Back to Journals » International Journal of Nanomedicine » Volume 16
Development of AE147 Peptide-Conjugated Nanocarriers for Targeting uPAR-Overexpressing Cancer Cells
Authors Park JY, Shin Y, Won WR, Lim C, Kim JC, Kang K, Husni P ![]() , Lee ES, Youn YS
, Lee ES, Youn YS ![]() , Oh KT
, Oh KT ![]()
Received 15 April 2021
Accepted for publication 2 July 2021
Published 12 August 2021 Volume 2021:16 Pages 5437—5449
DOI https://doi.org/10.2147/IJN.S315619
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Israel Rubinstein
June Yong Park,1,* Yuseon Shin,1,* Woong Roeck Won,1 Chaemin Lim,1,2 Jae Chang Kim,1 Kioh Kang,1 Patihul Husni,1 Eun Seong Lee,3 Yu Seok Youn,4 Kyung Taek Oh1,5
1Department of Global Innovative Drugs, The Graduate School of Chung-Ang University, Dongjak-gu, Seoul, 06974, Republic of Korea; 2Center for Nanotechnology in Drug Delivery and Division of Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA; 3Division of Biotechnology, The Catholic University of Korea, Bucheon, Gyeonggi-do, 14662, Republic of Korea; 4School of Pharmacy, Sungkyunkwan University, Suwon, Gyeonggi-do, 16419, Republic of Korea; 5College of Pharmacy, Chung-Ang University, Dongjak-gu, Seoul, 06974, Republic of Korea
*These authors contributed equally to this work
Correspondence: Kyung Taek Oh
College of Pharmacy, Chung-Ang University, 221 Heukseok-dong, Dongjak-gu, Seoul, 06974, Republic of Korea
Tel +82-2-824-5617
Email [email protected]
Purpose: An AE147 peptide-conjugated nanocarrier based on PEGylated liposomes was developed in order to target the metastatic tumors overexpressing urokinase-type plasminogen activator receptor (uPAR), which cancer progression via uPA signaling. Therefore, the AE147 peptide-conjugated nanocarrier system may hold the potential for active targeting of metastatic tumors.
Methods: The AE147 peptide, an antagonist of uPAR, was conjugated to the PEGylated liposomes for targeting metastatic tumors overexpressing uPAR. Docetaxel (DTX), an anticancer drug, was incorporated into the nanocarriers. The structure of the AE147-conjugated nanocarrier, its physicochemical properties, and in vivo biodistribution were evaluated.
Results: The DTX-loaded nanocarrier showed a spherical structure, a high drug-loading capacity, and a high colloidal stability. Drug carrying AE147 conjugates were actively taken up by the uPAR-overexpressing MDA-MB-231 cancer cells. In vivo animal imaging confirmed that the AE147-conjugated nanoparticles effectively accumulated at the sites of tumor metastasis.
Conclusion: The AE147-nanocarrier showed potential for targeting metastatic tumor cells overexpressing uPAR and as a nanomedicine platform for theragnosis applications. These results suggest that this novel nano-platform will facilitate further advancements in cancer therapy.
Keywords: tumor-targeting ligand, urokinase-type plasminogen activator, liposome, circulating tumor cell, metastatic tumor
Introduction
Cancer, a disease caused by the uncontrolled growth of abnormal cells in the body, is a major cause of death worldwide, accounting for approximately 9.6 million deaths in 2018.1 Although various anticancer therapies, including surgery at the primary tumor site, have decreased the mortality rate, the occurrence of cancer metastasis reflects treatment failure and a poor prognosis.2 In cancer metastasis, cancer cells disseminate from the primary tumor site to invade surrounding tissues, enter the vasculature as circulating tumor cells, and extravasate into distant sites to develop metastatic tumors.3–5 The cascading steps of cancer metastasis require stromal cells.6,7 Moreover, several protease systems including the urokinase-type plasminogen activator (uPA) and uPA receptor (uPAR) system have been recognized to play critical roles in the degradation of the basement membrane and the extracellular matrix, eventually resulting in cell invasion and metastasis.7 The uPAR, a glycolipid-anchored membrane glycoprotein, is overexpressed in several aggressive cancers, and its overexpression is linked to the regulation of tumorigenesis, invasion, metastasis, and cancer cell survival.8–12 As uPA binds to uPAR followed by the internalization of the uPA-uPAR complex, targeting uPAR with uPA antagonists may be a promising strategy to inhibit metastasis. Furthermore, incorporating cytotoxic payloads with uPAR targeting moieties can lead to the internalization of the complex and intracellular delivery of the chemotherapeutic drugs, siRNAs, and protein-based therapeutics.11,13–15
Several therapeutics based on nanomaterials, including liposomes, micelles, albumin nanoparticles, and metal nanoparticles, have been widely developed for treating metastatic cancers. In particular, nanocarriers modified with ligands targeting specific metastatic cancer cell types have been designed for effective tumor targeting. The AE147 peptide, composed of 13 amino acids, is an antagonist of uPAR.9,16,17 The AE147 binds to uPAR by occupying its central cavity, causing subsequent internalization via receptor-mediated endocytosis.18,19 Therefore, conjugating AE147 on therapeutic nanoformulations is expected to impart tumor specificity and improve cytotoxic drug delivery.
Liposomes are the most common and well-known nanocarriers used for anticancer drug delivery.20–23 Moreover, certain modification methods can stabilize encapsulated molecules, improve their cellular uptake by tumor cells via passive targeting, exhibit the enhanced permeability and retention (EPR) effect, and improve drug distribution at the target sites.23–28 Active cancer cell targeting has been achieved by the surface modification of nanoparticles. Attaching specific targeting moieties to the nanocarrier surface increases its accumulation and cellular uptake by the target cells.21,29–31 Targeted delivery of therapeutics not only reduces the non-specific cytotoxicity but also increases drug accumulation in cancer tissues, resulting in improved effects.18,23,32
In this study, we developed targeting nanocarrier liposomes to recognize uPAR expressed in metastatic cancer cells. The nanocarrier liposomes were prepared by the thin-film method using cholesterol, L-α-phosphatidylcholine, hydrogenated (Soy) (HSPC), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG). To target uPAR, AE147 was conjugated with DSPE-PEG via a stable amide group.33–35 Docetaxel (DTX), an anticancer drug currently used to treat metastasis, was encapsulated within the hydrophobic lipid bilayer of AE147-conjugated liposomes and evaluated for therapeutic efficacy. In addition, for the in vivo imaging study, the fluorescent dye chlorin e6 (Ce6) was conjugated with the liposomes. The AE147-conjugated PEGylated liposome targeted metastatic tumors overexpressing uPAR, the circulation tumor cells, as well as the soluble uPAR due to the increased systemic circulation time of the PEGylated liposomes.36,37 The liposomal formulation was characterized for its particle size, zeta potential, and morphology. In addition, we studied the targeting potential of AE147-conjugated liposomes in vitro and in vivo using fluorescence techniques. An outline of this study is depicted in Figure 1.
 |
Figure 1 Schematic conceptual representation of AE147-conjugated liposomes. Note: i.v. injection, intravenous injection. |
Materials and Methods
Materials
DSPE-PEG, DSPE-PEG-NH2, HSPC, and cholesterol were bought from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Dimethyl sulfoxide (DMSO) and dimethyl formamide (DMF) were purchased from Honeywell Burdick & Jackson (Muskegon, MI, USA). Triethylamine (TEA), 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDC), DMSO-d6, and N-hydroxysuccinimide (NHS) were purchased from Sigma–Aldrich (St. Louis, MO, USA). Fluorescein isothiocyanate (FITC) conjugated DSPE-PEG (DSPE-PEG-FITC) was acquired from Nanocs (New York, NY, USA). DSPE-PEG-NHS was obtained from Nanosoft Biotechnology LLC (Winston Salem, NC, USA). Ce6 was bought from Frontier Scientific (Logan, UT, USA). KSD-cha-FskYLWSSK (cha = L-cyclohexyl alanine, s = D-form Ser, k = D-form Lys, acetate salt) (AE147) was purchased from Bankpeptide Biological Technology (Hefei, Anhui, China). DTX was purchased from Samyang Bio Pharmaceutical (Seongnam-si, Gyeonggi-do, South Korea). Human breast cancer cell lines MDA-MB-231 (KCLB No 30026) and MCF-7 (KCLB No 30022) were obtained from the Korean Cell Line Bank (Seoul, South Korea). RPMI 1640 medium, fetal bovine serum, Dulbecco’s phosphate-buffered saline, trypsin-EDTA, and penicillin-streptomycin were acquired from Welgene (Gyeongsan-si, Gyeongsangbuk-do, South Korea). The Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Rockville, MD, USA). All other chemicals used were of analytical grade.
Synthesis of DSPE-PEG-AE147 and DSPE-PEG-Ce6
AE147 and DSPE-PEG-NHS were dissolved in DMSO at a molar ratio of 1:1 (total 5 mg/mL). After stirring for 1 h, TEA was added in excess, and the air was replaced with the nitrogen gas. After stirring at 25 °C for three days, the solution was dialyzed against distilled water (DW) for two days using a dialysis membrane (molecular weight cutoff [MWCO] 3.5 kDa; Spectrum Laboratories, Rancho Dominguez, CA, USA) to remove the by-products. DSPE-PEG-AE147 was lyophilized using an Operon FDB-5503 freeze dryer (Gyeonggi-do, Korea). The AE147 conjugation to DSPE-PEG was analyzed using a 1H NMR 600 MHz spectrometer (JNM-ECZ600R, JEOL, Tokyo, Japan) and confirmed by comparing the Fourier transform infrared (FT-IR) peaks measured by an FT-IR spectrometer (Nicolet 6700, Thermo Fisher Scientific) in the spectral range of 400–4000 cm−1. Each spectrum was obtained with four scans and a resolution of 2 cm−1, and peak fitting was performed using OMNIC software (Thermo Nicolet Corporation, Madison, WI, USA).38
For real-time in vivo tumor imaging experiments, DSPE-PEG-NH2 was reacted with the carboxyl group of Ce6 (excess amount) via EDC/NHS activation.39 Ce6 was dissolved in 10 mL DMF and activated with EDC and NHS for 30 min at room temperature. DSPE-PEG-NH2 was added to the solution, and a coupling reaction was carried out for 3 h at room temperature with N2 purging. Free reactants were removed by dialysis (MWCO 3.5 kDa) against DMSO for two days and then dialyzed against DW for two days. Finally, Ce6-conjugated DSPE-PEG was obtained by freeze-drying.
Preparation of Non-Targeting and Targeting Liposomes Encapsulating DTX
Liposomes were prepared using the thin-film hydration method followed by ultrasonication.20,40–42 For the preparation of non-targeting liposomes (Lipo), a mixture of HSPC:cholesterol:DSPE-PEG (molar ratio 11:8:1) was mixed with chloroform in a glass vial; the organic solvent was eliminated using a rotary evaporator and vacuum dried overnight. For preparing targeting liposomes (AE-Lipo), the same procedure, as for the Lipo preparation, was followed, except for the use of a mixture of DSPE-PEG and DSPE-PEG-AE147 at a molar ratio of 1:1. The lipid film was resuspended on DW, ultrasonicated for 30 min, and extruded 12 times through two stacked Nuclepore polycarbonate filters (400 nm) using a stainless-steel extruder (Avanti Polar Lipids, Alabaster, AL, USA). For the preparation of FITC or Ce6-conjugated liposomes, the same procedure, as described for AE-Lipo, was followed except for the use of DSPE-PEG-FITC or DSPE-PEG-Ce6. Table 1 summarizes the composition ratio of all liposomes.
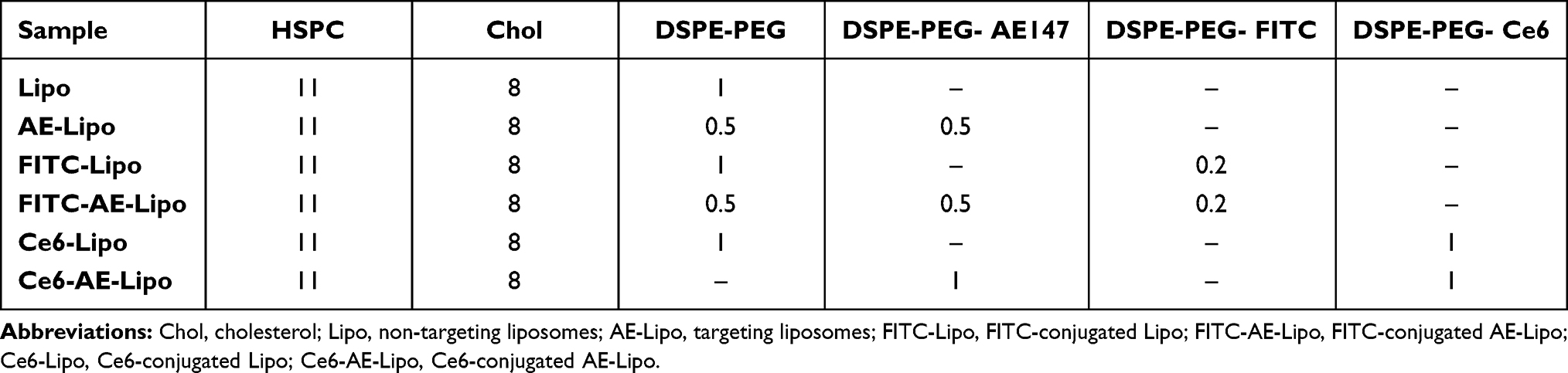 |
Table 1 Molar Ratio of Components Used in All Liposomal Formulations |
For the preparation of DTX-loaded liposomes (DTX/Lipo and DTX/AE-Lipo), DTX solution (1 mg/mL) in chloroform was mixed with the lipid stock solution (1 mg/mL) at a 1:9 volume ratio and stirred for one day. After a dry film of the mixed solution was formed, it was hydrated, ultrasonicated, and extruded. The liposome solution was filtered using a 0.2 μm-pore-size membrane filter to remove the unloaded drug and ensure the sterility of the liposome preparation.
Characterization of the Prepared Liposomes
To characterize liposome properties, we measured the effective hydrodynamic diameter, zeta potential, and particle size distribution by photon correlation spectroscopy using the Zetasizer Nano-ZS (Malvern instruments, Worcestershire, UK) equipped with the Multi Angle Sizing Option (BI-MAS).24,40 All samples were stabilized for 30 min before measurement. The zeta potential values, average particle size, and polydispersity index (PDI) were evaluated from three independent measurements for each sample (n = 3).
For the morphological evaluation, the prepared liposomes were investigated using field emission-scanning electron microscope (FE-SEM; Sigma, Carl Zeiss Meditec AG, Jena, Germany), which avoided damage to electron beam-sensitive samples.43 The samples of liposomal solutions were ground on a glass slide and dried in vacuo. Ion-beam sputtered platinum coating was applied to the sample surface before acquiring the microphotographs.44
The concentration of DTX in DTX/Lipo and DTX/AE-Lipo was determined by performing high-performance liquid chromatography (Agilent 1200 series, Agilent Technologies Santa Clara, CA, USA).45 A reverse phase C18 column (ZORBAX Eclipse Plus C18, 4.6×150 mm, pore size 5 μm, Agilent Tech., CA, USA) was used, and the flow rate of the mobile phase was set at 1 mL/min. The mobile phase included a mixture of acetonitrile and DW (55:45, v/v%). The column effluents were detected at 230 nm, and DTX concentrations were calculated using a linear calibration curve of the standard DTX solutions. The DTX loading capacity and efficiency of the prepared liposomes were calculated using the following equations:
Drug-loading capacity (wt %) = Weight of the drug in liposomes (mg)/Weight of the liposomes (mg) X 100 (Eq. 1)
Drug-loading efficiency (wt %) = Weight of the drug in liposomes (mg)/Weight of the drug initially added to the formulation (mg) X 100 (Eq. 2)
In vitro Cellular Uptake Study
The cellular uptake of liposomes by MDA-MB-231 and MCF-7 cells was evaluated by confocal microscopy and flow cytometry.12,13 To obtain confocal images, cells (4 × 105 cells/well) were seeded in each confocal dish, and after 24 h, treated with FITC-conjugated AE-Lipo (FITC-AE-Lipo) and FITC-conjugated Lipo (FITC-Lipo). After 4 h incubation, the cells were washed three times with cold PBS and incubated with Vectashield® mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA) for 15 min. The treated cells were covered with a confocal dish cover glass and analyzed under a confocal microscope (LSM 510 Meta, Carl Zeiss AG, Jena, Germany). For flow cytometry analysis, both cell lines (4 × 105 cells/well) were seeded in six-well plates. The next day, cells were treated with FITC-AE-Lipo and FITC-Lipo for 4 h. The cells were washed three times with cold PBS, harvested, and subjected to the fluorescence-activated cell sorting (FACS) analysis using FACSCalibur flow cytometer and Cell Quest Pro software (BD Biosciences, San Diego, CA, USA).46
In vitro Cytotoxicity Assay
MDA-MB-231 and MCF-7 cells (8 × 103 cells/well) were seeded in 96-well plates 24 h prior to the cytotoxicity test.12,13 Free DTX (in 0.1% DMSO aqueous solution47), DTX/Lipo, and DTX/AE-Lipo in RPMI 1640 medium were prepared immediately before use. The growth medium was removed from the 96-well plate and free DTX, DTX/Lipo, and DTX/AE-Lipo at different concentrations were added and incubated for 48 h. Cell cytotoxicity was assessed colorimetrically using the CCK-8, which contains a highly water-soluble tetrazolium salt that produces water-soluble formazan dye following reduction in the presence of electron mediators present in the viable cells. Briefly, 90 μL of fresh medium along with 10 μL of CCK-8 solution was added to each well, and the plates were incubated for an additional 2 h. The absorbance was read on the Flexstation 3 microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 450 nm.46
In vivo Fluorescence Imaging
In vivo studies were performed on four-week-old female nude mice (BALB/c, nu/nu mice; Nara Biotech, Seoul, South Korea). The mice were maintained according to the guidelines of the protocol approved by the Institutional Animal Care and Use Committee of Chung-Ang University of Korea, and all experiments were performed in accordance with the relevant laws and institutional guidelines (National Institutes of Health guidelines, “Principles of laboratory animal care”, approval number: 2019–00121). The uPAR-overexpressing MDA-MB-231 cells were inoculated into BALB/c nude mice by subcutaneous injection (5 × 105 cells per mouse) and Ce6-labeled liposomes (at a molar ratio of 1 of Ce6-conjugated DSPE-PEG in each liposome) were injected intravenously via the tail vein when the tumor volume reached 100 mm3. The fluorescence-labeled organism bioimaging instrument (FOBI) fluorescence live imaging system (NeoScience, Suwon, South Korea) was used to obtain fluorescent tumor images for the mice. The mice were euthanized 24 h post-injection, and organs were harvested for further analysis.44 The results were analyzed by Student’s t test at a significance level of p<0.01.
Results and Discussion
DSPE-PEG-AE147 Synthesis
To target uPAR-overexpressing tumors, AE147 peptide was conjugated to DSPE-PEG-NHS by a coupling reaction in the presence of TEA (Figure 2A).48,49 The imide bond of DSPE-PEG-NHS changed to the amide bond by reacting with the amine side group of the N-terminus Lys of the 13-mer peptide AE147. The conjugation of AE147 to DSPE-PEG-NHS was confirmed by the presence of a 1H nuclear magnetic resonance peak at δ 7.1, consistent with the benzene ring of AE147 (Figure 2B). In addition, the formation of a stable amide bond by coupling reaction was confirmed by FT-IR analysis (Figure 2C).33–35
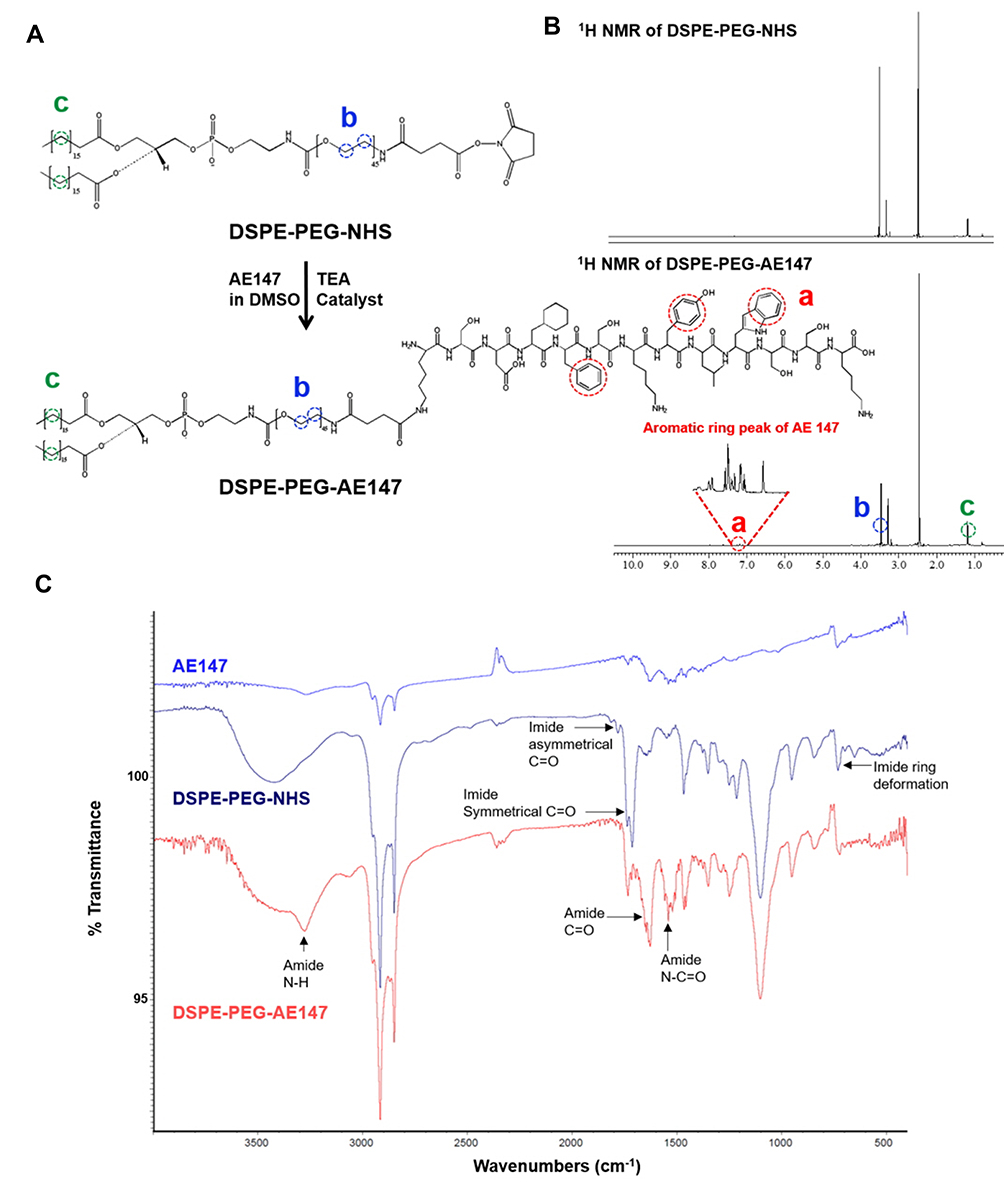 |
Figure 2 (A) Synthesis scheme of DSPE-PEG-AE147. (B) 1H NMR spectra of DSPE-PEG-NHS and DSPE-PEG-AE147. (C) FT-IR spectra of AE147, DPSE-PEG-NHS, and DSPE-PEG-AE147. |
The spectra of DSPE-PEG-NHS and DSPE-PEG-AE147 indicated that the 1713.73 cm−1 (imide bond) peak shifted to 1628.90 cm−1 (amide bond) due to the coupling reaction.38 Also, the conjugation of Ce6 to DSPE-PEG-NH2 was analyzed by the presence of a 1H nuclear magnetic resonance peak at δ 5.1, consistent with the vinyl of Ce6 (shown in Supplementary Figure S1).
Preparation and Characterization of Liposomes and DTX-Loaded Liposomes
PEGylated liposomes, also called stealth liposomes, exhibit improved physicochemical properties and performance in drug delivery applications, such as an increased hydrophilicity, a decreased opsonization, and an increased circulatory half-life.23,24,27,28 For example, Doxil®, a well-known doxorubicin incorporating PEGylated liposome, has a prolonged half-life and improved antitumor efficacy.28,50 In the present study, the targeting moiety, AE147 peptide, was anchored on the surface of the PEGylated liposomal carrier to target uPAR-overexpressing tumors, such as MDA-MB 231 derived tumors.12,13
The properties of Lipo and AE-Lipo are listed in Table 2. The Lipo with HSPC:cholesterol:DSPE-PEG (at a molar ratio of 55:40:5) showed a particle size of 92.6 ± 0.9 nm with approximately 0.2 PDI. The size of the AE-Lipo was slightly bigger (107.7 ± 1.3 nm) than that of Lipo, but had a similar PDI value. The zeta potentials were approximately −9.9 and −14.6 mV for Lipo and AE-Lipo, respectively. The difference in both the size and zeta potential of Lipo and AE-Lipo may be attributed to the anchored AE147 peptide and increased hydration of liposomal particles. DTX was selected as the anticancer drug for assessing payload delivery ability of targeting AE-Lipo. DTX, one of the most effective anticancer drugs, is widely used to treat tumors in multiple locations, including the stomach, breasts, prostate, and lungs.40,41,45 DTX-loaded liposomes had a slightly increased particle size and PDI compared to the unloaded liposomes (Table 2). The drug-loading efficiency of Lipo and AE-Lipo formulations was 80% and 71%, respectively. Reduced DTX-loading efficiency of AE-Lipo could be due to the increased hydrophilicity imparted by the hydrophilic AE147 peptides on the surface of liposomes. The particle distribution of DTX/AE-Lipo was mono-distributed, and the morphology indicated regular spherical particles of approximately 70 nm-diameter with smooth surfaces (Figure 3). This result is consistent with the previous hypothesis that AE147 conjugation could increase the hydration of liposomes in the aqueous solutions and dehydration under the condition of the FE-SEM experiment, leading to a considerably smaller particle size.51,52 Furthermore, the formulated DTX/AE-Lipo showed high stability in aqueous conditions, without undergoing a change in particle size or precipitation for four weeks (shown in Supplementary Figure S2).
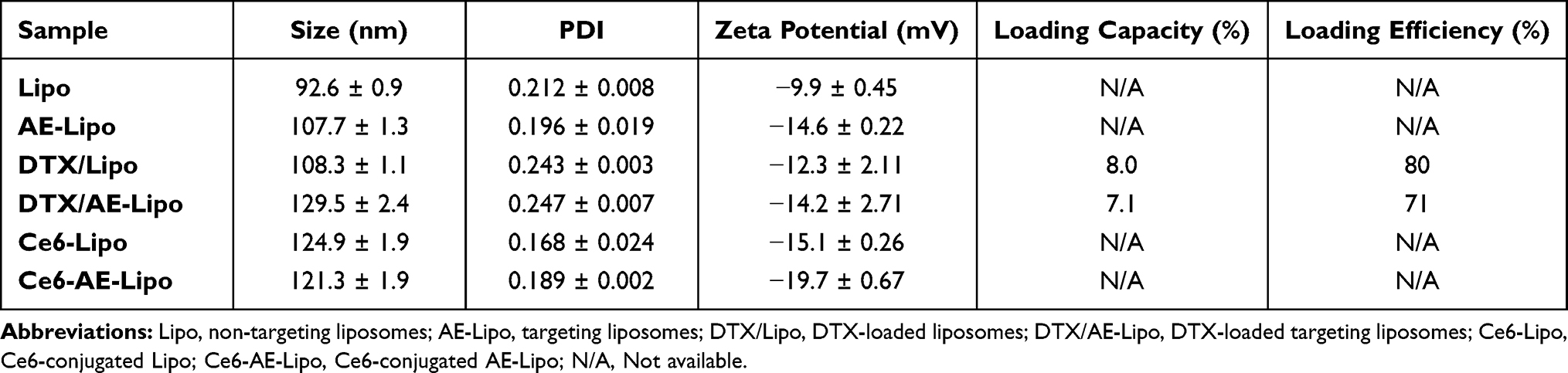 |
Table 2 Characterizations of All Liposome Formulations |
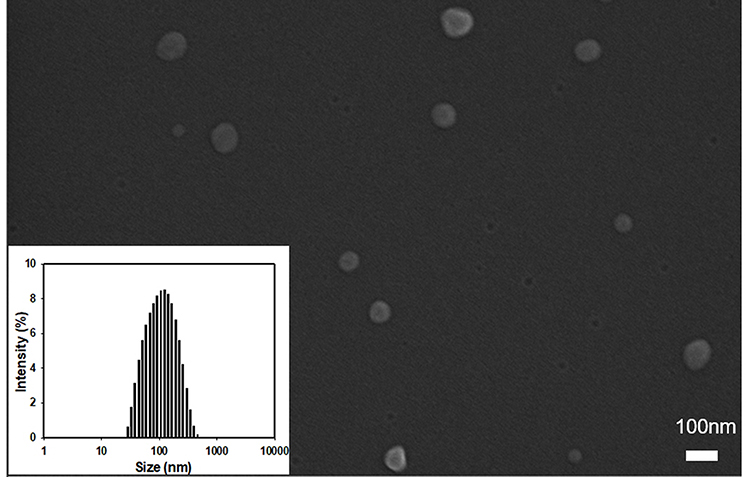 |
Figure 3 FE-SEM image and particle size distribution (see inset) of DTX/AE-Lipo. Abbreviation: DTX/AE-Lipo, DTX-loaded targeting liposomes. |
Cellular Uptake and in vitro Cytotoxicity of AE147-Conjugated Liposomes
The uPA-uPAR system plays a dominant role in tumorigenesis and metastasis and is overexpressed in metastatic tumor cells, such as breast cancer-derived MDA-MB-231 cells.8,53–56 This suggests that high uPAR expression on cancer cells can be a marker for metastatic tumors and used for targeted delivery of anticancer drugs to these cells. AE147 derived from a functionally identified uPA sequence binds to uPAR with a Kd of 16.4 nM.9 Targeting the cavity present in uPAR was suggested as a promising strategy for treating the tumors overexpressing uPAR. Moreover, the pathway of receptor-mediated endocytosis is more effective than the passive endocytosis for the intracellular uptake of liposomes.9,22,23 Thus, we attached AE147 on the liposomal surface via amide bond to enhance the efficacy of these liposomes as drug delivery vehicles.
Flow cytometry and confocal microscopy were used to determine the efficiency of the cellular delivery of AE147 conjugated liposomes to the MDA-MB-231 cells; MCF-7 cells, which do not express uPAR,13 were used as a control. Confocal fluorescence imaging showed no significant difference in FITC fluorescence intensity of MCF-7 cells treated with FITC-Lipo or FITC-AE-Lipo, while FITC fluorescence intensity of uPAR-overexpressing MDA-MB-231 cells treated with FITC-AE-Lipo was higher than FITC fluorescence intensity of cells incubated with FITC-Lipo (Figure 4A and B). FITC-AE-Lipo demonstrated a uniform distribution throughout cancer cells. Furthermore, the enhanced uptake of FITC-AE-Lipo was confirmed by flow cytometry analysis. Flow cytometry results demonstrated a similar uptake of FITC-Lipo and FITC-AE-Lipo by MCF-7 cells (Figure 4C). However, the cellular uptake of FITC-AE-Lipo in MDA-MB-231 cells was ~2.4 times higher than that of FITC-Lipo (Figure 4D). These results may be due to the AE147 moiety-conjugated delivery platform, which promotes cellular uptake via the ligand–receptor interaction in uPAR-overexpressing cancer cells. DTX-loaded liposomes of approximately 130 nm particle size (Table 2) were evaluated for their in vitro anticancer effect. AE147-conjugated liposomes are more likely to be taken up by the tumor cells via uPAR-mediated endocytosis, leading to enhanced anticancer activity.10,57,58
 |
Figure 4 Confocal microscopy images ((A) MCF-7 cell, (B) MDA-MB 231 cell) and flow cytometry analysis ((C) MCF-7 cell, (D) MDA-MB 231 cell) treated with FITC-Lipo or FITC-AE-Lipo for 4 h. Abbreviations: FITC-Lipo, FITC-conjugated Lipo; FITC-AE-Lipo, FITC-conjugated AE-Lipo. Note: *FL1-H indicates the relative intensity of fluorescence. |
The cytotoxicity of DTX/Lipo and DTX/AE-Lipo against MDA-MB-231 and MCF-7 cells was evaluated using a cell viability test. The cells were treated with equivalent concentrations of DTX, DTX/Lipo and DTX/AE-Lipo for 48 h and the viability of the treated cells was assessed using CCK-8. In MCF-7 cells, free DTX showed stronger anticancer activity than DTX/Lipo or DTX/AE-Lipo (shown in Supplementary Figure S3). However, in uPAR-overexpressing MDA-MB-231 cells, DTX/AE-Lipo (IC50 4.61 µg/mL) achieved better anticancer activity than free DTX (IC50 7.18 µg/mL) or DTX/Lipo (IC50 8.59 µg/mL), indicating that the addition of the targeting moiety AE147 could improve the therapeutic efficacy of the drug against uPAR-overexpressed tumors (Figure 5, and shown in Supplementary Table S1).
 |
Figure 5 Cell viability of MDA-MB-231 cells treated with free DTX (1 mg/mL DTX solution in DMSO), DTX/Lipo, and DTX/AE-Lipo when exposed to a series of equivalent concentrations of DTX for 48 h. Abbreviations: DTX/Lipo, DTX-loaded liposomes; DTX/AE-Lipo, DTX-loaded targeting liposomes. |
Collectively, these results indicate that AE147 conjugation to nanoparticles, such as liposomes, may be an effective strategy to improve the delivery of anticancer agents in uPAR-overexpressing tumors, especially those associated with metastatic cancer. Furthermore, this approach of active targeting via ligand–receptor mediation can be used in diagnostic applications, such as the detection of circulating tumor cells and metastasis.9,11,13,59
In vivo Imaging Analysis
To demonstrate tumor-targeting via the AE147-uPAR interaction, the time-dependent biodistribution of the prepared liposomes was evaluated in an MDA-MB-231 xenograft mouse model. Free Ce6, Ce6-labeled Lipo (Ce6-Lipo), and Ce6-labeled AE-Lipo (Ce6-AE-Lipo) were intravenously injected into uPAR-overexpressing, MDA-MB-231 cells-derived tumor bearing, nude mice. Ce6 is one of the most widely used second-generation photosensitizers in photodynamic therapy with high efficacy and low dark toxicity.60,61 As shown in Figure 6, Ce6-Lipo and Ce6-AE-Lipo gradually increased the fluorescence intensity at the tumor site, compared to the free Ce6. The PEGylated liposome has longer circulation time in the blood; moreover, these liposomes avoid the immune system due to the slightly negative charge and reduced degree of opsonization imparted by PEG62–64 and can accumulate at tumor sites due to the EPR effect.20,27,40 In addition, to determine the targeting ability of AE147, nude mice were euthanized, and the main organs as well as tumors were excised 24 h post-injection (Figure 7). The absolute fluorescence intensity of Ce6 of each system was remarkably higher in the liver compared to that in the other organs (shown in Supplementary Figure S4). However, when the intensity was normalized by the organ area, the relative fluorescence intensity of tumors treated with Ce6-AE-Lipo was 3.50 and 2.22 times higher than those treated with free Ce6 and Ce6-Lipo, respectively. This indicated that AE147-conjugated liposomes were more efficiently transported to the tumor site due to active targeting.
 |
Figure 6 Non-invasive in vivo fluorescent imaging of free Ce6, Ce6-Lipo, and Ce6-AE-Lipo after i.v. injection via the tail vein of MDA-MB-231 cells-derived tumor-bearing Balb/c nude mice. Whole body imaging at predetermined time points (0, 1, 3, 6, 12, and 24 h) after i.v. injection at the 24 h time point. Abbreviations: Ce6-Lipo, Ce6-conjugated Lipo; Ce6-AE-Lipo, Ce6-conjugated AE-Lipo; i.v. intravenous. |
 |
Figure 7 (A) Ex vivo optical and fluorescent imaging of (i) heart, (ii) lungs, (iii) kidneys, (iv) spleen, (v) liver, and (vi) tumor tissues obtained 24 h post-injection of liposomes (Free Ce6, Ce6-Lipo, and Ce6-AE-Lipo). (B) Quantitative FI of tumors and main organs (n = 3). (C) Relative FI of the tumor to liver. Abbreviations: Ce6-AE-Lipo, Ce6-conjugated AE-Lipo; Ce6-Lipo, Ce6-conjugated Lipo; FI, fluorescence intensity. Notes: **p < 0.01, Student’s t-test, Ce6-AE-Lipo versus Ce6-Lipo in the tumor. |
Conclusion
In this study, AE147-conjugated liposomes were prepared and evaluated for their potential as a drug delivery nano-platform to actively target uPAR-overexpressing metastatic tumors. HSPC, cholesterol, and DSPE-PEG at a molar ratio of 55:40:5 were used to improve the DTX-loading capacity and liposomal stability. The AE147 moiety was conjugated to the liposomes to enhance their targeting efficacy. The hydrophobic drug DTX was successfully loaded within the bilayer lipid membrane of liposomes with an average size of approximately 130 nm. Compared to the free DTX, DTX/AE Lipo demonstrated improved anticancer effects by showing a 64% lower IC50 value (4.61 µg/mL) against uPAR-overexpressing MDA-MB-231 cancer cells. Also, the AE147-conjugated liposomes showed improved tumor-targeting ability. Based on these findings, the AE147-conjugated liposomes are a potential delivery nano-platform for the treatment of metastatic tumors.
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2015R1A5A1008958 and No. 2021R1A2C2008519).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organization. Latest global cancer data: cancer burden rises to 18.1 million new cases and 9.6 million cancer deaths in 2018. International agency for research on cancer. Geneva: World Health Organization; 2018.
2. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi:10.1038/nm1469
3. Geho DH, Bandle RW, Clair T, Liotta LA. Physiological mechanisms of tumor-cell invasion and migration. Physiology. 2005;20(3):194–200. doi:10.1152/physiol.00009.2005
4. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi:10.1016/S0092-8674(00)81683-9
5. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4(6):448–456. doi:10.1038/nrc1370
6. Hynes RO. Metastatic potential: generic predisposition of the primary tumor or rare, metastatic variants—or both? Cell. 2003;113(7):821–823. doi:10.1016/S0092-8674(03)00468-9
7. Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25(1):9–34.
8. Montuori N, Pesapane A, Rossi FW, et al. Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Translat Med. 2016;15:15.
9. Llinas P, Helene Le Du M, Gårdsvoll H, et al. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005;24(9):1655–1663. doi:10.1038/sj.emboj.7600635
10. Wang M, Miller AD, Thanou M. Effect of surface charge and ligand organization on the specific cell-uptake of uPAR-targeted nanoparticles. J Drug Target. 2013;21(7):684–692. doi:10.3109/1061186X.2013.805336
11. Mekkawy AH, Pourgholami MH, Morris DL. Involvement of urokinase-type plasminogen activator system in cancer: an overview. Med Res Rev. 2014;34(5):918–956.
12. Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling*. J Biol Chem. 2009;284(34):22825–22833. doi:10.1074/jbc.M109.023960
13. Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev. 2008;34(2):122–136. doi:10.1016/j.ctrv.2007.10.005
14. Magdolen V, Rettenberger P, Koppitz M, et al. Systematic mutational analysis of the receptor‐binding region of the human urokinase‐type plasminogen activator. Eur J Biochem. 1996;237(3):743–751. doi:10.1111/j.1432-1033.1996.0743p.x
15. Ploug M. Identification of specific sites involved in ligand binding by photoaffinity labeling of the receptor for the urokinase-type plasminogen activator. Residues located at equivalent positions in uPAR domains I and III participate in the assembly of a composite ligand-binding site. Biochemistry. 1998;37(47):16494–16505.
16. Li D, Liu S, Shan H, Conti P, Li Z. Urokinase plasminogen activator receptor (uPAR) Targeted nuclear imaging and radionuclide therapy. Rev Theranostics. 2013;3(7):507–515. doi:10.7150/thno.5557
17. Li Santi A, Napolitano F, Montuori N, Ragno P. The urokinase receptor: a multifunctional receptor in cancer cell biology. therapeutic implications. Int J Mol Sci. 2021;22:8. doi:10.3390/ijms22084111
18. LeBeau AM, Duriseti S, Murphy ST, et al. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer Res. 2013;73(7):2070–2081. doi:10.1158/0008-5472.CAN-12-3526
19. Yuan C, Guo Z, Yu S, Jiang L, Huang M. Development of inhibitors for uPAR: blocking the interaction of uPAR with its partners. Drug Discov Today. 2021;26(4):1076–1085. doi:10.1016/j.drudis.2021.01.016
20. Sadzuka Y, Sugiyama I, Tsuruda T, Sonobe T. Characterization and cytotoxicity of mixed polyethyleneglycol modified liposomes containing doxorubicin. Int J Pharm. 2006;312(1–2):83–89. doi:10.1016/j.ijpharm.2005.12.043
21. Hua S, Wu SY. The use of lipid-based nanocarriers for targeted pain therapies. Front Pharmacol. 2013;4:143. doi:10.3389/fphar.2013.00143
22. Patil Y, Amitay Y, Ohana P, Shmeeda H, Gabizon A. Targeting of pegylated liposomal mitomycin-C prodrug to the folate receptor of cancer cells: intracellular activation and enhanced cytotoxicity. J Controlled Release. 2016;225:87–95. doi:10.1016/j.jconrel.2016.01.039
23. Nogueira E, Gomes AC, Preto A, Cavaco-Paulo A. Design of liposomal formulations for cell targeting. Colloids Surf B Biointerfaces. 2015;136:514–526. doi:10.1016/j.colsurfb.2015.09.034
24. Muthu MS, Kulkarni SA, Xiong J, Feng -S-S. Vitamin E TPGS coated liposomes enhanced cellular uptake and cytotoxicity of docetaxel in brain cancer cells. Int J Pharm. 2011;421(2):332–340. doi:10.1016/j.ijpharm.2011.09.045
25. Singh S. Liposome encapsulation of doxorubicin and celecoxib in combination inhibits progression of human skin cancer cells. Int J Nanomedicine. 2018;13:11. doi:10.2147/IJN.S124701
26. Metselaar JM, Storm G. Liposomes in the treatment of inflammatory disorders. Expert Opin Drug Deliv. 2005;2(3):465–476. doi:10.1517/17425247.2.3.465
27. Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of pegylation. Drug Deliv. 2006;13(6):399–409. doi:10.1080/10717540600814402
28. Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–436. doi:10.2165/00003088-200342050-00002
29. Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front Pharmacol. 2015;6:286. doi:10.3389/fphar.2015.00286
30. Sahoo SK, Labhasetwar V. Nanotech approaches to drug delivery and imaging. Drug Discov Today. 2003;8(24):1112–1120. doi:10.1016/S1359-6446(03)02903-9
31. Monteiro N, Martins A, Reis RL, Neves NM. Liposomes in tissue engineering and regenerative medicine. J R Soc Interface. 2014;11(101):20140459. doi:10.1098/rsif.2014.0459
32. Wu J, Liu Q, Lee RJ. A folate receptor-targeted liposomal formulation for paclitaxel. Int J Pharm. 2006;316(1–2):148–153. doi:10.1016/j.ijpharm.2006.02.027
33. Wang B, Cao Z. Acid-catalyzed reactions of twisted amides in water solution: competition between hydration and hydrolysis. Chemistry. 2011;17(42):11919–11929. doi:10.1002/chem.201101274
34. Kemnitz CR, Loewen MJ. “Amide resonance” correlates with a breadth of C−N rotation barriers. J Am Chem Soc. 2007;129(9):2521–2528. doi:10.1021/ja0663024
35. Mujika JI, Mercero JM, Lopez X. Water-promoted hydrolysis of a highly twisted amide: rate acceleration caused by the twist of the amide bond. J Am Chem Soc. 2005;127(12):4445–4453. doi:10.1021/ja044873v
36. Mustjoki S, Sidenius N, Sier CFM, et al. Soluble urokinase receptor levels correlate with number of circulating tumor cells in acute myeloid leukemia and decrease rapidly during chemotherapy. Cancer Res. 2000;60(24):7126.
37. Llinas P, Hélène Le Du M, Gårdsvoll H. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005;24(9):1655–1663.
38. Saedi H. Intercalated polyamide-imide nanocomposite with montmorillonite. Am J Polymer Sci. 2015;5(2):47–53.
39. Zhang Q, Zhao J, Hu H, et al. Construction and in vitro and in vivo evaluation of folic acid-modified nanostructured lipid carriers loaded with paclitaxel and chlorin e6. Int J Pharm. 2019;569:118595. doi:10.1016/j.ijpharm.2019.118595
40. Qu M-H, Zeng R-F, Fang S, Dai Q-S, Li H-P, Long J-T. Liposome-based co-delivery of siRNA and docetaxel for the synergistic treatment of lung cancer. Int J Pharm. 2014;474(1–2):112–122. doi:10.1016/j.ijpharm.2014.08.019
41. Immordino ML, Brusa P, Arpicco S, Stella B, Dosio F, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Controlled Release. 2003;91(3):417–429. doi:10.1016/S0168-3659(03)00271-2
42. Yang -Z-Z, Li J-Q, Wang -Z-Z, Dong D-W, Qi X-R. Tumor-targeting dual peptides-modified cationic liposomes for delivery of siRNA and docetaxel to gliomas. Biomaterials. 2014;35(19):5226–5239. doi:10.1016/j.biomaterials.2014.03.017
43. Rautela A, Rani J, Debnath M. Green synthesis of silver nanoparticles from Tectona grandis seeds extract: characterization and mechanism of antimicrobial action on different microorganisms. J Analytical Sci Tech. 2019;10(1):5. doi:10.1186/s40543-018-0163-z
44. Lim C, Moon J, Sim T, et al. Cyclic RGD-conjugated Pluronic® blending system for active, targeted drug delivery. Int J Nanomedicine. 2018;13:4627. doi:10.2147/IJN.S171794
45. Chang M, Lu S, Zhang F, et al. RGD-modified pH-sensitive liposomes for docetaxel tumor targeting. Colloids Surf B Biointerfaces. 2015;129:175–182. doi:10.1016/j.colsurfb.2015.03.046
46. Lim C, Kang JK, Won WR, et al. Co-delivery of D-(KLAKLAK) 2 peptide and chlorin e6 using a liposomal complex for synergistic cancer therapy. Pharmaceutics. 2019;11(6):293. doi:10.3390/pharmaceutics11060293
47. Nguyen ST, Nguyen HT-L, Truong KD. Comparative cytotoxic effects of methanol, ethanol and DMSO on human cancer cell lines. Biomed Res Therapy. 2020;7(7):3855–3859. doi:10.15419/bmrat.v7i7.614
48. Wang J-L, Liu Y-L, Li Y, et al. EphA2 targeted doxorubicin stealth liposomes as a therapy system for choroidal neovascularization in rats. Invest Ophthalmol Vis Sci. 2012;53(11):7348–7357. doi:10.1167/iovs.12-9955
49. Zhang N, Li C, Zhou D, et al. Cyclic RGD functionalized liposomes encapsulating urokinase for thrombolysis. Acta Biomaterialia. 2018;70:227–236. doi:10.1016/j.actbio.2018.01.038
50. Sharpe M, Easthope SE, Keating GM, Lamb HM. Polyethylene glycol-liposomal doxorubicin. Drugs. 2002;62(14):2089–2126. doi:10.2165/00003495-200262140-00012
51. Hammond N. Retraction: the next generation cell-penetrating peptide and carbon dot conjugated nano-liposome for transdermal delivery of curcumin. Biomaterials Sci. 2018;7(1):442. doi:10.1039/C8BM90064G
52. Wang Y, Su W, Li Q, et al. Preparation and evaluation of lidocaine hydrochloride-loaded TAT-conjugated polymeric liposomes for transdermal delivery. Int J Pharm. 2013;441(1–2):748–756. doi:10.1016/j.ijpharm.2012.10.019
53. Gårdsvoll H, Jacobsen B, Kriegbaum MC, et al. Conformational regulation of urokinase receptor function: IMPACT OF RECEPTOR OCCUPANCY AND EPITOPE-MAPPED MONOCLONAL ANTIBODIES ON LAMELLIPODIA INDUCTION. J Biol Chem. 2011;286(38):33544–33556. doi:10.1074/jbc.M111.220087
54. Mekkawy AH, Morris DL, Pourgholami MH. Urokinase plasminogen activator system as a potential target for cancer therapy. Future Oncol. 2009;5(9):1487–1499. doi:10.2217/fon.09.108
55. Zhang Y, Kenny HA, Swindell EP, et al. Urokinase plasminogen activator system–targeted delivery of nanobins as a novel ovarian cancer therapy. Mol Cancer Ther. 2013;12(12):2628–2639. doi:10.1158/1535-7163.MCT-13-0204
56. Bifulco K, Longanesi-Cattani I, Liguori E, et al. A Urokinase receptor–derived peptide inhibiting VEGF-dependent directional migration and vascular sprouting. Mol Cancer Ther. 2013;12(10):1981–1993. doi:10.1158/1535-7163.MCT-13-0077
57. Mahmood N, Mihalcioiu C, Rabbani SA. Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): diagnostic, prognostic, and therapeutic applications. Front Oncol. 2018;8:24. doi:10.3389/fonc.2018.00024
58. Su S-C, Lin C-W, Yang W-E, Fan W-L, Yang S-F. The urokinase-type plasminogen activator (uPA) system as a biomarker and therapeutic target in human malignancies. Expert Opin Ther Targets. 2016;20(5):551–566. doi:10.1517/14728222.2016.1113260
59. Li Y, Cozzi P. Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev. 2007;33(6):521–527. doi:10.1016/j.ctrv.2007.06.003
60. Martynenko IV, Kuznetsova VA, Orlova АO, et al. Chlorin e6–ZnSe/ZnS quantum dots based system as reagent for photodynamic therapy. Nanotechnology. 2015;26(5):055102. doi:10.1088/0957-4484/26/5/055102
61. Du D, Wang K, Wen Y, Li Y, Li YY. Photodynamic graphene quantum dot: reduction condition regulated photoactivity and size dependent efficacy. ACS Appl Mater Interfaces. 2016;8(5):3287–3294. doi:10.1021/acsami.5b11154
62. Lim C, Sim T, Hoang NH, et al. A charge-reversible nanocarrier using PEG-PLL (-g-Ce6, DMA)-PLA for photodynamic therapy. Int J Nanomedicine. 2017;12:6185. doi:10.2147/IJN.S142912
63. He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. doi:10.1016/j.biomaterials.2010.01.065
64. Zhu S, Hong M, Zhang L, Tang G, Jiang Y, Pei Y. PEGylated PAMAM dendrimer-doxorubicin conjugates: in vitro evaluation and in vivo tumor accumulation. Pharm Res. 2010;27(1):161–174. doi:10.1007/s11095-009-9992-1
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.