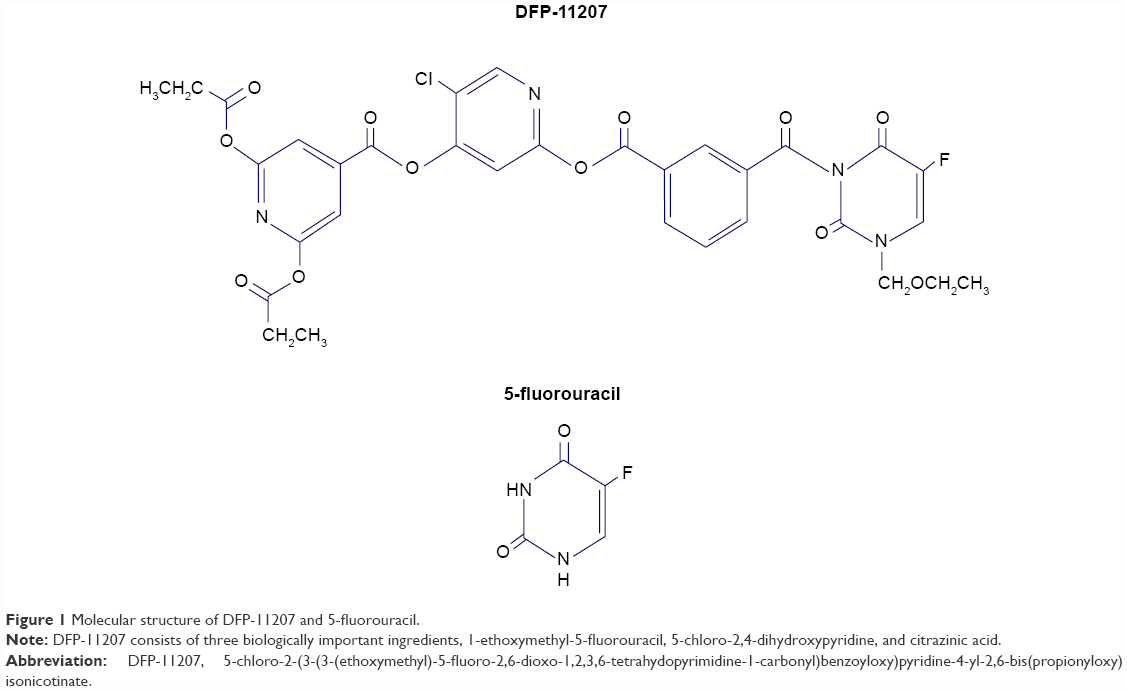 | Figure 1 Molecular structure of DFP-11207 and 5-fluorouracil. |
Materials and methods
Materials
5-FU and CTA were purchased from Sigma Chemical Co. (St Louis, MO, USA). CDHP (gimeracil) was supplied from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). EM-FU and DFP-11207 were synthesized in our laboratory (Tokushima, Japan). [6-3H]-5-FU (710 GBq.mmol) was obtained from New England Nuclear Co. (Tokyo, Japan). All other chemical and biochemical materials were standard commercial products.
Animals and tumor cells
F344/N-nu nude rats (5-week old) were purchased from CLEA Japan Inc. (Tokyo, Japan) and were fed with a sterilized pellet diet and autoclaved water ad libitum. Rats were kept in laminar air-flow units throughout experiments performed.
Human colorectal cancer cells (CoLo320DM, KM12C, and HT-29), pancreatic cancer cells (PANC-1 and BxPC-3), and gastric cancer cell (AZ521) were obtained from ATCC (The Global BioSource Center, Manassas, VA, USA) and maintained in vitro as a monolayer culture in a RPMI-1640 medium supplemented with heat-inactivated fetal calf serum containing penicillin (100 U/mL), streptomycin (100 μg/mL), and L-glutamine (2 mM) until used for in vitro and in vivo experiments.
Antitumor experiments
The care and treatment of the animals were in accordance with the guidelines issued by the Science and International Affairs Bureau of the Japanese Ministry of Education, Science, Culture, and Sports. The experimental protocol was performed after approval from the Institutional Animal Ethical Committee in Delta-Fly Pharma Inc (Tokushima, Japan).
Groups of six nude rats were used. Human tumor xenografts were prepared by subcutaneous implantation of cultured tumor cells (1–5×106 cells) into the right axilla of rats. When each tumor volume reached ~100–200 mm3, DFP-11207 and S-1 (combination of 1 M tegafur, 0.4 M gimeracil, and 1 M oteracil) were administered orally for 14–21 consecutive days. 5-FU and gemcitabine were intraperitoneally (IP) injected daily for 5 days and weekly for 3 weeks, respectively. The tumor volume (1/2 × [the major axis] × [the minor axis]2) was measured twice a week throughout the experiments, relative tumor volume (RTV) was calculated as follows: RTV = (mean tumor volume during therapy)/(mean tumor volume at the start of therapy). The antitumor effects of drugs tested were estimated by the following equation: mean inhibition rate of tumor growth (IR, %) = (1 − [mean RTV of drug-treated group/mean RTV of control group] ×100).
Enzyme assay
Activity of dihydropyrimidine dehydrogenase (DPD) catalyzing 5-FU degradation and OPRT catalyzing 5-FU phosphorylation were determined according to the method of Shirasaka et al.16
Intracellular phosphorylation of 5-FU and its subsequent incorporation into RNA in intact cells in vitro
According to the method described previously,16 CoLo320DM cells (2×107) suspended in a RPMI-1640 medium were incubated with 1 μM [6-3H] 5-FU (37 kBq) alone or in the presence of DFP-11207 and CTA in a final volume of 2 mL at 37°C for 45 min and then 2 mL of 10% trichloroacetic acid (TCA) were added. The mixtures were centrifuged at 2,000× g for 5 min, and the TCA-soluble fraction was neutralized with KOH and 50 μL aliquots were subjected to silica gel thin layer chromatography. The spot of 5-fluorouridine-5′-monophosphate (FUMP) was scrapped off for measurement of its radioactivity. The radioactivity incorporated into RNA present in the TCA-precipitated material was extracted by the method of Schneider17 for determination of the amount of 5-FU incorporated into RNA.
In vitro hydrolysis of DFP-11207
DFP-11207 (1 mM) was incubated with rat serum, and 20% (w/v) homogenates extracted from rat liver and small intestine at 37°C for 10–60 min and then 10% TCA was added to the reaction mixture followed by centrifugation at 3,000× g for 10 min. The resultant supernatant was neutralized with 2 M KOH solution and subjected to high-performance liquid chromatography (HPLC) to determine the contents of EM-FU, CDHP, and CTA produced.
Extraction and determination of 5-FU and CTA in the blood and tissues
DFP-11207 was orally administered to AZ521 tumor-bearing nude rats. The animals were sacrificed at the times indicated, and their blood and tissues were rapidly removed. The tumors and small intestines were homogenized with three volumes of ice-cold saline and centrifuged at 10,000× g for 30 min. The supernatant obtained was used as crude extracts containing 5-FU and CTA. The extraction and determination of 5-FU in the blood and tissues were performed according to the method reported previously.18
Extraction and determination of 5-FU concentration in the blood of rats
Rats were treated with DFP-11207 or S-1, then sacrificed at various indicated times and their blood were rapidly removed and centrifuged to obtain the serum samples. One milliliter of the serum was added to 0.1 mL of the known amount of the internal standard solution (5-bromouracil) for 5-FU and shaken with 5 mL of ethyl acetate (EA) twice. The two EA layers were combined and evaporated at 40° under a gentle stream of N2 gas. The residue was dissolved in distilled water, passed through a 0.45 μm filter and the 5-FU content of the filtrate was determined by the method described previously.19
Pathological evaluation of injury of the digestive tracts
The degree of GI injury of drug-treated rats was evaluated pathologically as described in the previous paper.16 In this experiment, rough microscopic changes in GI tract of rats treated with DFP-11207, S-1, and tegafur-gimeracil (molar ratio; 1:0.4) as negative control were observed.
Statistical analysis
The significant difference between groups and/or drugs with or without treatment was assessed using Dunnett’s test and the Student’s t-test.
Results
Enzymatic hydrolysis of DFP-11207 in vitro
DFP-11207 consists of three important components, EM-FU, CDHP, and CTA, and needs to be cleaved by the hydrolysis to functional metabolites which then exhibit biological activities. Using rat plasma and crude homogenates from the rat liver and small intestine, time-dependent hydrolysis of DFP-11207 was investigated as shown in Figure 2. Three metabolites, EM-FU, CDHP, and CTA were formed in a time dependent manner, and the majority of metabolites was produced within 60 min in this assay system. However, 5-FU was not produced from EM-FU, the prodrug of 5-FU by this enzymatic reaction. However, DFP-11207 was found to be stable and not to be hydrolyzed in phosphate-buffered saline solution alone (data not shown).
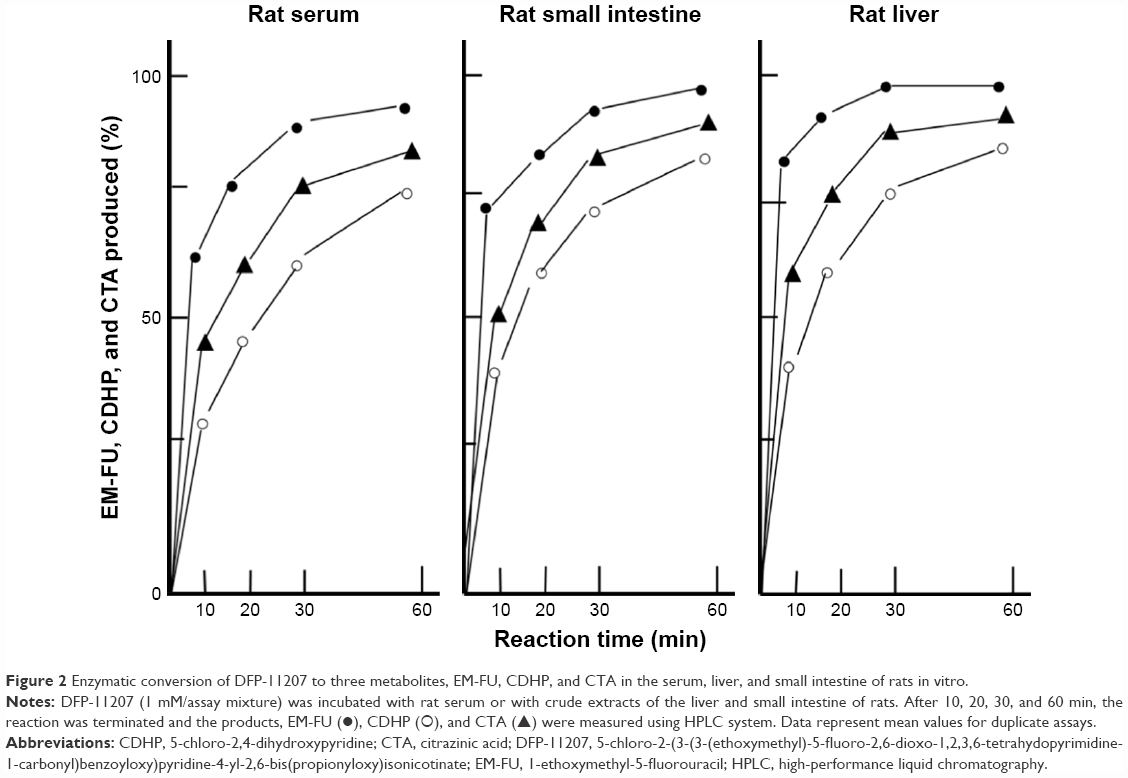 | Figure 2 Enzymatic conversion of DFP-11207 to three metabolites, EM-FU, CDHP, and CTA in the serum, liver, and small intestine of rats in vitro. |
Inhibition by DFP-11207 of DPD and OPRT activities in vitro
The degree of inhibition activity of DFP-11207 was evaluated using crude extracts of the rat liver for DPD and of SC-2 tumors for OPRT as shown in Table 1. DFP-11207 inhibited both DPD and OPRT activities dose-dependently, indicating that DFP-11207 was promptly hydrolyzed to each component, CDHP and CTA, and exhibited their inhibitory activities to both DPD and OPRT. However, DFP-11207 did not inhibit both DPD and OPRT activities when heat-denatured crude extracts was used in the assay, suggesting that the intact DFP-11207 has no inhibitory activity (data not shown).
 | Table 1 Inhibitory effect of DFP-11207 on the activities of DPD and OPRT |
Effect of DFP-11207 on intracellular phosphorylation of 5-FU and its subsequent incorporation into RNA
CTA has been reported to strongly inhibit OPRT activity (5-FU phosphorylation) in cell-free tumor extract but not intact cells in vitro, suggesting no transport of it into the cells.16 When incubated with 1 μM [6-3H] 5-FU, DFP-11207 inhibited dose-dependently the intracellular phosphorylation of 5-FU and its subsequent incorporation into RNA in Colo320DM cells while CTA alone did not inhibit as, expected, shown in Figure 3. This result indicates that after incorporated into the cells, DFP-11207 is rapidly cleaved, and resultant CTA freed from intact DFP-11207 could inhibit the intracellular formation of F-nucleotides from 5-FU incorporated into the cells.
 | Figure 3 Inhibitory effects of DFP-11207 and citrazinic acid on intracellular phosphorylation of 5-FU and its subsequent incorporation into RNA in human colorectal tumor cells. |
Conversion of EM-FU, prodrug of 5-FU, to active 5-FU by various liver microsomes
Tegafur (FT), prodrug of 5-FU, is well known to be hydrolyzed to 5-FU by liver microsomes, mainly by CYP2A6 species. As EM-FU shows a similar chemical structure of FT, we investigated the conversion of EM-FU to 5-FU in vitro by intact liver microsomes derived from various species. As shown in Figure 4A, EM-FU was hydrolyzed similarly in rat, monkey, and human microsomes (~30–60 pmol/min/mg) except for dog microsomes which showed the lowest conversion rate of EM-FU to 5-FU. Next, to specify a subgroup of CYPs catalyzing the conversion of EM-FU to 5-FU, an inhibitory activity for the activation of EM-FU was examined using chemical compounds inhibiting each CYP enzyme. As shown in Figure 4B, the conversion of EM-FU to 5-FU was strongly inhibited by coumarin, α-naphthoflavone, p-nitrophenol, and toleandomycin, indicating that EM-FU is converted to 5-FU by CYP1A2, CYP2A6, CYP2E1, and CYP3A4, respectively. And these data seem to be quite different from tegafur being hydrolyzed by only CYP2A6 (data not shown).
 | Figure 4 Conversion of EM-FU, a major metabolite of DFP-11207, to 5-FU by various liver microsomes. |
Blood and tissue levels of CTA and 5-FU in tumor-bearing rats after oral administration of DFP-11207
DFP-11207 (53.4 mg/kg) was orally given to AZ521 tumor-bearing rats and the distribution of CTA and 5-FU in the blood, tumor, and small intestine was investigated. As presented in Figure 5A, CTA was mainly retained in the small intestine (Cmax; ~100 ng/g tissue) and persisted longer over 12 h than that in the blood (Cmax; 32 ng/mL) and AZ521 tumor (Cmax; below 5 ng/g tissue). At 4 h following administration of DFP-11207, the levels of 5-FU in the blood and tissues were compared with that of CTA. As shown in Figure 5B, 5-FU levels in the blood and tumor were much higher than CTA, suggesting little effect of CTA on the 5-FU phosphorylation to exhibit its antitumor activity in the tumor. In striking contrast, almost equal level of 5-FU and CTA remained in the small intestine. This may well explain with the selective inhibition of 5-FU phosphorylation in the digestive tract resulting in little or no GI toxicity in those rats.
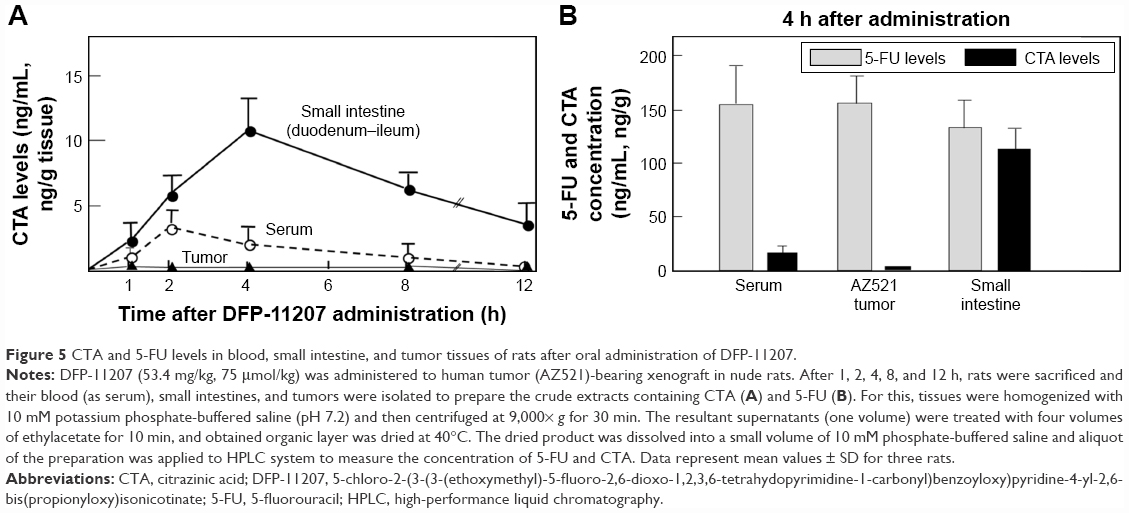 | Figure 5 CTA and 5-FU levels in blood, small intestine, and tumor tissues of rats after oral administration of DFP-11207. |
Comparative 5-FU concentration in blood of rats after administration of DFP-11207 and FT-based prodrug S-1
To distinguish the difference of PK profile between DFP-11207 as the single molecule and S-1 as the cocktail formulation with 1 M FT, 0.4 M gemeracil, and 1 M oteracil, an equimolar amount of DFP-11207 and S-1 (both 50 μmol/kg) was orally given to rats and their plasma 5-FU levels were analyzed.
As shown in Figure 6, DFP-11207 had significantly different PK profile from S-1 with low Cmax, longer Tmax, and T1/2 values although the total AUC with 5-FU from S-1 was twofold higher than that from DFP-11207 due to a high spike Cmax of 5-FU from S-1. The desired PK profile with DFP-11207 simulates the 5-FU level in plasma by CI of 5-FU and may well be resulted in the low incidence of myelosuppression by DFP-11207.
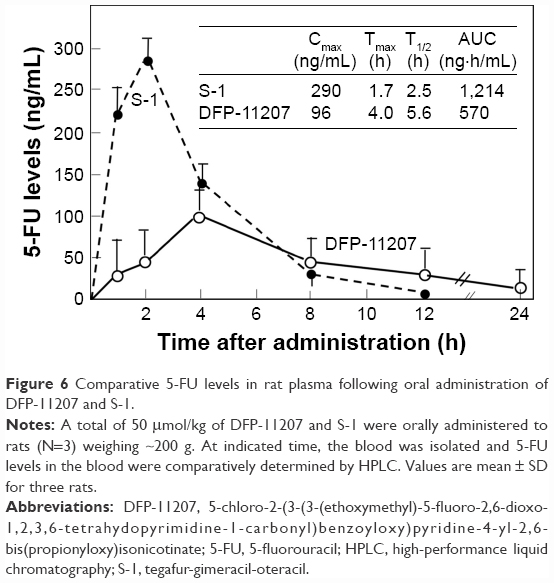 | Figure 6 Comparative 5-FU levels in rat plasma following oral administration of DFP-11207 and S-1. |
Comparative antitumor and toxic effects of DFP-11207 and S-1 on human tumor xenografts in nude rats
Antitumor activity and toxicity represented by the body weight change, visible diarrhea, and change of hematological parameters (white blood cell [WBC], red blood cell [RBC], and platelet [PLT]) were evaluated in KM12C tumor-bearing nude rats after administering equimolar doses of DFP-11207 and S-1 (both 75–112.5 μmol/kg) once daily and 14-day consecutive administration as shown in Figure 7A–D.
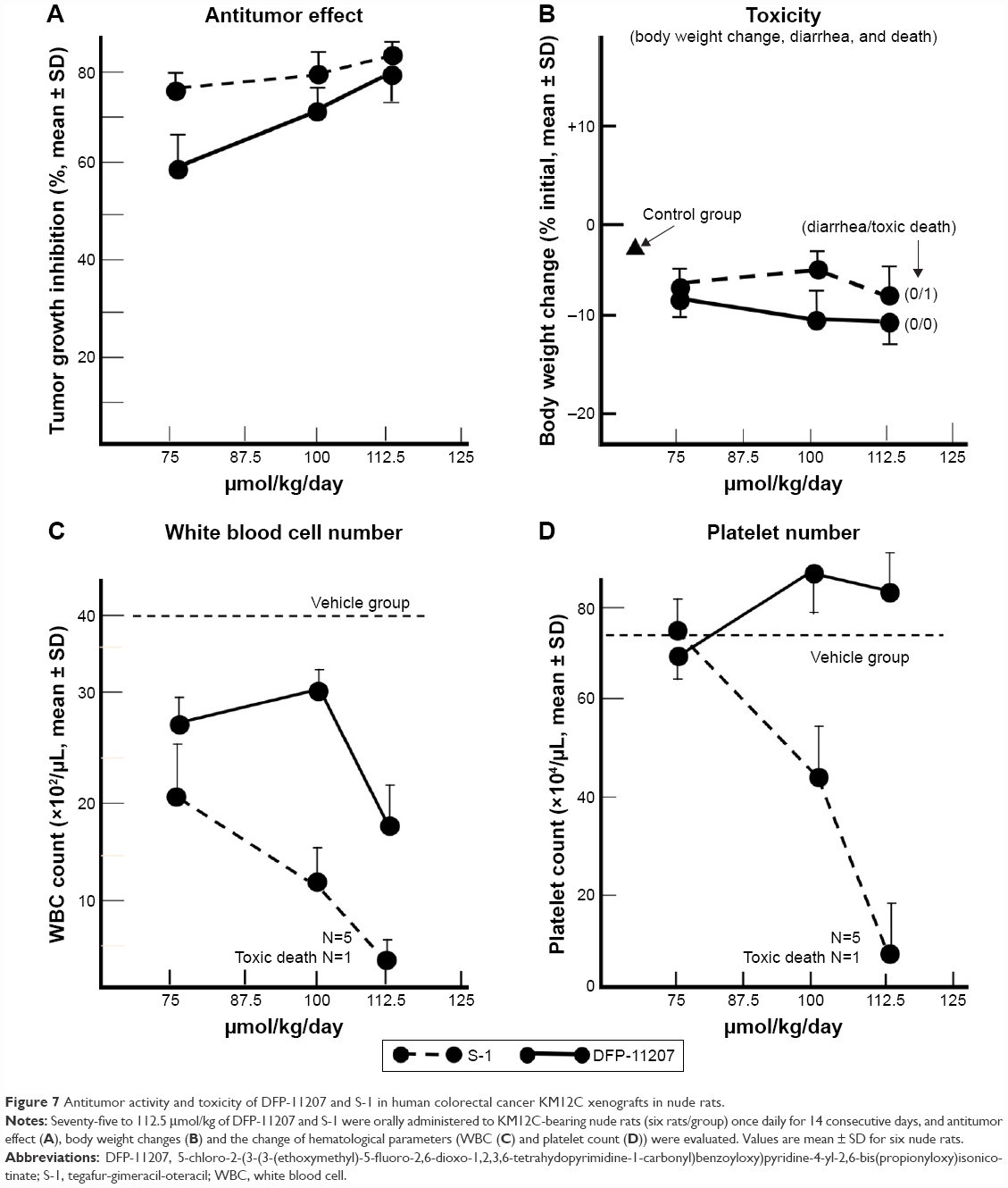 | Figure 7 Antitumor activity and toxicity of DFP-11207 and S-1 in human colorectal cancer KM12C xenografts in nude rats. |
Both DFP-11207 and S-1 at 100–112.5 μmol/kg showed a similar and potent inhibition of the tumor growth (Figure 7A) without a significant decrease in body weights (Figure 7B). However, in S-1 group, one out of six rats died after last administration of the drug, suggesting that a hematological change but not GI damage might occur in S-1 group as further described in Figure 7C and D. Figure 7C and D shows the significant decrease in WBC and PLT counts in S-1 groups whereas little or no change in the PLT number with a mild decrease in WBC number at the highest dose is evident in DFP-11207 groups. In this pharmacology experiment, no diarrhea was noticed in rats treated with the highest dose of both DFP-11207 and S-1 by the cage-side observation. However, to confirm the evidence of no GI damage in drug-treated rats, a portion of the duodenum was evaluated histochemically as shown in Figure 8. Both DFP-11207 and S-1 did not cause a severe damage of mucosal cells in the GI tract while tegafur plus gimeracil alone (S-1 without oteracil) induced the damage of mucosal layer as shown in the right bottom panel. In another pathological study, tegafur plus gimeracil without oteracil has also resulted in 5-FU-induced the GI damage as reported previously.16 These findings suggest that CTA released from DFP-11207 in the GI mucosal cells prevents the risk from the 5-FU-induced GI toxicity through its inhibition of 5-FU phosphorylation.
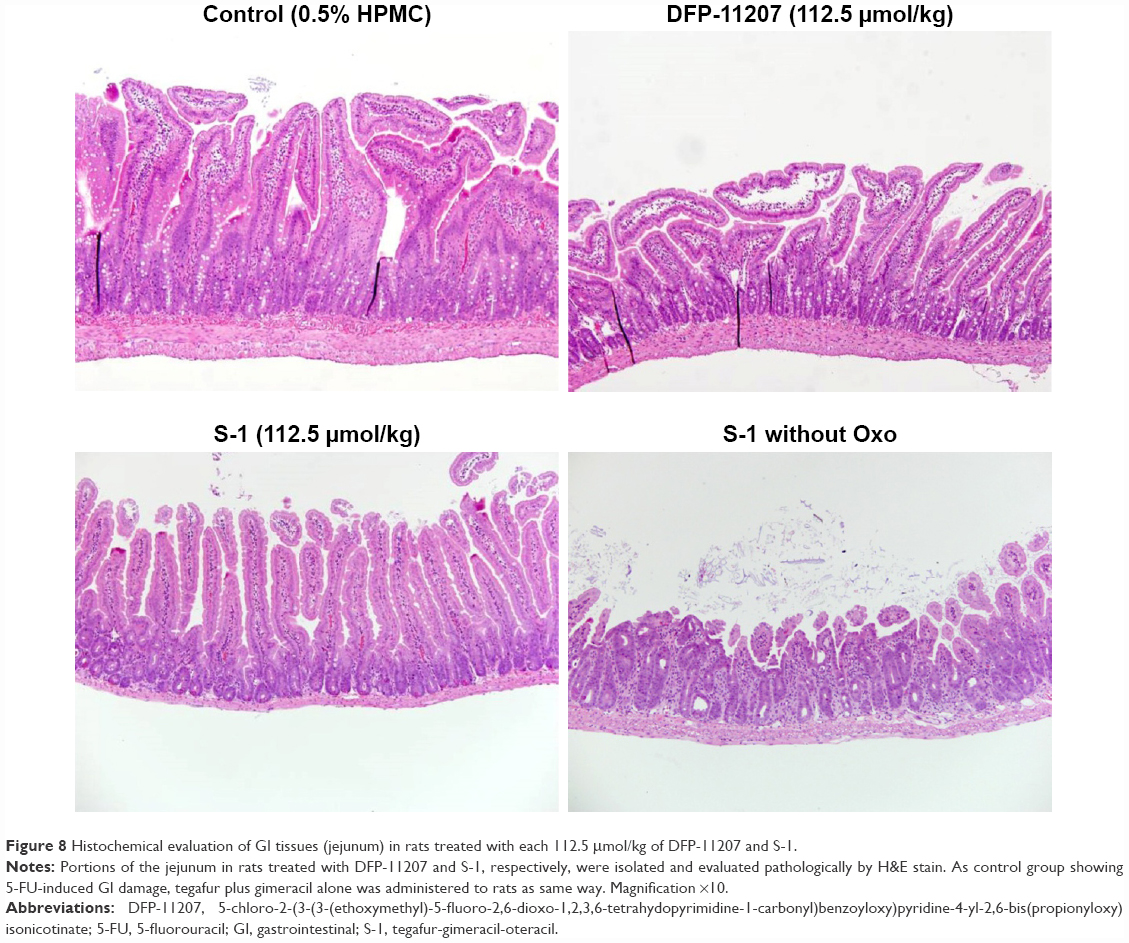 | Figure 8 Histochemical evaluation of GI tissues (jejunum) in rats treated with each 112.5 μmol/kg of DFP-11207 and S-1. |
Antitumor activity of DFP-11207 compared with other antimetabolites on GI cancer xenografts in nude rats
Antitumor activity of DFP-11207 was further evaluated in human colorectal (HT-29), gastric (MKN-45), and pancreatic (BxPC-3 and PANC-1) cancer xenografts in nude rats, and compared its efficacy with 5-FU (IP) or gemcitabine (intravenous [IV]) using maximum safety doses of the standard care drugs without severe decrease in body weights. As summarized in Table 2, DFP-11207 showed significantly higher antitumor activity with ~50%–65% of tumor growth inhibition (TGI) against HT-29, MKN-45, and BxPC-3 tumor models compared with ~30% TGI by 5-FU and gemcitabine, whereas both DFP-11207 and gemcitabine showed a lower activity, 27% versus 23% in the PANC-1 tumor model. These results strongly support the notion that DFP-11207 can be a new version of clinically development candidates of 5-FU derivatives for the treatment of human GI cancers without a burden of commonly seen side effects including GI toxicity and bone marrow suppression by current standard care drugs, such as 5-FU and gemcitabine.
 | Table 2 Antitumor activity of DFP-1207 compared with 5-FU or gemcitabine on human gastrointestinal tumor xenografts in nude rats |
Discussion
For over 50 years, 5-FU has been playing a critical role in the systemic chemotherapy for patients with various cancer types, especially GI cancers. However, remarkably different from other cytotoxic drugs, 5-FU exerts its antitumor activity and also toxic effect through an intracellular metabolism to form its active nucleotide, 5-fluoro-2′-deoxyuridine monophosphate (FdUMP) which inhibits the activity of thymidylate synthase and subsequently inhibits the biosynthesis of DNA in tumor or rapid-growing normal cells. However, 5-FU is rapidly degraded to inactive catabolites by DPD in the liver and interestingly, resultant hydrolysates from 5-FU have been suggested resulting in adverse events, such as cardiotoxicity, neurotoxicity, and skin-toxicity to patients. Also it has been well known that 5-FU-induced efficacy and toxicity are altered by clinical doses and cumulative schedules of 5-FU to cancer patients.9 Considering such in vivo events of 5-FU in cancer patients, it is an urgent unmet medical need to develop a novel 5-FU derivative which provides a maximal antitumor activity of 5-FU with less adverse events in patients. On the viewpoint of such thought, previously Shirasaka et al developed S-1, the oral combination drug consisting of 1 M tegafur (prodrug of 5-FU), 0.4 M gimeracil (inhibitor of DPD), and 1 M oteracil (OPRT inhibitor), with protection of GI toxicity.12,19 Due to the nature of a cocktail formulation, each component in S-1 exhibits independent PK profile, respectively. Especially the higher Cmax level of 5-FU derived from tegafur in the blood resulted in hematological and/or GI toxicities in patients for a long-term use.15,20 Accordingly, it is desired to have 5-FU level in blood maintained for a long-time at a relatively low Cmax level for a new oral candidate of fluoropyrimidine derivatives.
Based on the past preclinical and clinical experiences to pharmacological and PK properties of 5-FU and its oral prodrugs, we newly developed a conceptual oral 5-FU derivative with self-controlled toxicity, DFP-11207 (Figure 1). DFP-11207 consists of three important chemical components, EM-FU, CDHP, and CTA as a single molecule. As expected, DFP-11207 protected the 5-FU-induced GI track, hematological, cardiac and neuro toxicities and HFS with favorable PK profiles, the low Cmax and long half-life in the plasma 5-FU level. To ensure that these components in DFP-11207 work each other to attain a functional role, we investigated enzymatic hydrolysis and inhibitory activity in both 5-FU-degradation and phosphorylation enzymes in vitro, inhibitory effect on the intracellular phosphorylation and subsequent metabolism of 5-FU in intact cells, and a metabolism in liver microsomes (CYP species). As shown in Figure 2 and Table 1, DFP-11207 was found to be rapidly hydrolyzed to produce EM-FU, CDHP, and CTA, and strongly inhibit both DPD and OPRT activities in cell-free system using rat plasma and 20% homogenates of the rat liver and small intestine. Especially, their inhibitory activity of DFP-11207 in DPD and OPRT was almost equivalent to those by CDHP and CTA alone.16,21 However, it is necessary to confirm whether DFP-11207 inhibits an intracellular phosphorylation and subsequent metabolism of 5-FU in intact cells because of no inhibition by CTA alone for the intracellular phosphorylation of 5-FU as reported previously.16 As presented in Figure 3, only DFP-11207 did inhibit dose-dependently the intracellular metabolism of 5-FU in intact tumor cells, suggesting a possible inhibition of 5-FU phosphorylation by CTA produced in GI mucosal cells after an oral administration of DFP-11207. Unfortunately we could not confirm such intracellular inhibition by DFP-11207 using innate normal mucosal cells due to the technical challenge for a stable isolation of mucosal cells from intact GI tissues. Therefore, we investigated the distribution of CTA and 5-FU in GI tissues in tumor-bearing rats followed by an oral administration of 53.4 mg/kg DFP-11207 to ensure whether CTA derived from DFP-11207 inhibits the intracellular phosphorylation of 5-FU (formation of 5-fluoronucleotides) which is sufficient to induce GI injury including diarrhea. As shown in Figure 5A, CTA was found to be mainly retained in GI tissues compared with that in plasma and tumor tissues. Furthermore, 5-FU levels were almost same to the CTA level in GI tissues whereas 5-FU was highly distributed in the plasma and tumors. The results strongly support the notion that CTA protects the incidence of GI injury via the inhibition of 5-FU phosphorylation without compromising the antitumor activity by 5-FU (Figure 5B).
As described above, EM-FU is a prodrug and shows the antitumor activity by its active form, 5-FU. However, an active 5-FU was not produced from EM-FU in a 20% tissue homogenate system, suggesting the formation of 5-FU from EM-FU by drug-metabolizing enzymes (namely microsomes) in the liver. And we confirmed that EM-FU was specifically hydrolyzed to 5-FU by various CYP subtypes (CYP1A2, 2A6, 2E1, and 3A4) as shown in Figure 4 although microsomal activation rate of EM-FU was lower than that by tegafur. In the case of tegafur, it has been reported to be activated by only CYP2A6.22
Based on the data described above, we postulate that DFP-11207 exhibits its unique function, the tumor-selective cytotoxicity via its metabolic pathway as illustrated in Figure 9.
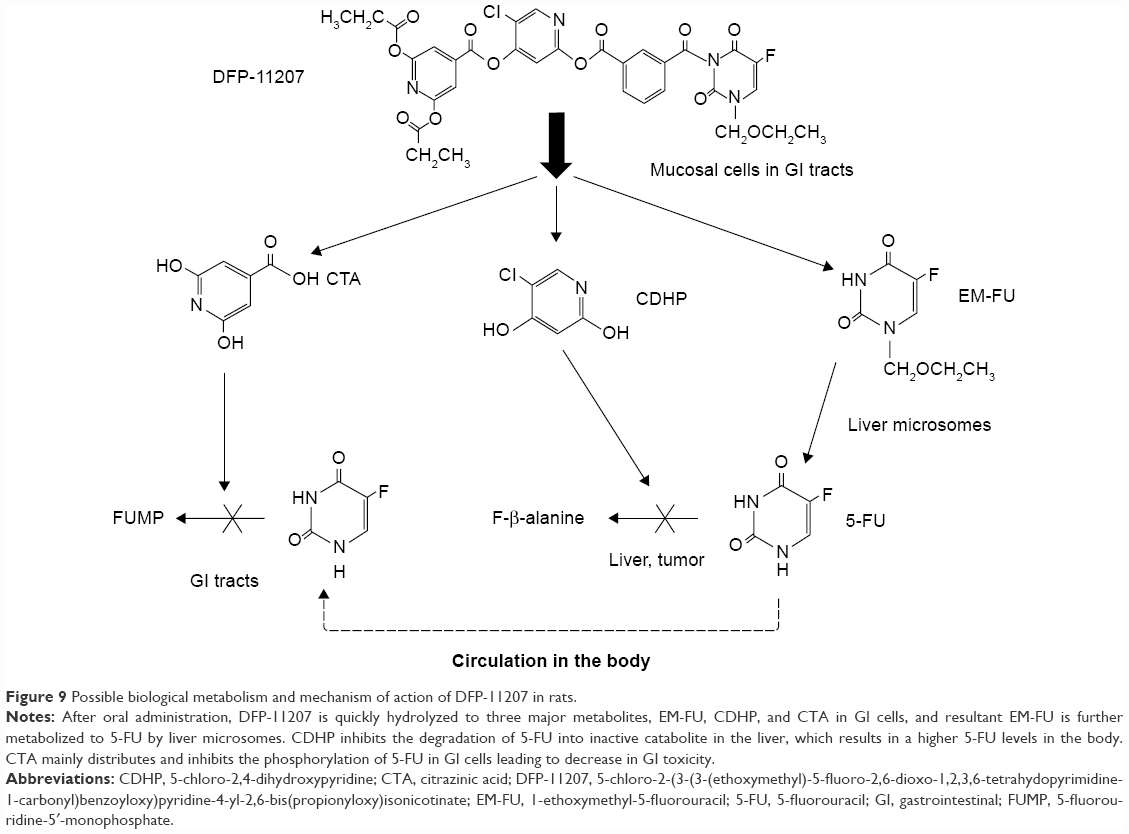 | Figure 9 Possible biological metabolism and mechanism of action of DFP-11207 in rats. |
It has been demonstrated in clinical studies that the efficacy and toxicity of 5-FU depend on its concentration and duration time in the blood of patients,9 and recently, Lee et al have proposed the importance for therapeutic drug monitoring of 5-FU to maximize its efficacy and to reduce 5-FU-induced adverse events in their review article.23 DFP-11207 is the oral prodrug of 5-FU minimizing the 5-FU-induced toxicities without compromising its antitumor activity. The desired feature is different from that by the IV 5-FU or other oral 5-FU prodrugs and supported by its favored PK profile of plasma 5-FU following administration of DFP-11207 to rats. We measured 5-FU levels in the blood and compared with that after the administration of another oral prodrug S-1, possessing the product from a similar drug concept. As shown in Figure 6, DFP-11207 resulted in lower Cmax and AUC values but longer Tmax and T1/2 values of 5-FU, respectively than S-1, which suggests that DFP-11207 would be superior to avoid the 5-FU-induced severe hematological toxicity including neutropenia, in particular, thrombocytopenia on the top of the protection of 5-FU-induced GI toxicities.
In pharmacology study with S-1 in a side-by-side comparison, DFP-11207 showed an antitumor activity in a dose-dependent manner without loss of host body weights or pathological GI damages as same as S-1. However, DFP-11207 resulted in a mild WBC count decrease at a higher dose but no effect on the PLT number, whereas the hematological toxicities were evident with the treatment with S-1. Although the mechanism remains to be cleared, the desired PK profile of DFP-11207 as described above may well contribute to the advantage over S-1 for the bone marrow effect that could be a good benefit for a population of patients who is prone to and compromised with bone marrow suppression or recovery.
Regarding to 5-FU-induced cardiotoxicity and/or neurotoxicity, it has been speculated that the catabolites of 5-FU would contribute to an incidence of those toxicities. In safety pharmacology study of DFP-11207, it showed no neurotoxicity in rats and no cardiovascular toxicity in cynomolgus monkeys (data not shown), suggesting that inhibition of 5-FU degradation may be concerning to decrease in the incidence of such toxicities.
Cumulative dosage and schedule of 5-FU in combination with other cytotoxic drugs are variable in clinical setting to treat patients with GI cancers; bolus injection of 5-FU is applied in FP regimen for advanced gastric cancer, and both 46-h continuous and sequential bolus injection of it are used in FOLFOX and FOLFIRI regimens for metastatic colorectal cancer. However, these combination regimens certainly accompany severe adverse events for most patients although the therapeutic response assessed by response rate, progression-free survival, and overall survival. The advantage of DFP-11207 such as self-controlled toxicity sheds light on a new potential applied to clinical setting and combined with other cytotoxic drugs to improve QOL in cancer patients and for long survival of patients.
Phase I/II study of oral DFP-11207 has been underway to evaluate adverse events and PK profile of this drug. A preliminary result has suggested that DFP-11207 is well tolerated and no severe drug-limiting toxicity including severe diarrhea and thrombocytopnia. It is hopeful to have a further clinical investigation to confirm the advantage of DFP-11207 in preclinical settings.
Conclusion
We have developed DFP-11207, the promising oral fluoropyrimidine prodrug, which consists of a biologically important three components in one molecule in preclinical settings. Following the oral administration, DFP-11207 is immediately separated to EM-FU, CDHP, and CTA in GI cells, and EM-FU is further converted to the active form of 5-FU by the liver microsomes specifically. However, released CDHP prevents 5-FU from a rapid degradation (inactivation) in the liver which maintains a persistent plasma 5-FU concentration, and CTA mainly retained in GI tract cells protects the GI tract from the injury by inhibiting the phosphorylation of 5-FU. In pharmacology study using human tumor xenografts in nude rats, DFP-11207 showed remarkable antitumor activity without any drug-related GI toxicity or thrombocytopenia. Taken together, our pre-clinical evaluation of DFP-11207, strongly supports the notion that DFP-11207 as self-controlled toxicity drug may well contribute to the advance of treatment for GI cancer patients as a monotherapy or combination therapy.
Disclosure
All authors are employees of Delta-Fly Pharma Inc. The authors report no other conflicts of interest in this work.
References
Heidelbelger C, Ansfield FJ. Experimental and clinical use of fluorinated pyrimidines in cancer chemotherapy. Cancer Res. 1963;23:1226–1243. | ||
De Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18(16):2938–2947. | ||
Andre T, Louvet C, Maindrault-Goebel F, et al. CPT-11 (irinotecan) addition to bimonthly, high-dose leucovorin and bolus and continuous-infusion 5-fluorouracil (FOLFIRI) for pretreated metastatic colorectal cancer, GERCOR. Eur J Cancer. 1999;35(9):1343–1347. | ||
Kim NK, Park YS, Heo DS, et al. A Phase III study of 5-fliorouracil and cisplatin versus 5-fluorouracil, doxorubicin, and mitomycin versus 5-fluorouracil alone in the treatment of advanced gastric cancer. Cancer. 1993;71(12):3813–3818. | ||
Caballero GA, Ausman RK, Quebbeman EJ. Long-term, ambulatory, continuous iv infusion of 5-FU for the treatment of advanced adenocarcinomas. Cancer Treat Rep. 1985;69(1):13–15. | ||
Quebbeman EJ, Ausman RK, Hansen R, et al. Long-term ambulatory treatment of metastatic colorectal adenocarcinoma by continuous intravenous infusion of 5-fluorouracil. J Surg Oncol. 1985;30(1):60–65. | ||
Moynihan T, Hansen R, Anderson T, et al. Continuous 5-fluorouracil infusion in advanced gastric adenocarcinoma. Am J Clin Oncol. 1988;11(4):461–464. | ||
Barbounis VP, Kalofonos HP, Munro AJ, Mckenzie CG, Sackier JM, Epenetos AA. Treatment of colorectal cancer and other malignancies with continuous infusion of 5-fluorouracil. Anticancer Res. 1989;9(1):33–40. | ||
Lokich JJ, Ahigren JD, Gullo JJ, Philips JA, Fryer JA. A prospective randomized comparison of continuous infusion fluorouracil with a conventional bolus schedule in colorectal adenocarcinoma: a mid-Atlantic oncology program study. J Clin Oncol. 1989;7(4):425–432. | ||
Ishikawa T, Utoh M, Sawada N, et al. Tumor selective delivery of 5-fluorouracil by capecitabine, a new fluoropyrimidine carbamate, in human cancer xenografts. Biochem Pharmacol. 1998;55(7):1091–1097. | ||
Fujii S, Ikenaka K, Fukushima M, Shirasaka T. Effect of uracil and its derivatives on antitumor activity of 5-fluorouracil and 1-(2-tetrahydrofuryl)-5-fluorouracil. Gann. 1978;69(6):763–772. | ||
Shirasaka T, Nakano K, Takechi T, et al. Antitumor activity of 1M tegafur-0.4M 5-chloro-2,4-dihydroxypyridine-1M patasium oxonate (S-1) against human colon carcinoma orthotopically implanted into nude rats. Cancer Res. 1996;56(11):2602–2606. | ||
Yen-Revollo JL, Goldberg RM, McLeod HL. Can inhibiting dihydropyrimidine dehydrogenase limit hand-foot syndrome caused by fluoropyrimidines? Clin Cancer Res. 2006;14(1):8–13. | ||
Taguchi T. Experience with UFT in Japan. Oncology. 1997;11(9 Suppl 10):30–34. | ||
Sugimachi K, Maehara Y, Horikoshi N, et al. The S-1 Gastrointestinal Cancer Study Group. An early phase II study of oral S-1, a newly developed 5-fluorouracil derivative for advanced and recurrent gastrointestinal cancers. Oncology. 1999;57(3):202–210. | ||
Shirasaka T, Shimamoto Y, Fukushima M. Inhibition by oxonic acid of gastrointestinal toxicity of 5-fluorouracil without loss of its antitumor activity in rats. Cancer Res. 1993;53(17):4004–4009. | ||
Schneider WC. Phosphorus compounds in animal tissues. III. Comparison of methods for the estimation of nucleic acid. J Biol Chem. 1946;164:747–751. | ||
Fukushima M, Satake H, Uchida J, et al. Preclinical antitumor efficacy of S-1: a new oral formulation of 5-fluorouracil on human tumor genografts. Int J Oncol. 1998;13(4):691–698. | ||
Shirasaka T, Shimamoto Y, Ohshima H, et al. Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anticancer Drugs. 1996;7(5):548–557. | ||
Ikagawa M, Kimura M, Iwai M, Usami E, Yoshimura T, Yasuda K. Neutropenia as a prognostic factor and safety of second-line therapy with S-1 for advanced or recurrent pancreatic cancer. Mol Clin Oncol. 2016;5(3):281–288. | ||
Tatsumi K, Fukushima M, Shirasaka T, Fujii S. Inhibitory effects of pyrimidine, barbituric acid and pyridine derivatives on 5-fluorouracil degradation in rat liver extracts. Gann. 1987;78:748–755. | ||
Ikeda K, Yoshisue K, Matsushima E, et al. Bioactivation of tegafur to 5-fluorouracil is catalyzed by cytochrome-450 2A6 in human liver microsomes in vitro. Clin Cancer Res. 2000;6(11):4409–4415. | ||
Lee JJ, Beumer JH, Chu E. Therapeutic drug monitoring of 5-fluorouracil. Cancer Chemother Pharmacol. 2016;78(3):447–464. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.