Back to Journals » International Journal of Nanomedicine » Volume 16
Development of a Solid Supersaturable Micelle of Revaprazan for Improved Dissolution and Oral Bioavailability Using Box-Behnken Design
Authors Goo YT, Sa CK ![]() , Choi JY, Kim MS, Kim CH, Kim HK, Choi YW
, Choi JY, Kim MS, Kim CH, Kim HK, Choi YW ![]()
Received 23 December 2020
Accepted for publication 23 January 2021
Published 17 February 2021 Volume 2021:16 Pages 1245—1259
DOI https://doi.org/10.2147/IJN.S298450
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Yoon Tae Goo,1,* Cheol-Ki Sa,1,* Ji Yeh Choi,2 Min Song Kim,1 Chang Hyun Kim,1 Hyeon Kyun Kim,1 Young Wook Choi1
1College of Pharmacy, Chung-Ang University, Seoul, Republic of Korea; 2Department of Psychology, York University, Toronto, Ontario, Canada
*These authors contributed equally to this work
Correspondence: Young Wook Choi
College of Pharmacy, Chung-Ang University, 84 Heuksuk-Ro, Dongjak-Gu, Seoul, 06974, Korea
Tel +82 2 820 5609
Email [email protected]
Purpose: To enhance the oral bioavailability of revaprazan (RVP), a novel solid, supersaturable micelle (SSuM) was developed.
Methods: Surfactants and solid carriers were screened based on a solubility and a flowability test, respectively. Supersaturating agents, including Poloxamer 407 (P407), were screened. The SSuM was optimized using a Box-Behnken design with three independent variables, including Gelucire 44/14:Brij L4 (G44/BL4; X1) and the amounts of Florite PS-10 (FLO; X2) and Vivapur 105 (VP105; X3), and three response variables, ie, dissolution efficiency at 30 min (Y1), dissolution enhancing capacity (Y2), and Carr’s index (Y3). The solid state property was evaluated, and a dissolution test was conducted. RVP, Revanex®, solid micelle (P407-free from the composition of SSuM), and SSuM were orally administrated to rats (RVP 20 mg equivalent/kg) for in vivo pharmacokinetic study.
Results: G44 and BL4 showed great solubility, with a critical micelle concentration range of 119.2– 333.0 μg/mL. P407 had an excellent supersaturating effect. FLO and VP105 were selected as solid carriers, with a critical solidifying ratio (g/mL) of 0.30 and 0.91, respectively. With optimized values of X1 (– 0.41), X2 (0.31), and X3 (– 0.78), RVP (200 mg)-containing SSuM consisting of G44 (253.8 mg), BL4 (106.2 mg), FLO (99.3 mg), VP105 (199.8 mg), and P407 (40 mg) was developed, resulting in Y1 (40.3%), Y2 (0.008), and Y3 (12.3%). RVP existed in an amorphous state in the optimized SSuM, and the SSuM formed a nanosized dispersion in the aqueous phase, with approximately 71.7% dissolution at 2 h. The optimized SSuM improved the relative bioavailability of RVP in rats by approximately 478%, 276%, and 161% compared to raw RVP, Revanex®, and solid micelle, respectively.
Conclusion: The optimized SSuM has great potential for the development of solidified formulations of poorly water-soluble drugs with improved oral absorption.
Keywords: revaprazan, supersaturation, solid micelle, Box-Behnken design, dissolution, oral bioavailability
Introduction
Revaprazan (RVP), a new acid-suppressive agent, reversibly inhibits H+/K+-ATPase.1 Because of its prolonged action as an alternative proton pump inhibitor, RVP has been recently adopted for the treatment of gastroesophageal reflux disease and gastric ulcer.2 After absorption in the gastrointestinal (GI) tract, RVP accumulates in the canaliculi of parietal cells.2 In the acidic environment around the parietal cells, RVP activates and exerts its pharmacological activity. RVP shows linear pharmacokinetics after oral administration at doses of 2–30 mg/kg and increases the intragastric pH in a dose-dependent manner.3,4 RVP, classified as a class II drug according to the biopharmaceutical classification system (BCS), has limited water solubility and therefore low oral bioavailability (BA).5,6 To improve the solubility and oral BA of RVP, various approaches, such as solid dispersion, microspheres, and self-microemulsifying drug delivery systems, have been used.7–9
Lipid-based formulations have been spotlighted as a means to enhance the dissolution and oral absorption of poorly water-soluble drugs.10 Among them, micelles have been recognized as one of the most promising drug delivery systems.11 In the aqueous phase, amphipathic surfactants spontaneously assemble at concentrations above the critical micelle concentration (CMC), forming micellar structures that can accommodate hydrophobic molecules in their cores. Thus, micelles can enhance the dissolution and oral absorption of poorly water-soluble drugs.12 Further solubilization of poorly soluble drugs can be achieved by incorporating additional strategies, such as supersaturation.13
Supersaturating agents render water-insoluble drugs soluble at concentrations above equilibrium solubility, without precipitation.14 Achieving an increased drug concentration in the supersaturated state in the intestinal lumen would enhance absorption. A number of hydrophilic supersaturating agents, including polyvinylpyrrolidones and poloxamers, have been introduced as precipitation inhibitors.15 These supersaturating agents have been verified to kinetically and/or thermodynamically prolong the supersaturated state by inhibiting crystal growth or altering the microclimate conditions, where drug molecules are enveloped with an aqueous phase.16 Theoretically, optimal formulations should maintain the supersaturated state for a sufficient period of time to allow enhanced absorption. Silybin-loaded, supersaturable, polymeric micelle using Soluplus and Copovidone showed 4.5-fold increased oral BA compared to free silybin in rats.17
Most conventional micellar formulations are in the liquid form and have limitations of drug leakage and capsule aging.18,19 Recently, solid carriers have been introduced to generate solidified micelles, in which water-insoluble drug molecules exist in an amorphous state, resulting in a high dissolution rate.20 After reaching the GI tract, solid micelles release the micellar components that form micellar structures due to peristalsis of the GI tract thus enhancing the absorption of poorly water-soluble drugs.20,21 When developing a solidified micelle, various variables, such as micelle composition and mass of the solid carrier, have to be optimized. The response surface methodology, which combines mathematical and statistical approaches to suggest optimized response variables, has been used to optimize pharmaceutical formulations.22,23 In particular, the Box-Behnken design is a well-known experimental method for formulation optimization because it efficiently suggests an accurate response with fewer combinations.24
In the present study, we aimed to develop a novel RVP-loaded solid supersaturable micelle (SSuM). To this end, RVP-dissolved supersaturable micelle (SuM), composed of surfactant mixtures, was solidified with selected solid carriers. A three-level Box-Behnken design was employed to optimize the composition, using three independent variables, ie, the ratio of surfactants and the amounts of silica-based adsorbent and cellulose-based diluent. Optimized SSuM revealed excellent characteristics in terms of flowability, dissolution, and oral BA in rats. Thus, we expect that this SSuM system would be useful for practical development of a solid dosage form with enhanced dissolution of poorly soluble drugs.
Materials and Methods
Materials
RVP powder (purity >99%) and Revanex® tablets containing 200 mg RVP were supplied by Yuhan (Seoul, Korea). Aerosil® 200 was supplied by Evonik Degussa GmbH (Frankfurt am Main, Germany). Avicel PH 101, Brij L4 (BL4), Tween® 20, and Tween® 80 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Florite PS-10 (FLO; calcium silicate) was purchased from Tomita Pharmaceutical Co., Ltd. (Tokushima, Japan). Gelucire 44/14 (G44), Labrafil® M 1944 CS, Labrasol®, and Lauroglycol® 90 were gifted by Gattefossé (Saint Priest, France). Kolliphor® EL, Kolliphor® RH40, Kolliphor® RH60, Kollidon K30, Kollidon K90, Poloxamer (P)188, and P407 were obtained from BASF (Ludwigshafen, Germany). Low-substituted hydroxypropyl cellulose B1 (L-HPC) was supplied by Shin-Etsu Chemical Co., Ltd. (Tokyo, Japan). Neusilin® US2 (magnesium aluminometasilicate) was supplied by Fuji Chemical Industry Company (Toyama, Japan). Sylysia® 350 (porous silica) was supplied by Fuji Silysia Chemical Co., Ltd. (Aichi, Japan). Vivapur (VP)12 and VP105 were supplied by JRS Pharma (Rosenberg, Germany). High-performance liquid chromatography (HPLC)-grade methanol was purchased from JT Baker (Phillipsburg, NJ, USA). All other chemicals used were of analytical grade.
Animals
Sprague-Dawley rats (Male, 200–250 g, 7–9 weeks) were purchased from Orient Bio (Gyeonggi-do, Korea). Prior to the experiments, all rats were allowed to adapt to our legal animal care facility for one week. All rats were fasted overnight for 12–18 h before drug administration, while they freely accessed to water. All animal trials were carried out in compliance with the National Institutes of Health guidelines on the care and use of laboratory animals and were permitted by the Institutional Animal Care and Use Committee of Chung-Ang University (protocol no. 2020–00062), Seoul, Korea.
Solubility Test
The equilibrium solubility of RVP in various vehicles was measured to screen a suitable surfactant. Briefly, an excess of RVP was added into 1 mL of 1% surfactant solutions. Each solution was shaken on a mechanical shaker (CM-1000; EYELA, Tokyo, Japan) at 25°C for 24 h. After reaching equilibrium, the mixtures were centrifuged (Smart R17; Hanil Science Industrial, Incheon, Korea) at 12,000 ×g for 10 min to exclude undissolved RVP. The supernatant was filtered through a 0.45-μm nylon membrane filter (Whatman International, Kent, UK) and appropriately diluted with methanol. Each sample was analyzed by HPLC to determine the concentration of RVP. To determine the CMC, aqueous solutions of G44, BL4, and a G44/BL4 mixture (50:50 [w/w]) were prepared and serially diluted. Then, the solubility of RVP at each concentration was measured as described above.
HPLC Analysis of RVP
RVP was quantified by HPLC as previously reported.6 Twenty microliters of each sample was injected into the HPLC system (e2695; Waters, Milford, MA, USA) consisting of a pump (W2690/5; Waters), an ultraviolet detector (W2489; Waters), and a data station (Empower 3; Waters). An isocratic mobile phase consisting of 93% of methanol in distilled water and a C18 column (150 × 4.6 mm, 5 μm; Agilent Technologies, Santa Clara, CA, USA) was used for chromatographic separation. The flow rate was 1.0 mL/min and the column temperature was set to 35°C, then RVP detection was achieved at 270 nm.
Preparation of RVP-Loaded SuM and SSuM
Based on a previous report,20 drug-free micelles were prepared by mixing G44 and BL4 at 60°C. Various supersaturating agents (10%, w/w) were added to the micelles and homogeneously mixed to prepare SuM, in which RVP (200 mg) was dissolved. To prepare SSuM, RVP-loaded SuM was adsorbed into the powder blend of FLO and VP105, and thoroughly kneaded. Then, the paste was granulated using a high-speed kneading granulator (Triple Master TMGV-1; Shinagawa Machinery Works, Nara, Japan) at a revolution speed of 40 rpm for 20 min, and the granules were passed through a 1000 μm mesh sieve. To avoid unintentional water involvement during granulation process, the obtained fine granules were dried in an oven at 60°C for 12 h (no weight loss was observed).
In vitro Dissolution Study
According to the USP apparatus II (paddle) method, dissolution tests for each sample were performed using a Vision Classic 6TM dissolution tester and a Vision heater. Each formulation containing 200 mg RVP equivalent was dispersed in 900 mL of distilled water at 37 ± 0.5°C and stirred at 50 rpm. Five milliliters of sample were taken at predetermined time points (5, 15, 30, 60, 90, and 120 min) and filtered through a 0.45 μm nylon membrane filter. An equivalent volume of fresh medium was added to maintain a constant dissolution environment. The filtrates were appropriately diluted with mobile phase, and the amount of dissolved RVP was quantified by HPLC as described above. To compare the dissolution behavior between formulations, the dissolution efficiency (DE) and dissolution enhancing capacity (DEC) were calculated using the trapezoidal rule as follows: DE (%) = 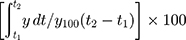 , where y is the percentage of dissolved product; DEC = (DESSuM-DERVP)/WSSuM, where DESSuM and DERVP are the DE of RVP-loaded SSuM and raw RVP, respectively, and WSSuM is the amount (mg) of SSuM.25
, where y is the percentage of dissolved product; DEC = (DESSuM-DERVP)/WSSuM, where DESSuM and DERVP are the DE of RVP-loaded SSuM and raw RVP, respectively, and WSSuM is the amount (mg) of SSuM.25
Screening of Solid Carriers
To obtain SSuM, two types of solid carriers were screened at different weight ratios: Aerosil® 200, Neusilin® US2, Sylysia® 350, and FLO as silica-based adsorbents (0.25–0.5 weight ratio to SuM); VP12, VP105, L-HPC, and Avicel PH 101 as cellulose-based diluents (0.77–2.0 weight ratio to SuM). Solidifying behavior was determined by the levigation method as previously reported.26,27 Briefly, solid carriers were added and mixed with a fixed amount of drug-free SuM (G44/BL4 mixture, 50:50 [w/w]) in a mortar, and blending was discontinued when a non-flowing clot was formed. The physical property of the solidified products was evaluated in terms of flow behavior.
Determination of Flow Behavior
The flow property of the solidified mass was evaluated using Carr’s index (CI), which was calculated using the following equation: CI (%) = [ρ(tapped)-ρ(bulk)]·100/ρ(tapped), where ρ(tapped) and ρ(bulk) refer to the tapped and bulk densities, respectively. Apparent bulk and tapped bulk densities were measured using the cylinder method. Briefly, accurately weighed granules were poured into a cylinder and the volume was read to calculate the apparent bulk density. Another sample was tapped 100 times for tapped bulk density using a powder tester (ABD-100; Tsutsui Scientific Instruments, Tokyo, Japan). The critical solidifying ratio (CSR; g/mL), defined by the minimum required mass (g) of solid carrier to solidify a 1 mL of drug-free SuM, was obtained using a critical constant value of CI.28
Optimization of RVP-Loaded SSuM Using a Box-Behnken Design
To optimize the composition of the SSuM, the Box-Behnken design was employed. Three components were included as independent variables: the ratio of surfactants (G44/BL4; X1) and the amounts of FLO (X2) and VP105 (X3), which were set within ranges of 1:0–0:1, 60–120 mg, and 180–360 mg, respectively. To obtain the optimized formulation with high desirability function, three response variables including DE at 30 min (DE30; Y1), DEC (Y2), and CI (Y3) were adopted. The design consisted of 15 experimental points to find a model fit, and mathematical correlations between inputs and outputs were evaluated using Minitab software (ver. 18.0; Minitab, State College, PA, USA).
Droplet Size Measurement
The droplet size of SSuM dispersed in distilled water was measured using a dynamic light-scattering particle size analyzer (Zetasizer Nano-ZS; Malvern Instruments, Worcestershire, UK). SSuM containing equivalent to 200 mg RVP was accurately weighted and dispersed in 250 mL of distilled water and gently vortexed to obtain a homogeneous dispersion. The mixture was centrifuged at 14,000 ×g for 5 min to completely remove undissolved solid carriers. Then, all samples were transferred to a transparent cuvette, placed in a thermostatic chamber, and light scattering was monitored with a 50-mV laser at an angle of 90° at 25°C.
Solid-State Property Assessment
The solid-state properties of free RVP, FLO, VP105, a physical mixture (PM) of RVP and solid carriers, and optimized SSuM were inquired using scanning electron microscopy (SEM), differential scanning calorimetry (DSC), and powder X-ray diffractometry (PXRD). Morphological features of each sample were visualized using a scanning electron microscope (Sigma 300; Carl Zeiss Meditec AG, Jena, Germany). The samples were put on a brass disc using double-sided adhesive carbon tape. Employing a Hitachi ion sputter (E-1030), each sample was coated with platinum under vacuum for 120 s at a current of 4 mA. The samples were scanned at 5 kV. The thermal characteristics of each powder were evaluated using a DSC-Q20 calorimeter (TA Instruments, New Castle, DE, USA). After sealing in an aluminum pan, each sample (2–3 mg) was subjected to heating in the range of 50°C to 260°C at a rate of 5°C/min under a constant nitrogen flow at 25 mL/min. PXRD patterns of the samples were determined using an X-ray diffractometer (D8 Advance; Bruker, Mannheim, Germany) with nickel-filtered Cu Kα radiation. The diffractogram was acquired by scanning at the 2θ range of 5–60º, with a step angle of 0.02º and scanning speed of 5º/min.
In vivo Oral Absorption Study
Oral Administration and Plasma Sampling
Rats were randomly 4-grouped (n = 6–8 rats per group): group 1 for raw RVP, group 2 for Revanex® (reference drug), group 3 for solid micelle (P407-free from the composition of SSuM), and group 4 for the optimized SSuM. The tablet form of Revanex® was ground into powder for oral intake. Of all preparations, an equivalent dose of 20 mg/kg RVP was accurately weighed and filled into hard gelatin capsules (Torpac® capsule size 9; Torpac, Fairfield, NJ, USA). Each capsules were administered directly into the stomach lumen using a dosing syringe plunger (Torpac® Kit, Torpac) and then, 1 mL of distilled water was injected orally to encourage the micelle formation. Blood samples of approximately 0.3 mL were collected from the retro-orbital plexus using heparinized capillary tubes at fixed time points (0.33, 0.67, 1, 1.5, 2, 4, 6, 8, 12, and 24 h) and underwent centrifugation at 16,000 ×g for 10 min for plasma separation. The plasma was transferred to a microtube and rapidly frozen at –80°C until liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) analysis.
Determination of RVP in Plasma Samples by LC-ESI-MS/MS
Fifty microliters of plasma samples were mixed with 1 mL of internal standard solution (50 ng/mL pioglitazone in methanol) and the mixture was vortexed for 3 min. After centrifugation at 16,000 ×g for 5 min, 50% methanol (800 μL) was added to 200 μL of the supernatant. The mixture was vortexed and centrifuged at 16,000 ×g for 5 min. Finally, supernatant (100 μL) was transferred into a vial for introduction into the LC-ESI-MS/MS system. LC separations were achieved using a Vanquish Duo UPLC (Thermo Scientific, MA, USA). One microliter of each sample was separated using a Hypersil GOLD dC18 column (100 × 2.1 mm, 1.9 μm; Thermo Scientific) at 40°C. An isocratic mobile phase consisting of distilled water, methanol, and formic acid (30:70:0.05, [v/v/v]) was used at a flow rate of 0.2 mL/min. Eluted components were delivered into an Orbitrap Q Exactive Plus mass spectrometer (Q-EXACTIVE PLUS orbitrap; Thermo Scientific) equipped with a heated ionization source operated in positive ion mode. The operating parameters were as follows: electrospray voltage, 3.5 kV; sheath gas flow rate, 35 (arbitrary units); auxiliary gas, 10 (arbitrary units); sweep gas, 0 (arbitrary units); auxiliary gas heater temperature, 350°C; and capillary temperature, 350°C.
Pharmacokinetic (PK) Assessment
PK parameters including area under the curve from 0 to 24 h (AUC0–24h), maximum plasma concentration (Cmax), and time to reach the Cmax (Tmax) were analyzed using the BA Calc 2007 PK analysis program (Ministry of Food and Drug Safety, Chungcheongbuk-do, Korea). AUC0–24h was calculated by the linear trapezoidal rule and both Cmax and Tmax were calculated directly from the plasma concentration-time data. To compare oral absorption between formulations, relative BA was computed by dividing the AUC0–24h of the samples by that of the reference.
Statistical Analysis
All data are represented as the mean ± standard deviation (SD). Statistical significance was evaluated using Student’s t-test, and p <0.05 was considered statistically significant.
Results and Discussion
Screening of Solubilizing Vehicles
The selection of a suitable solubilizing vehicle is critical for micelle formation, as RVP has poor water solubility (approximately 230 μg/mL in distilled water). The solubility of RVP in various surfactant solutions is shown in Figure 1A. Among the surfactants tested, G44 showed the greatest solubility (1,489 μg/mL) followed by BL4 (1,342 μg/mL). G44 alone did not form a homogenous dispersion (polydispersity index (PDI) >0.6, data not shown); however, the addition of BL4 resulted in a homogeneous and nano-sized dispersion. This might be attributed to the difference in the hydrophilic–lipophilic balance value between G44 (14) and BL4 (9). As BL4 is more hydrophobic than G44, it could efficiently enclose RVP. Both surfactants are biocompatible and have been widely used in nanoemulsion formulations.29,30 Therefore, a mixture of G44 and BL4 was selected as a vehicle for micellization.
 |
Figure 1 Solubility of RVP in various surfactant solutions (1% [w/w]) (A) and CMC determination via the solubilization method (B). Abbreviations: LG, Lauroglycol 90; LF, Labrafil M 1944 CS; BL4, Brij L4; LS, Labrasol; RH60, Kolliphor RH60; RH40, Kolliphor RH40; G44, Gelucire 44/14; EL, Kolliphor EL; T80, Tween 80; T20, Tween 20. |
To determine the amount of mixed surfactants required for optimal micelle formation, the CMC of the mixture was measured by the solubilization method.31 As depicted in Figure 1B, at low concentration (~100 μg/mL), the surfactants did not increase the solubility of RVP; however, the solubility sharply increased at high surfactant concentrations (>300 μg/mL), indicating the formation of surfactant micelles. The CMC values were determined as the intersection of the regression lines obtained from the two linear portions of the plot:32 119.2 μg/mL for G44 alone, 333.0 μg/mL for BL4 alone, and 208.5 μg/mL for the G44/BL4 mixture. Based on these values, to guarantee a sufficiently high concentration above the CMC, G44/BL4 was used at 400 μg/mL in the dissolution experiment.
Screening of Supersaturating Agents
To further enhance RVP solubilization, supersaturating agents were screened. Based on previous reports, the amount of polymers was set to 10% [w/w] of the total amount of vehicle.25,33 Hydrophilic polymers can induce supersaturation and maintain the supersaturated state by inhibiting nucleation and crystallization via steric stabilization and/or specific interactions, such as hydrogen bonding, hydrophobic interactions, and complex formation.15,34 Poloxamers and polyvinylpyrrolidones have been used as effective supersaturating agents in various formulations, including lipid-based systems.25,35 Dissolution profiles of G44/BL4 micelles in the presence or absence of various supersaturating agents are shown in Figure 2. All formulations tested showed rapid dissolution during the first 30 min and plateaued thereafter. Compared to micelle alone, the addition of supersaturating polymers somewhat increased RVP dissolution, in the order P407 >K90 ≥K188 ≥K30. In particular, the dissolution level of P407-added micelle (approximately 70% for 2 h) was significantly higher than those of other polymer-added micelles and polymer-free micelle. P407 consists of hydrophobic block (polypropylene glycol) and hydrophilic block (polyethylene glycol). Because of its amphiphilic nature, P407 is able to act as a surfactant and squeeze in the colloidal interface.36 Hydrophobic moieties interact with hydrophobic tails of drug-loaded colloid, rigidifying hydrocarbon layer of the interface, resulting in the action of supersaturation.37 Meanwhile, hydrophilic moieties shield the surface of the micelle, generating hydrophilic barrier which hamper the aggregation and/or destruction of the colloid.37 P407 also spontaneously forms micelle and solubilizes water-insoluble drugs, thereby contributing to supersaturation.38 Thus, P407 was selected as a supersaturating agent for SuM formulation.
 |
Figure 2 Dissolution profiles of micelle with or without various supersaturating agents. Abbreviations: P407, Poloxamer 407; K90, Kollidon K90; P188, Poloxamer 188; K30, Kollidon K30. Note: *p <0.05 versus Micelle only. |
Selection of Solid Carriers
To develop a solid micelle, it is important to select appropriate solid carriers. We screened two types of solid carriers: silica-based adsorbents (Aerosil® 200, Sylysia® 350, Neusilin® US2, and FLO) and cellulose-based diluents (VP105, VP12, L-HPC, and Avicel PH 101). Silica-based adsorbents have a small particle size and high surface area, a porous structure, and high oil-absorption capacity, thereby minimizing the amount required for solidification.39 However, they sometimes entail incomplete desorption of micelle components because of strong interaction between the drug and silicates.40 In comparison, cellulose-based diluents are hydrophilic and completely dissolve in aqueous phase, avoiding the problems of incomplete desorption.28 In this regard, a combination of these solid carriers would be preferred. The flow properties of the solidified granules were investigated based on plots of the CI against the ratio of solid carrier to SuM (Figure 3). Silica-based adsorbents were superior to cellulose-based diluents, showing smaller CSR values (range, 0.30–0.44) than cellulose-based diluents (range, 0.91–1.47). As FLO and VP105 showed the lowest CSR values among the silica-based adsorbents and cellulose-based diluents, respectively, they were selected as solid carriers for SSuM formulation.
 |
Figure 3 Plots of the CI against the ratio of solid carrier to SuM to evaluate flow property changes in SuM formulations with silica-based adsorbents (A) and cellulose-based diluents (B). Abbreviations: FLO, Florite PS-10; VP105, Vivapur 105; VP12, Vivapur 12; L-HPC, low-substituted hydroxypropyl cellulose B1. Note: Arrows indicate the critical solidifying ratio. |
Statistical Analysis Using Box-Behnken Design
To optimize the RVP-loaded SSuM formulation, a three-level Box-Behnken design was employed. In Box-Behnken design, optimal responses are suggested by calculating the effect of each independent factor and their interactions.24 Based on a preliminary study, three factors were selected as independent variables: the ratio of G44 to BL4 (X1), the amount of FLO (X2), and the amount of VP105 (X3). As shown in Table 1, X1 was set to range from 1:0 to 0:1. The upper limits (1) of X2 and X3 were based on their CSR values (120 mg and 360 mg, respectively) and the lower limits (–1) were set to halves thereof. To find an optimal SSuM formulation, three response variables, ie, DE30 (%; Y1), DEC (Y2), and CI (%; Y3) were employed, with Y1 ranging from 21.23 to 45.56, Y2 from 0.050 to 0.097, and Y3 from 10.2 to 20.6 (Table 2). To estimate the effects of single terms and/or interactions between factors, each response was fitted to a linear and polynomial regression (up to quadratic) models, and statistical parameters of each model were evaluated (Table 3). The standard error of regression (S) represents the quantitative average distance between the experimental values and regression curve and provides information on whether the experimental values were adequately fitted to the adopted model.41 The model exhibiting the lowest S value is recommended. The prediction accuracy of the model was inferred from the R2 value, where the model with the highest R2 value is suggested. A good model should turn out to be statistically significant (p <0.05). On the other hand, a lack of fit measure should be statistically non-significant, because the measure indicates large deviation of the data from the fitted model.42 Based on the statistical parameter results, quadratic models were selected for further optimization.
 |
Table 1 Independent and Response Variables Used in Box-Behnken Design |
 |
Table 2 Combinations of Independent Variable and Experimental Responses from Runs |
 |
Table 3 Summary of the Results of Statistical Analysis and Model Equations for the Measured Responses |
Effects of Independent Variables on the Responses in Experimental Design
A normal distribution is considered a crucial assumption for statistical analysis. Normal probability plots for standardized residuals were linear, indicating that the experimental run data were normally distributed (Supplementary Figure S1A).4,43 4 Pareto charts indicating the relative importance of the individual and interaction effects based on Student’s t-tests are shown in Supplementary Figure S1B. Statistically significant independent factors were extended the dotted bar which positioned at α = 0.05.44 The results of the overall analysis of variances for the three response variables with the sum of squares are presented in Table 4. The amount of variability accounted for by the quadratic model response was statistically significant (p <0.05) for all of the response variables. A lack of fit also signified a small deviation of the data from the fitted model, indicating the predicted values were close to observed values of the response variable. The result of the analysis of variance for each predictor is further provided in Supplementary Table S1. Overall, all linear effect parameters (X1, X2, and X3) had a significant impact on each response. Moreover, some polynomial effect parameters were found to be statistically significant as well. Considering linear and polynomial effect parameters, quadratic model was selected for three responses and their polynomial regression equations calculated as follows:
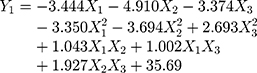
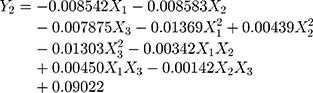
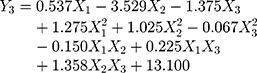
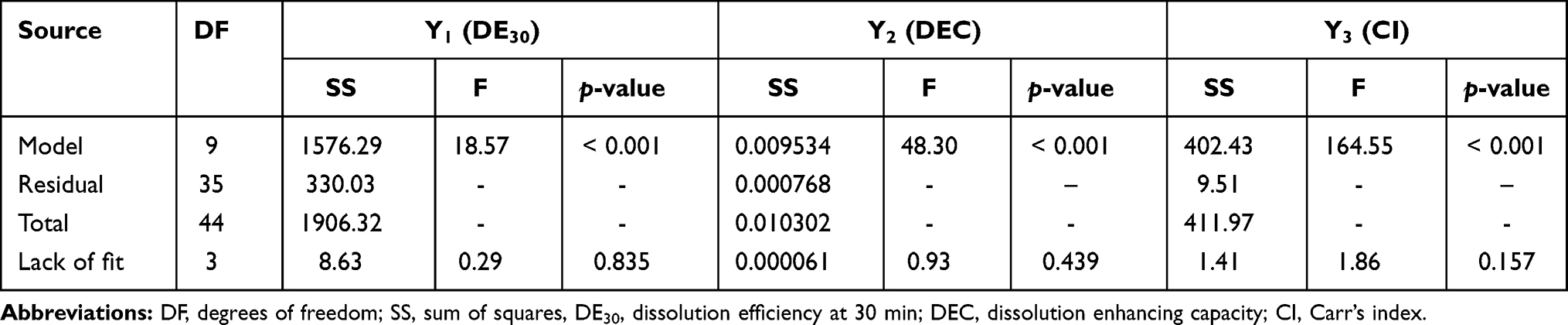 |
Table 4 Analysis of Variance of the Experimental Responses for Quadratic Model |
Each linear or polynomial regression coefficient reflects not only the magnitude of its effect on the response variables but also the proportional relationship between the input and responses. Positive coefficients indicate positive relationships to each response, whereas negative coefficients indicate negative relationships between the factors and each response.45 As shown in Figure 4, both X2 and X3 had negative impacts on Y1 and Y2. Once liquid formulations underwent solidification, the dissolution rate decreased, as micelle components tend to be incompletely desorbed from the pores of solid carriers.46 Especially, both Y1 and Y2 changed drastically when X2 shifted from the lower limit to the upper limit compared to X3, because of the insolubility of FLO. Two different solid carriers were employed to obtain SSuM. The silica-based adsorbent, FLO, has a large surface area and high oil-absorption capacity because of its mesoporous structure.47 Water-insoluble silicates exert disadvantage on the dissolution rate due to molecular adhesion (eg, hydrophobic interaction) between the silanol group and micelle components, which resulted in a negative association with both Y1 and Y2.40 In contrast, VP105 is completely soluble in water and enables complete desorption of the micelle components into the aqueous medium.28 Nevertheless, X3 was negatively associated with Y1 because Y1 reflected dissolution behavior in the early phase only. Combinations with a high VP105 content showed a gradual increase in dissolution throughout the period, showing a similar dissolution rate as P407-added SuM at 2 h. Moreover, X3 had a negative impact on Y2 because of the amount of VP105 added. To obtain fine granules, it would be necessary to add a large quantity of solid carriers, thus increasing the total system quantity. The large amount of solid carriers entailed a large unit quantity, but this would be disadvantageous to DEC, leading to a negative association between Y2 and X3. The addition of both solid carriers had a positive effect on granulation, whereby CI values steeply decreased when the amount of solid carriers increased. As a result, negative effects on Y3 were found for both X2 and X3.
 |
Figure 4 Effects of independent factors on response variables: three-dimensional response surface plots of Y1 (A), Y2 (B), and Y3 (C). |
The ratio of surfactants also had a statistically significant impact on each response. G44 showed a higher hydrophilic–lipophilic balance value and better RVP solubility than BL4.48,49 Therefore, the dissolution rate increased with an increasing amount of G44 and hence, X1 showed a negative correlation with Y1. Because the unit quantity was not changed by X1, X1 was negatively associated with Y2 as well (Figure 4C). In addition, because G44 is semi-solid at room temperature, solidification is expected to become easier when the proportion of G44 increases.50 This explains the positive correlation between Y3 and X1.
Optimization of RVP-Loaded SSuM Using a Desirability Function
The three responses were optimized using appropriate targets. Micelle components should be released from SSuM as soon as it reaches the GI tract. A high dissolution rate in the early phase contributes to the rapid absorption of poorly soluble drugs. At this point, DE30, reflecting the early dissolution behavior, was set to be maximized. Y2 represents the ratio of dissolution to total mass, and a product exhibiting high dissolution with low nit quantity is desirable; thus, Y2 was set to be maximized. And Y3 (CI) was set to be minimized to acquire free-flowing granules.
According to the goals of each response, three independent variables were optimized using a desirability function. As shown in Table 5, X1, X2, and X3 were optimized as –0.41, 0.31, and –0.78, respectively, and the desirability function was 0.748, resulting in a composition of 253.8 mg G44, 106.2 mg BL4, 99.3 mg FLO, 199.8 mg VP105, 40 mg P407, and 200 mg RVP. The granules obtained were easily reconstituted in distilled water, resulting in size and PDI values of 181.3 ± 5.8 nm and 0.218 ± 0.027, respectively, indicating a homogeneous, nanosized micelle formation. To evaluate how well the experiment was designed, prediction percentage errors were calculated: the values were lower than 5% for all responses, indicating that optimized SSuM was successfully prepared using the Box-Behnken design.
 |
Table 5 Predicted Values and Experimental Values for the Optimized RVP-Loaded SSuM Formulation |
Solid-State Property Evaluation
The solid-state properties of RVP, FLO, VP105, PM, and SSuM were evaluated by SEM, DSC, and PXRD. The morphological aspects of each sample were visualized using SEM (Figure 5A). Raw RVP had a small, irregular, and asymmetrical shape, as previously reported.9 Solid carriers appeared to have indefiniteness of their shape and size. In PM, typical morphologies of both solid carriers and raw RVP were present. However, no distinct crystallinity of RVP was observed in SSuM that existed in a lumped mass, indicating that RVP was completely solubilized and absorbed onto the solid carriers. Further, DSC and PXRD analyses were conducted to evaluate the crystalline property of RVP. DSC thermograms are shown in Figure 5B. RVP showed a sharp endothermic peak at near 220°C, which corresponded to its melting point, and was crystalline in nature (205–208°C).8,9 There were no specific endothermic peaks for both solid carriers, FLO and VP105, as reported.28 PM exhibited a small endothermic peak near the melting point of RVP; however, SSuM yielded no endothermic peak at the melting point of RVP. In PXRD observation (Figure 5C), intrinsic sharp peaks of the free drug were found at several diffraction angles, indicating the crystallinity of RVP.8 The solid carriers produced characteristic intrinsic peaks: 29º for FLO and 21º for VP105.51 PM yielded similar major peaks of RVP, demonstrating that RVP maintained its crystallinity in PM. In contrast, no intrinsic peaks were found for SSuM. This result is consistent with an earlier report that solubilized drugs within a micelle structure exist in an amorphous state, even though the micelle is solidified.20
 |
Figure 5 Solid-state properties of powders. (A) Scanning electron microscopy images. (B) Differential scanning calorimetry thermograms. (C) Powder X-ray diffractometry patterns. Abbreviations: RVP, revaprazan; FLO, Florite PS-10; VP105, Vivapur 105; PM, physical mixture of RVP and solid carriers; SSuM, solid supersaturable micelle. Note: Scale bar = 10 μm. |
In vitro Dissolution Study
Figure 6 shows the dissolution profiles of raw RVP, Revanex® as a reference product, RVP-loaded solid micelle, and RVP-loaded SSuM. As RVP is insoluble in the aqueous phase, raw RVP scarcely dissolved in the medium, exhibiting an extremely low dissolution rate throughout the test period. Revanex® was ground into powder for direct comparison with SSuM granules. Revanex® powder showed improved dissolution (28.0% at 2 h) compared to raw RVP; however, its dissolution rate was not sufficiently high. In contrast, solid micelle and SSuM elevated the dissolution level, showing approximately 48.7% and 71.7% dissolution at 2 h, respectively. These values were quite consistent with the dissolution results of G44/BL4 micelle and SuM, suggesting that micelle components were completely released from the solid carriers and solubilized the poorly soluble drug. The dissolution level of SSuM was significantly higher (p <0.05) than those of other samples, showing >50% dissolution within 30 min, after which it gradually increased. The SSuM formulation successfully increased the dissolution rate of RVP via spontaneous generation of a micelle structure as well as the supersaturating action of P407. Together, these results suggested that SSuM provides a promising strategy to improve the oral BA of drugs with poor aqueous solubility.
 |
Figure 6 Dissolution profiles of raw RVP, Revaprazan® (powder), solid micelle, and the optimized SSuM in distilled water. Abbreviations: RVP; revaprazan; SSuM, solid supersaturable micelle. Note: Values are the mean ± standard error (n = 3). |
In vivo PK Behavior
To evaluate the effect of the optimized SSuM on oral absorption behavior, a PK study was conducted in rats orally administered raw RVP, Revanex® powder, solid micelle, and the optimized SSuM. The plasma concentration of RVP was plotted against time (Figure 7). At an equivalent dose of 20 mg/kg of RVP, the plasma concentrations of all four formulations gradually increased for 6 h. However, both solid micelle and optimized SSuM showed greater absorption behavior than Revanex® and raw RVP. Enhanced intestinal permeability to the nano-sized dispersion may have contributed to the increased plasma RVP concentration.52 In particular, the absorption pattern of both micellar formulations was biphasic: a sharp increase within 1 h, then gradual absorption up to 6 h. This behavior was consistent with the results of the dissolution study, suggesting that the micelle components rapidly solubilized RVP in the GI tract and continuously provided a favorable condition for absorption. This may be attributed to the absorption-enhancing effect of the surfactants used, BL4 and G44, which have been widely used to enhance the oral BA of hydrophobic materials.29,30 Specifically, throughout the test period, the PK profile of the optimized SSuM was superior to that of the solid micelle, indicating the usefulness of P407 to achieve supersaturation. P407 has been shown to enhance the oral absorption of water-insoluble drugs in numerous lipid formulations.33,53 Further, because of its amphipathic nature, many pharmaceutical formulations have exploited P407 as an intestinal permeability enhancer.54,55
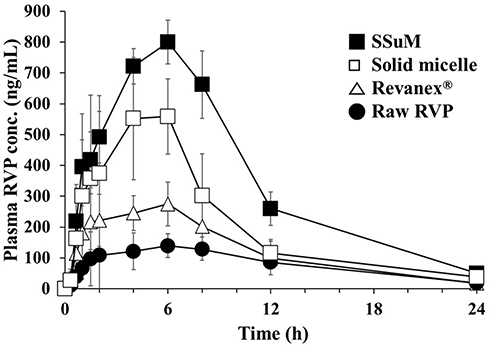 |
Figure 7 Plasma RVP concentration profiles after oral administrations of different formulations to Sprague-Dawley rats at an equivalent dose of 20 mg/kg of RVP. Abbreviations: RVP, revaprazan; SSuM, solid supersaturable micelle. Note: Values are the mean ± standard deviation (n = 6–8). |
The PK parameters of each sample are listed in Table 6. Compared with raw RVP and Revanex®, the solid micelle and optimized SSuM formulations showed greater Cmax and AUC0–24h values, which were in the order SSuM > solid micelle > Revanex® > raw RVP. Based on the AUC0–24h values, the solid micelle improved the oral BA of RVP by approximately 3.0- and 1.7-fold versus raw RVP and Revanex®, respectively. This result was comparable with an earlier report that orally administered sulpiride-loaded micelle composed of Tween 80 and propylene glycol increased the intestinal permeability in rats, showing approximately 4-fold greater permeability coefficient compared to that of sulpiride solution.56 Furthermore, relative BA of the optimized SSuM was approximately 4.8-, 2.8-, and 1.6-fold greater than those of raw RVP, Revanex®, and solid micelle, respectively, indicating the importance of supersaturation in addition to micelle formation in the GI tract. Lipid-based formulations containing the supersaturating agent were superior in drug absorption to conventional formulation. For instance, oral BA of indirubin-loaded self-microemulsifying drug delivery system was increased 1.3-fold by the addition of 0.5% PVP K17 as a supersaturating agent.14 For BSC class II drugs, including RVP, dissolution is a rate-limiting step in drug absorption.57 The improved dissolution of RVP in the micelle formulations enabled enhanced oral absorption. In addition, middle chain fatty acid enhances the permeability of particles across the cell layers.58 Both surfactants, G44 and BL4, contain lauric acid moieties that enhance intestinal permeability.59 Meanwhile, the Tmax of SSuM was slightly delayed, indicating that continuous intestinal absorption of RVP occurred via supersaturated micelle formation. Therefore, we suggest that SSuM would be a promising candidate for the clinical development of the BCS II drug such as RVP. However, as the present study designed in a rodent model only, further investigations on allometric scaling and/or human PK evaluation are necessary for clinical application.
 |
Table 6 PK Parameters of RVP Formulations in Rats |
Conclusion
We successfully developed a novel RVP-containing SSuM using a Box-Behnken design with three independent variables, X1 (G44/BL4), X2 (FLO), and X3 (VP105), resulting in DE30 (40.3%), DEC (0.088), and CI (12.3%). With a high desirability function value (0.748), the optimized SSuM reflected a negligible percentage prediction error (<5%). No distinct crystalline of RVP was found indicating that RVP existed in an amorphous state in the optimized SSuM and showed substantially improved dissolution behavior compared to raw RVP. PK study in rats revealed that the SSuM improved relative BA by approximately 4.8- and 2.8-fold versus raw RVP and Revanex®, respectively. Thus, SSuM shows promising potential for the development of a solid formulation of the poorly water-soluble RVP.
Acknowledgments
This work was supported by a grant from the National Research Foundation of Korea (NRF) funded by the Korea government (MSIP) (No. 2019R1A2C2002510). The work was also supported by the Brain Korea (BK) 21 FOUR through the NRF funded by the Ministry of Education of Korea. We also thank to Editage (www.editage.co.kr) for English language editing (CAUNE_6678).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Inatomi N, Matsukawa J, Sakurai Y, Otake K. Potassium-competitive acid blockers: advanced therapeutic option for acid-related diseases. Pharmacol Ther. 2016;168:12–22. doi:10.1016/j.pharmthera.2016.08.001
2. Boeckxstaens G. Emerging drugs for gastroesophageal reflux disease. Expert Opin Emerg Drugs. 2009;14(3):481–491. doi:10.1517/14728210903133807
3. Suzuki H. New pharmacological treatments for gastroesophageal reflux: potassium-competitive acid blockers and bile acid sequestrants. NeuroGastroLatam Rev. 2018;2:18–27. doi:10.24875/NGL.18000007
4. Vakil N. New pharmacological agents for the treatment of gastro‐oesophageal reflux disease. Aliment Pharmacol Ther. 2004;19(10):1041–1049. doi:10.1111/j.1365-2036.2004.01957.x
5. Shahid SK. Novel anti-Helicobacter pylori therapies. Pharm Pat Anal. 2014;3(4):411–427. doi:10.4155/ppa.14.27
6. Li W, Yang Y, Tian Y, et al. Preparation and in vitro/in vivo evaluation of revaprazan hydrochloride nanosuspension. Int J Pharm. 2011;408(1–2):157–162. doi:10.1016/j.ijpharm.2011.01.059
7. Park JH, Cho JH, Kim DS, et al. Revaprazan-loaded surface-modified solid dispersion: physicochemical characterization and in vivo evaluation. Pharm Dev Technol. 2019;24(6):788–793. doi:10.1080/10837450.2019.1597114
8. Kim JS, Park JH, Jeong SC, et al. Novel revaprazan-loaded gelatin microsphere with enhanced drug solubility and oral bioavailability. J Microencapsul. 2018;35(5):421–427. doi:10.1080/02652048.2018.1515997
9. Park JH, Kim DS, Mustapha O, et al. Comparison of a revaprazan-loaded solid dispersion, solid SNEDDS and inclusion compound: physicochemical characterisation and pharmacokinetics. Colloids Surf B Biointerfaces. 2018;162:420–426. doi:10.1016/j.colsurfb.2017.12.017
10. Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25(1):103–128. doi:10.1016/S0169-409X(96)00494-2
11. Ozeki T, Tagami T. Functionally engineered nanosized particles in pharmaceutics: improved oral delivery of poorly water-soluble drugs. Curr Pharm Des. 2013;19(35):6259–6269. doi:10.2174/1381612811319350004
12. Rangel-Yagui CO, Pessoa A, Tavares LC. Micellar solubilization of drugs. J Pharm Pharm Sci. 2005;8(2):147–163.
13. Suys EJ, Chalmers DK, Pouton CW, Porter CJ. Polymeric precipitation inhibitors promote fenofibrate supersaturation and enhance drug absorption from a type IV lipid-based formulation. Mol Pharm. 2018;15(6):2355–2371. doi:10.1021/acs.molpharmaceut.8b00206
14. Chen ZQ, Liu Y, Zhao JH, Wang L, Feng NP. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int J Nanomedicine. 2012;7:1115. doi:10.2147/IJN.S28761
15. Xu S, Dai WG. Drug precipitation inhibitors in supersaturable formulations. Int J Pharm. 2013;453(1):36–43. doi:10.1016/j.ijpharm.2013.05.013
16. Warren DB, Benameur H, Porter CJ, Pouton CW. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: a mechanistic basis for utility. J Drug Target. 2010;18(10):704–731. doi:10.3109/1061186X.2010.525652
17. Zhu C, Gong S, Ding J, et al. Supersaturated polymeric micelles for oral silybin delivery: the role of the Soluplus–PVPVA complex. Acta Pharm Sin B. 2019;9(1):107–117. doi:10.1016/j.apsb.2018.09.004
18. Wang Z, Sun J, Wang Y, et al. Solid self-emulsifying nitrendipine pellets: preparation and in vitro/in vivo evaluation. Int J Pharm. 2010;383(1–2):1–6. doi:10.1016/j.ijpharm.2009.08.014
19. Lv QY, Li XY, Dai L, et al. A solid phospholipid-bile salts-mixed micelles based on the fast dissolving oral films to improve the oral bioavailability of poorly water-soluble drugs. J Nanopart Res. 2014;16(6):2455. doi:10.1007/s11051-014-2455-6
20. Goo YT, Park SY, Chae BR, et al. Optimization of solid self-dispersing micelle for enhancing dissolution and oral bioavailability of valsartan using box-behnken design. Int J Pharm. 2020;585:119483. doi:10.1016/j.ijpharm.2020.119483
21. Hou Y, Wang H, Zhang F, et al. Novel self-nanomicellizing solid dispersion based on rebaudioside A: a potential nanoplatform for oral delivery of curcumin. Int J Nanomedicine. 2019;14:557. doi:10.2147/IJN.S191337
22. Bezerra MA, Santelli RE, Oliveira EP, Villar LS, Escaleira LA. Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta. 2008;76(5):965–977. doi:10.1016/j.talanta.2008.05.019
23. Liu Y, Zhang P, Feng N, Zhang X, Wu S, Zhao J. Optimization and in situ intestinal absorption of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2009;365(1–2):136–142. doi:10.1016/j.ijpharm.2008.08.009
24. Villar AMS, Naveros BC, Campmany ACC, Trenchs MA, Rocabert CB, Bellowa LH. Design and optimization of self-nanoemulsifying drug delivery systems (SNEDDS) for enhanced dissolution of gemfibrozil. Int J Pharm. 2012;431(1–2):161–175. doi:10.1016/j.ijpharm.2012.04.001
25. Yeom DW, Chae BR, Son HY, et al. Enhanced oral bioavailability of valsartan using a polymer-based supersaturable self-microemulsifying drug delivery system. Int J Nanomedicine. 2017;12:3533. doi:10.2147/IJN.S136599
26. Shao A, Chen G, Jiang N, et al. Development and evaluation of self-microemulsifying liquid and granule formulations of Brucea javanica oil. Arch Pharm Res. 2013;36(8):993–1003. doi:10.1007/s12272-013-0113-7
27. Mura P, Valleri M, Cirri M, Mennini N. New solid self-microemulsifying systems to enhance dissolution rate of poorly water soluble drugs. Pharm Dev Technol. 2012;17(3):277–284. doi:10.3109/10837450.2010.535825
28. Yeom DW, Chae BR, Kim JH, et al. Solid formulation of a supersaturable self-microemulsifying drug delivery system for valsartan with improved dissolution and bioavailability. Oncotarget. 2017;8(55):94297. doi:10.18632/oncotarget.21691
29. Acharya A, Sanyal S, Moulik S. Formation and characterization of a pharmaceutically useful microemulsion derived from isopropylmyristate, polyoxyethylene (4) lauryl ether (Brij-30), isopropyl alcohol and water. Curr Sci. 2001;362–370.
30. Wehrung D, Geldenhuys WJ, Bi L, Oyewumi MO. Biocompatibility, efficacy and biodistribution of Gelucire-stabilized nanoparticles engineered for docetaxel delivery. J Nanosci Nanotechnol. 2012;12(3):2901–2911. doi:10.1166/jnn.2012.5789
31. Shimizu K, Iwatsuru M. Measurement of the critical micelle concentration of ionic-nonionic mixed micelles by the first derivative absorption spectrum method. Chem Pharm Bull. 1990;38(3):744–747. doi:10.1248/cpb.38.744
32. Bakkour Y, Darcos V, Li S, Coudane J. Diffusion ordered spectroscopy (DOSY) as a powerful tool for amphiphilic block copolymer characterization and for critical micelle concentration (CMC) determination. Polym Chem. 2012;3(8):2006–2010. doi:10.1039/c2py20054f
33. Tung NT, Tran CS, Nguyen HA, et al. Formulation and biopharmaceutical evaluation of supersaturatable self-nanoemulsifying drug delivery systems containing silymarin. Int J Pharm. 2019;555:63–76. doi:10.1016/j.ijpharm.2018.11.036
34. Kojima T, Higashi K, Suzuki T, Tomono K, Moribe K, Yamamoto K. Stabilization of a supersaturated solution of mefenamic acid from a solid dispersion with EUDRAGIT® EPO. Pharm Res. 2012;29(10):2777–2791. doi:10.1007/s11095-011-0655-7
35. Nan Z, Lijun G, Tao W, Dongqin Q. Evaluation of carbamazepine (CBZ) supersaturatable self-microemulsifying (S-SMEDDS) formulation in-vitro and in-vivo. Iran J Pharm Res. 2012;11(1):257.
36. Kim MS, Baek IH. Fabrication and evaluation of valsartan–polymer–surfactant composite nanoparticles by using the supercritical antisolvent process. Int J Nanomedicine. 2014;9:5167. doi:10.2147/IJN.S71891
37. Lee DR, Ho MJ, Jung HJ, et al. Enhanced dissolution and oral absorption of tacrolimus by supersaturable self-emulsifying drug delivery system. Int J Nanomedicine. 2016;11:1109.
38. Shen J, Jin Y, Tian H, et al. Enhancement of oral bioavailability of akebia saponin D by destroying self-micelles and inhibiting multidrug resistance-associated protein mediated efflux. RSC Adv. 2016;6(76):72439–72446. doi:10.1039/C6RA14427F
39. Yeom DW, Son HY, Kim JH, et al. Development of a solidified self-microemulsifying drug delivery system (S-SMEDDS) for atorvastatin calcium with improved dissolution and bioavailability. Int J Pharm. 2016;506(1–2):302–311. doi:10.1016/j.ijpharm.2016.04.059
40. Na YG, Byeon JJ, Wang M, et al. Statistical approach for solidifying ticagrelor loaded self-microemulsifying drug delivery system with enhanced dissolution and oral bioavailability. Mater Sci Eng C. 2019;104:109980. doi:10.1016/j.msec.2019.109980
41. Danmaliki GI, Saleh TA, Shamsuddeen AA. Response surface methodology optimization of adsorptive desulfurization on nickel/activated carbon. Chem Eng J. 2017;313:993–1003. doi:10.1016/j.cej.2016.10.141
42. Praveen R, Verma PRP, Singh SK, George JK. Cross linked alginate gel beads as floating drug delivery system for cefdinir: optimization using Box–Behnken design. J Pharm Investig. 2015;45(2):187–199. doi:10.1007/s40005-014-0164-x
43. Kim MS, Yeom DW, Kim SR, et al. Development of a chitosan based double layer-coated tablet as a platform for colon-specific drug delivery. Drug Des Devel Ther. 2017;11:45. doi:10.2147/DDDT.S123412
44. Leonard SA, Stegemann JA. Stabilization/solidification of petroleum drill cuttings: leaching studies. J Hazard Mater. 2010;174(1–3):484–491. doi:10.1016/j.jhazmat.2009.09.078
45. Karnachi AA, Khan MA. Box-Behnken design for the optimization of formulation variables of indomethacin coprecipitates with polymer mixtures. Int J Pharm. 1996;131(1):9–17. doi:10.1016/0378-5173(95)04216-4
46. Kang MJ, Jung SY, Song WH, et al. Immediate release of ibuprofen from Fujicalin®-based fast-dissolving self-emulsifying tablets. Drug Dev Ind Pharm. 2011;37(11):1298–1305. doi:10.3109/03639045.2011.571695
47. Florite®: new technology for innovative formulation design. Maruyama: Tomita Pharmaceutical Co Ltd; 2018. Available from: http://www.tomitaph.co.jp/english/data/FLORITE.pdf.
48. Karataş A, Yüksel N, Baykara T. Improved solubility and dissolution rate of piroxicam using gelucire 44/14 and labrasol. Farmaco. 2005;60(9):777–782. doi:10.1016/j.farmac.2005.04.014
49. Jeong M, Lee JW, Lee SJ, Kang YT. Mass transfer performance enhancement by nanoemulsion absorbents during CO2 absorption process. Int J Heat Mass Transf. 2017;108:680–690. doi:10.1016/j.ijheatmasstransfer.2016.12.073
50. Zhao K, Yuan Y, Wang H, Li P, Bao Z, Li Y. Preparation and evaluation of valsartan by a novel semi-solid self-microemulsifying delivery system using Gelucire 44/14. Drug Dev Ind Pharm. 2016;42(10):1545–1552. doi:10.3109/03639045.2016.1151034
51. Shin DJ, Chae BR, Goo YT, et al. Improved dissolution and Oral bioavailability of valsartan using a solidified Supersaturable self-microemulsifying drug delivery system containing Gelucire® 44/14. Pharmaceutics. 2019;11(2):58. doi:10.3390/pharmaceutics11020058
52. Li F, Hu R, Wang B, et al. Self-microemulsifying drug delivery system for improving the bioavailability of huperzine A by lymphatic uptake. Acta Pharm Sin B. 2017;7(3):353–360. doi:10.1016/j.apsb.2017.02.002
53. Sureshkumar RA. Review on Precipitation inhibitors in supersaturable self emulsifying drug delivery system. Int J Res Pharm Sci. 2020;11(2):2481–2488. doi:10.26452/ijrps.v11i2.2242
54. Sultan AA, El-Gizawy SA, Osman MA, El Maghraby GM. Self dispersing mixed micelles forming systems for enhanced dissolution and intestinal permeability of hydrochlorothiazide. Colloids Sur B Biointerfaces. 2017;149:206–216. doi:10.1016/j.colsurfb.2016.10.028
55. Zhou J, Dai J, Fu H, et al. Solubilization and intestinal permeability studies of poloxamers in the absence and presence of sodium deoxycholate on a poorly water-soluble compound. Lat Am J Pharm. 2016;35(2):312–329.
56. Chitneni M, Peh KK, Darwis Y, Abdulkarim M, Abdullah GZ, Qureshi MJ. Intestinal permeability studies of sulpiride incorporated into self-microemulsifying drug delivery system (SMEDDS). Pak J Pharm Sci. 2011;24(2):113–121.
57. Emami J. In vitro-in vivo correlation: from theory to applications. J Pharm Pharm Sci. 2006;9(2):169–189.
58. Xiong J, Guo J, Huang L, Meng B, Ping Q. Self-micelle formation and the incorporation of lipid in the formulation affect the intestinal absorption of Panax notoginseng. Int J Pharm. 2008;360(1–2):191–196. doi:10.1016/j.ijpharm.2008.04.016
59. Pabla D, Akhlaghi F, Zia H. Intestinal permeability enhancement of levothyroxine sodium by straight chain fatty acids studied in MDCK epithelial cell line. Eur J Pharm Sci. 2010;40(5):466–472. doi:10.1016/j.ejps.2010.05.002
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.