Back to Journals » ImmunoTargets and Therapy » Volume 4
Development of antibody-based c-Met inhibitors for targeted cancer therapy
Authors Lee D, Sung E, Ahn J, An S, Huh J, You W
Received 31 October 2014
Accepted for publication 12 December 2014
Published 9 February 2015 Volume 2015:4 Pages 35—44
DOI https://doi.org/10.2147/ITT.S37409
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Dongheon Lee, Eun-Sil Sung, Jin-Hyung Ahn, Sungwon An, Jiwon Huh, Weon-Kyoo You
Hanwha Chemical R&D Center, Biologics Business Unit, Daejeon, Republic of Korea
Abstract: Signaling pathways mediated by receptor tyrosine kinases (RTKs) and their ligands play important roles in the development and progression of human cancers, which makes RTK-mediated signaling pathways promising therapeutic targets in the treatment of cancer. Compared with small-molecule compounds, antibody-based therapeutics can more specifically recognize and bind to ligands and RTKs. Several antibody inhibitors of RTK-mediated signaling pathways, such as human epidermal growth factor receptor 2, vascular endothelial growth factor, epidermal growth factor receptor or vascular endothelial growth factor receptor 2, have been developed and are widely used to treat cancer patients. However, since the therapeutic options are still limited in terms of therapeutic efficacy and types of cancers that can be treated, efforts are being made to identify and evaluate novel RTK-mediated signaling pathways as targets for more efficacious cancer treatment. The hepatocyte growth factor/c-Met signaling pathway has come into the spotlight as a promising target for development of potent cancer therapeutic agents. Multiple antibody-based therapeutics targeting hepatocyte growth factor or c-Met are currently in preclinical or clinical development. This review focuses on the development of inhibitors of the hepatocyte growth factor/c-Met signaling pathway for cancer treatment, including critical issues in clinical development and future perspectives for antibody-based therapeutics.
Keywords: hepatocyte growth factor, ligands, receptor tyrosine kinase, signaling pathway, therapeutic agent
Introduction
In living organisms, communication between individual cells and between cells and the environment plays essential roles in various cellular processes including growth, differentiation, migration, and apoptosis.1 These cellular processes are mediated in large part by signaling pathways triggered by interactions between receptor tyrosine kinases (RTKs) and their ligands.1 Upon ligand binding, RTKs on the cell surface activate downstream signaling cascades and regulate target gene expression in a paracrine or autocrine manner.1 These signaling pathways mediated by RTKs and their ligands are critical for both regulation of diverse cellular processes and development and progression of cancers.2–4 RTK-mediated signaling pathways are tightly regulated according to the physiological status of normal cells. In contrast, RTK signaling pathways are dysregulated or hyperactivated in a wide range of cancers via gain-of-function mutations, gene rearrangements, gene amplifications, and overexpression or abnormal stimulation of receptors/ligands.1–4 Furthermore, it has been suggested that loss-of-function by deletion or mutation of RTKs, including fibroblast growth factor receptor 1, EphA, and c-Met, is associated with several diseases, including cancers.5–7 Thus, RTK-mediated signaling pathways have become promising therapeutic targets for treating cancer.2–4
Individual RTK-mediated signaling cascades can be targeted at several levels to develop anti-cancer therapeutic agents that inhibit the signaling pathways via disruption of interactions between RTKs and their ligands, dimerization and phosphorylation of RTKs, and activation of downstream elements.2,4 For monoclonal antibody-based therapeutics, RTK-mediated signaling pathways are mainly inhibited by disrupting interactions between RTKs and their ligands.4 Compared with small-molecule compounds that inhibit the kinase activity and autophosphorylation of a broad spectrum of RTKs, antibody inhibitors have greater target specificity for particular RTKs and/or ligands. Several antibody-based therapeutic agents that inhibit RTK-mediated signaling pathways have already been approved for the treatment of human cancers.4 Trastuzumab (Herceptin™, Genentech/Roche, South San Francisco, CA, USA; approved by the US Food and Drug Administration [FDA] in 1998) binds to human epidermal growth factor receptor 2 (HER2)/neu receptor and is used to treat breast cancer.8 Bevacizumab (Avastin™, Genentech/Roche; approved by the FDA in 2004) binds to vascular endothelial growth factor (VEGF) and is used to treat colorectal cancer, metastatic renal cell carcinoma, recurrent glioblastoma, and non-small cell lung cancer (NSCLC).9–12 Both cetuximab (Erbitux™; developed by Bristol-Myers Squibb, New York, NY, USA/Eli Lilly, Indianapolis, IN, USA and Merck KGaA, Darmstadt, Germany and approved in 2004) and panitumumab (Vectibix™, developed by Amgen (Thousand Oaks, CA, USA) and approved in 2006) bind to epidermal growth factor receptor (EGFR) and are used to treat colorectal and head/neck cancers.13,14 Ramucirumab (Cyramza™, developed by Eli Lilly and approved in 2014) binds to VEGF receptor 2 and is used to treat gastric cancer.15 Although these FDA-approved therapeutic antibodies have contributed to improving clinical outcomes, there are still unmet needs for difficult-to-treat cancers. Identification of additional RTK/ligand targets may enable more effective treatment of these cancers.2–4 Currently, a variety of novel antibody-based inhibitors targeting RTKs such as c-Met, fibroblast growth factor receptor, HER1, HER3, insulin-like growth factor-1 receptor, platelet-derived growth factor receptor, and RON (recepteur d’origine nantais) are in preclinical and clinical development.4
c-Met, the RTK oncogene, was first cloned in 1984.16 The ligand of c-Met, hepatocyte growth factor (HGF; also known as scatter factor) was identified in 1991 as a potent mitogen/morphogen.17,18 The HGF/c-Met signaling pathway has been demonstrated to play important roles in the development and progression of various cancers.19–21 Dysregulation and hyperactivation of HGF or c-Met have been reported in human cancers and linked to poor prognosis.22,23 It has been suggested that the HGF/c-Met signaling pathway may be a promising target for drugs designed to overcome development of resistance to inhibition of other ligand/RTK signaling pathways.19–21 A large number of inhibitors of the HGF/c-Met signaling pathway are under development to treat human cancers.19–21 Similar to other inhibitors of RTK-mediated signaling pathways, agents targeting HGF or c-Met are categorized into two groups, ie, small-molecule drugs and biologics such as monoclonal antibodies.19–21 Currently, multiple therapeutic antibodies targeting the HGF/c-Met signaling pathway are in preclinical or clinical development.24 In this review, we summarize the role of HGF and c-Met in normal tissues and cancers. We then discuss the development of HGF/c-Met signaling pathway inhibitors for cancer treatment, including critical issues in clinical development and future perspectives focusing on monoclonal antibody-based therapeutics.
Physiological role of HGF and c-Met
The HGF/c-Met signaling pathway influences a variety of cellular functions to control diverse biological processes such as embryonic development, epithelial branching morphogenesis, postnatal organ regeneration, and wound healing (Figure 1).19–21 HGF is a paracrine signaling molecule produced and secreted from mesenchymal cells to affect neighboring epithelial cells expressing c-Met. Protective roles of HGF have been reported in tissue fibrosis, liver cirrhosis, endothelial injury, and lung fibrosis.25–29
 | Figure 1 HGF/c-Met signaling cascades. |
HGF is produced as an inactive single-chain precursor that is processed to yield an active heterodimer of one alpha and one beta chain linked by a disulfide bond.30,31 c-Met is also produced as a single-chain precursor and processed to yield a mature receptor composed of a glycosylated, extracellular alpha subunit disulfide-bonded to a transmembrane beta subunit.32 The extracellular portion of c-Met is composed of a Sema domain (homologous to semaphorins), a cysteine-rich, Met-related-sequence domain, and four immunoglobulin (Ig)-like modules (IgG domains) responsible for binding HGF.32 The intracellular portion of c-Met is composed of a juxtamembrane domain, a tyrosine kinase domain, and a C-terminal regulatory tail responsible for signal transduction.33 Tyrosine residues Tyr 1234 and Tyr 1235 in the tyrosine kinase domain regulate the kinase activity of c-Met, and tyrosine residues Tyr 1349 and Tyr 1356 in the C-terminal regulatory tail (the multisubstrate docking site) are important for recruitment of downstream adapters, including growth factor receptor-bound protein 2 (GRB2) protein and GRB2-associated binding protein 1 (GAB1) (Figure 1).34,35 HGF/c-Met signaling through these downstream effectors stimulates diverse cellular processes such as cell proliferation, differentiation, migration, invasion, and survival (Figure 1).19–21
Mice lacking either HGF or c-Met exhibit embryonic lethality due to incomplete liver development.36,37 In addition, loss of HGF signaling in mice decreases proliferation of gastric mucosal cells and delays recovery from mucosal injury.38 Depletion of c-Met in cancer cells results in the inhibition of cell proliferation, invasion, and survival, suggesting an essential role of c-Met in the development, progression, and invasion of cancers.39–42
Role of HGF and c-Met in cancer
In addition to the developmental roles described above, the HGF/c-Met signaling pathway is highly activated in human cancers via overexpression, amplification, or mutation, and promotes development, progression, invasive growth, and metastasis of cancers.43 Abnormal expression of HGF and c-Met has been reported in various solid tumors, including breast, colon, lung, ovary, kidney, and liver cancers (Table 1).44,45 HGF secretion from tumor stromal cells has been correlated with overexpression of c-MET. Overexpression of HGF/c-Met has been reported in breast, colon, lung, ovarian, and renal cancer.44,45 HGF/c-Met overexpression has also been correlated with metastasis and poor survival in hepatocellular carcinoma, renal cell carcinoma, and breast cancer.46–50
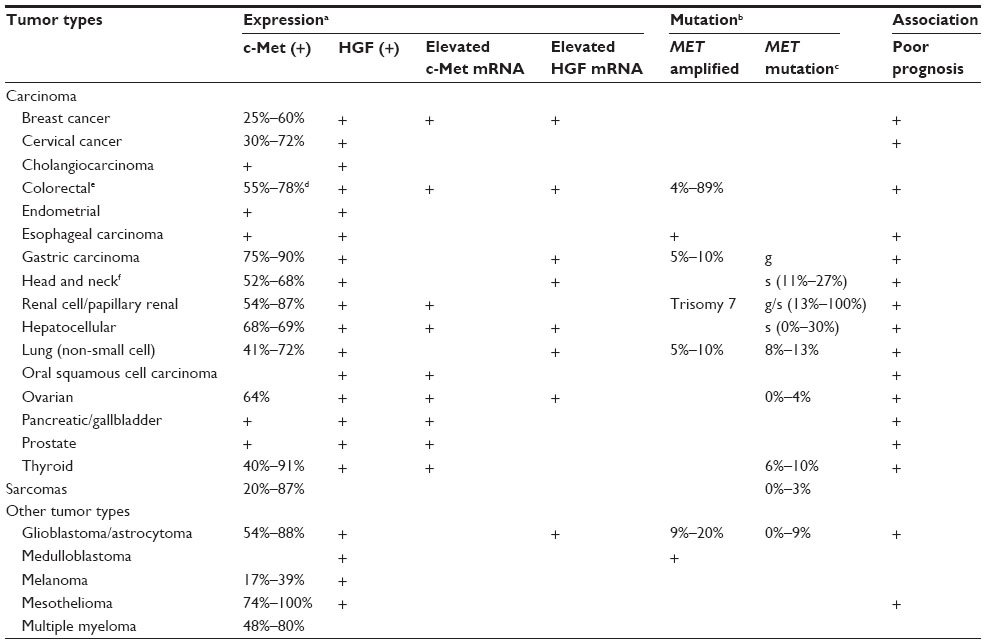 | Table 1 Expression and mutation pattern of HGF/c-Met in human cancers |
Both germline and somatic mutations in c-Met have been reported in various cancers. These mutations increase tumorigenic potential via constitutive activation of the c-Met receptor.51 The mutations are found in the tyrosine kinase domain, juxtamembrane domain, and extracellular domain of c-Met. The activating mutations in the tyrosine kinase domain of c-Met induce different downstream cascades and biological processes. D1228H/N and M1250T mutations increase c-Met phosphorylation and Ras activation, while L1195V and Y1230C mutations activate phosphatidylinositol-4, 5-bisphosphate 3-kinase to promote invasive and anchorage-independent growth.52
Amplification of c-Met receptor and overexpression of HGF are associated with resistance to inhibitors targeting other RTKs. Amplification of c-Met activates ERBB3 (HER3)-dependent phosphatidylinositol-4,5-bisphosphate 3-kinase signaling, leading to resistance to gefitinib (a small-molecule inhibitor of EGFR) in lung cancer.53 In addition, HGF is involved in decreased susceptibility to irreversible EGFR tyrosine kinase inhibition in lung cancer with the EGFR T790M mutation.54 Similarly, amplification of c-Met is involved in resistance to the anti-EGFR monoclonal antibodies, cetuximab and panitumumab, in metastatic colorectal cancer.55 Recent studies have demonstrated that the copy number of HGF and MET genes correlates with sensitivity to treatment with trastuzumab in HER2-positive metastatic breast cancer. An increased copy number for the MET gene has been linked to a higher failure rate of trastuzumab treatment and to a shorter time to progression, which means the length of time from the date of diagnosis or the start of treatment for a breast cancer patient until the breast cancer starts to get worse or spreads to other parts of the body. Increased copy number for the HGF gene is also linked to a higher failure rate of trastuzumab treatment.56 These studies on mutation and amplification of HGF and MET genes provide important information for the development of therapeutic agents targeting the HGF/c-Met signaling pathway.57,58 These results indicate that the c-Met receptor, together with other RTK signaling pathways such as ERBB3 (HER3), EGFR, and ERBB2 (HER2), has a synergic role in tumor progression in certain types of cancers. Therefore, combination therapy targeting both c-Met and other RTKs may be more effective for cancer treatment compared with monotherapy.
Antibody-based therapeutics targeting HGF and c-Met
Inhibitors of the HGF/c-Met signaling pathway are divided into two groups: while small-molecule compounds block the signaling pathway by inhibiting tyrosine kinase activity and autophosphorylation of c-Met, biologics including truncated HGF, N-terminal Sema domain of HGF, soluble extracellular domain of c-Met (decoy Met), and antibodies against HGF and c-Met suppress the signaling pathway by inhibiting interactions between HGF and c-Met. Compared with small-molecule compounds that often target multiple RTKs, biologics more specifically inhibit the HGF/c-Met signaling pathway. Multiple therapeutic antibodies targeting the HGF/c-Met signaling pathway are currently in preclinical and clinical development (Table 2).
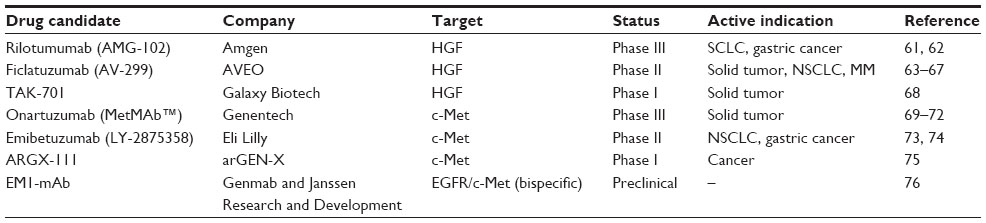 | Table 2 Monoclonal antibody therapeutics targeting HGF or c-Met under development |
Anti-HGF monoclonal antibodies
Rilotumumab (AMG102, Amgen) is a human monoclonal antibody against HGF that blocks interactions between HGF and its receptor c-Met, thereby inhibiting cellular processes driven by the HGF/c-Met signaling pathway.59 In a Phase II clinical trial in gastric and esophagogastric junction cancers, rilotumumab in combination with epirubicin, cisplatin, and capecitabine (ECX) has been shown to improve both progression-free survival and overall survival in patients with tumors expressing high levels of c-Met.60,61 In this study, patients were given placebo or rilotumumab at 15 mg/kg or 7.5 mg/kg on day 1 in addition to ECX (50 mg/m2 epirubicin and 60 mg/m2 cisplatin on day 1, and 625 mg/m2 capecitabine twice a day on days 1–21) every 3 weeks. Median progression-free survival was 5.1 months for patients treated with rilotumumab 15 mg/kg and 6.8 months for patients treated with rilotumumab 7.5 mg/kg, compared with 4.2 months for the placebo group. Objective response rates reported for patients treated with rilotumumab 15 mg/kg, rilotumumab 7.5 mg/kg and placebo were 31%, 48%, and 21%, respectively. Median overall survival times for patients treated with rilotumumab 15 mg/kg, rilotumumab 7.5 mg/kg, and placebo were 9.7, 11.1, and 8.9 months, respectively. Adverse events, including hematologic adverse events, peripheral edema, and venous thromboembolism, were reported for both the placebo and rilotumumab groups, but were more common in the rilotumumab group. Based on the safety profile and results indicating better efficacy of rilotumumab in combination with ECX, a Phase III study is ongoing in c-Met-positive gastric and gastroesophageal junction cancers (RILOMET-1).61 The efficacy of rilotumumab has also been demonstrated in metastatic colorectal cancer with wild-type KRAS. In a Phase II clinical trial of rilotumumab in combination with panitumumab (a fully human anti-EGFR monoclonal antibody), the median progression-free survival was 5.2 months for the combination treatment and 3.7 months for treatment with panitumumab alone.62
Ficlatuzumab (AV-299; SCH 900105, AVEO Pharmaceuticals, Cambridge, MA, USA) is a humanized monoclonal antibody against HGF that inhibits the HGF-induced c-Met signaling pathway by neutralizing HGF/c-Met binding.63 Preclinical studies carried out in the H596 NSCLC xenograft model have demonstrated increased anti-cancer activity of ficlatuzumab in combination with an EGFR inhibitor (erlotinib or cetuximab) compared with single agents.64 In a Phase I study of advanced solid tumors including sarcoma, ovarian cancer, mesothelioma, and glioblastoma multiforme, ficlatuzumab was intravenously administered at 2, 5, 10, or 20 mg/kg once every 2 weeks. At the recommended Phase II dose, ficlatuzumab was administered in combination with erlotinib (150 mg/day). Adverse events including fatigue, peripheral edema, headache, hematologic problems, and pruritus have been reported in the monotherapy group, and common adverse events of rash and diarrhea have been reported in the combination therapy group. From the Phase I study, it has been demonstrated that the selected dose of ficlatuzumab is safe and well tolerated when used in combination with erlotinib at the standard dose.65,66 This was followed by a randomized Phase II study with gefitinib alone or in combination with ficlatuzumab to treat NSCLC patients.67
TAK-701 (Galaxy Biotech) is a humanized monoclonal antibody that binds to HGF with high affinity.68 Combination treatment with TAK-701 and gefitinib (a small-molecule inhibitor of EGFR) inhibits the phosphorylation of both c-Met and EGFR and downstream signaling cascades in HCC827-HGF tumor cells (engineered human NSCLC cells that contain an activating EGFR mutation and stably express HGF).68 Combination treatment with TAK-701 and gefitinib also markedly inhibits tumor growth in HCC827-HGF xenograft models.68 These results suggest that combination treatment with TAK-701 and gefitinib may provide a means of overcoming resistance to EGFR-tyrosine kinase inhibitor therapy in HGF-induced NSCLC. Phase I studies are ongoing for TAK-701 as a single-agent treatment for advanced solid tumors.
Anti-c-Met monoclonal antibodies
Onartuzumab (MetMAb, Genentech) is a humanized, monovalent monoclonal antibody against c-Met. Onartuzumab was developed with the knob-into-hole technology, which allows one-to-one interaction between the antibody and the receptor.69 Onartuzumab potently inhibits binding of HGF, phosphorylation of c-Met, and downstream signaling in the HGF/c-Met pathway with antibody-like pharmacokinetics. Strong anti-cancer activity has been reported for onartuzumab in preclinical xenograft studies.69 Activated HGF/c-Met signaling has been associated with a poor prognosis and with resistance to EGFR inhibitors in NSCLC. In a Phase II study, prolonged progression-free survival (2.9 months versus 1.5 months) and overall survival (12.6 months versus 3.8 months) have been reported in c-Met-positive patients treated with erlotinib and onartuzumab compared with patients treated with erlotinib alone.70 However, in a randomized Phase III trial, combination therapy with onartuzumab and erlotinib failed to confirm the efficacy demonstrated in the Phase II study, as no improvement in overall survival (6.8 months versus 9.1 months) or progression-free survival (2.7 months versus 2.6 months) was observed in c-Met-positive patients.71 Despite the failure of the Phase III trial, Genentech has continued to develop onartuzumab in two additional Phase III trials in different subgroups of NSCLC, ie, c-Met-positive stage IIIB or IV NSCLC with activating EGFR mutation. Subgroup analyses may provide a means of targeting patients more selectively.72 Another Phase III clinical trial is ongoing in gastric cancer for evaluation of the efficacy and safety of onartuzumab. In this study, onartuzumab is administered in combination with 5-fluorouracil, folinic acid, and oxaliplatin (mFOLFOX6) to treat metastatic HER2-negative and c-Met-positive gastroesophageal cancer.72
Emibetuzumab (LY-2875358, Eli Lilly) is a humanized, bivalent anti-c-Met antibody that inhibits both ligand-dependent and ligand-independent activation of c-Met.73 In the case of HGF-dependent c-Met activation, emibetuzumab inhibits HGF binding to c-Met, c-Met phosphorylation, and tumor growth both in vitro and in vivo, similar to a humanized, one-armed 5D5 anti-c-Met antibody (precursor of onartuzumab). In the case of HGF-independent c-Met activation by MET gene amplification in tumors, emibetuzumab promotes internalization and degradation of c-Met. Decreases in phosphorylated and total c-Met after treatment with emibetuzumab induces inhibition of cell proliferation and tumor growth in the gastric cancer cell lines, MKN-45 and SNU-5, and in the NSCLC cell lines, EBC-1 and H1993. However, the one-armed 5D5 antibody has exhibited no anti-tumor activity in the case of HGF-independent c-Met activation.73 In a Phase I study, treatment with emibetuzumab alone or in combination with erlotinib resulted in a durable partial response in NSCLC and was also shown to be safe and well tolerated. Based on the pharmacokinetic/pharmacodynamic data, the recommended Phase II dose of emibetuzumab for intravenous administration is 750 mg once every 2 weeks as a single agent or in combination with erlotinib.74
ARGX-111 (arGEN-X) is a defucosylated antagonistic anti-c-Met antibody with potent anti-cancer activity based on enhanced antibody-dependent cellular cytotoxicity. A Phase Ib study was initiated in January 2014 to evaluate ARGX-111 in advanced cancers with c-Met overexpression.75 EM1-mAb (Genmab™, Janssen Research and Development, San Diego, CA, USA) is a bispecific anti-EGFR/c-Met antibody that inhibits both EGFR and c-Met signaling pathways. EM1-mAb has exhibited more potent inhibition of downstream signaling cascades compared with the combination of monospecific antibodies.76
Critical analysis for potential of antibody-based HGF/c-Met inhibitors in human cancer
Several antibody-based inhibitors of the HGF/c-Met signaling pathway are under active preclinical/clinical development as novel therapeutic agents to treat cancers. There are important aspects of HGF/c-Met biology that need to be carefully addressed for successful development of these therapeutic antibodies targeting the HGF/c-Met signaling pathway. These include unwanted activation of c-Met by bivalent anti-c-Met antibodies and the recent failure of the Phase III study of onartuzumab in combination with erlotinib in NSCLC.
A monovalent antibody, onartuzumab (MetMAb™), was designed to address issues reported for several bivalent anti-c-Met reagents that induce unwanted stimulation of the c-Met signaling by mimicking c-Met dimerization.69,77 Onartuzumab, the one-armed humanized antibody against c-Met, blocks the interaction between the HGF-α chain and the Sema domain (homologous to semaphorins) of the c-Met receptor, demonstrated by crystallographic, structural, and biochemical analysis.69 However, despite concerns regarding c-Met activation by bivalent anti-c-Met antibodies and the development of the one-armed antibody, onartuzumab, clinical trials are still ongoing for two bivalent antibodies against c-Met (emibetuzumab and ARGX-111).74,75 More important aspects to consider for the development of HGF/c-Met inhibitors include patient stratification and identification of effective combinations of inhibitors targeting the HGF/c-Met pathway along with other interacting signaling pathways such as the EGFR pathway. Since alterations in HGF/c-Met signaling are closely related to tumorigenesis, tumor progression, and metastasis, it is critical to more accurately detect c-Met expression levels and alterations. In the Phase II study of onartuzumab in NSCLC, it has been demonstrated that the intensity of c-Met labeling detected via immunohistochemistry can successfully identify target patient groups for treatment.70 Immunohistochemistry-based detection of c-Met expression levels was also important for patient stratification in the Phase I and II trials of rilotumumab.61,62 However, the same strategy has failed to meet the initial goal in the Phase III study of onartuzumab in combination with erlotinib in NSCLC, possibly resulting from the recruitment of nonspecific patient groups rather than from ineffectiveness of the antibodies.71,72
In addition to immunohistochemistry, different detection methods and other specific targets have also been examined. Elevated c-Met expression levels have been detected via Western blotting of serum proteins from patients with hepatocellular carcinoma and tissues from patients with colorectal cancer and metastasis to the liver.78,79 Phosphorylated c-Met (the active form of c-Met) has been intensively examined in preclinical and clinical studies and via in silico modeling. Antibodies that specifically recognize phosphorylated c-Met can be used to detect this pharmacodynamic biomarker.80–82 In addition to the development of better detection methods and detecting antibodies, well defined scoring systems and minimization of interobserver differences are all required for effective patient diagnosis and stratification.
Although no significant correlation between a high MET gene copy number and patient response to onartuzumab has been reported from clinical trials, results from both preclinical and clinical studies with small-molecule inhibitors have demonstrated a correlation between a high copy number or specific mutation of MET gene and increased clinical response.83–85 Due to the association between MET gene amplification/mutation and poor clinical outcomes, various technologies including next-generation sequencing are under development.86
As reported from studies in NSCLC, inhibitors of the HGF/c-Met pathway may become more potent when combined with antagonists of other signaling pathways. Most clinical trials with anti-c-Met monoclonal antibodies were designed in combination with chemical inhibitors of other signaling pathways. Clinical trials of onartuzumab were conducted in combination with erlotinib, an EGFR inhibitor, to overcome EGFR-mediated resistance.70,71 Synergistic effects of dual inhibition of the HGF/c-Met and VEGF signaling pathways have been reported in preclinical studies, suggesting that combination of HGF/c-Met and VEGF/VEGFR inhibition may allow more effective treatment of human cancers.87–89 The efficacy of onartuzumab and rilotumumab are being evaluated in combination with bevacizumab or panitumumab in the clinical settings of glioblastoma multiforme and metastatic colorectal cancer.86 Successful clinical development of candidates requires a better understanding of the interactions between signaling pathways. The use of two or more therapeutic agents in combination should be carefully designed because this combination treatment may elevate the risk of adverse effects and increase the treatment cost.
Results from clinical studies reflect the highly complex nature of HGF/c-Met signaling in human cancers and suggest that successful development of antibody-based therapeutics targeting the HGF/c-Met signaling pathway requires a better understanding of the pathways involved in the diseases, along with more careful analyses of the clinical data.
Conclusion
Studies carried out over the past two decades have demonstrated that the HGF/c-Met signaling pathway is a promising therapeutic target for treating cancers. Therapeutic agents designed to target HGF/c-Met-mediated signaling cascades have been developed and evaluated in preclinical and clinical studies. In clinical trials in lung, gastric, prostate and renal cancers, inhibitors of the HGF/c-Met signaling pathway have yielded promising results. Like inhibitors of other RTK signaling pathways, inhibitors of the HGF/c-Met signaling pathway are divided into two groups, ie, small-molecule compounds suppressing the tyrosine kinase activity of c-Met and biologics such as monoclonal antibodies that specifically bind to HGF or c-Met.
Among the therapeutic agents targeting HGF or c-Met, a small-molecule inhibitor, cabozantinib (XL184, Exelixis, South San Francisco, CA, USA) became the first agent approved by the FDA for the clinical treatment of progressive metastatic medullary thyroid cancer (in November 2012). In addition, several small-molecule inhibitors of c-Met, including tivantinib (ARQ 197, ArQule, Woburn, MA, USA), golvatinib (E7050, Eisai, Tokyo, Japan), and foretinib (GSK 1363089, GlaxoSmithKline, Brentford, UK) are currently being evaluated in clinical studies for treatment of hepatocellular carcinoma.90
In contrast with the success of cabozantinib, an antibody-based inhibitor of c-Met, onartuzumab failed to improve the clinical benefit in a recent Phase III study in NSCLC. However, the failure in this Phase III trial suggests several key issues to consider for successful clinical development of antibody-based HGF or c-Met inhibitors. Challenges to face for effective clinical application of HGF/c-Met-targeted therapeutics include advances in analytical methods to identify specific patient groups, more careful selection of target populations, development of biomarkers for HGF/c-Met signaling, and identification of effective combination therapies with other RTK inhibitors.
Acknowledgments
The authors thank Sang Hoon Lee and Sang K Park at the Hanwha Chemical Corporation and William B Stallcup at the Sanford-Burnham Medical Research Institute for their valuable advice and discussion during preparation for this review.
Disclosure
The authors report no conflicts of interest in this review.
References
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. | |
Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4(5):361–370. | |
Xu AM, Huang PH. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010;70(10):3857–3860. | |
Fauvel B, Yasri A. Antibodies directed against receptor tyrosine kinases: current and future strategies to fight cancer. MAbs. 2014;6(4):838–851. | |
Dodé C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33(4):463–465. | |
Feldheim DA, Nakamoto M, Osterfield M, et al. Loss-of-function analysis of EphA receptors in retinotectal mapping. J Neurosci. 2004;24(10):2542–2550. | |
Kong-Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66(1):283–289. | |
Thor A. HER2-a discussion of testing approaches in the USA. Ann Oncol. 2001;12 Suppl 1:S101–S107. | |
Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus FOLFOX4 as second-line treatment of colorectal cancer. Oncologist. 2007;12(3):356–361. | |
Summers J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma. Oncologist. 2010;15(1):104–111. | |
Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14(11):1131–1138. | |
Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) plus carboplatin and paclitaxel as first-line treatment of advanced/metastatic recurrent nonsquamous non-small cell lung cancer. Oncologist. 2007;12(6):713–718. | |
Garrett CR, Eng C. Cetuximab in the treatment of patients with colorectal cancer. Expert Opin Biol Ther. 2011;11(7):937–949. | |
Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: panitumumab (Vectibix). Oncologist. 2007;12(5):577–583. | |
Poole RM, Vaidya A. Ramucirumab: first global approval. Drugs. 2014;74(9):1047–1058. | |
Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311(5981):29–33. | |
Gherardi E, Stoker M. Hepatocytes and scatter factor. Nature. 1990;346(6281):228. | |
Naldini L, Vigna E, Narsimhan RP, et al. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene. 1991;6(4):501–504. | |
You WK, McDonald DM. The hepatocyte growth factor/c-Met signaling pathway as a therapeutic target to inhibit angiogenesis. BMB Rep. 2008;41(12):833–839. | |
Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(6):553–572. | |
Blumenschein GR Jr, Mills GB, Gonzalez-Angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol. 2012;30(26):3287–3296. | |
Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 2002;109(7):863–867. | |
Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–925. | |
Scagliotti GV, Novello S, von Pawel JV. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. 2013;39(7):793–801. | |
Biswas P, Roy A, Gong R, et al. Hepatocyte growth factor induces an endothelin-mediated decline in glomerular filtration rate. Am J Physiol Renal Physiol. 2005;288(1):F8–F15. | |
Ueki T, Kaneda Y, Tsutsui H, et al. Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat Med. 1999;5(2):226–230. | |
Kusumoto K, Ido A, Moriuchi A, et al. Repeated intravenous injection of recombinant human hepatocyte growth factor ameliorates liver cirrhosis but causes albuminuria in rats. Int J Mol Med. 2006;17(3):503–509. | |
Nakamura Y, Morishita R, Nakamura S, et al. A vascular modulator, hepatocyte growth factor, is associated with systolic pressure. Hypertension. 1996;28(3):409–413. | |
Mizuno S, Matsumoto K, Li MY, Nakamura T. HGF reduces advancing lung fibrosis in mice: a potential role for MMP-dependent myofibroblast apoptosis. FASEB J. 2005;19(6):580–582. | |
Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342(6248):440–443. | |
Gohda E, Tsubouchi H, Nakayama H, et al. Purification and partial characterization of hepatocyte growth factor from plasma of a patient with fulminant hepatic failure. J Clin Invest. 1988;81(2):414–419. | |
Rodrigues GA, Naujokas MA, Park M, et al. Alternative splicing generates isoforms of the met receptor tyrosine kinase which undergo differential processing. Mol Cell Biol. 1991;11(6):2962–2970. | |
Bardelli A, Ponzetto C, Comoglio PM, et al. Identification of functional domains in the hepatocyte growth factor and its receptor by molecular engineering. J Biotechnol. 1994;37(2):109–122. | |
Longati P, Bardelli A, Ponzetto C, Naldini L, Comoglio PM. Tyrosines 1234–1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene. 1994;9(1):49–57. | |
Ponzetto C, Bardelli A, Zhen Z, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994;77(2):261–271. | |
Uehara Y, Minowa O, Mori C, et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373(6515):702–705. | |
Huh CG, Factor VM, Sánchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101(13):4477–4482. | |
Nakahira R, Mizuno S, Yoshimine T, Nakamura T. The loss of local HGF, an endogenous gastrotrophic factor, leads to mucosal injuries in the stomach of mice. Biochem Biophys Res Commun. 2006;341(4):897–903. | |
Zhang SZ, Pan FY, Xu JF, et al. Knockdown of c-Met by adenovirus-delivered small interfering RNA inhibits hepatocellular carcinoma growth in vitro and in vivo. Mol Cancer Ther. 2005;4(10):1577–1584. | |
Que W, Chen J. Knockdown of c-Met inhibits cell proliferation and invasion and increases chemosensitivity to doxorubicin in human multiple myeloma U266 cells in vitro. Mol Med Rep. 2011;4(2):343–349. | |
Sun B, Liu R, Xiao ZD, Zhu X. c-MET protects breast cancer cells from apoptosis induced by sodium butyrate. PLoS One. 2012;7(1):e30143. | |
Que W, Chen J, Chuang M, Jiang D. Knockdown of c-Met enhances sensitivity to bortezomib in human multiple myeloma U266 cells via inhibiting Akt/mTOR activity. APMIS. 2012;120(3):195–203. | |
Gherardi E, Birchmeier W, Birchmeier C, Van de Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103. | |
Smyth EC, Sclafani F, Cunningham D. Emerging molecular targets in oncology: clinical potential MET/hepatocyte growth-factor inhibitors. OncoTargets Ther. 2014;7:1001–1014. | |
Ma PC, Tretiakova MS, MacKinnon AC, et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer. 2008;47(12):1025–1037. | |
Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002;13(1):41–59. | |
Ueki T, Fujimoto J, Suzuki T, Yamamoto H, Okamoto E. Expression of hepatocyte growth factor and its receptor c-met proto-oncogene in hepatocellular carcinoma. Hepatology. 1997;25(4):862–866. | |
Daveau M, Scotte M, Francois A, et al. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog. 2003;36(3):130–141. | |
Schutz FA, Pomerantz MM, Gray KP, et al. Single nucleotide polymorphisms and risk of recurrence of renal-cell carcinoma: a cohort study. Lancet Oncol. 2013;14(1):81–87. | |
Lengyel E, Prechtel D, Resau JH, et al. C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer. 2005;113(4):678–682. | |
Graveel CR, Tolbert D, Van de Woude GF. MET: a critical player in tumorigenesis and therapeutic target. Cold Spring Harb Perspect Biol. 2013;5(7):a009209. | |
Giordano S, Maffe A, Williams TA, et al. Different point mutations in the met oncogene elicit distinct biological properties. FASEB J. 2000;14(2):399–406. | |
Engelman JA, Zejnullahu K, Mitsudomi T, et al. Met amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | |
Yamada T, Matsumoto K, Wang W, et al. Hepatocyte growth factor reduces susceptibility to an irreversible epidermal growth factor receptor inhibitor in EGFR-T790M mutant lung cancer. Clin Cancer Res. 2010;16(1):174–183. | |
Bardelli, A, Corso S, Bertotti, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–673. | |
Minuti G, Cappuzzo F, Duchnowska R, et al. Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. Br J Cancer. 2012;107(5):793–799. | |
Wang W, Li Q, Takeuchi S, et al. Met kinase inhibitor E7050 reverses three different mechanisms of hepatocyte growth factor-induced tyrosine kinase inhibitor resistance in EGFR mutant lung cancer. Clin Cancer Res. 2012;18(6):1663–1671. | |
Nakagawa T, Takeuchi S, Yamada T, et al. Combined therapy with mutant-selective EGFR inhibitor and Met inhibitor for overcoming erotinib resistance in EGFR-mutant lung cancer. Mol Cancer Ther. 2012;11(10):2149–2157. | |
Gordon MS, Sweeney CS, Mendelson DS, et al. Safety, pharmacokinetics, and pharmacodynamics of AMG-102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res. 2010;16(2):699–710. | |
Min Z, Sameer D, Gisleskog PO, et al. Population pharmacokinetics of rilotumumab, a fully human monoclonal antibody against hepatocyte growth factor, in cancer patient. J Pharm Sci. 2014;103(1):328–336. | |
Iveson T, Donehower RC, Davidenko I, et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double blind, randomized phase 2 study. Lancet Oncol. 2014;15(9):1007–1018. | |
Van Cutsem E, Eng C, Nowara E, et al. Randomized phase Ib/II trials of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin Cancer Res. 2014;20(16):4240–4250. | |
D’Arcangelo M, Cappuzzo F. Focus on the potential role of ficlatuzumab in the treatment of non-small cell lung cancer. Biologics. 2013;7:61–68. | |
Meetze KA, Boudrow A, Connoly K, et al. Anti-tumor activity of SCH 900105 (AV299), an anti-HGF antibody, in non-small cell lung cancer models. Mol Cancer Ther. 2009;8 Suppl 12:Abstr C173. | |
Patnaik A, Weiss GJ, Papadopoudos K, et al. Phase I study of SCH 900105 (SC), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb), as a single agent and in combination with erlotinib (E) in patients (pts) with advanced solid tumors. J Clin Oncol. 2010;28 Suppl:Abstr 2525. | |
Patnaik A, Weiss GJ, Papadopoulos KP, et al. Phase I ficlatuzumab monotherapy or with erlotinib for refractory advanced solid tumours and multiple myeloma. Br J Cancer. 2014;111(2):272–280. | |
Mok TSK, Park K, Geater SL, et al. A randomized phase (ph) 2 study with exploratory biomarker analysis of ficlatuzumab (F) a humanized hepatocyte growth factor (HGF) inhibitory monoclonal antibody (MAb) in combination with gefitinib (G) versus gefitinib alone in Asian patients with lung adenocarcinoma (LA). Presented at the European Society for Medical Oncology annual meeting, September 28 to October 2, 2012, Vienna, Austria. | |
Okamoto W, Okamoto I, Tanaka K, et al. TAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol Cancer Ther. 2010;9(10):2785–2792. | |
Merchant M, Ma X, Maun HR, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A. 2013;110(32):E2987–E2996. | |
Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small cell lung cancer. J Clin Oncol. 2013;31(32):4105–4114. | |
Spigel DR, Edelman MJ, O’Byrne K, et al. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb and IV NSCLC: Results from the pivotal phase III randomized, multicenter, placebo-controlled METLung (OAM4971g) global trial. J Clin Oncol. 2014;32:5 Suppl:Abstr 8000. | |
Cormac S. Genentech to salvage anti-MET antibody with subgroup analysis. Nat Biotechnol. 2014;32(5):399–400. | |
Zeng W, Peek V, Wortinger M, et al. LY2875358, a bivalent MET antibody with anti-tumor activity through blocking HGF as well as inducing degradation of MET, differentiates from a one-armed 5D5 MET antibody. Cancer Res. 2013;73 Suppl 8:5465. | |
Goldman JW, Rosen LS, Algazi AP, et al. First-in-human dose escalation study of LY2875358 (LY), a bivalent MET antibody, as monotherapy and in combination with erlotinib (E) in patients with advanced cancer. J Clin Oncol. 2013;31 Suppl 15:Abstr 8093. | |
Hultberg A, Huyghe L, de Jonge N, et al. ARGX-111, a defucosylated antagonistic anti-MET antibody, displays potent anti-tumor activity through enhanced ADCC. Presented at the 12th International Congress on Targeted Anticancer Therapies, March 5–7, 2014, Washington, DC, USA. | |
Moores S, Chiu M, Bushey B, et al. Bispecific antibody targeting EGFR and cMet demonstrates superior activity compared to the combination of single pathway inhibitors. Mol Cancer Ther. 2013;12(11 Suppl):B241. | |
Martens T, Schmidt NO, Eckerich C, et al. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res. 2006;12(20 Pt 1):6144–6152. | |
Osada S, Kanematsu M, Imai H, et al. Clinical significance of serum HGF and c-Met expression in tumor tissue for evaluation of properties and treatment of hepatocellular carcinoma. Hepatogastroenterology. 2008;55(82–83):544–549. | |
Osada S, Matsui S, Komori S, Goshima S. Effect of hepatocyte growth factor on progression of liver metastasis in colorectal cancer. Hepatogastroenterology. 2010;57(97):76–80. | |
Srivastava AK, Hollingshead MG, Weiner J, et al. Development and validation of biomarker assays to assess pharmacodynamic modulation of MET. Presented at the American Society of Clinical Oncology Annual Meeting, June 3–7, 2011, Chicago, IL, USA. | |
Yamazaki S, Vicini P, Shen Z, et al. Pharmacokinetic/pharmacodynamic modeling of crizotinib for anaplastic lymphoma kinase inhibition and antitumor efficacy in human tumor xenograft mouse models. J Pharmacol Exp Ther. 2010;340(3):549–557. | |
Arriola E, Cañadas I, Arumí-Uría M, et al. MET phosphorylation predicts poor outcome in small cell lung carcinoma and its inhibition blocks HGF-induced effects in MET mutant cell lines. Br J Cancer. 2011;105(6):814–823. | |
Bellon SF, Kaplan-Lefko P, Yang Y, et al. c-Met inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations. J Biol Chem. 2008;283(5):2675–2683. | |
Berthou S, Aebersold DM, Schmidt LS, et al. The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene. 2004;23(31):5387–5393. | |
Sequist LV, von Pawel J, Garmey EG, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29(24):3307–3315. | |
Maroun CR, Rowlands T. The Met receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacol Ther. 2014;142(3):316–338. | |
Sulpice E, Ding S, Muscatelli-Groux B, et al. Cross-talk between the VEGF-A and HGF signaling pathways in endothelial cells. Biol Cell. 2009;101(9):525–539. | |
You WK, Sennino B, Williamson CW, et al. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71(14):4758–4768. | |
Roy S, Narang BK, Rastogi SK, Rawal RK. A novel multiple tyrosine-kinase targeted agent to explore the future perspectives of anti-angiogenic therapy for the treatment of multiple solid tumors: cabozantinib. Anticancer Agents Med Chem. 2014;15(1):37–47. | |
Goyal L, Muzumdar MD, Zhu AX, Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res. 2013;19(9):2310–2318. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.