Back to Journals » Drug Design, Development and Therapy » Volume 20
Development of Membrane-Targeting Cannabigerol Derivatives as Potent Broad-Spectrum Antibacterial Agents
Authors Yin Z, Zhang T, Wang Y, Wang Y, Xie X, Zhang F, Wang C ![]() , Hu W
, Hu W
Received 5 August 2025
Accepted for publication 7 January 2026
Published 23 January 2026 Volume 2026:20 558606
DOI https://doi.org/10.2147/DDDT.S558606
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leonidas Panos
Zhicheng Yin, Tingting Zhang, Yipeng Wang, Yan Wang, Xiaoqing Xie, Fengyu Zhang, Chuandong Wang, Wei Hu
State Key Laboratory of Microbial Technology, Microbial Technology Institute, Shandong University, Qingdao, People’s Republic of China
Correspondence: Wei Hu, State Key Laboratory of Microbial Technology, Microbial Technology Institute, Shandong University, Qingdao, People’s Republic of China, Email [email protected] Chuandong Wang, State Key Laboratory of Microbial Technology, Microbial Technology Institute, Shandong University, Qingdao, People’s Republic of China, Email [email protected]
Purpose: This research aimed to develop novel membrane-targeting antibacterial agents via rational design and synthesis of hybrid compounds derived from cannabigerol (CBG) and antimicrobial peptide (AMP) motifs. This approach targeted key limitations of CBG, specifically its poor aqueous solubility, restricted activity against Gram-negative pathogens, and low bioavailability. By incorporating AMP domains, we intended to exploit their membrane-disruptive capability, thereby achieving enhanced broad-spectrum activity, diminished propensity for resistance, and improved pharmacological properties.
Methods: A library of membrane-active cannabigerol derivatives was designed and synthesized through conjugation of antimicrobial peptides structural motifs to the cannabigerol core scaffold by a flexible chemical linker. All compounds were characterized by 1H, 13C NMR, and high-resolution mass spectrometry. Antibacterial activity was evaluated using a broth microdilution method. Hemolytic activity was assessed against sheep erythrocytes, and cytotoxicity was determined using human cell lines. Mechanistic studies included molecular dynamics simulations, scanning electron microscopy, membrane depolarization and permeabilization assays, and quantification of reactive oxygen species generation. In vivo efficacy was validated in a murine peritonitis-sepsis model.
Results: The compound 5d demonstrated potent broad-spectrum activity against both Gram-positive and Gram-negative bacteria with low hemolytic toxicity and negligible cytotoxicity. Moreover, 5d exhibited rapid bactericidal action, resistance development prevention, and robust antibiofilm efficacy. Molecular dynamics simulations revealed the selective affinity of 5d for bacterial membranes. Mechanistic studies indicated membrane disruption via specific binding to phosphatidylglycerol and cardiolipin, leading to reactive oxygen species accumulation, DNA/proteins leakage, and bacterial death. In a murine peritonitis-sepsis model, 5d achieved superior survival rates and bacterial clearance compared to vancomycin.
Conclusion: Compound 5d demonstrated potent broad-spectrum antibacterial activity, an advantageous safety margin, and a low resistance propensity. Its membrane-disruptive mode of action underpins both efficacy and the reduced potential for resistance development. These attributes identify 5d as a promising lead for novel membrane-targeted anti-infective agents.
Keywords: cannabigerol derivatives, antimicrobial peptide, broad-spectrum antimicrobials, drug-resistant, membrane-targeting mode
Introduction
Antibiotics have long served as critical clinical tools for combating bacterial infections.1 However, the widespread overuse and misuse of these agents have accelerated the emergence of antimicrobial resistance (AMR), posing substantial challenges in infectious disease management and driving global healthcare expenditures.2–4 The severity of AMR is highlighted through epidemiological data. In 2021 alone, approximately 4.71 million deaths worldwide were linked to bacterial infections resistant to antimicrobial medications, with 1.14 million of these directly attributable to AMR.5 Current projections suggest annual mortality rates could escalate to 10 million deaths by 2050, with economic losses exceeding $100 trillion if unmitigated.6,7
The World Health Organization (WHO) has highlighted several Gram-positive (G+) bacteria as priorities for research and control.8 Among these, methicillin-resistant Staphylococcus aureus (MRSA), classified as a high-priority pathogen, is of particular concern due to its high prevalence and associated mortality.9 Streptococci also contribute significantly to the global burden, causing diseases ranging from pharyngitis and pneumonia to invasive conditions such as sepsis and meningitis.10 Notably, Streptococcus pneumoniae, Group A Streptococcus (GAS), and Group B Streptococcus (GBS) are all designated as medium‑priority pathogens by the WHO.11 Meanwhile, Gram-negative (G−) bacteria, including Escherichia coli, Pseudomonas aeruginosa, and Klebsiella pneumoniae, are frequently responsible for various infections such as urinary tract infections, hospital-acquired pneumonia, and bloodstream infections.12 Despite this growing threat, the conventional antibiotic development pipelines have failed to match the accelerating pace of AMR.13 This critical gap underscores the urgent need for innovative antimicrobial agents featuring novel chemical architectures and non-traditional mechanisms of action to bypass existing resistance mechanisms.
Antimicrobial peptides (AMPs), evolutionarily conserved components of the innate immune system, constitute pivotal defense mechanisms against microbial invasion.14–16 These molecules exhibit potent antimicrobial activity through membrane-disruptive action mechanisms, making them promising alternatives to traditional antibiotics.17,18 Distinct from traditional antibiotics that target specific molecular pathways, AMPs primarily compromise bacterial cell membranes, which is a non-specific mode that dramatically reduces the resistance development potential.19,20 In contrast to mammalian cell membranes which predominantly contain phosphatidylcholine, phosphatidylethanolamine (PE), and phosphatidylinositol, bacterial cell membranes are primarily composed of PE along with anionic lipids such as phosphatidylglycerol (PG) and cardiolipin (CL).21,22 The cationic domains of AMPs selectively accumulate on bacterial membrane surfaces via electrostatic interactions with negatively charged phospholipids (eg, PG and CL), whereupon their hydrophobic residues insert into the phospholipid bilayer, thereby disrupting membrane integrity and ultimately leading to bacterial cell death.23–25 The selective targeting of bacterial membranes through electrostatic interactions effectively minimizes the potential mammalian cell toxicity associated with AMPs. Nevertheless, AMPs face significant translational barriers, including suboptimal in vivo stability, unfavorable pharmacokinetic properties, and prohibitively high production costs.26,27 To circumvent these limitations, researchers have developed biomimetic strategies to design synthetic membrane-targeting agents that recapitulate AMPs’ structural motifs and functional characteristics.28–31 These efforts have achieved clinical relevance: small-molecule peptidomimetics including Lytixar (LTX-109) and Brilacidin (PMX30063), engineered to mimic AMP functionality while addressing their limitations, have progressed to clinical trials.32–34
Cannabigerol (CBG, 1, Figure 1), a geranylated phenol derived from Cannabis sativa,35 demonstrates diverse pharmacological potential, encompassing antitumor,36 anti-inflammatory,37,38 neuroprotective,39 and antibacterial activities.40 Emerging evidence highlights its efficacy against G+ pathogens such as Streptococcus mutans and MRSA, which may involve membrane targeting or biofilm inhibition.41,42 For instance, CBG can disrupt the bacterial cell membrane,42 and it also inhibits S. mutans biofilm formation through dual mechanisms: directly as an antibacterial agent and indirectly by interfering with metabolic pathways that regulate biofilm development.43 However, CBG shows limited activity against G− bacteria (eg, E. coli and Acinetobacter baumannii), attributed to the low permeability of their outer membranes.36 Additionally, its therapeutic utility is hampered by poor aqueous solubility, rapid first-pass metabolism, and low oral bioavailability.44 These pharmacological limitations necessitate strategic structural modifications to improve solubility and expand its antibacterial spectrum.
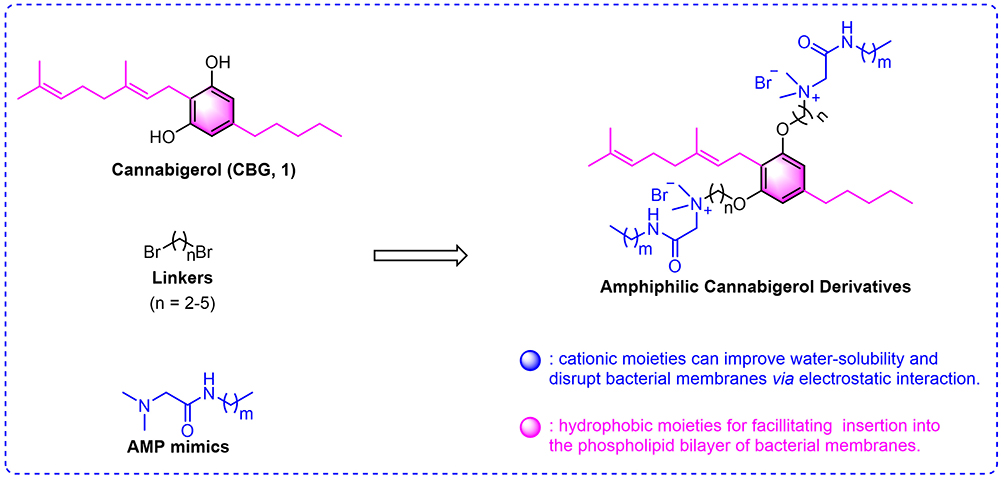 |
Figure 1 Design of amphiphilic Antimicrobial peptide-Cannabigerol (AMP-CBG) derivatives. |
Drawing inspiration from the amphiphilic architecture of AMPs,45 this study aimed to develop novel membrane-targeting antibacterial agents through the rational modification of CBG derivatives. Our central hypothesis was that conjugating cationic AMP motifs to the lipophilic CBG core via a flexible chemical linker (Figure 1) would yield hybrid compounds capable of synergistic membrane disruption. We postulated that the cationic AMP moiety would enhance aqueous solubility and mediate electrostatic interactions with negatively charged bacterial membranes, while the CBG core would facilitate integration into the phospholipid bilayer. This dual-action design was expected to collectively enhance membrane destabilization and improve bactericidal efficacy. To test this hypothesis, we pursued the following objectives: design and synthesize a library of AMP‑CBG hybrid derivatives to improve solubility and broaden antibacterial activity; evaluate the antibacterial profile of the resulting compounds, including bactericidal kinetics, potential for resistance development, and biofilm inhibition/disruption; investigate the mechanism of action, with a focus on membrane targeting and destabilization; and assess in vivo antibacterial efficacy in a relevant infection model. Through this systematic approach, we seek to provide a rational design strategy and actionable insights for developing novel membrane-targeting antibacterials, thereby laying a foundation for future agents with clinical translation potential.
Materials and Methods
Chemistry
Unless otherwise stated, the synthetic chemicals were purchased from Shanghai Macklin Biochemical Technology Co., Ltd. (Shanghai, China) and used as received. Column chromatography was performed using commercially available silica gel (200–300 mesh). Reactions were monitored by thin-layer chromatography (TLC) on glass silica gel plates (0.25 mm, GF254), visualized under UV light (254 nm). 1H and 13C NMR spectra were acquired on a Bruker Avance 600 NMR instrument (Germany) with DMSO-d6 or CDCl3 as deuterated solvents. High-resolution mass spectral (HRMS) data were acquired on an Impact HD Q-TOF (Bruker, Germany) equipped with an ESI source.
Synthesis of CBG (1)
Olivetol (1.5 mmol, 270.2 mg) and geraniol (1.0 mmol, 173.5 μL) were dissolved in anhydrous 1,2-dichloroethane (12 mL) in a round flask, and acidic alumina (75–150 mesh, 2.0 g) was added as a catalyst. The mixture was stirred magnetically and refluxed for 6 h with continuous TLC monitoring. Completion of the reaction was confirmed by TLC analysis, showing disappearance of starting materials. After cooling to room temperature, the acidic alumina was removed by vacuum filtration, and the filter cake was thoroughly washed with ethyl acetate (3 × 15 mL) to ensure complete recovery of the product. The combined filtrates were concentrated under reduced pressure (7.0 mbar) and purified by column chromatography on silica gel (200–300 mesh) using an eluent system of hexane/ethyl acetate (20/1, v/v). The pure CBG (1) was obtained as a pale yellow solid with a yield of 53%.46
Synthesis of Intermediates 2A-D
CBG (1) (1.0 mmol, 316.2 mg) was dissolved in 10 mL of dry acetone with potassium carbonate (5.0 mmol, 691.1 mg) and the corresponding brominated alkanes (1,2-dibromoethane; 1,3-dibromopropane; 1,4-dibromobutane and 1,5-dibromopentane; 15.0 mmol). The mixture was subjected to reflux conditions for 24 h, with progress monitored by TLC until completion. After cooling, the mixture was filtered and concentrated. The resulting residue was diluted with ethyl acetate (20 mL) and washed with water twice. The organic solvent was evaporated in vacuo, and the crude product was purified by silica gel column chromatography using petroleum ether/ethyl acetate (200/1, v/v) as eluents to afford intermediates 2A-D in moderate yields (19–77%).
Synthesis of Compounds 4E-I
The corresponding amine (10.0 mmol) and potassium carbonate (15.0 mmol, 2.1 g) were dissolved in a mixed solvent of CH2Cl2/H2O (1:1, 200 mL). Bromoacetyl bromide (15.0 mmol, 3.0 g) was added dropwise to the mixture at 0°C under stirring. After continuous stirring for 30 min, the reaction was warmed to ambient temperature, and stirred further for 3–5 h. Progress was monitored by TLC. The organic layer was isolated, and aqueous phase was extracted with CH2Cl2 (three times). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure (7.0 mbar) to yield intermediates 3E-I, which were used directly for the next reaction. Subsequently, intermediates 3E-I (5.0 mmol) and aqueous solution of dimethylamine (20.0 mmol, 40%, m/m) were dissolved in anhydrous ethanol (30 mL) and subjected to reflux at 100°C for 10 h under a nitrogen atmosphere. After cooling, the reaction mixture concentrated under reduced pressure (7.0 mbar). The crude product was dissolved in water (30 mL), and the aqueous layer was extracted with CH2Cl2 (three times). The organic extracts were combined, dried, and concentrated. The resulting residue was purified by silica gel column chromatography with dichloromethane/methanol (50/1, v/v) as eluents to produce compounds 4E-I.47
Synthesis of Amphiphilic CBG Derivatives 5a-t
A mixture of intermediates 2A-D (0.2 mmol) and compounds 4E-I (0.6 mmol) in anhydrous ethanol (6 mL) was placed in a screw-top pressure tube. The reaction mixture was heated to 85°C under stirring for 48 h. After cooling, the mixture was concentrated under reduced pressure (7.0 mbar). The crude product was purified by silica gel column chromatography using dichloromethane/methanol (30:1 to 15:1, v/v) as eluents to afford the target products (yield: 35–80%). Structures of 5a-t were characterized by 1H NMR, 13C NMR and HRMS. The spectral data of 5a-t were provided in the Supplementary Materials (Supplementary Data 1−20) 1H NMR, 13C NMR and HRMS spectra of 5a-t are provided in the Supplementary Materials (Figures S1-S60).
Biological Evaluation
In vitro Antibacterial Assay
The antibacterial activity of all compounds was determined using the broth microdilution method in accordance with the CLSI guidelines. Four G+ bacterial strains (S. aureus ATCC 29213, MRSA ATCC 43300, Enterococcus faecalis ATCC 29212, and Bacillus subtilis ATCC 6051) and four G− bacterial strains (A. baumannii ATCC 19606, K. pneumoniae ATCC 13883, E. coli ATCC 25922 and P. aeruginosa PAO1) were maintained in our laboratory. Bacterial strains were cultured overnight in Mueller Hinton Broth (MHB) medium (Oxoid, UK) at 37°C and 200 rpm. The formulation of MHB medium per liter is as follows: beef, dehydrated infusion from 300.0 g; casein hydrolysate 17.5 g; starch 1.5 g. The bacterial concentration was adjusted to 1×106 CFU/mL. All of the tested compounds were dissolved in DMSO or H2O to prepare stock solutions at a concentration of 5120 μg/mL. The stock solutions were diluted to 256 μg/mL by serial two-fold dilution method using MHB medium. These solutions were placed in 96-well plates, and then serially diluted twice to reach concentrations ranging from 256 to 0.5 μg/mL. A 100 μL aliquot of bacterial suspension was transferred into 96-well plates and mixed with 100 μL of serially diluted compounds. After incubation at 37°C for 24 h, the minimum inhibitory concentrations (MICs) of the compounds were determined as the lowest concentration that completely inhibited bacterial growth. The experiment was performed in triplicate.
Hemolysis Assay
The assay was performed as previously described with minor modifications.30 Red blood cells (RBCs) were isolated via centrifugation (3500 rpm, 5 min) of sheep blood. The RBCs were washed three times with phosphate buffered saline (PBS) and resuspended at a concentration of 5% (v/v). All compounds were dissolved in DMSO and then serially diluted twice with PBS to obtain various concentrations. A total of 150 μL of RBC suspension was mixed with 50 μL of compound solution in a 96-well plate. After incubation at 37°C for 1 h, the mixture was centrifuged at 3500 rpm for 3 min. The absorbance of the supernatant was measured at 540 nm to evaluate hemolysis. The hemolysis percentage was calculated as: Hemolysis (%) = (Abstest - Absnegative control)/(Abspositive control - Absnegative control) × 100%. Triton X-100 (1%) and 1% DMSO in PBS were used as positive and negative controls, respectively. The experiment was performed in triplicate.
Time-Kill Kinetics Assay
The assay was performed as previously described with minor modifications.48 The bacterial suspension (1 × 106 CFU/mL) was treated with compound 5d at concentrations of 1×, 2×, and 4× MIC. Aliquots 100 μL were taken at different time points (0, 0.5, 1, 2, 4, 8, and 24 h), serially diluted, and spread on MHA agar plates. The colonies were counted after incubation for 24 h. Vancomycin and colistin (final concentrations of 4×, 8× MIC) served as positive controls against S. aureus ATCC 29213 and E. coli ATCC 25922, respectively. The experiment was performed in triplicate.
Resistance Development Assay
The assay was performed as previously described with minor modifications.31 The initial MIC of compound 5d against S. aureus ATCC 29213 and E. coli ATCC 25922 were determined by the method described above. Bacterial suspensions were exposed to sublethal concentrations of the compound to investigate potential resistance development. Vancomycin and colistin were used as positive controls. After each exposure, the bacteria were collected, re-suspended in fresh medium, and subjected to subsequent MIC determinations. This process was repeated over 20 consecutive passages to assess the stability of compound activity. The experiment was performed in quintuplicate.
Biofilm Inhibition Assay
The assay was performed as previously described with modifications.49 Single colonies of S. aureus ATCC 29213 and E. coli ATCC 25922 were picked, suspended in MHB medium, and cultured overnight at 37°C and 200 rpm. The bacterial suspension was adjusted to a final concentration of 1×106 CFU/mL using fresh MHB medium. A 100 μL aliquot of the bacterial solution was mixed with an equal volume of serially diluted compound 5d in a 96-well plate. After incubation at 37°C for 24 h, the plates were gently washed three times with PBS buffer to remove planktonic cells. The biofilms were then fixed with 150 μL of methanol for 30 min and stained with 200 μL of 0.1% crystal violet for 10 min. The excess dye was removed by washing three times with PBS buffer, and the plates were air-dried. Biofilm-associated crystal violet was dissolved using 200 μL of 33% acetic acid. After mixing 50 μL of the resulting solution with 150 μL of acetic acid, the absorbance was measured at 575 nm to quantify biofilm formation. The experiment was performed in quintuplicate.
Biofilm Disruption Assay
The assay was performed as previously described with modifications.50 A 200 μL aliquot of bacterial suspension (1× 106 CFU/mL) was dispensed into 96-well plates and incubated at 37°C for 24 h without shaking to allow biofilm formation. After incubation, the supernatant was carefully removed, and the plates were gently washed three times with PBS buffer to remove unattached cells. Subsequently, 200 μL of compound 5d at various concentrations was added to the wells containing the biofilms. The plates were then incubated at 37°C for an additional 24 h. Following this, the treated biofilms were quantified using a crystal violet staining assay as described above. The experiment was performed in quintuplicate.
Confocal Laser Scanning Microscopy (CLSM) Observation
Mature biofilms of S. aureus ATCC 29213 and E. coli ATCC 25922 were prepared using the method described above. The biofilms were treated with compound 5d at a concentration of 8× MIC for 24 h. PBS buffer was used as a negative control. After treatment, CLSM (LSM900, ZEISS, Germany) was employed to analyze the biofilms stained by LIVE/DEAD® BacLight Bacterial Viability Kit (Invitrogen, Molecular Probes Inc., USA) according to the manufacturer’s instructions. Quantitative analysis was carried out in accordance with the previously reported procedure.51
Scanning Electron Microscopy (SEM) Observation
Overnight cultures of S. aureus ATCC 29213 and E. coli ATCC 25922 were resuspended in PBS buffer and diluted to a final OD600 of 0.5. The bacterial suspensions were incubated with compound 5d at a final concentration of 8× MIC for 4 h at 37°C. Subsequently, the mixture was centrifuged at 3500 rpm and 4°C for 10 min, and the supernatant was removed. The bacterial cells were washed three times with PBS buffer. Following this, 1 mL of 2.5% glutaraldehyde was used to fix the bacterial cells at 4°C overnight. After washing with PBS buffer, the samples were dehydrated using a graded series of ethanol solutions, dried, and coated with gold. Finally, the cell morphology was observed and imaged using a SEM (Quanta 250 FEG, Field Electron and Ion Company, USA).
Molecular Dynamics (MD) Simulations
All-atom MD simulations were conducted based on three membrane systems (G− bacterial outer membrane, G+ bacterial membrane, and mammalian plasma membrane) obtained from CHARMM-GUI (http://www.charmm-gui.org).52,53 Details of their lipid compositions are shown in Tables S1–S3. The membranes were oriented such that they lay in the xy-plane, with the z-axis perpendicular to the membrane surface. Construction and simulation of the membrane systems employed the CHARMM36 force field54 for the lipid molecules, with a simulation box size of approximately 100×100×90 Å3. The small molecule 5d was positioned above the upper leaflet of each membrane using GROMACS (version 2023.2) and its gmx editconf command. The force-field parameters and topology for 5d were generated by the SwissParam 2023 online tool (https://www.swissparam.ch).55
For the free-diffusion simulation, 5d was positioned on the membrane surface. Extended MD simulations were performed with GROMACS to observe its spontaneous transmembrane diffusion and assess energetic changes in the absence of external pulling. The simulation protocol began with energy minimization via the steepest descent method, followed by six equilibration steps during which positional restraints on lipid molecules were gradually removed. The production run was carried out under a semi-isotropic NPT ensemble. The temperature was maintained at 313 K using the v-rescale thermostat (coupling constant = 1.0 ps), with separate coupling groups for membrane lipids, solvent, and the small molecule. Pressure was controlled at 1.0 bar via the C-rescale algorithm with a coupling constant of 5.0 ps, allowing independent adjustment of the membrane plane and normal direction to preserve area density and thickness. Hydrogen-involving bonds were constrained using the linear constraint solver (LINCS) algorithm with constraints applied to h-bonds. The integration time step was set to 0.002 ps/step, with a total of ~1.0×109 steps corresponding to approximately 2 μs of simulation time under periodic boundary conditions in all spatial dimensions. Van der Waals interactions were handled using a force-switch scheme between 1.0 and 1.2 nm, while electrostatic interactions were treated with the PME method at a cutoff distance of 1.2 nm. System energies and log information were recorded every 2 ps, and coordinates/velocities were saved every 100 ps. Throughout the simulations, 5d was free to diffuse without additional restraints, enabling observations of its spontaneous transmembrane movement over microsecond timescales. This setup allowed for both qualitative analysis of the translocation mechanism and quantitative assessment of energetics associated with transmembrane diffusion.
In the replica-exchange umbrella sampling (REUS), also known as bias-exchange US (BEUS), umbrella sampling was systematically performed on the same three membrane systems to determine the free energy profile of 5d translocation across each membrane. Specifically, along the membrane normal (z-axis), a set of initial distances (DIST) was selected for each system, and a harmonic restraint with a force constant of ~5000 kJ·mol−1·nm−2 was applied. Each window underwent a short equilibration (eq) run followed by a production (prd) run. For G+ bacterial inner membranes and mammalian membranes, the distances ranged from −5.5 nm to 5.0 nm with an interval of 0.05 nm, while for the G− bacterial outer membrane, the distance ranged from −5.5 nm to 8.0 nm at the same interval (0.05 nm). The equilibration phase employed an integration time step of 0.001 ps/step (~500 ps in duration), and the production phase used a larger time step of 0.002 ps/step (~5 ns). Neighbor lists were updated every 20 steps, and Van der Waals interactions were smoothly switched between 1.0 and 1.2 nm to minimize artifacts. Electrostatic interactions were handled using the PME algorithm with a cutoff distance of 1.2 nm. Bonds involving hydrogens were constrained using LINCS to reduce computational cost while maintaining numerical stability.
The system was simulated under a semi-isotropic NPT ensemble, where C-Rescale was used to maintain a pressure of 1 bar (coupling constant of 5.0 ps), and v-rescale was employed to control the temperature at 313 K for the membrane, solvent, and the small molecule (coupling constant of 1.0 ps). To ensure accurate calculation of free energy profiles despite multi-state sampling challenges, the multistate Bennett acceptance ratio estimator (MBAR) method56 was utilized for post-processing the umbrella windows due to its ability to provide an unbiased estimator with reduced variance. This approach allowed us to obtain the free energy surfaces from individual windows and ultimately combine them into a complete potential of mean force (PMF) describing 5d’s transmembrane pathway. By merging the results from windows on both the upper and lower leaflets, the free energy landscape of 5d was thoroughly captured, spanning from the extracellular to the intracellular side of each membrane.
Membrane Depolarization Assay
The assay was performed as previously described with minor modifications.57,58 Bacterial cells of S. aureus ATCC 29213 and E. coli ATCC 25922 were resuspended in HEPES buffer (5 mM, pH = 7.4) to an OD600 of 0.05. A 150 μL aliquot of bacterial suspension was mixed with 50 μL of 3,3′-dipropylthiadicarbocyanine iodide [DiSC3(5)] (10 μM) in a 96-well black plate. The plates were then incubated in the dark for 30 min (for S. aureus ATCC 29213) and 40 min (for E. coli ATCC 25922). Fluorescence intensity was measured at an excitation wavelength of 622 nm and an emission wavelength of 670 nm using a fluorescent microplate reader (SpectraMax Gemini XPS, Molecular Devices, USA). After 8 min, 10 μL of compound 5d (1×, 2×, 4×, and 8× MIC) was added to the wells. Fluorescence intensity was monitored at intervals of 2 min for a total duration of 40 min. The experiment was performed in triplicate.
Bacterial Membrane Permeabilization Assay
The assay was performed as previously described with minor modifications.59 Bacterial cells of S. aureus ATCC 29213 and E. coli ATCC 25922 were resuspended in HEPES buffer (5 mM, pH 7.4) to an OD600 of 0.3. A 150 μL aliquot of bacterial suspension was mixed with 50 μL of 10 μM propidium iodide (PI) in a 96-well black plate. The plates were then incubated in the dark for 30 min (for S. aureus) and 40 min (for E. coli). Fluorescence intensity was measured at an excitation wavelength of 535 nm and an emission wavelength of 617 nm using a fluorescent microplate reader. After 8 min, 10 μL of compound 5d (at concentrations of 1×, 2×, 4×, and 8× MIC) was added to the wells. Fluorescence intensity was monitored at intervals of 2 min for a total duration of 40 min. The experiment was performed in triplicate.
Membrane Component Competition Assays
The checkerboard assay was performed to determine the effect of exogenous phospholipids and lipopolysaccharide (LPS) on the antibacterial activity of compound 5d.60 Bacterial cells (S. aureus ATCC 29213 and E. coli ATCC 25922) were cultured for 24 h and diluted in MHB medium to obtain a bacterial suspension at a concentration of 1×105 CFU/mL. All lipids including PG (Sigma-Aldrich), CL (Sigma-Aldrich), PE (Sigma-Aldrich) were dissolved in methanol to prepare stock solutions at a concentration of 5120 μg/mL. LPS (Sigma-Aldrich) was resuspend in sterile double distilled water to prepare stock solutions at a concentration of 5120 μg/mL. A 50 μL aliquot of PG, CL, PE, or LPS (0–32 μg/mL) were mixed with 50 μL of compound 5d (0–32 μg/mL) in 96-well plates. Afterward, 100 μL bacterial suspension was added to each well. The plates were incubated at 37°C for 18 h, and the MIC value was interpreted. The experiment was performed in triplicate.
Intracellular DNA and Protein Leakage
Mid-logarithmic phase bacterial cells of S. aureus ATCC 29213 and E. coli ATCC 25922 were resuspended in PBS buffer and diluted to an OD600 of 0.3. The bacterial suspensions were treated with different concentrations of compound 5d (1×, 2×, 4×, and 8× MIC) for 4 h. The concentration of DNA and protein in the supernatant was measured at wavelengths of 260 nm and 280 nm using a microspectrophotometer, respectively. The experiment was performed in triplicate.
Reactive Oxygen Species (ROS) Determination
The assay was performed as previously described with minor modifications.29,61 The intracellular ROS level was assessed using the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) from a Reactive Oxygen Species Assay Kit (Beyotime, China), with the kit-provided Rosup used as the positive control to induce oxidative stress.62 S. aureus ATCC 29213 and E. coli ATCC 25922 cells, grown for 6 h, were washed and resuspended in PBS buffer to prepare bacterial solutions with an OD600 of 0.5. The bacterial suspension was then incubated with 100 μM DCFH-DA for 30 min. After incubation, the supernatant was removed, and the residual probe was washed three times with PBS buffer. Subsequently, 180 μL of the bacterial suspension was transferred to black 96-well plates and mixed with 20 μL of compound 5d (final concentrations: 1×, 2×, 4×, and 8× MIC) at 37°C for 40 min. The fluorescence intensity was measured using a microplate reader with an excitation wavelength of 488 nm and an emission wavelength of 530 nm. The experiment was performed in quintuplicate.
In vitro Cytotoxicity Assay
The cytotoxicity of compound 5d toward human liver cancer HepG2 and lung adenocarcinoma A549 cell lines was determined by the Cell Counting Kit-8 (CCK-8) assay.63 Human liver cancer HepG2 and lung adenocarcinoma A549 cell lines were all purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Briefly, 100 μL of cell suspension (final density 2×104 cells/well) was inoculated into sterile 96-well plates and cultured at 37°C for 24 h with 5% CO2. After the cells adhered to the plate surface, the medium was carefully removed. Then, 100 μL of compound 5d at two-fold serial dilutions was added to each well, and the plates were incubated at 37°C for 24 h. Subsequently, 10 μL of CCK-8 solution was added to each well, and the plates were further incubated at 37°C for 1 h. The absorbance of each well was detected using a microplate reader at 450 nm. The cell survival rate was calculated using the following formula: Cell viability (%) = [(Abstest − Absnegative control)/(Abspositive control − Absnegative control)] × 100%. The experiment was performed in triplicate.
In vivo Antibacterial Assay
Female BALB/c mice were used for evaluating the in vivo efficacy of compound 5d. These mice were purchased from Jinan Pengyue Experimental Animal Breeding Co., Ltd. (Jinan, China), with a weight of approximately 18–20 g and an age of 6–8 weeks. All mice were housed under specific pathogen-free conditions in individually ventilated cages and provided ad libitum access to food and water. The animal house conditions maintained a controlled environment at 22 ± 2°C and 40–60% humidity under a 12 h light/dark cycle.
For the survival assay, mice were randomly divided into seven groups (6 mice/group). The sample size was determined based on a previous mouse sepsis model, as no formal power calculation was performed.64 Each mouse received intraperitoneal injection of 100 μL S. aureus ATCC 29213 at a concentration of 1×107 CFUs. After 1 h post-infection, mice were treated with a single intraperitoneal dose of compound 5d (5, 10, or 15 mg/kg) or the positive control vancomycin (5, 10, or 15 mg/kg). The blank control group received PBS buffer alone. Survival status was monitored for 7 days, and survival curves were plotted.
For bacterial load detection in various organs, mice were randomly assigned to five groups (6 mice/group). Each mouse underwent intraperitoneal injection of 100 μL S. aureus ATCC 29213 at a concentration of 5×106 CFUs. After 1 h post-infection, mice were treated with a single intraperitoneal dose of compound 5d (10 or 15 mg/kg) or vancomycin (10 or 15 mg/kg). The negative control group received PBS buffer only. Forty-eight hours after administration, all mice were euthanized by cervical dislocation and dissected. Liver, spleen, and kidneys were collected from each mouse, homogenized in sterile PBS buffer, and subjected to 10-fold serial dilution. Dilutions were plated onto MHA plates for CFU counting.
Statistical Analysis
All data are reported as mean ± standard deviation (SD). Each experiment comprised at least three independent biological replicates (n ≥ 3), and inferential statistics were computed only when n ≥ 5. Group comparisons were carried out with the Student’s t-test (for two groups) or one-way analysis of variance (ANOVA, for multiple groups) using GraphPad Prism software (version 10.0). Prior to parametric testing, the normality of data distribution was assessed using the Shapiro–Wilk test, and homogeneity of variances was evaluated with Levene’s test. If the data violated either the normality assumption (Shapiro–Wilk test, p < 0.05) or the homogeneity of variances assumption (Levene’s test, p < 0.05), non-parametric tests were applied accordingly: the Mann–Whitney U-test was used in place of the Student’s t-test, and the Kruskal–Wallis H-test replaced one-way ANOVA. A two-sided p-value < 0.05 was considered statistically significant.
Results and Discussion
Chemistry
The synthetic routes of amphiphilic CBG derivatives are illustrated in Scheme 1. Initially, commercially available olivetol was reacted with geraniol in the presence of acidic alumina to generate CBG (1).46 Subsequent alkylation of 1 using α,ω-dibromoalkanes in basic conditions (K2CO3) yielded intermediates 2A-D with variable alkyl chain lengths (n). In parallel, bromoacetyl bromide was reacted with primary amines to produce intermediates 3E-I, which underwent dimethylamine treatment to afford cationic AMP fragments 4E-I featuring tunable alkyl chain lengths (m). Finally, amphiphilic CBG derivatives 5a-t were obtained via coupling intermediates 2A-D with cationic AMP fragments 4E-I under reflux conditions. All final compounds 5a-t were characterized by 1H NMR, 13C NMR and HRMS.
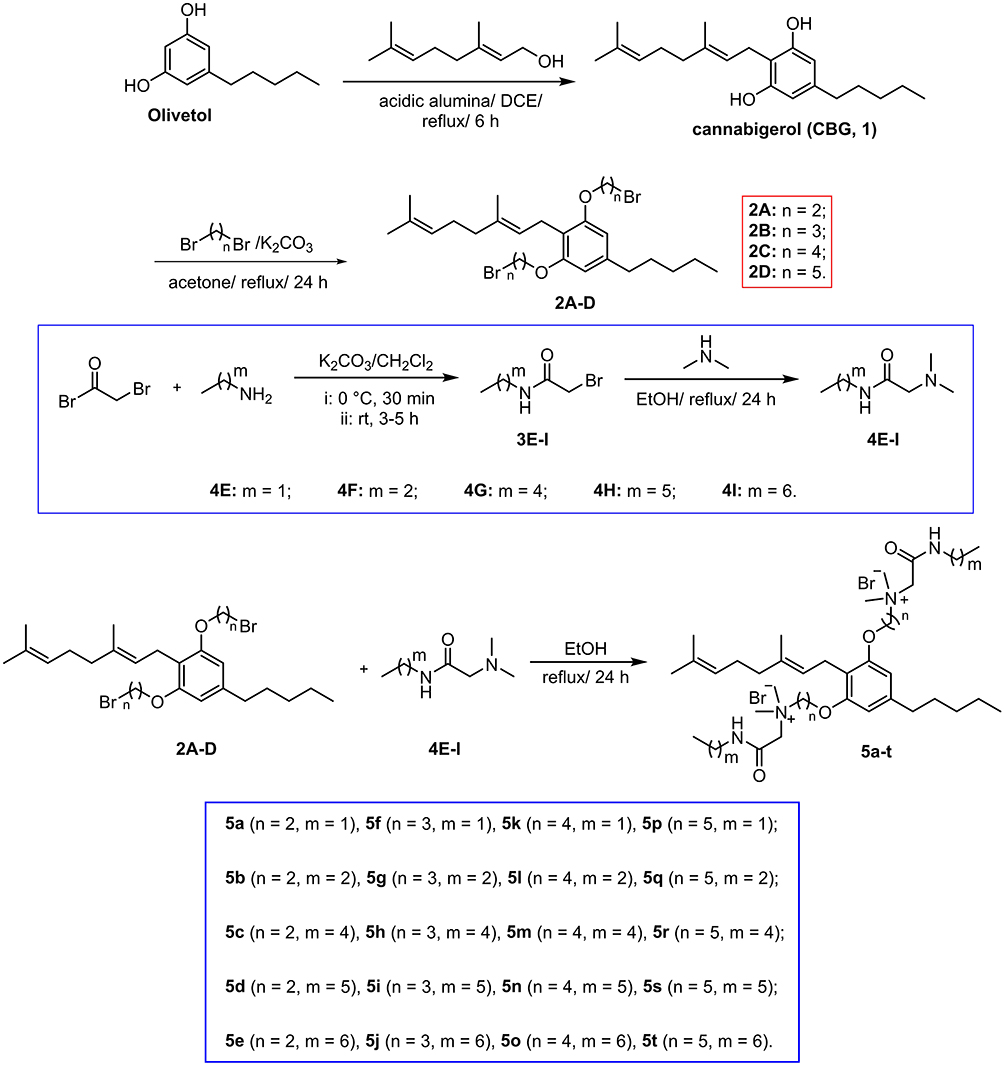 |
Scheme 1 Synthetic routes for CBG derivatives 5a-t. The linker spacer length (n) is consistent between compounds 2A-D and 5a-t, while the alkyl chain length (m) is shared by compounds 4E-I and 5a-t. |
Biological Evaluation
In vitro Antibacterial and Hemolytic Analysis
Antimicrobial susceptibility testing was performed using a broth microdilution method against eight clinically relevant strains listed in Table 1. Reference antibiotics vancomycin, levofloxacin, and colistin served as comparators. Hemolytic activity (HC50) was quantified against sheep erythrocytes to assess mammalian cell toxicity.
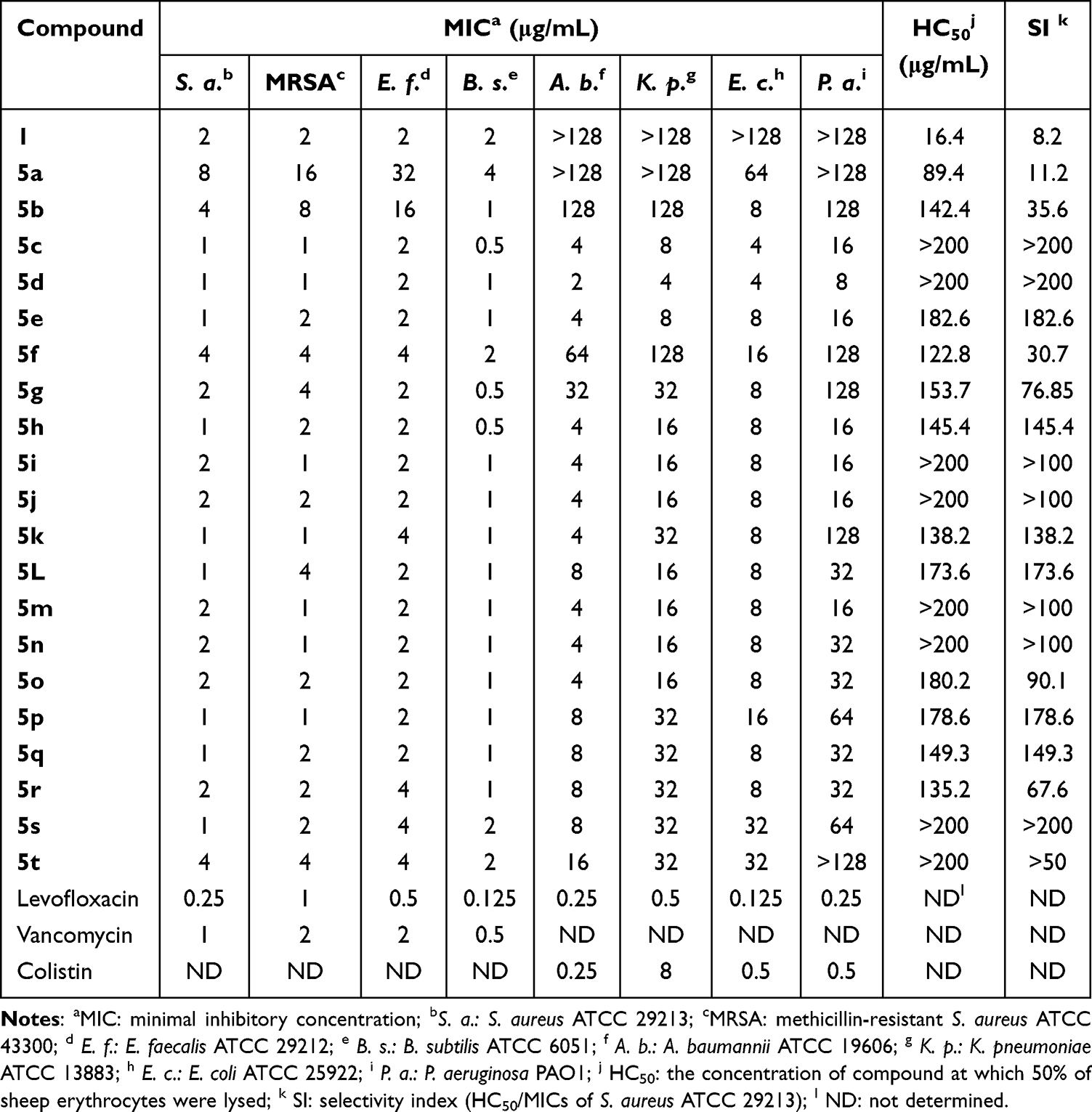 |
Table 1 In vitro Antibacterial Activities Against G+ and G− Bacterial Pathogens (MIC) and Hemolytic Activities (HC50) on Sheep Erythrocytes of Compound 1 and 5a-t |
As summarized in Table 1, unmodified CBG (1) exhibited selective activity against G+ bacteria (MICs = 2 μg/mL) but was inactive against G− strains (MICs > 128 μg/mL). This spectrum of activity aligns with that reported for other major cannabinoids. For example, cannabidiol (CBD) has been shown to exhibit potent activity against G+ bacteria (MICs = 1.56–3.125 μg/mL) yet demonstrates only weak activity against G− bacteria (MICs > 50 μg/mL).18 Strikingly, conjugation of AMP-mimetic fragments to CBG’s phenolic hydroxyl groups produced derivatives 5c-t with enhanced potency (MICs = 0.5–4 μg/mL against G+ pathogens). Notably, derivatives 5c-e, 5h-j, and 5m-o achieved broad-spectrum activity (MICs = 2–32 μg/mL against G− bacteria), representing a significant improvement over compound 1 (MICs > 128 μg/mL) and CBD (MICs > 50 μg/mL). Compound 5d emerged as the optimal candidate, exhibiting dual efficacy (G+: 1–2 μg/mL; G−: 2–8 μg/mL) with minimal hemolysis (HC50 > 200 μg/mL). Therapeutic selectivity, quantified via SI (HC50/MIC), revealed SI > 200 for 5d against S. aureus, surpassing the parent compound 1 (SI = 8.2) and CBD (SI > 64).18 This enhancement correlates with 5d’s optimized amphiphilic balance, positioning it as a viable candidate for further development.
Structure-Activity Relationship (SAR) Analysis
A regular pattern of SAR was observed in the amphiphilic CBG derivatives. As shown in Table 1, both the alkyl chain length (m) and the intermediate linker (n) profoundly influenced the MICs against G⁺ and G− bacteria. Principal component analysis (PCA) of MIC data revealed negative correlations between antibacterial activity and m or n (Figure 2A). For derivatives 5a-e (n = 2), increasing m from 1 to 6 reduced MICs against G+ bacteria from 4–32 μg/mL to 1–2 μg/mL, while G− MICs dropped from 64->128 μg/mL to 4–16 μg/mL. Similarly, elongating n (m = 1, n = 2 to 5) in 5a, 5f, 5k, and 5p improved G+ activity from 4–32 μg/mL to 1–2 μg/mL, and G− activity from 64->128 μg/mL to 8–64 μg/mL. Notably, PCA analysis highlighted a stronger negative correlation between MIC and m than between MIC and n, suggesting m exerts a dominant influence on activity, particularly against G− bacteria. For instance, 5a (m = 1, n = 2) showed weak G− activity (MICs = 64->128 μg/mL), and elongating n to 5 (5p) yielded only moderate improvement (MICs = 8–64 μg/mL). In contrast, extending m from 2 to 5 enhanced G− activity: 5b (m = 2, n = 2) and 5d (m = 5, n = 2) achieved MICs of 8–128 μg/mL and 2–8 μg/mL, respectively. A parallel trend was observed for derivatives 5g and 5i (n = 3), where antibacterial activity correlated with alkyl chain length elongation. In contrast, derivatives 5k-o (n = 4) exhibited minimal variation in efficacy against both G⁺ and G− bacteria as m increased. Derivatives 5p-t (n = 5) demonstrated a gradual decline in activity against both bacterial classes with progressive alkyl chain extension.
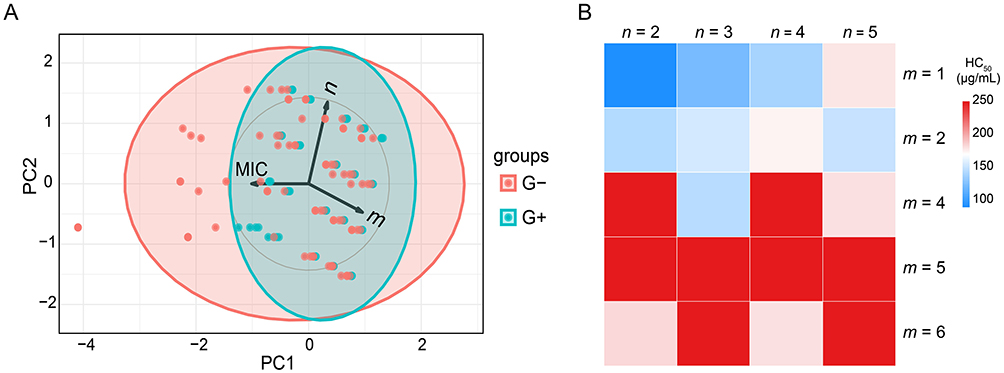 |
Figure 2 Structure-activity relationship (SAR) analysis. (A) Principal component analysis (PCA) of minimum inhibitory concentration (MIC) values against Gram-positive (G⁺) and Gram-negative (G−) bacterial strains, plotted as a function of alkyl chain length (m) on the amide nitrogen and linker spacer length (n). PC, principal component. (B) Correlation heatmap illustrating relationships between hemolytic activity (HC50) and structural parameters m (alkyl chain length) and n (linker length). |
Furthermore, the partition coefficient (clog P) analysis was conducted to evaluate the hydrophilicity of CBG (1) and its AMP-conjugated derivatives (5a-t). As summarized in Table S4, the clog P values ranged from 0.06 to 7.50, indicating enhanced hydrophilicity for most derivatives compared to the parent compound 1 (clog P = 7.35). This improvement was achieved through strategic incorporation of hydrophilic AMP motifs, effectively addressing CBG’s inherent solubility limitations and consequently enhancing bioavailability. Notably, experimental solubility profiles in MIC assays correlated strongly with theoretical clog P predictions, validating the design rationale. These results underscore the necessity of balancing m and n lengths to maintain optimal amphiphilic equilibrium for antibacterial efficacy. Collectively, derivatives 5a-t displayed superior antibacterial efficacy against G⁺ bacteria relative to G− strains. Consistent with prior studies on G− bacteria,65 the observed activity disparity of between G⁺ and G− is attributed to the impermeable LPS layer in G− bacteria, which impedes amphiphilic compound penetration.
Hemolytic activity analysis further revealed strong inverse correlation between alkyl chain length (m) and toxicity (Figure 2B). For derivatives with fixed n, progressive elongation of m from 1 to 6 resulted in HC50 values increasing from 89.4 μg/mL (5a, m = 1) to >200 μg/mL (5c-e, m = 4–6). Notably, all hexyl-substituted derivatives (m = 5; 5d, 5i, 5n, 5s) maintained HC50 > 200 μg/mL regardless of n variation, demonstrating the dominant influence of m on hemocompatibility. These findings reinforce the critical importance of alkyl chain optimization for balancing antimicrobial potency and biosafety.
Bactericidal Time-Kill Kinetics
Time-kill assays were conducted to quantify the concentration-dependent bactericidal activity of 5d against planktonic cultures of S. aureus ATCC 29213 and E. coli ATCC 25922 at 1×, 2×, and 4× MIC. As illustrated in Figure 3, 5d exhibited rapid dose-responsive killing kinetics, achieving complete death of both strains within 0.5 h at 4× MIC with no detectable regrowth over 24 h. Notably, this bactericidal velocity surpassed clinical benchmarks: vancomycin required 4 h at 8× MIC to eradicate S. aureus, while colistin needed 8 h at 4× MIC for E. coli clearance. The stark temporal contrast underscores 5d’s rapid-action profile, a critical attribute for managing acute infections.
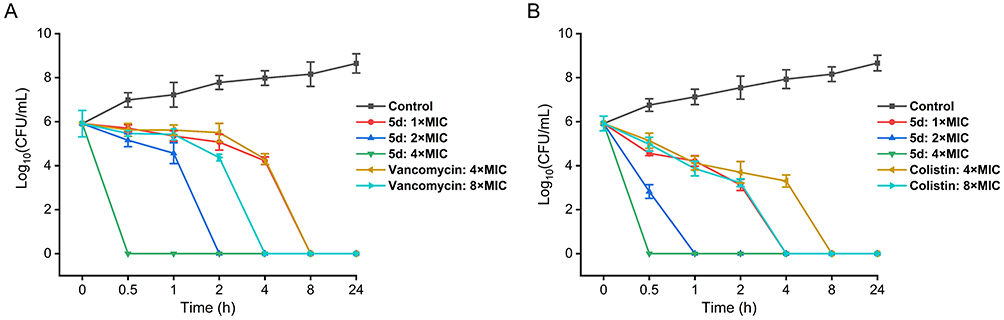 |
Figure 3 Dose- and time- dependent killing curves of compound 5d against S. aureus ATCC 29213 (A) and E. coli ATCC 25922 (B). Vancomycin and colistin served as positive controls for G+ and G− bacterium, respectively. The error bars represent standard deviation (n = 3). Abbreviation: CFU, colony-forming units. |
Resistance Development
The propensity for resistance development constitutes a critical challenge in antimicrobial drug development.66,67 To systematically evaluate this risk, serial passages (20 cycles) were conducted using S. aureus ATCC 29213 and E. coli ATCC 25922 under non-lethal concentrations (0.5× MIC) exposure to 5d, with vancomycin and colistin serving as reference controls. As depicted in Figure 4, vancomycin maintained stable MIC values (1–2 μg/mL) against S. aureus, indicating no detectable resistance emergence. However, colistin exhibited a 64-fold MIC escalation (from 0.5 to 32 μg/mL) against E. coli, demonstrating rapid resistance selection. Remarkably, 5d demonstrated complete resistance stability, maintaining baseline MICs throughout the experimental regimen. This resistance-suppressing characteristic is mechanistically attributed to 5d’s membrane-targeting mode of action, a strategy evolutionarily conserved in AMPs, which circumvents single-target mutations and thereby minimizes resistance development pathways.68
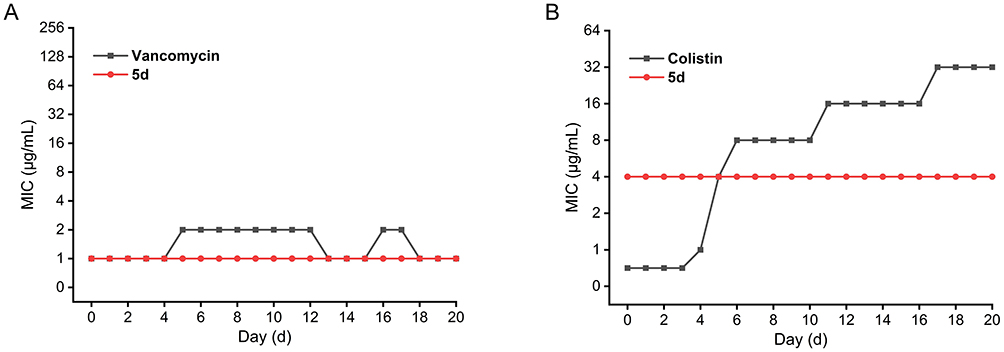 |
Figure 4 Changes in minimum inhibitory concentration (MIC) values during serial passages of S. aureus ATCC 29213 (A) and E. coli ATCC 25922 (B) exposed to subinhibitory concentrations of compound 5d. Vancomycin and colistin were used as comparators. |
Antibiofilm Activity
Bacterial biofilms, characterized by extracellular matrix-encased microbial communities, confer enhanced antimicrobial tolerance through impaired drug penetration and persisted cell formation.69 Developing novel agents capable of disrupting biofilm formation and maintenance is critical to overcoming antibiotic tolerance.70 Crystal violet assays were employed to quantify both biofilm formation inhibition and mature biofilm disruption in S. aureus and E. coli. At 0.5× MIC (0.5 μg/mL for S. aureus; 2 μg/mL for E. coli), 5d achieved 71.04% and 42.27% biofilm inhibition, respectively (Figure 5A and B). Elevating the concentration to 1× MIC enhanced inhibition to 82.39% and 90.98%, demonstrating concentration-dependent efficacy. Against 24-h mature biofilms, 5d exhibited 66.51% (S. aureus) and 57.72% (E. coli) disruption at 1× MIC, with plateaued elimination rates reaching 73.12% and 82.64% at 8× MIC (Figure 5C and D), which aligns with established biofilm recalcitrance to single-agent eradication.71 Notably, 5d exhibited superior antibiofilm activity against S. aureus compared to E. coli.
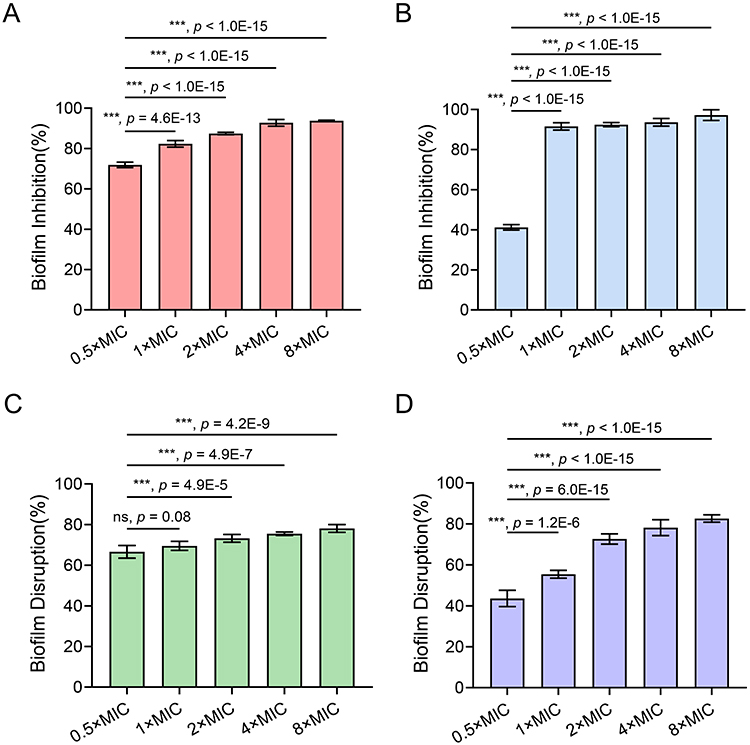 |
Figure 5 Dose-responsive antibiofilm activity of compound 5d against S. aureus ATCC 29213 (A and C) and E. coli ATCC 25922 (B and D). Panels A and B show biofilm inhibition rates, while panels C and D illustrate biofilm disruption efficacy against 24-h mature biofilms. The error bars represent standard deviation (n = 5). ***, p < 0.001, one-way analysis of variance. Abbreviation: ns, non-significant. |
CLSM analysis with SYTO9/PI dual staining72–75 confirmed 5d’s biofilm-damaging capacity. After 24 h of exposure to 5d at 8× MIC, mature biofilms showed dominant red fluorescence (Figure 6A), indicating extensive cell death. Quantification revealed significant increases in dead/live ratios (80.7 ± 4.6% versus 14.2 ± 2.5% for S. aureus; 93.3 ± 2.8% versus 7.7 ± 2.1% for E. coli) versus controls (Figure 6B). Concomitant reductions in bio-volume (0.2 ± 0.1 μm3/μm2 versus 1.9 ± 0.1 μm3/μm2 for S. aureus; 0.6 ± 0.1 μm3/μm2 versus 1.4 ± 0.2 μm3/μm2 for E. coli), mean thickness (1.3 ± 0.1 μm versus 4.3 ± 0.1 μm for S. aureus; 1.7 ± 0.2 μm versus 3.1 ± 0.3 μm for E. coli), and roughness coefficient (0.5 ± 0.1 versus 4.0 ± 0.2 for S. aureus; 0.3 ± 0.2 versus 2.1 ± 0.3 for E. coli) further corroborated structural biofilm disintegration, with S. aureus biofilms exhibiting greater susceptibility. These findings align with prior observations of 5d’s stronger antibiofilm efficacy against G+ bacteria.
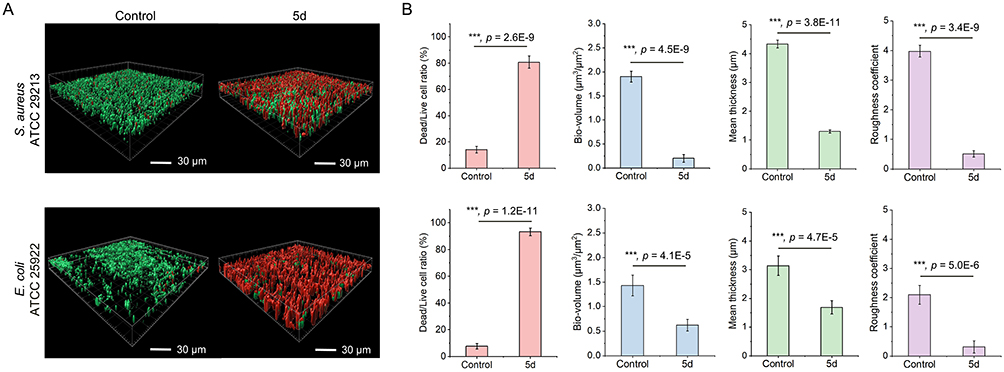 |
Figure 6 Three-dimensional biofilm architecture analysis via CLSM (A) and quantitative metrics (B) after 5d treatment (8× MIC, 24 h). Control groups received PBS. (A) SYTO9/PI-stained Z-stacks (scale bar = 30 μm): Red dominance indicates membrane-compromised cells (PI), while residual green marks viable populations (SYTO9). (B) Key parameters: Dead/live ratio (%), bio-volume (μm3/μm2), thickness (μm), and surface roughness. Bio-volume represents the overall volume of the biofilm, which is calculated as voxel size [(pixel size)x × (pixel size)y × (pixel size)z]/substratum area of the image stack. The error bars represent standard deviation (n = 5). ***p < 0.001, Student’s t-test. |
SEM Observation
SEM imaging revealed profound 5d-induced ultrastructural damage in both S. aureus and E. coli (Figure 7). Untreated S. aureus exhibited pristine coccal morphology with smooth surfaces and intact cellular architecture, whereas 5d-treated cells displayed collapsed structures with membrane debris and cytoplasmic leakage. Similarly, untreated E. coli maintained characteristic rod-shaped morphology with filamentous extracellular appendages, while 5d-exposed cells underwent loss of structural integrity and appendage detachment. These observations confirm 5d’s capacity to compromise bacterial envelope integrity through potential membrane-disruptive action.
 |
Figure 7 SEM ultrastructural analysis of S. aureus ATCC 29213 (upper panels) and E. coli ATCC 25922 (lower panels) treated with 5d (8× MIC, 4 h). Untreated controls display intact cellular morphology with smooth surfaces. Abbreviation: MIC, minimum inhibitory concentration. |
MD Simulations of Compound 5d
To decipher 5d’s membrane selectivity, all-atom MD simulations were performed to track its spontaneous partitioning across bacterial (G−/G⁺) versus plasma membranes of mammals (PMm). As illustrated in Figure 8A–C, snapshots at 0 μs, 1 μs, and 2 μs were captured for the G−, G⁺, and PMm systems, respectively. 5d was able to progressively embed into the hydrophobic core of bacterial bilayers within 2 μs, whereas in mammalian systems, it remained surface-adsorbed before aqueous phase expulsion. Quantitative trajectory analysis (Figure 8D) revealed stable positioning at 2.48 nm (G−) and 1.05 nm (G⁺) from the bilayer center, versus 2.406 nm (PMm), demonstrating stronger bacterial membrane affinity. Hydrogen-bond analysis (Figure 8E) revealed that 5d formed more hydrogen bonds with lipid headgroups or water molecules in the G−/G⁺ systems, facilitating its stable embedding within the bacterial bilayer.
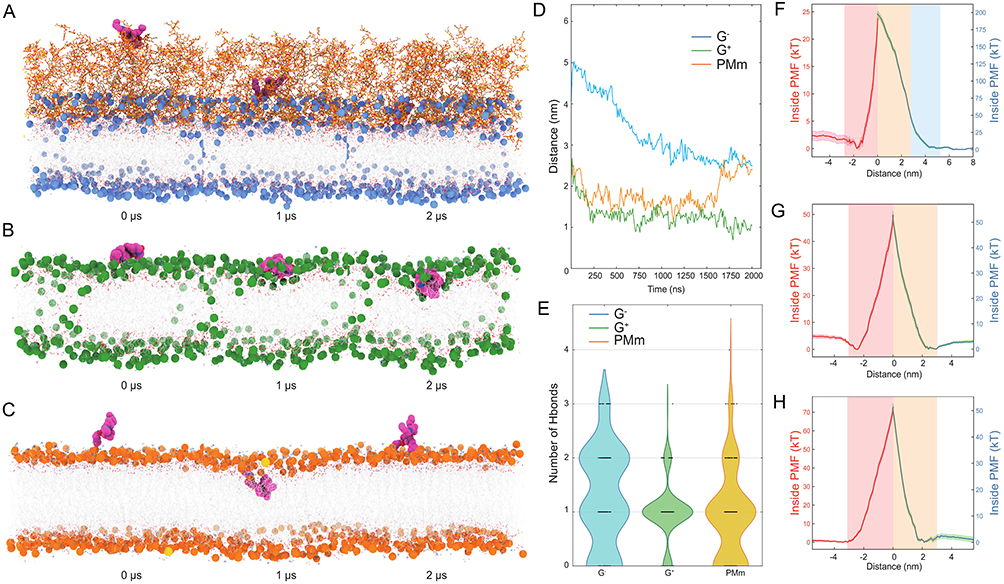 |
Figure 8 Molecular dynamics simulations of 5d-membrane interactions. (A–C) Time-resolved insertion profiles in Gram-negative (G−) bacteria outer membrane (A, blue spheres for polar heads, Orange sticks for lipids), Gram-positive (G⁺) bacteria membrane (B, green for lipids), and plasma membranes of mammals (C, PMm; orange for lipids) membranes. Representative snapshots at 0 μs, 1 μs, and 2 μs illustrating the spontaneous diffusion and insertion of compound 5d (magenta) in three membrane models. Water molecules are omitted for clarity. (D) Time evolution of the distance between 5d and the membrane center (z = 0) for each system. Lower distance values indicate deeper insertion. (E) Hydrogen-bond network analysis. Violin plots showing the distribution of the number of hydrogen bonds formed by 5d with surrounding lipids or water in each membrane model. (F–H) Potential of mean force (PMF) profiles for transmembrane translocation of 5d in G− (F), G⁺ (G), and mammalian (H) membranes. PMF curves obtained via umbrella sampling, where the horizontal axis is the distance of 5d from the membrane center and the vertical axes show inside (red line, left y-axis) versus outside (blue line, right y-axis) free energies in units of kT. |
To quantitatively delineate the thermodynamic landscape of transmembrane translocation, enhanced sampling via replica exchange umbrella sampling was implemented to construct potential of mean force (PMF) along the insertion pathway. As shown in Figure 8F–H, comparative PMF analysis of 5d in G−, G⁺, and PMm membranes revealed system-dependent energy barriers. In G− membranes, the upper leaflet lipids posed a substantial initial barrier (~180 kT), impeding surface-to-core transition, while lower leaflet penetration required the lowest activation energy (~25 kT) among all systems, indicating facilitated translocation post-insertion. G⁺ membranes exhibited uniform energy demands for both insertion and penetration (~50 kT). Notably, mammalian membranes displayed prohibitive late-stage barriers (~70 kT post-insertion), 70% higher than G− and 35% higher than G⁺, thermodynamically favoring 5d’s expulsion to the aqueous phase, a phenomenon consistent with both surface diffusion MD trajectories and hydrogen-bond topology analyses.
Bacterial Membrane Depolarization and Permeabilization
Using DiSC3(5) as a transmembrane potential-sensitive probe, we quantified 5d-induced depolarization through fluorescence dequenching kinetics. Upon 5d exposure (8 min), dose-responsive fluorescence surges were observed (Figure 9A and B), with S. aureus exhibiting rapid depolarization (550 a.u. at 4–8× MIC within 2 min) versus E. coli’s delayed response (400 a.u. after 10 min), indicating 5d induces faster depolarization in G+ bacteria. Concomitant PI influx assays cells76,77 revealed dose-dependent membrane poration (Figure 9C and D). In S. aureus, PI fluorescence peaked at 450 a.u. within 15 min (8× MIC), whereas E. coli showed limited permeabilization (130 a.u. after 32 min). The 3.5-fold kinetic disparity between G⁺ and G− strains correlate with 5d’s enhanced efficacy against G⁺ pathogens, directly linking membrane destabilization to bactericidal outcome.
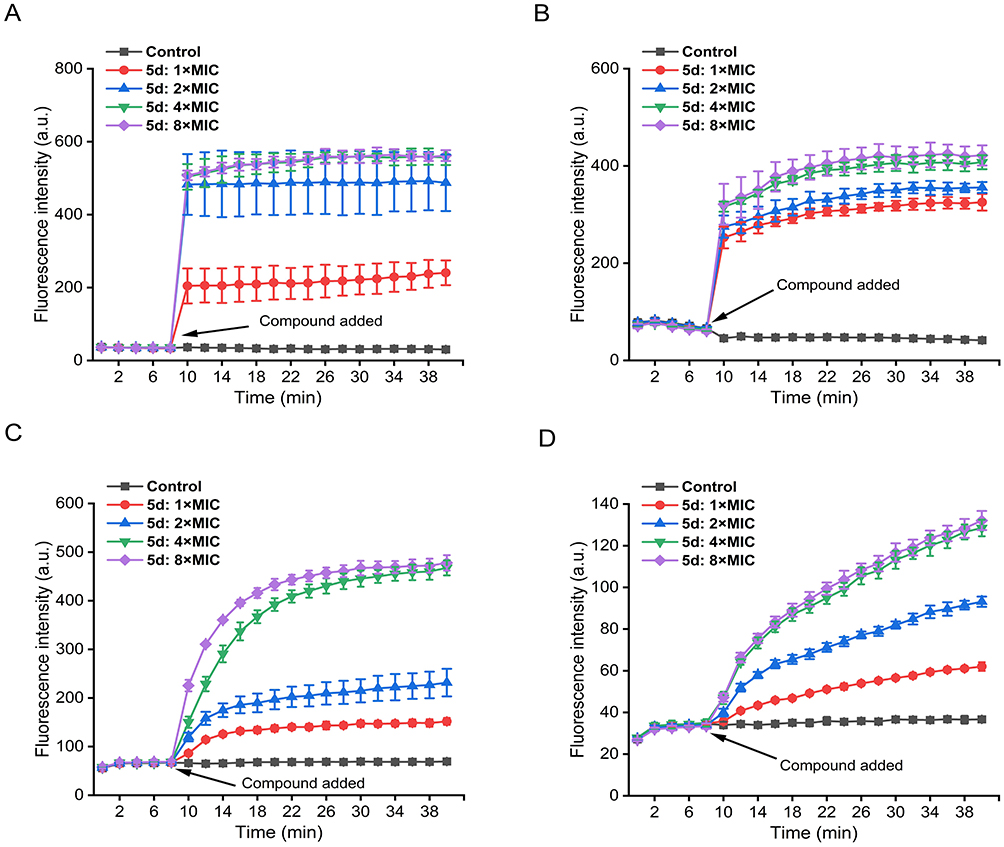 |
Figure 9 Real-time membrane perturbation kinetics assessed via DiSC3(5) depolarization (A and B) and PI permeabilization (C and D) in S. aureus ATCC 29213 and E. coli ATCC 25922. Negative controls received PBS. The error bars represent standard deviation (n = 3). Abbreviations: a.u., arbitrary units; MIC, minimum inhibitory concentration; PBS, phosphate buffered saline. |
Membrane Component Competition
To elucidate 5d’s molecular targeting specificity, MIC determinations were conducted in the presence of exogenous membrane components. Given the PE, PG, and CL predominance in bacterial inner membranes versus LPS in G− outer membranes,22,78 their competitive effects on 5d’s activity were systematically quantified (Figure 10A and B). Dose-dependent MIC elevation was observed for PG (16-fold in S. aureus at 2–32 μg/mL versus 16-fold in E. coli at 32 μg/mL), CL (16-fold in S. aureus versus 4-fold in E. coli at 32 μg/mL) and PE (2-fold in S. aureus at 8–32 μg/mL versus 4-fold in E. coli at 8–32 μg/mL), whereas LPS showed no interference. Notably, PG emerged as the most potent antagonist, exhibiting a 16-fold reduction in the antimicrobial efficacy of 5d against S. aureus and E. coli at 32 μg/mL. Parallel profiling of membrane-targeting antibiotics (daptomycin, colistin79,80) revealed similar PG desensitization patterns (Figure 10C and D; Table S5), while non-membrane agents (ceftazidime, gentamicin, etc.) maintained unaltered efficacy. This target-specific competition implicates PG and CL as primary interaction partners, mechanistically aligning 5d with established membrane-targeting pharmacophores.
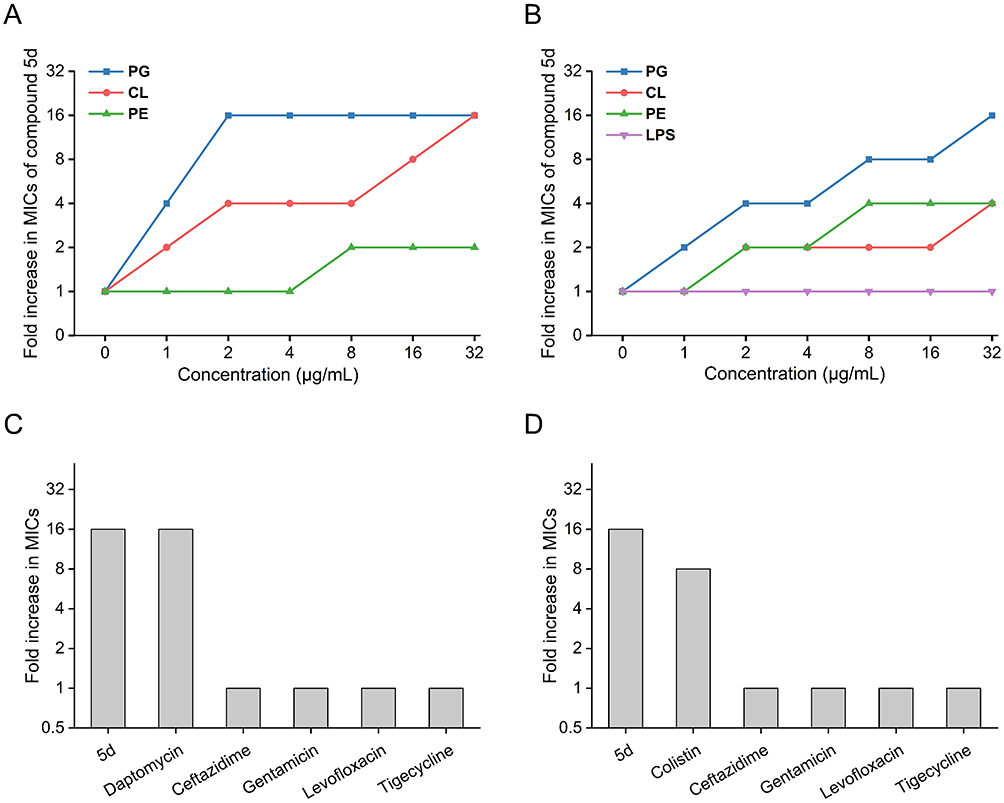 |
Figure 10 Membrane component competition assays evaluating 5d’s antibacterial activity against S. aureus ATCC 29213 (A) and E. coli ATCC 25922 (B), and comparative pharmacodynamic profiles with conventional antibiotics in the presence of 32 μg/mL PG against S. aureus ATCC 29213 (C) and E. coli ATCC 25922 (D). Abbreviations: CL, cardiolipin; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; LPS, lipopolysaccharide; MIC, minimum inhibitory concentration. |
Cytoplasmic Component Leakage
Membrane integrity loss is directly correlated with the expulsion of intracellular macromolecules such as DNA and proteins.81–83 Quantification of cytoplasmic component efflux following 5d treatment revealed dose-dependent biomolecule release in both S. aureus and E. coli (Figure 11). At 8× MIC, total DNA efflux reached 85.2 ± 0.9 ng/μL (S. aureus) versus 73.5 ± 1.1 ng/μL (E. coli), while protein release attained 2.2 ± 0.1 mg/mL versus 1.1 ± 0.1 mg/mL. The observed 1.2- to 2-fold higher leakage in G⁺ bacteria mechanistically explains 5d’s enhanced bactericidal efficacy against these pathogens, definitively establishing membrane disruption as the primary killing mechanism.
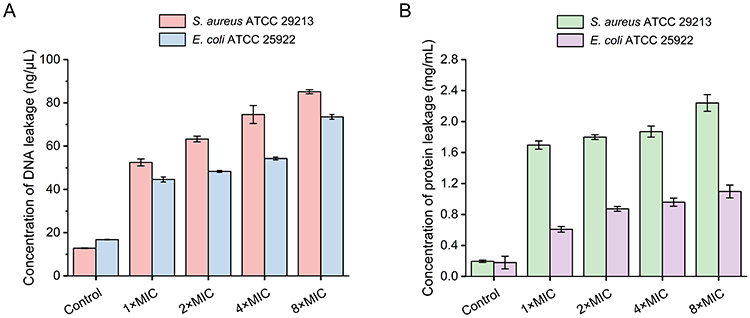 |
Figure 11 Quantification of DNA (A) and protein (B) efflux from S. aureus ATCC 29213 and E. coli ATCC 25922 following 5d treatment. Untreated controls received PBS. The error bars represent standard deviation (n = 3). Abbreviation: PBS, phosphate buffered saline. |
ROS Generation
Elevated intracellular ROS levels compromise membrane integrity and potentiate bacterial lethality through oxidative damage cascades.84,85 To assess 5d’s oxidative stress induction capacity, DCFH-DA fluorescence assays were employed to monitor intracellular ROS accumulation in real-time. Positive controls treated with Rosup exhibited 2-fold higher fluorescence intensity (~100 a.u.) versus PBS-treated cells (~50 a.u.), validating assay sensitivity (Figure 12). Notably, 5d provoked dose-dependent ROS escalation in both strains, with S. aureus displaying supraphysiological levels (172.3 ± 4.7 a.u. at 8× MIC) exceeding Rosup-induced values. In E. coli, ROS peaked at 104.4 ± 3.7 a.u. (8× MIC), equivalent to Rosup but 39.4% lower than G⁺ counterparts. This strain-specific ROS amplification mechanistically correlates with 5d’s enhanced bactericidal efficacy against G⁺ pathogens, suggesting synergistic interplay between membrane disruption and oxidative stress in the killing process.
 |
Figure 12 Intracellular reactive oxygen species (ROS) accumulation profiles in S. aureus ATCC 29213 (A) and E. coli ATCC 25922 (B) treated with 5d. Rosup (ROS inducer) and PBS served as controls. The error bars represent standard deviation (n = 5). ***p < 0.001, one-way analysis of variance. Abbreviations: a.u., arbitrary units; PBS, phosphate buffered saline; MIC, minimum inhibitory concentration. |
Cytotoxicity Assessment
To evaluate 5d’s therapeutic index, its cytotoxic effects were systematically analyzed in human non-small cell lung cancer (A549) and hepatocellular carcinoma (HepG2) cell lines using CCK-8 assay. As shown in Figure 13, cell viability remained >95% at antibacterial concentrations (≤4 μg/mL; MIC = 1 μg/mL for S. aureus, 4 μg/mL for E. coli), demonstrating negligible cytotoxicity at therapeutic doses. Dose-dependent toxicity emerged at ≥8 μg/mL, with 50% cytotoxic concentration (CC50) values of 27.1 μg/mL (A549) and 31.1 μg/mL (HepG2), yielding a favorable therapeutic window (27- and 31-fold over S. aureus MICs, and 6.8- and 7.8-fold over E. coli MICs). These findings correlate with molecular dynamics simulations demonstrating 5d’s selective bacterial membrane targeting, thereby validating its membrane-specific action and low mammalian cell toxicity at bactericidal concentrations.
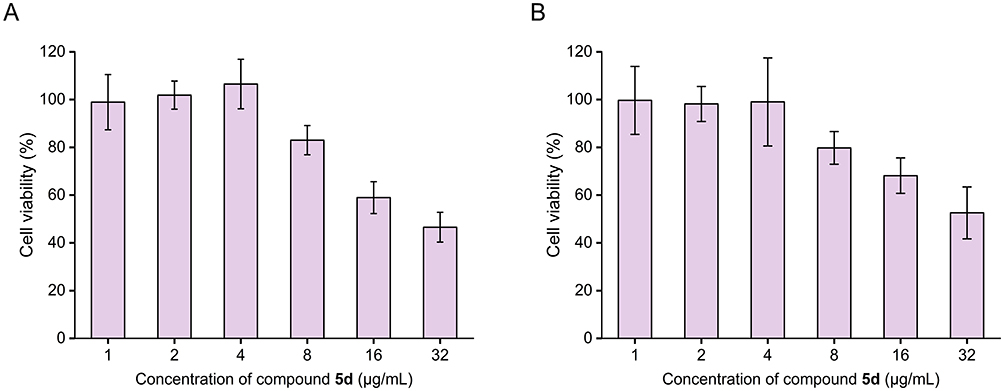 |
Figure 13 In vitro cytotoxicity profiling of 5d against human non-small cell lung cancer (A549) cells (A) and hepatocellular carcinoma (HepG2) cells (B), evaluated via CCK-8 cell viability assay. The error bars represent standard deviation (n = 3). |
In vivo Therapeutic Efficacy
Given the compound’s potent antibacterial activity and favorable safety profile, its translational potential was validated in a S. aureus-induced sepsis model (Figure 14A). Lethally infected mice receiving 5d (5–15 mg/kg, i.p.) exhibited dose-dependent survival improvement, with 100% survival at 15 mg/kg versus 66.6% for vancomycin at equivalent doses (Figure 14B). Notably, 5d achieved superior protection (50–100% survival) over vancomycin (33.3–66.6%) across all tested doses, surpassing the reference drug by 16.7–33.4%.
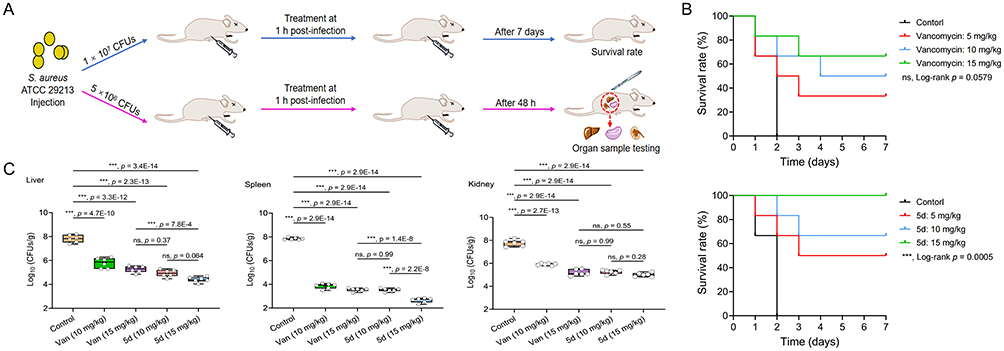 |
Figure 14 In vivo therapeutic evaluation of 5d in a murine sepsis model. (A) Experimental timeline. (B) Kaplan–Meier curves of BALB/c mice (6 mice/group) infected with lethal dose S. aureus ATCC 29213 (1 × 107 CFUs) treated with single-dose 5d or vancomycin (5–15 mg/kg, i.p.). The p value was determined using the Log rank test. (C) Organ-specific bacterial burden in sublethally infected mice (5 × 106 CFUs) after 48 h treatment. Data points indicate individual biological replicates. Horizontal bars indicate group means. The error bars represent standard deviation (n = 6). ***p < 0.001, one-way analysis of variance. Abbreviations: ns, non-significant; CFU, colony-forming units; PBS, phosphate buffered saline. |
Bacterial clearance analysis in sublethally infected mice revealed significant organ burden reduction (p < 0.001) by 5d versus PBS controls (Figure 14C). At 10 mg/kg, 5d matched vancomycin’s efficacy at 15 mg/kg, demonstrating its higher potency. Maximum efficacy at 15 mg/kg 5d yielded 2.8- to 5.2-log reductions in splenic (2.6 log10 CFUs/g), hepatic (4.4 log10 CFUs/g), and renal (5.0 log10 CFUs/g) burdens versus controls (7.8 log10 CFUs/g). These data collectively confirm 5d’s superior in vivo performance in both survival and pathogen clearance.
Conclusion
This work establishes a novel class of CBG-based amphiphilic derivatives through a biomimetic design strategy emulating AMPs. Systematic SAR optimization identified compound 5d as the lead candidate, exhibiting clinically relevant broad-spectrum activity against both G+ (MICs = 1–2 μg/mL, including drug-resistant MRSA) and G− pathogens (MICs = 2–8 μg/mL). Beyond its direct antimicrobial effects, 5d demonstrated good translational promise, characterized by low hemolytic toxicity (HC50 > 200 μg/mL), rapid bactericidal kinetics (complete eradication within 0.5 h at 4× MIC), resistance prevention, and robust biofilm disruption capacity. Mechanistic dissection revealed a multimodal bactericidal mechanism: selective targeting of bacterial membranes via PG and CL interactions; concerted membrane destabilization through depolarization-permeabilization cascades; intracellular oxidative stress amplification via ROS overproduction; massive cytoplasmic component efflux (DNA/proteins). This multipronged attack circumvents conventional resistance pathways, explaining the observed lack of MIC escalation during serial passage. In vivo validation in a murine sepsis model confirmed 5d’s superiority over vancomycin, achieving 100% survival at 15 mg/kg with 2.8–5.2 log10 CFUs/g reductions in splenic, hepatic, and renal bacterial burdens. Critically, the compound maintained negligible cytotoxicity toward human cells (A549 and HepG2) at therapeutic concentrations, yielding an exceptional therapeutic index. These collective attributes thus support the further development of 5d as a promising preclinical candidate. Consequently, it represents a potential strategy to address the urgent need for new agents against biofilm-associated and multidrug-resistant infections. Future investigations will prioritize characterizing its pharmacokinetic profile, evaluating its in vivo efficacy against biofilm-based infections, and exploring its potential for synergy with established antibiotics.
Ethical Approval
Animal experiments were approved by the Animal Research Ethical Review Committee of Shandong University (approval No. SYDWLL-2023-091) and were conducted in full accordance with the National Standard GB/T35892-2018 (Laboratory Animal-Guideline for Ethical Review of Animal Welfare) of the People’s Republic of China.
Acknowledgments
We sincerely acknowledge Haiyan Sui, Zhifeng Li, Haiyan Yu, Yuyu Guo, Xiangmei Ren and Guannan Lin from the Shandong University Core Facilities for Life and Environmental Sciences for their technical assistance with NMR, SEM, CLSM and HRMS analyses.
Funding
This research was supported by the National Key R&D Program of China (2021YFC2101000 to W. Hu), National Natural Science Foundation of China (32400148 to F. Zhang), and Shandong Provincial Natural Science Foundation (ZR2021QC087 to C. Wang).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that might appear to influence the work reported in this paper.
References
1. Aljeldah MM. Antimicrobial resistance and its spread is a global threat. Antibiotics. 2022;11(8):1084. doi:10.3390/antibiotics11081082
2. Friedman ND, Temkin E, Carmeli Y. The negative impact of antibiotic resistance. Clin Microbiol Infect. 2016;22(5):416–24. doi:10.1016/j.cmi.2015.12.002
3. Dhingra S, Rahman NAA, Peile E, et al. Microbial resistance movements: an overview of global public health threats posed by antimicrobial resistance, and how best to counter. Front Public Health. 2020;8:535668. doi:10.3389/fpubh.2020.535668
4. Su WY, Wang WJ, Li L, et al. Mechanisms of tigecycline resistance in gram-negative bacteria: a narrative review. Eng Microbiol. 2024;4(3):100165. doi:10.1016/j.engmic.2024.100165
5. Sati H, Carrara E, Savoldi A, et al. The who bacterial priority pathogens list 2024: a prioritisation study to guide research, development, and public health strategies against antimicrobial resistance. Lancet Infect Dis. 2025;25(9):1033–1043. doi:10.1016/S1473-3099(25)00118-5
6. Meade E, Slattery MA, Garvey M. Bacteriocins, potent antimicrobial peptides and the fight against multi drug resistant species: resistance is futile? Antibiotics. 2020;9(1):32. doi:10.3390/antibiotics9010032
7. Diseases GBD, Injuries C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet. 2020;396(10258):1204–1222. doi:10.1016/S0140-6736(20)30925-9
8. Craft KM, Nguyen JM, Berg LJ, Townsend SD. Methicillin-resistant Staphylococcus aureus (MRSA): antibiotic-resistance and the biofilm phenotype. Medchemcomm. 2019;10(8):1231–1241. doi:10.1039/c9md00044e
9. Xu B, Wang L, Yang C, et al. Specifically targeted antimicrobial peptides synergize with bacterial-entrapping peptide against systemic MRSA infections. J Adv Res. 2025;67:301–315. doi:10.1016/j.jare.2024.01.023
10. Hsu CY, Moradkasani S, Suliman M, et al. Global patterns of antibiotic resistance in group B Streptococcus: a systematic review and meta-analysis. Front Microbiol. 2025;16:1541524. doi:10.3389/fmicb.2025.1541524
11. Shivaee A, Kazemi F, Navidifar T, et al. Global trends in macrolide and lincosamide resistance in Streptococcus species: a comprehensive systematic review and meta-analysis. BMC Infect Dis. 2025;25(1):1577. doi:10.1186/s12879-025-11884-5
12. Niu Z, Lei P, Wang Y, Wang J, Yang J, Zhang J. Small molecule LpxC inhibitors against gram-negative bacteria: advances and future perspectives. Eur J Med Chem. 2023;253:115326. doi:10.1016/j.ejmech.2023.115326
13. Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature. 2016;529(7586):336–343. doi:10.1038/nature17042
14. Lin S, Li H, Tao Y, et al. In vitro and in vivo evaluation of membrane-active flavone amphiphiles: semisynthetic kaempferol-derived antimicrobials against drug-resistant gram-positive bacteria. J Med Chem. 2020;63(11):5797–5815. doi:10.1021/acs.jmedchem.0c00053
15. Xu T, Yan X, Kang A, et al. Development of membrane-targeting fluorescent 2-phenyl-1h-phenanthro[9,10-d]imidazole-antimicrobial peptide mimic conjugates against methicillin-resistant Staphylococcus aureus. J Med Chem. 2024;67(11):9302–9317. doi:10.1021/acs.jmedchem.4c00436
16. Nguyen LT, Haney EF, Vogel HJ. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011;29(9):464–472. doi:10.1016/j.tibtech.2011.05.001
17. Lin S, Koh JJ, Aung TT, et al. Semisynthetic flavone-derived antimicrobials with therapeutic potential against methicillin-resistant Staphylococcus aureus (MRSA). J Med Chem. 2017;60(14):6152–6165. doi:10.1021/acs.jmedchem.7b00380
18. Fang S, Kang WT, Li H, et al. Development of cannabidiol derivatives as potent broad-spectrum antibacterial agents with membrane-disruptive mechanism. Eur J Med Chem. 2024;266:116149. doi:10.1016/j.ejmech.2024.116149
19. Mahlapuu M, Bjorn C, Ekblom J. Antimicrobial peptides as therapeutic agents: opportunities and challenges. Crit Rev Biotechnol. 2020;40(7):978–992. doi:10.1080/07388551.2020.1796576
20. Magana M, Pushpanathan M, Santos AL, et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect Dis. 2020;20(9):e216–e230. doi:10.1016/S1473-3099(20)30327-3
21. Vance JE. Phospholipid synthesis and transport in mammalian cells. Traffic. 2015;16(1):1–18. doi:10.1111/tra.12230
22. Sohlenkamp C, Geiger O. Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiol Rev. 2016;40(1):133–159. doi:10.1093/femsre/fuv008
23. Hancock RE, Brown KL, Mookherjee N. Host defence peptides from invertebrates--emerging antimicrobial strategies. Immunobiology. 2006;211(4):315–322. doi:10.1016/j.imbio.2005.10.017
24. Bhopale GM. Antimicrobial peptides: a promising avenue for human healthcare. Curr Pharm Biotechnol. 2020;21(2):90–96. doi:10.2174/1389201020666191011121722
25. Wang G, Vaisman II, van Hoek ML. Machine learning prediction of antimicrobial peptides. Methods Mol Biol. 2022;2405:1–37. doi:10.1007/978-1-0716-1855-4_1
26. Barman S, Konai MM, Samaddar S, Haldar J. Amino acid conjugated polymers: antibacterial agents effective against drug-resistant Acinetobacter baumannii with no detectable resistance. ACS Appl Mater Interfaces. 2019;11(37):33559–33572. doi:10.1021/acsami.9b09016
27. Xie J, Zhou M, Qian Y, et al. Addressing MRSA infection and antibacterial resistance with peptoid polymers. Nat Commun. 2021;12(1):5898. doi:10.1038/s41467-021-26221-y
28. Liang W, Yu Q, Zheng Z, et al. Design and synthesis of phenyl sulfide-based cationic amphiphiles as membrane-targeting antimicrobial agents against gram-positive pathogens. J Med Chem. 2022;65(20):14221–14236. doi:10.1021/acs.jmedchem.2c01437
29. Cheng W, Xu T, Cui L, et al. Discovery of amphiphilic xanthohumol derivatives as membrane-targeting antimicrobials against methicillin-resistant Staphylococcus aureus. J Med Chem. 2023;66(1):962–975. doi:10.1021/acs.jmedchem.2c01793
30. Guo Y, Hou E, Wen T, et al. Development of membrane-active honokiol/magnolol amphiphiles as potent antibacterial agents against methicillin-resistant Staphylococcus aureus (MRSA). J Med Chem. 2021;64(17):12903–12916. doi:10.1021/acs.jmedchem.1c01073
31. Li S, Wang M, Chen S, et al. Development of biaromatic core-linked antimicrobial peptide mimics: substituent position significantly affects antibacterial activity and hemolytic toxicity. Eur J Med Chem. 2023;247:115029. doi:10.1016/j.ejmech.2022.115029
32. Mookherjee N, Anderson MA, Haagsman HP, Davidson DJ. Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discov. 2020;19(5):311–332. doi:10.1038/s41573-019-0058-8
33. Lin SM, Liu JY, Li HX, et al. Development of highly potent carbazole amphiphiles as membrane-targeting antimicrobials for treating gram-positive bacterial infections. J Med Chem. 2020;63(17):9284–9299. doi:10.1021/acs.jmedchem.0c00433
34. Chen Y, Li H, Liu J, et al. Synthesis and biological evaluation of indole-based peptidomimetics as antibacterial agents against gram-positive bacteria. Eur J Med Chem. 2021;226:113813. doi:10.1016/j.ejmech.2021.113813
35. Salha M, Adenusi H, Dupuis JH, et al. Bioactivity of the cannabigerol cannabinoid and its analogues - the role of 3-dimensional conformation. Org Biomol Chem. 2023;21(22):4683–4693. doi:10.1039/d3ob00383c
36. Lah TT, Novak M, Pena Almidon MA, et al. Cannabigerol is a potential therapeutic agent in a novel combined therapy for glioblastoma. Cells. 2021;10(2):340. doi:10.3390/cells10020340
37. Borrelli F, Fasolino I, Romano B, et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem Pharmacol. 2013;85(9):1306–1316. doi:10.1016/j.bcp.2013.01.017
38. Kogan NM, Lavi Y, Topping LM, et al. Novel CBG derivatives can reduce inflammation, pain and obesity. Molecules. 2021;26(18):5601. doi:10.3390/molecules26185601
39. Valdeolivas S, Navarrete C, Cantarero I, Bellido ML, Munoz E, Sagredo O. Neuroprotective properties of cannabigerol in huntington’s disease: studies in r6/2 mice and 3-nitropropionate-lesioned mice. Neurotherapeutics. 2015;12(1):185–199. doi:10.1007/s13311-014-0304-z
40. Karas JA, Wong LJM, Paulin OKA, et al. The antimicrobial activity of cannabinoids. Antibiotics. 2020;9(7):406. doi:10.3390/antibiotics9070406
41. Aqawi M, Sionov RV, Gallily R, Friedman M, Steinberg D. Anti-bacterial properties of cannabigerol toward Streptococcus mutans. Front Microbiol. 2021;12:656471. doi:10.3389/fmicb.2021.656471
42. Farha MA, El-Halfawy OM, Gale RT, et al. Uncovering the hidden antibiotic potential of cannabis. Acs Infect Dis. 2020;6(3):338–346. doi:10.1021/acsinfecdis.9b00419
43. Aqawi M, Sionov RV, Gallily R, Friedman M, Steinberg D. Anti-biofilm activity of cannabigerol against Streptococcus mutans. Microorganisms. 2021;9(10):2031. doi:10.3390/microorganisms9102031
44. Lorincz EB, Tóth G, Spolárics J, et al. Mannich-type modifications of (-)-cannabidiol and (-)-cannabigerol leading to new, bioactive derivatives. Sci Rep-Uk. 2023;13(1):19618. doi:10.1038/s41598-023-45565-7
45. Dong W, Liu Z, Sun L, et al. Antimicrobial activity and self-assembly behavior of antimicrobial peptide chensinin-1b with lipophilic alkyl tails. Eur J Med Chem. 2018;150:546–558. doi:10.1016/j.ejmech.2018.03.025
46. Jentsch NG, Zhang X, Magolan J. Efficient synthesis of cannabigerol, grifolin, and piperogalin via alumina-promoted allylation. J Nat Prod. 2020;83(9):2587–2591. doi:10.1021/acs.jnatprod.0c00131
47. Chu W, Yang Y, Cai J, et al. Synthesis and bioactivities of new membrane-active agents with aromatic linker: high selectivity and broad-spectrum antibacterial activity. Acs Infect Dis. 2019;5(9):1535–1545. doi:10.1021/acsinfecdis.9b00078
48. Zhao Y, Zhang TT, Liang YP, et al. Combination of aloe emodin, emodin, and rhein from Aloe with EDTA sensitizes the resistant Acinetobacter baumannii to polymyxins. Front Cell Infect Mi. 2024;14:1467607. doi:10.3389/fcimb.2024.1467607
49. Wang YH, Wu P, Liu FQ, et al. Design, synthesis, and biological evaluation of membrane-active honokiol derivatives as potent antibacterial agents. Eur J Med Chem. 2022;240:114593. doi:10.1016/j.ejmech.2022.114593
50. Zhang JY, Ouyang X, Zhang FY, et al. Structure-activity relationship study of antimicrobial peptide PE2 delivered novel linear derivatives with potential of eradicating biofilms and low incidence of drug resistance. J Med Chem. 2023;66(13):8526–8544. doi:10.1021/acs.jmedchem.3c00181
51. Wang CD, Zhang TT, Wang Y, et al. Proguanil and chlorhexidine augment the antibacterial activities of clarithromycin and rifampicin against Acinetobacter baumannii. Int J Antimicrob Agents. 2024;63(2):107065. doi:10.1016/j.ijantimicag.2023.107065
52. Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865. doi:10.1002/jcc.20945
53. Pogozheva ID, Armstrong GA, Kong L, et al. Comparative molecular dynamics simulation studies of realistic eukaryotic, prokaryotic, and archaeal membranes. J Chem Inf Model. 2022;62(4):1036–1051. doi:10.1021/acs.jcim.1c01514
54. Klauda JB, Venable RM, Freites JA, et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114(23):7830–7843. doi:10.1021/jp101759q
55. Bugnon M, Goullieux M, Rohrig UF, et al. Swissparam 2023: a modern web-based tool for efficient small molecule parametrization. J Chem Inf Model. 2023;63(21):6469–6475. doi:10.1021/acs.jcim.3c01053
56. Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys. 2008;129(12):124105. doi:10.1063/1.2978177
57. Bai PY, Qin SS, Chu WC, et al. Synthesis and antibacterial bioactivities of cationic deacetyl linezolid amphiphiles. Eur J Med Chem. 2018;155:925–945. doi:10.1016/j.ejmech.2018.06.054
58. Cai QN, Yu Q, Liang WX, et al. Membrane-active nonivamide derivatives as effective broad-spectrum antimicrobials: rational design, synthesis, and biological evaluation. J Med Chem. 2022;65(24):16754–16773. doi:10.1021/acs.jmedchem.2c01604
59. Anantharaman A, Sahal D. Reverse engineering truncations of an antimicrobial peptide dimer to identify the origins of potency and broad spectrum of action. J Med Chem. 2010;53(16):6079–6088. doi:10.1021/jm100483y
60. Shi JR, Chen C, Wang DJ, Wang ZQ, Liu Y. The antimicrobial peptide LI14 combats multidrug-resistant bacterial infections. Commun Biol. 2022;5(1):926. doi:10.1038/s42003-022-03899-4
61. Sui YF, Ansari MF, Fang B, Zhang SL, Zhou CH. Discovery of novel purinylthiazolylethanone derivatives as anti- agents through possible multifaceted mechanisms. Eur J Med Chem. 2021;221:113557. doi:10.1016/j.ejmech.2021.113557
62. Liu Y, Li J, Ma Y, et al. Discovery of potent ClpX modulators with pronounced antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur J Med Chem. 2026;301:118207. doi:10.1016/j.ejmech.2025.118207
63. Yu Q, Cai QN, Liang WX, et al. Design of phenothiazine-based cationic amphiphilic derivatives incorporating arginine residues: potential membrane-active broad-spectrum antimicrobials combating pathogenic bacteria in vitro and in vivo. Eur J Med Chem. 2023;260:115733. doi:10.1016/j.ejmech.2023.115733
64. Yang S, Miao G, Wang X, et al. Development of membrane-targeting chalcone derivatives as antibacterial agents against multidrug-resistant bacteria. Eur J Med Chem. 2024;280:116969. doi:10.1016/j.ejmech.2024.116969
65. Maher C, Hassan KA. The gram-negative permeability barrier: tipping the balance of the in and the out. mBio. 2023;14(6):e0120523. doi:10.1128/mbio.01205-23
66. Vivas R, Barbosa AAT, Dolabela SS, Jain S. Multidrug-resistant bacteria and alternative methods to control them: an overview. Microb Drug Resist. 2019;25(6):890–908. doi:10.1089/mdr.2018.0319
67. O’Connell KM, Hodgkinson JT, Sore HF, Welch M, Salmond GP, Spring DR. Combating multidrug-resistant bacteria: current strategies for the discovery of novel antibacterials. Angew Chem Int Ed Engl. 2013;52(41):10706–10733. doi:10.1002/anie.201209979
68. Zhou M, Zheng M, Cai J. Small molecules with membrane-active antibacterial activity. ACS Appl Mater Interfaces. 2020;12(19):21292–21299. doi:10.1021/acsami.9b20161
69. Saini V, Mehta D, Gupta S, et al. Targeting vancomycin-resistant Enterococci (VRE) infections and van operon-mediated drug resistance using dimeric cholic acid-peptide conjugates. J Med Chem. 2022;65(22):15312–15326. doi:10.1021/acs.jmedchem.2c01293
70. Melander RJ, Basak AK, Melander C. Natural products as inspiration for the development of bacterial antibiofilm agents. Nat Prod Rep. 2020;37(11):1454–1477. doi:10.1039/d0np00022a
71. Su Y, Yrastorza JT, Matis M, et al. Biofilms: formation, research models, potential targets, and methods for prevention and treatment. Adv Sci. 2022;9(29):e2203291. doi:10.1002/advs.202203291
72. Yarlagadda V, Akkapeddi P, Manjunath GB, Haldar J. Membrane active vancomycin analogues: a strategy to combat bacterial resistance. J Med Chem. 2014;57(11):4558–4568. doi:10.1021/jm500270w
73. Ghosh C, Manjunath GB, Akkapeddi P, et al. Small molecular antibacterial peptoid mimics: the simpler the better! J Med Chem. 2014;57(4):1428–1436. doi:10.1021/jm401680a
74. Hu XL, Sedgwick AC, Mangel DN, et al. Tuning the solid- and solution-state fluorescence of the iron-chelator deferasirox. J Am Chem Soc. 2022;144(16):7382–7390. doi:10.1021/jacs.2c01155
75. Huang YJ, Zang YP, Peng LJ, Yang MH, Lin J, Chen WM. Cajaninstilbene acid derivatives conjugated with siderophores of 3-hydroxypyridin-4(1h)-ones as novel antibacterial agents against gram-negative bacteria based on the trojan horse strategy. Eur J Med Chem. 2024;269:116339. doi:10.1016/j.ejmech.2024.116339
76. Hoenes K, Bauer R, Spellerberg B, Hessling M. Microbial photoinactivation by visible light results in limited loss of membrane integrity. Antibiotics. 2021;10(3):341. doi:10.3390/antibiotics10030341
77. Liu F, Yang S, Zhang L, et al. Design, synthesis and biological evaluation of amphiphilic benzopyran derivatives as potent antibacterial agents against multidrug-resistant bacteria. Eur J Med Chem. 2024;277:116784. doi:10.1016/j.ejmech.2024.116784
78. Song MR, Liu Y, Huang XY, et al. A broad-spectrum antibiotic adjuvant reverses multidrug-resistant gram-negative pathogens. Nat Microbiol. 2020;5(8):1040–1050. doi:10.1038/s41564-020-0723-z
79. Hansen MH, Stegmann E, Cryle MJ. Beyond vancomycin: recent advances in the modification, reengineering, production and discovery of improved glycopeptide antibiotics to tackle multidrug-resistant bacteria. Curr Opin Biotechnol. 2022;77:102767. doi:10.1016/j.copbio.2022.102767
80. Nang SC, Azad MAK, Velkov T, Zhou QT, Li J. Rescuing the last-line polymyxins: achievements and challenges. Pharmacol Rev. 2021;73(2):679–728. doi:10.1124/pharmrev.120.000020
81. Hong Y, Zeng J, Wang X, Drlica K, Zhao X. Post-stress bacterial cell death mediated by reactive oxygen species. Proc Natl Acad Sci U S A. 2019;116(20):10064–10071. doi:10.1073/pnas.1901730116
82. Zheng L, Li J, Yu M, et al. Molecular sizes and antibacterial performance relationships of flexible ionic liquid derivatives. J Am Chem Soc. 2020;142(47):20257–20269. doi:10.1021/jacs.0c10771
83. Galbadage T, Liu D, Alemany LB, et al. Molecular nanomachines disrupt bacterial cell wall, increasing sensitivity of extensively drug-resistant Klebsiella pneumoniae to meropenem. ACS Nano. 2019;13(12):14377–14387. doi:10.1021/acsnano.9b07836
84. Kong Q, Li G, Zhang F, et al. N-arylimidazoliums as highly selective biomimetic antimicrobial agents. J Med Chem. 2022;65(16):11309–11321. doi:10.1021/acs.jmedchem.2c00818
85. Zhou M, Qian Y, Xie J, et al. Poly(2-oxazoline)-based functional peptide mimics: eradicating MRSA infections and persisters while alleviating antimicrobial resistance. Angew Chem Int Ed Engl. 2020;59(16):6412–6419. doi:10.1002/anie.202000505
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.