Back to Journals » Drug Design, Development and Therapy » Volume 11
Development of a chitosan based double layer-coated tablet as a platform for colon-specific drug delivery
Authors Kim MS, Yeom DW, Kim SR, Yoon HY ![]() , Kim CH, Son HY, Kim JH, Lee S, Choi YW
, Kim CH, Son HY, Kim JH, Lee S, Choi YW ![]()
Received 28 September 2016
Accepted for publication 26 November 2016
Published 21 December 2016 Volume 2017:11 Pages 45—57
DOI https://doi.org/10.2147/DDDT.S123412
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Min Soo Kim,1,* Dong Woo Yeom,1,* Sung Rae Kim,1 Ho Yub Yoon,1 Chang Hyun Kim,1 Ho Yong Son,1 Jin Han Kim,1 Sangkil Lee,2 Young Wook Choi1
1College of Pharmacy, Chung-Ang University, Seoul, 2College of Pharmacy, Keimyung University, Daegu, South Korea
*These authors contributed equally to this work
Abstract: A double layer-coated colon-specific drug delivery system (DL-CDDS) was developed, which consisted of chitosan (CTN) based polymeric subcoating of the core tablet containing citric acid for microclimate acidification, followed by an enteric coating. The polymeric composition ratio of Eudragit E100 and ethyl cellulose and amount of subcoating were optimized using a two-level factorial design method. Drug-release characteristics in terms of dissolution efficiency and controlled-release duration were evaluated in various dissolution media, such as simulated colonic fluid in the presence or absence of CTNase. Microflora activation and a stepwise mechanism for drug release were postulated. Consequently, the optimized DL-CDDS showed drug release in a controlled manner by inhibiting drug release in the stomach and intestine, but releasing the drug gradually in the colon (approximately 40% at 10 hours and 92% at 24 hours in CTNase-supplemented simulated colonic fluid), indicating its feasibility as a novel platform for CDD.
Keywords: chitosan, colon-specific delivery, acidification, microflora, factorial design, controlled release
Introduction
Developing a colon-specific drug delivery system (CDDS) has garnered significant attention, owing to the distinct characteristics of the colon, such as neutral pH, longer transit time, reduced digestive enzymatic activity, and a greater responsiveness to absorption enhancers.1 A CDDS possesses practical implications for the treatment of bowel diseases, such as Crohn’s disease, ulcerative colitis, and colon cancer, and the systemic delivery of protein and peptide drugs that are labile and poorly absorbed in the gastrointestinal tract.2
Various approaches for targeting drugs to the colon have been widely investigated, including the pH-sensitive polymer-based system, the time-dependent system, and the pressure-dependent release system.3–5 However, the similarity in pH between the small intestine and colon and high variations in gastrointestinal retention time rendered the aforementioned approaches less reliable for colon targeting.6 In contrast, the colonic microflora-activated system has been considered as an alternative approach, since this strategy exploits the distinctive colonic characteristic of abrupt increase in bacterial population and associated enzyme activities, thus accomplishing greater site specificity of drug release.7,8 The microflora population of the colon is normally in the range of 1011–1012 CFU/mL, while that of the intestine is fewer than 103–104 CFU/mL.9 The use of polysaccharides that are biodegraded by colonic microflora could result in comparatively safer and more effective delivery of drugs to the colon.9,10
Chitosan (CTN) has been evaluated and reported to possess the highest potential as a biodegradable polymer and has been formulated in several dosage forms, such as capsules, matrices, hydrogels, and microparticles.11–16 It is completely digested by the colonic microflora and is highly stable, safe, nontoxic, and inexpensive.17 However, the utilization of CTN has been limited because of its unfavorable properties. For example, it is solubilized in acidic pH and easily digested by lysozymes, nonspecific cellulases, and enzymes secreted by intestinal bacteria.18,19 Enteric coating with hydroxypropyl methylcellulose phthalate and Eudragit® RS has been efficiently employed to avoid undesirable exposure to these conditions, representing bifunctional release characteristics, ie, time-dependent and site-specific.11,17 In addition, several trials to modulate microflora activation have been attempted. Polyelectrolyte complexes composed of pectin and CTN improved vancomycin availability at alkaline pH in the presence of β-glucosidase by enzyme-dependent degradation.20 A multiparticulate system of CTN hydrogel beads was shown to be degraded by rat cecal and colonic enzymes, resulting in a marked acceleration in the release of the model protein.15
In the present study, as a novel platform, a double layer-coated CDDS (DL-CDDS) was developed, consisting of a CTN-dispersed polymeric subcoating of the core tablet containing citric acid (CA) as an acidifying agent, followed by an enteric coating. Loxoprofen sodium (LXP) was used as a model drug, and the polymeric composition for the subcoating layer was optimized. Figure 1 shows a stepwise schematic representation of LXP release from the DL-CDDS. Drug-release characteristics were evaluated in various dissolution media, including simulated colonic fluid (SCF), in the presence or absence of chitosanase (CTNase). Dissolution efficiency (DE) and controlled-release duration (CRD) were used for further evaluation.
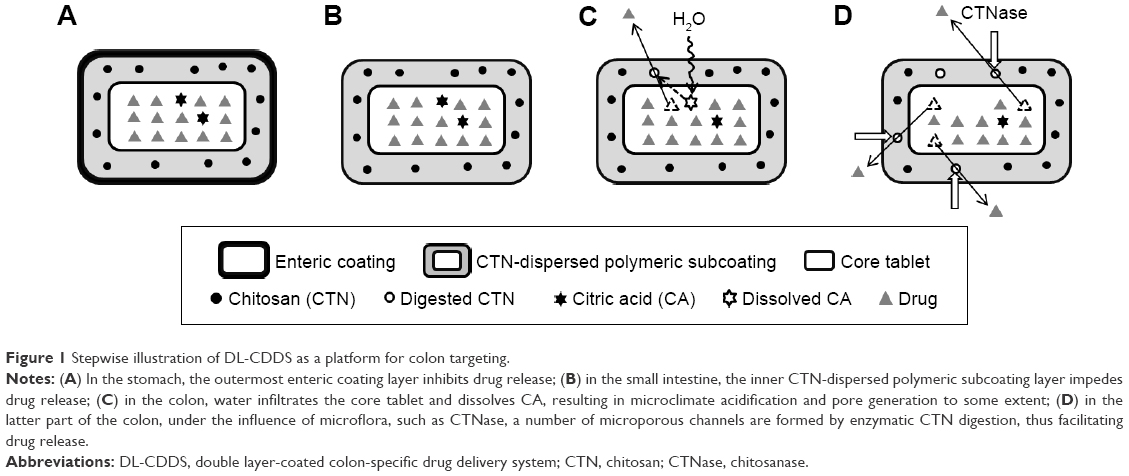 | Figure 1 Stepwise illustration of DL-CDDS as a platform for colon targeting. |
Materials and methods
Materials
LXP was purchased from Kolon Life Science Inc (Gwacheon, South Korea). CTN (deacetylation degree 93%) was purchased from Boao Bioscience (Huazhou, China). CA was purchased from Samchun Chemical Co (Pyeongtaek, South Korea). Ethyl cellulose (EC; Aqualon™ N7, 7 cP, 48%–49.5% ethoxyl substitution) was purchased from Ashland (Lexington, KY, USA). Eudragit E100 (EE), Eudragit L100-55 (EL), and colloidal silicon dioxide were supplied by Evonik (Essen, Germany). Triethyl citrate was purchased from Morimura Bros (Tokyo, Japan). Talc was obtained from Nippon Talc (Osaka, Japan). Hydrated lactose was obtained from DFE Pharma (Goch, Germany). Low-substituted hydroxypropyl cellulose (L-HPC) was obtained from Shin-Etsu (Tokyo, Japan). Polyvinylpyrrolidone K30 was supplied by BASF (Ludwigshafen, Germany). Croscarmellose sodium was supplied by JRS Pharma (Rosenberg, Germany). Magnesium stearate was purchased from FACI (Genoa, Italy). Recombinant CTNase (OHK, 5,000 units/g) was purchased from Kyowa Chemical Industry (Sakaide, Japan). Ethanol and methylene chloride were purchased from Duksan Chemical Co (Ansan, South Korea). Other reagents were of reagent grade and used as received from commercial sources.
Preparation of DL-CDDS tablets
Preparation of core tablets
The core tablets were prepared using a conventional wet-granulation and direct-compression method. For wet granulation, LXP was dried at 90°C using a desiccator (Thermo Fisher Scientific, Waltham, MA, USA) and pre-mixed with lactose hydrate and L-HPC as diluent and lubricant, respectively, using a V-mixer (VB-3; Erweka GmbH, Heusenstamm, Germany) for 5 minutes with or without CA. The resultant powder mixture was mixed with polyvinylpyrrolidone K30 solution (25% w/w in ethanol) as a binder, and a damp mass was granulated manually using a high-speed mixer (Xena-II, Raonxena, Shiheung, South Korea). The granules were dried in an oven at 60°C for 3 hours and passed through a 20-mesh sieve. For direct compression, the prepared granules were mixed with croscarmellose sodium, colloidal silicon dioxide, and magnesium stearate using a cube mixer for 5 minutes. The resultant powder mixture was compressed into tablets on a single-punch tablet machine at compression force 2 kN using 8.92 mm normal circular concave punches.
Physical properties of core tablets
Physical testing of core tablets was performed after a relaxation period of at least 24 hours. Weight-variation tests were performed with 20 individually weighed tablets using a balance (XS603S analytical balance; Mettler-Toledo, Columbus, OH, USA). The thickness and diameter of ten tablets were measured individually using Vernier calipers (CD-15APX; Mitutoyo, Kawasaki, Japan). Crushing strength was determined using a hardness tester (PTB111E; Pharma Test, Hainburg, Germany). Tablet friability was calculated as the percentage of weight loss (4 minutes, 25 rpm, 20 tablets) using a “friabilator” (PTF 20E; Pharma Test). Separately, to determine drug content by high-performance liquid chromatography (HPLC) assay, 20 tablets were weighed individually, crushed into a fine powder, and combined to give a sample containing 60 mg of LXP. The powdered sample was extracted with water, and the solution was filtered through a membrane filter (0.45 μm) prior to injection into the column.
CTN-dispersed subcoating
The core tablets were film-coated with CTN-dispersed polymeric coating solution using an Accela coater (Thomas Engineering, Hoffman Estates, IL, USA). A coating solution of an ethanol–methylene chloride mixture (6:4, v:v) was sprayed onto the core drug tablets at a flow rate of 12 mL/min; rotation speed was adjusted to 12 rpm with an inlet air temperature of 50°C, an outlet air temperature of 40°C, and an atomizing air pressure of 1.5 kgf/cm2. The coating solution contained the coating agents (10%, w/w) CTN, triethyl citrate, talc, and polymers. As a polymer, EE alone or a combination of EE and EC was used, designated as CTN/EE subcoating or CTN/EE/EC subcoating, respectively. The compositions are listed in Table 1.
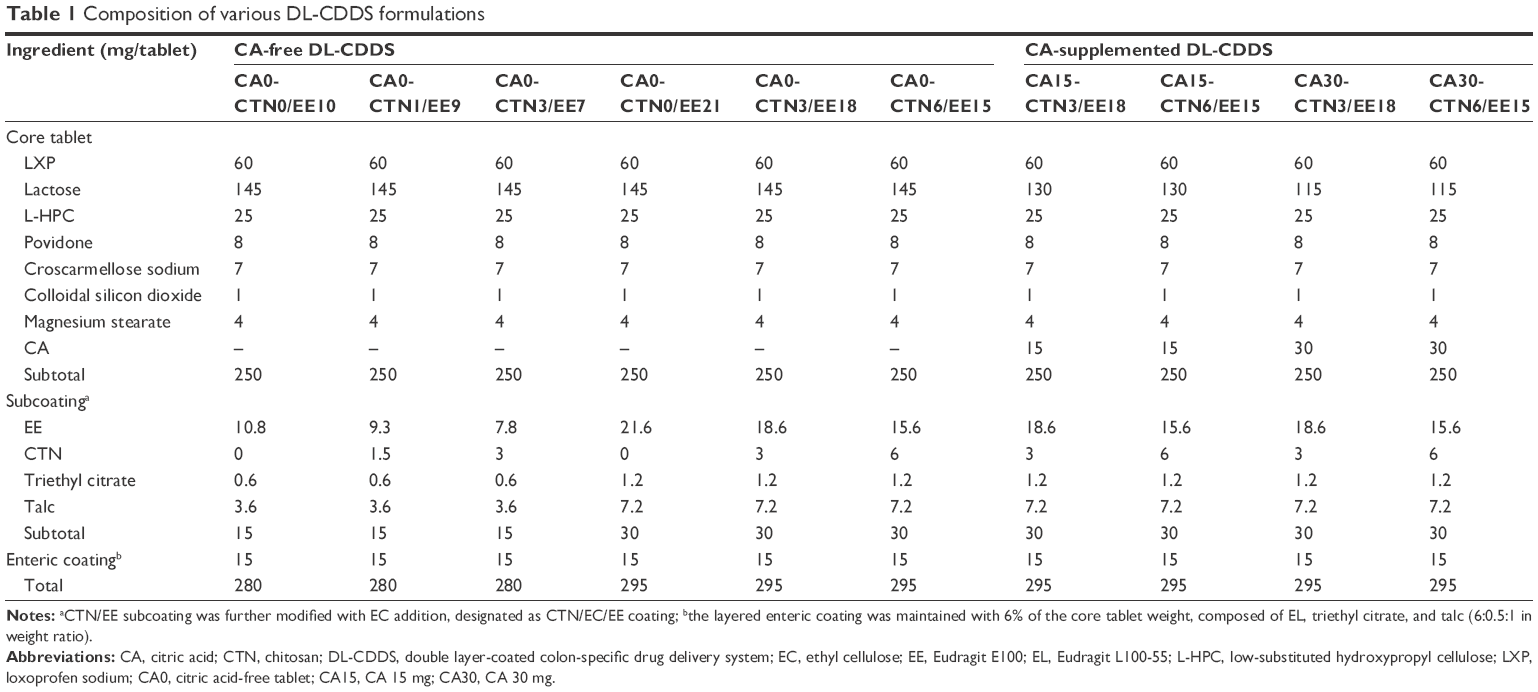 | Table 1 Composition of various DL-CDDS formulations |
Enteric coating
After the subcoating, the tablets were subjected to enteric coating under the same conditions as described for subcoating, except for the inlet and outlet temperatures, which were altered to 45°C and 35°C, respectively. The coating solution contained the coating agents (10%, w/w) EL, triethyl citrate, and talc (6:0.5:1 in weight ratio). The amount of enteric coating used was fixed at 6% of the core tablet’s weight.
In vitro dissolution test
Dissolution tests were performed with USP Apparatus 1 using a dissolution tester (VK7025; Agilent Technologies, Santa Clara, CA, USA) for 24 hours, as described in the literature.19 The revolution speed of paddles and the volume of dissolution medium were set to 100 rpm and 900 mL, respectively, at 37°C±0.5°C. As soon as the dissolution test started, an equivalent of 60 mg of LXP was introduced into the dissolution medium. Drug-release studies were initially performed in pH 1.2 simulated gastric fluid (SGF) for 2 hours, which was then replaced with pH 7.4 simulated intestinal fluid (SIF) for the next 3 hours. Finally, pH 6.8 SCF was used for the following 19 hours. SGF was prepared by dissolving 2 g of sodium chloride in 7 mL of 0.1 M hydrochloric acid and diluting with distilled water to a volume of 1,000 mL. SIF was prepared by dissolving 9.6 g of Dulbecco’s phosphate-buffered saline (Sigma-Aldrich, St Louis, MO, USA) in 1,000 mL of distilled water. SCF was prepared by mixing 250 mL of 0.2 M potassium phosphate monobasic solution and 118 mL of 0.2 M sodium hydroxide solution, and diluting with distilled water to a volume of 1,000 mL. Separately, to mimic the colonic microflora environment, CTNase (20 mg) was added as an enzyme to one batch of SCF (900 mL), designated as CTNase-supplemented SCF (CTNase-SCF). At predetermined sampling points, 5 mL of the sample was taken and filtered through a membrane filter (0.45 μm, polyvinylidene difluoride, SmartPor®). The filtrate was diluted with the same volume of methanol prior to analysis. The amount of dissolved LXP in each sample was determined by HPLC.
HPLC analysis of LXP
The concentrations of LXP were determined by HPLC. The HPLC systems consisted of a pump (W2690/5; Waters Corp., Milford, MA, USA), ultraviolet detector (W2489, Waters Corp.), and a data station (Empower 3; Waters Corp.). Chromatographic separation was performed using a Kromasil 100-5-C18 column (150×4.6 mm, 5 μm; AkzoNobel, Bohus, Sweden) at a flow rate of 1 mL/min at 40°C. The mobile phase consisted of methanol, water, acetic anhydride, and trimethylamine (600:400:1:1 in volume ratio). Of each sample, 10 μL was injected into the column, and ultraviolet detection was performed at 222 nm.
Comparison of in vitro dissolution data
For comparison of dissolution profiles, model-independent analyses based on DE in the colon (DEcolon) and CRD in the colon (CRDcolon) were conducted.
DEcolon
The area under the dissolution curve was calculated by the trapezoidal rule, as reported elsewhere.21,22 DEcolon was defined as a percentage of area under the curve for the specified time period (from 5 hours to 24 hours) against the rectangular area, representing 100% dissolution. To observe the effect of CTNase on dissolution, DEcolon values measured in both CTNase-SCF medium and CTNase-free SCF medium were compared.
CRDcolon
To compare the controlled-release property of various DL-CDDS formulations, CRD was used to evaluate time periods required to reach 90% of the cumulative amount of drug released at the terminal time point (24 hours). CRDcolon was observed and measured in CTNase-SCF medium, which simulated the colonic microflora activation.
Design of the experiments
CTN/EE subcoating was further modified with EC addition to control drug release. A two-level factorial design (2-LFD) was built up for optimizing subcoating composition using (version 15; Minitab Inc., State College, PA, USA). A typical 2-LFD is displayed in Table 2. The study design involved the investigation of the effect of two independent variables: polymer composition (X1; wt%), studied by changing the polymer ratio of EE:EC at low level (1:2) and high level (2:1), added center points (1:1), and the amount of subcoating (X2; mg), studied at 7.5, 18.75, and 30 mg. The dependent variables surveyed were DEcolon (SCF) (Y1), DEcolon (CTNase-SCF) (Y2), and CRDcolon (Y3). To analyze the factorial design, the original measurement units for the experimental factors (uncoded units) were transformed into coded units. The factor levels were coded as −1 (low), 0 (center), and 1 (high). A full 2×2 design was generated by setting the number of replicates for corner points as three and the number of center points per block as three, subsequently resulting in 15 runs in total.
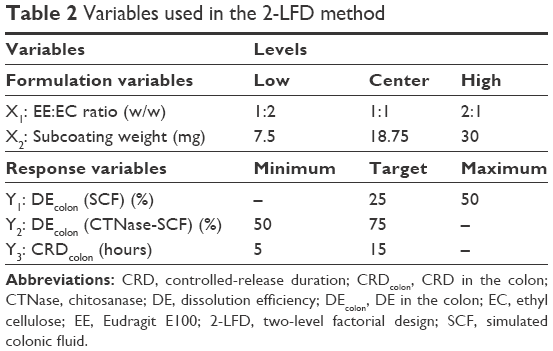 | Table 2 Variables used in the 2-LFD method |
Statistical analyses
All data are expressed as mean ± standard deviation. Statistical significance was verified using Student’s t-test, and differences were considered significant at P<0.05, unless otherwise indicated.
Results and discussion
Core tablet formulation
The core tablets prepared were classified into two categories, based on the presence or absence of CA as an acidifying agent: CA-free tablets (CA0) and CA-added tablets (15 mg [CA15] and 30 mg [CA30]). Tablets were prepared successfully by the conventional procedure, and the physical properties of core tablets are summarized in Table 3. Drug content was in the range of 99.96%–100.32%. Weight variation was within 2% of the total weight. Hardness was in the range of 7.7–8.4 kg/cm2. The inclusion of CA did not alter the tablets’ physical properties. All the core tablets were suitable for a further coating process, since friability was in the range of 0.08%–0.11%. Weight loss of less than 1% in the friability test is generally acceptable.23
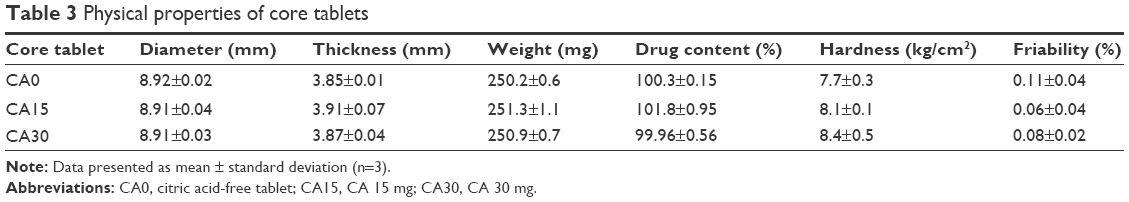 | Table 3 Physical properties of core tablets |
Degree of enteric coating
To find a sufficient degree of enteric coating, core tablets were film-coated with different amounts of a water-insoluble polymer. EL is usually used as a pH-dependent coating polymer that dissolves at over pH 5.5.24 Figure 2A shows the dissolution profiles of uncoated and enterically coated CA0 tablets. Enterically coated CA0 tablets revealed a very low level of dissolution in SGF for a 2-hour period, while uncoated CA0 tablets displayed rapid dissolution of up to about 100% for the initial period of 30 minutes. After 2 hours, regardless of the amount of enteric coating, all enterically coated CA0 tablets showed a dissolution level of about 100% in SIF. In addition, enterically coated CA15 and CA30 tablets represented the same pattern in acid resistibility (data not shown). Liu et al8 reported that the thickness of enteric coating from 5% to 10% in weight ratio led to insignificant differences in drug-release profiles of a CDDS. Although mean gastric emptying time was reported as approximately 2 hours on average, the high variation in gastric retention time in the human population can affect the acid resistibility of enteric coating.25 Therefore, to ensure full protection, the highest level of 6% enteric coating was selected for further experiments.
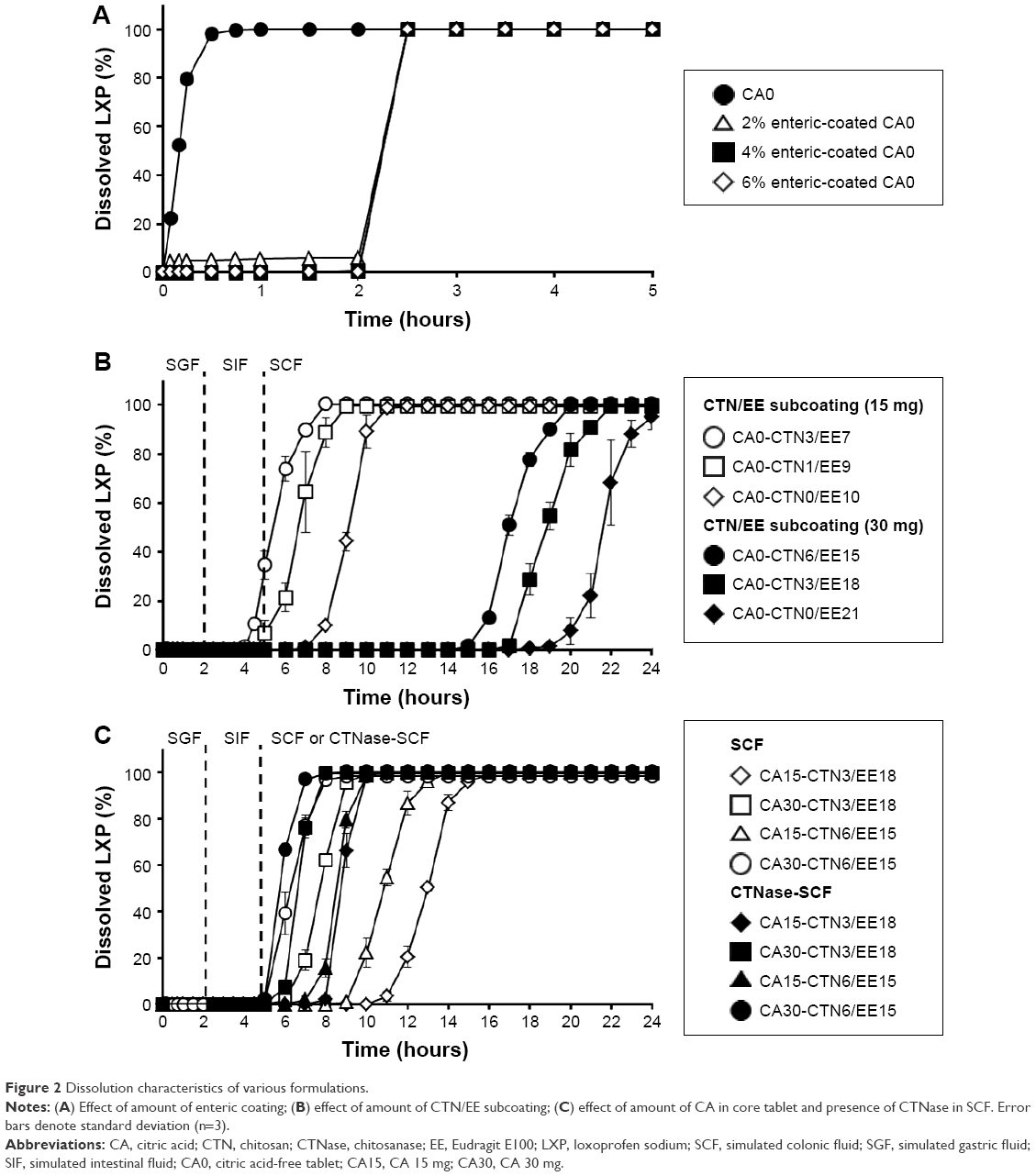 | Figure 2 Dissolution characteristics of various formulations. |
CTN-dispersed EE coating (CTN/EE subcoating)
The core tablets were coated with CTN/EE to retard drug release in SIF and to control drug release in SCF. Figure 2B illustrates the LXP-dissolution profiles of various CA-free DL-CDDSs with different amounts of CTN/EE subcoating. During 5 hours in SGF and SIF media, drug release was not observed, although CA0-CTN3/EE7 containing the lowest amount of EE showed drug release to a lesser extent. After 5 hours in SCF medium, rapid release was observed at a sufficient rate, reaching a plateau at 100%. However, the time to reach the plateau was dependent on the amount of subcoating and composition, ie, initiation of drug release was delayed as the amount of subcoating and ratio of EE content increased: 5–8 hours for 15 mg subcoating versus 16–20 hours for 30 mg subcoating; when arranged by length of delay of drug release, the order was EE7 < EE9 < EE10 < EE15 < EE18 < EE21. For the CA-free DL-CDDS, the content of CTN would not have been a key factor in controlling drug release because the acidifying process was not involved. In contrast, EE contents could have played a major role in release control. This result is consistent with an earlier report that higher amounts of coating containing EE reduced permeability of EE, resulting in the inhibition of water infiltration into the core tablet.26
Meanwhile, to modify the release pattern, CA was included in the core tablet as an acidifying agent. As illustrated in Figure 1 (step C), CA decreased the microenvironmental pH of the subcoating layer and dissolved CTN, thus facilitating drug release through micropore generation. Drug = release profiles from CA-added DL-CDDS are depicted in Figure 2C. Two types of DL-CDDS (CTN3/EE18 and CTN6/EE15) were investigated for further comparison by varying the amount of CA: 15 mg added versus 30 mg added. With regard to SCF, the initiation of drug release was dependent not only on EE and CTN content but also on the amount of CA included. Doubling the amount of CA moved the release curve to the left by about 5 hours, indicating earlier initiation of drug release. Therefore, we suggest that drug release from a DL-CDDS might be controlled by the microclimate-acidification process via dissolution of CTN in the subcoating layer.
Microflora activation by CTNase
CTNase was added to SCF medium to simulate a colonic microflora environment, and dissolution studies were separately performed for the same DL-CDDS. The human colon has an ecologically diverse environment containing over 400 distinct species of bacteria.27 Tozaki et al11,13 reported that CTN could be degraded by rat cecal contents, which are known to contain various microorganisms, due to CTNase secreted by them. In this study, the level of CTNase was maintained at 0.1 units/mL. As shown in Figure 2C, the initiation of drug release in CTNase-SCF medium (closed symbols) was earlier than in SCF (open symbols). This proved the involvement of microflora activation for drug release. As illustrated in Figure 1 (step D), under conditions displayed by the microflora, such as CTNase, digestion of CTN took place by enzyme attack, followed by the generation of microporous channels to facilitate drug diffusion through the layer of polymeric subcoating. This kind of drug release mechanism has been reported in numerous studies.13,28 CTNase hydrolyzes partially acetylated CTN, in which the reducing- and nonreducing-end residues are N-acetyl-glucosamine (GlcNAc) and glucosamine (GlcN), respectively, resulting in the endohydrolysis of β-1,4-glycosidic linkage GlcNAc–GlcN in addition to GlcN–GlcN.29 When CTN–polymer conjugates were incubated with CTNase for 1 hour, complete hydrolysis of CTN into monomers, dimers, and oligomers was accomplished.30
For further comparison, the effect of the CTN/EE content and the amount of CA on drug release was analyzed in terms of DEcolon in the medium of either SCF or CTNase-SCF (Figure 3). As a whole, DEcolon was greatly influenced by the amount of CA and CTN, but independent of the total CTN/EE contents. The formulation CA0-CTN0/EE21 that was coated with EE only showed a poor DEcolon of approximately 10% in both media. However, by increasing CTN content in this formulation, as in the case of CA0-CTN3/EE18 and CA0-CTN6/EE15, DEcolon was improved to 20%–30% and 45%–60% in SCF and CTNase-SCF media, respectively. In comparison, CA-added formulations (CA15 and CA30) showed DEcolon values of more than 60% in all conditions. Unlike the CA30 series, which showed no difference between SCF and CTNase-SCF media-based dissolution, the CA15 series showed a significant difference (P<0.05), indicating a dependence on the microflora environment. Based on the difference in DEcolon values of formulations based in the two different media, which suggested a significant role for microflora activation in drug release, CA15-CTN3/EE18 was selected for further experiments.
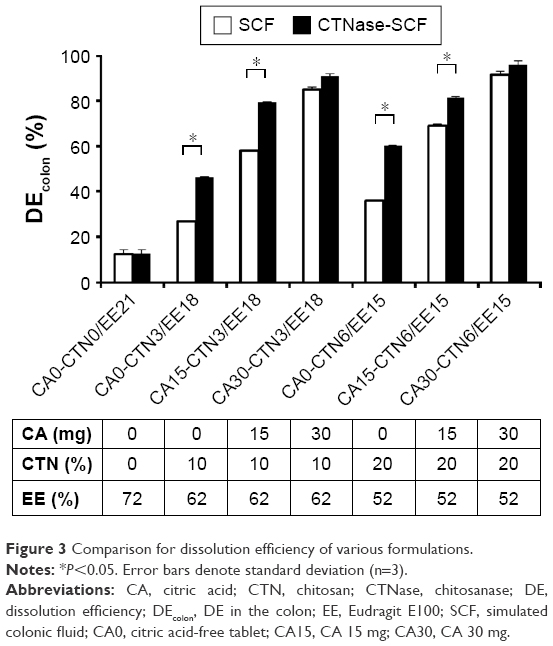 | Figure 3 Comparison for dissolution efficiency of various formulations. |
Modification of CTN/EE coating with EC (CTN/EE/EC subcoating)
Application of 2-LFD
As shown in Figure 2B and C, all DL-CDDSs with CTN/EE subcoating showed a steep sigmoidal curve, indicating rapid drug release once the release was initiated. This pattern was the same in both SCF and CTNase-SCF media, which is undesirable for controlled release for colon targeting. A controlled-release system can reduce dosing frequency and decrease adverse events by providing a reduced maximum concentration, thus resulting in a greater fraction of the drug being delivered to the colon.31 To fulfill this ideal property, the CTN/EE subcoating layer was further modified with EC, which acts as a hydrophobic release retardant, and the resulting design was designated as CTN/EE/EC subcoating. EC is a water-insoluble polymer used extensively as coating material for the preparation of matrix-type controlled-release tablets.32,33 Response variables of DEcolon (SCF) (Y1), DEcolon (CTNase-SCF) (Y2), and CRDcolon (Y3) play a key role in determining the microflora-activated and controlled-release drug delivery. As a result, for the 15 experimental runs, Y1, Y2, and Y3 were 0.5%–96.2%, 2.7%–100%, and 11.8–18.8 hours, respectively (Table 4). Statistical parameters analyzed using the Minitab software are listed in Table 5. All P-values were <0.05, indicating that the effects of the responses were statistically significant up to a 95% confidence level. Multiple regression analyses of the responses are expressed as squared correlation coefficient (R2), adjusted R2, and predicted R2 for validating the analysis.34,35 All the R2, adjusted R2, and predicted R2 values were >94%, indicating satisfactory analysis quality. Substituting the regression coefficients, we derived model equations regarding the level of factors and responses as follows:
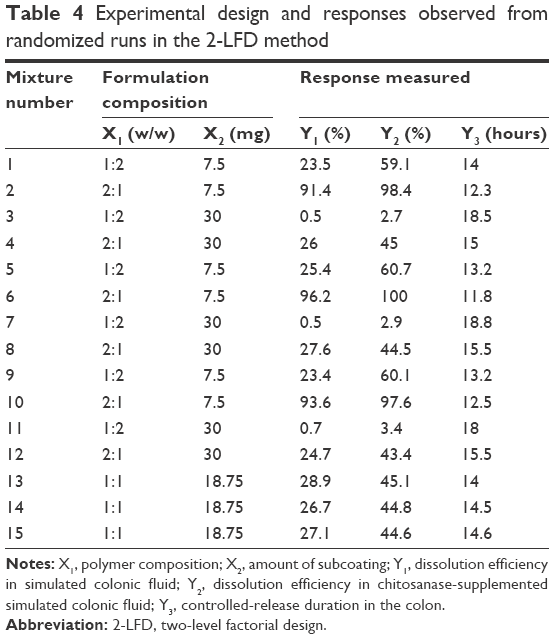 | Table 4 Experimental design and responses observed from randomized runs in the 2-LFD method |
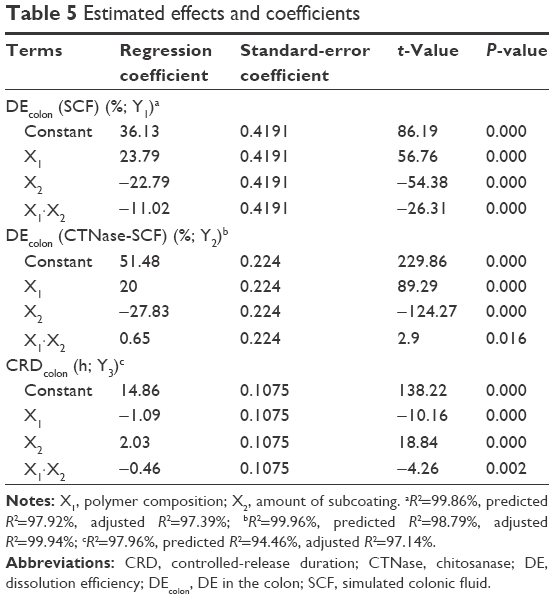 | Table 5 Estimated effects and coefficients |
DEcolon (SCF) (Y1) = 36.13 + 23.79 X1 − 22.79 X2 − 11.02 X1·X2 | (1) |
DEcolon (CTNase-SCF) (Y2) = 51.48 + 20.00 X1 − 27.83 X2 + 0.65 X1·X2 | (2) |
CRDcolon (Y3) = 14.86 − 1.09 X1 + 2.03 X2 − 0.46 X1·X2 | (3) |
An important assumption for statistical analysis of data is a normal distribution.36 As shown in Figure 4, the normal probability plots for standardized residuals formed a straight line, indicating that the results of the experimental runs were distributed normally. The results of Student’s t-test are illustrated in the Pareto charts to represent the relative importance of the individual and interaction effects. The vertical line in the Pareto chart was equal to 2.23, indicating the minimum statistically significant effect magnitude for 95% confidence level (α=0.05). All terms surpassed the critical value, and thus were considered to be statistically significant.
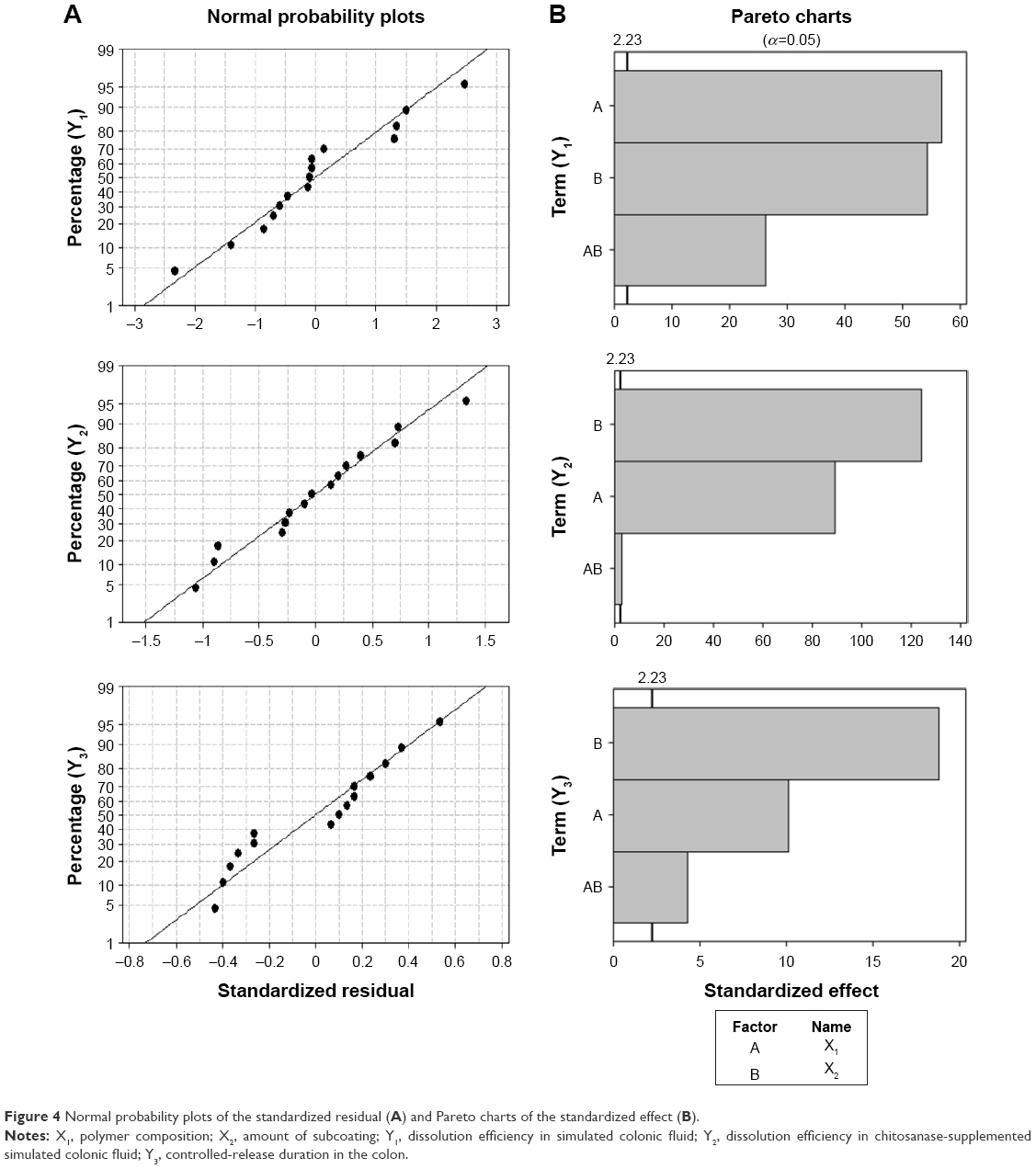 | Figure 4 Normal probability plots of the standardized residual (A) and Pareto charts of the standardized effect (B). |
Effect of independent variables on Y1 and Y2
Figure 5 showed that there were significant main and interaction effects of the polymer ratio of EE:EC (X1) and subcoating weight (X2). Increasing the polymer ratio of EE:EC from 1:2 to 2:1 significantly increased DEcolon (SCF) from 12% to 60%, while increasing the subcoating weight caused DEcolon (SCF) to drop from 59% to 14% (Figure 5A). The negative effect of increasing EC ratio on DEcolon (SCF) may have been due to EC having no permeability or expandability in aqueous medium. The negative effect of increasing the subcoating weight also resulted from an increase in the amount of EC. Meanwhile, the polymer ratio of EE:EC and the subcoating weight produced significant interaction effects on the DEcolon (SCF) (P<0.001) as shown in Table 5 and Figure 5B. If the lines of the interaction-effect plot are not parallel, it is an indication of interactions between the two factors.36 The formulations with 7.5 mg CTN/EE/EC subcoating represented significantly higher increments of DEcolon (SCF) after increasing the EE:EC polymer ratio from 1:2 to 2:1 when compared with 30 mg CTN/EE/EC subcoated formulations. Therefore, at the low weight of CTN/EE/EC subcoating, DEcolon (SCF) was able to be easily controlled by the polymer ratio of EE:EC.
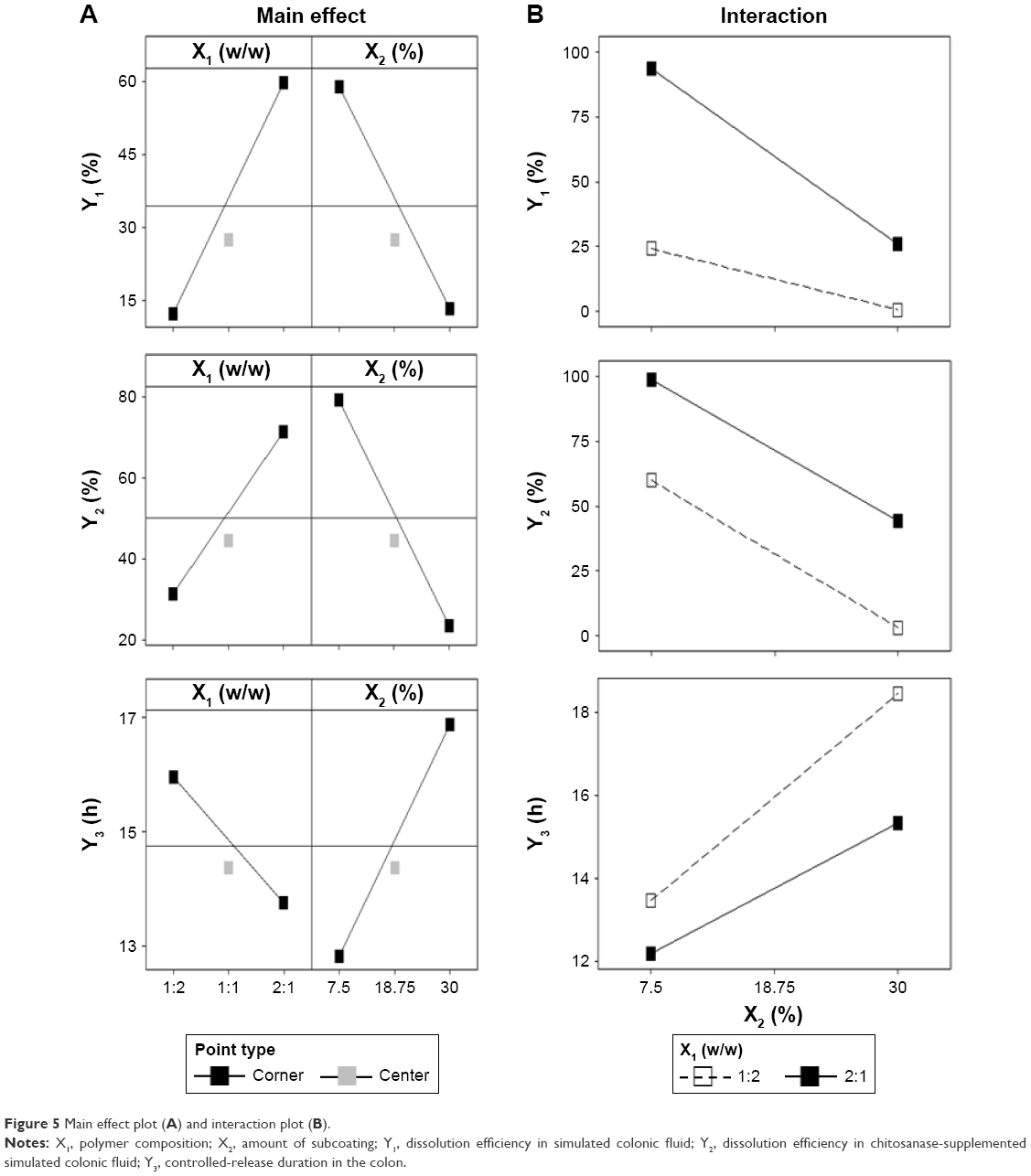 | Figure 5 Main effect plot (A) and interaction plot (B). |
On the other hand, increasing the EE:EC polymer ratio from 1:2 to 2:1 significantly increased DEcolon (CTNase-SCF) from 31% to 71%, while increasing the subcoating weight resulted in a decrease in DEcolon (CTNase-SCF) from 80% to 22%. Under the same polymer ratio and subcoating-weight conditions, the response value of DEcolon (CTNase-SCF) was significantly higher than that of DEcolon (SCF). This might have been due to the fact that CTNase-induced digestion of CTN and microporous channel formation facilitated drug release, regardless of the addition of EC to CTN/EE subcoating. Meanwhile, it was clear that there was no significant interaction effect (P>0.01) between polymer ratio and subcoating weight on DEcolon (CTNase-SCF). This means that the EE:EC polymer ratio and the subcoating weight both affected DEcolon (CTNase-SCF) in a similar manner.
Effect of independent variables on Y3
The main effects and interactions between the polymer ratio and subcoating weight are depicted in Figure 5. In contrast with DEcolon, increasing the polymer ratio of EE:EC from 1:2 to 2:1 significantly decreased CRDcolon from 16 to 13.7 hours, while increasing the subcoating weight caused CRDcolon to increase from 12.9 to 17 hours (Figure 5A). However, the interaction effect between the polymer ratio and subcoating weight was minor (P=0.001). As the EE:EC polymer ratio was varied from 2:1 to 1:2, the formulations containing 30 mg CTN/EE/EC subcoating showed a slightly higher increment in CRDcolon when compared to the 7.5 mg CTN/EE/EC subcoated formulations (Figure 5B). This means that both the subcoating weight and the polymer ratio of EE:EC showed a synergistic effect, while the effect of the subcoating weight was more prominent. By increasing the subcoating weight from 7.5 mg to 30 mg, the amount of EC varied: 5–20 mg and 2.5–10 mg for 1:2 and 2:1 ratios, respectively. Therefore, we suggest that controlled release drug delivery could effectively be regulated by CTN/EE subcoating because of the impermeable property of EC.
Optimized CTN/EE/EC subcoating
Independent variables were simultaneously optimized for responses by using the desirability function. In this study, Y1 was set to be minimized for targeting 25%, whereas Y2 and Y3 were set to be maximized for targeting 75% and 15 hours, respectively. The optimized EE:EC polymer ratio (X1) and the subcoating weight (X2) were 1:1.9 and 7.5 mg, respectively, with a corresponding desirability value of 0.71. The experimental values for all the responses were very close to the predicted values: 24.1% versus 27.6% for DEcolon (SCF), 60% versus 61.9% for DEcolon (CTNase-SCF), and 13.6 hours versus 13.4 hours for CRDcolon, respectively. This similarity proves that the 2-LFD method was accurate and reliable for optimizing CTN/EE/EC subcoating.
Dissolution profiles of the optimized DL-CDDS tablets composed of the inner core tablet (250 mg) containing CA 15 mg, the subcoating layer (7.5 mg) with CTN 1.5 mg, EE 6.4 mg, and EC 12.2 mg, and the outermost enteric coating layer (15 mg) are shown in Figure 6. Drug release was not found in either SGF or SIF media. After replacement of the medium with CTNase-SCF, it showed a controlled-release pattern: approximately 40% at 10 hours and 92% at 24 hours, which was obviously different from the steep sigmoidal release pattern observed in CTN/EE subcoated formulations. In contrast, in SCF the drug-release rate was relatively low: approximately 10% at 10 hours and 42% at 24 hours. Therefore, we conclude that the optimized DL-CDDS could be an efficient platform that incorporates responsiveness to microflora activation in the colon, in addition to microclimate acidification.
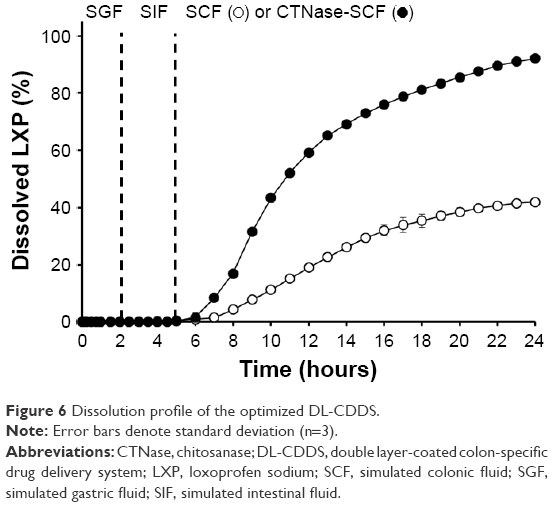 | Figure 6 Dissolution profile of the optimized DL-CDDS. |
Conclusion
A CTN-based DL-CDDS was successfully designed as a novel platform for controlled drug delivery to the colon. The microclimate-acidification technique was employed by introducing CA to the core tablet, and the microflora activation by CTNase was attained using CTN-based polymeric subcoating. Polymeric composition in the ratio of EE:EC and the amount of subcoating were optimized using the 2-LFD method. Consequently, the optimized DL-CDDS showed drug release in a controlled manner by inhibiting drug release in the stomach and intestine, but releasing the drug gradually in the colon.
Acknowledgment
This work was partially supported by the Advanced Technology Center program (10051950), funded by the Ministry of Trade, Industry, and Energy.
Disclosure
The authors report no conflicts of interest in this work.
References
Sinha VR, Kumria R. Polysaccharides in colon-specific drug delivery.Int J Pharm. 2001;224(1–2):19–38. | ||
Pawar PK, Gautam C. Design, optimization and evaluation of mesalamine matrix tablet for colon drug delivery system. J Pharm Invest. 2016;46(1):67–78. | ||
Steed KP, Hooper G, Monti N, Benedetti MS, Fornasini G, Wilding IR. The use of pharmacoscintigraphy to focus the development strategy for a novel 5-ASA colon targeting system (Time Clock system). J Control Release. 1997;49(2–3):115–122. | ||
Muraoka M, Hu Z, Shimokawa T, et al. Evaluation of intestinal pressure-controlled colon delivery capsule containing caffeine as a model drug in human volunteers. J Control Release. 1998;52(1–2):119–129. | ||
You YC, Dong LY, Dong K, et al. In vitro and in vivo application of pH-sensitive colon-targeting polysaccharide hydrogel used for ulcerative colitis therapy. Carbohydr Polym. 2015;130:243–253. | ||
Yang L, Watanabe S, Li J, et al. Effect of colonic lactulose availability on the timing of drug release onset in vivo from a unique colon-specific drug delivery system (Codes). Pharm Res. 2003;20(3):429–434. | ||
McConnell EL, Liu F, Basit AW. Colonic treatments and targets: issues and opportunities. J Drug Target. 2009;17(5):335–363. | ||
Liu H, Yang XG, Nie, SF, et al. Chitosan-based controlled porosity osmotic pump for colon-specific delivery system: screening of formulation variables and in vitro investigation. Int J Pharm. 2007;332(1–2):115–124. | ||
Moore WE, Holdeman LV. Discussion of current bacteriological investigations of the relationships between intestinal flora, diet, and colon cancer. Cancer Res. 1975;35(11 Pt 2):3418–3420. | ||
Simon GL, Gorbach SL. Intestinal flora in health and disease. Gastroenterology. 1984;86(1):174–193. | ||
Tozaki H, Komoike J, Tada C, et al. Chitosan capsules for colon-specific drug delivery: improvement of insulin absorption from the rat colon. J Pharm Sci. 1997;86(9):1016–1021. | ||
Tozaki H, Fujita T, Odoriba T, et al. Colon-specific delivery of R68070, a new thromboxane synthase inhibitor, using chitosan capsules: therapeutic effects against 2,4,6-trinitrobenzene sulfonic acid-induced ulcerative colitis in rats. Life Sci. 1999;64(13):1155–1162. | ||
Tozaki H, Odoriba T, Okada N, et al. Chitosan capsules for colon-specific drug delivery: enhanced localization of 5-aminosalicylic acid in the large intestine accelerates healing of TNBS-induced colitis in rats. J Control Release. 2002;82(1):51–61. | ||
Zambito Y, Di Colo G. Preparation and in vitro evaluation of chitosan matrices for colonic controlled drug delivery. J Pharm Pharm Sci. 2003;6(2):274–281. | ||
Zhang H, Alsarra IA, Neau SH. An in vitro evaluation of a chitosan-containing multiparticulate system for macromolecule delivery to the colon.Int J Pharm. 2002;239(1–2):197–205. | ||
Cerchiara T, Abruzzo A, Parolin C, et al. Microparticles based on chitosan/carboxymethylcellulose polyelectrolyte complexes for colon delivery of vancomycin. Carbohydr Polym. 2016;143:124–130. | ||
Shimono N, Takatori T, Ueda M, Mori M, Higashi Y, Nakamura Y. Chitosan dispersed system for colon-specific drug delivery. Int J Pharm. 2002;245(1–2):45–54. | ||
Xia W, Liu P, Liu J. Advance in chitosan hydrolysis by non-specific cellulases. Bioresour Technol. 2008;99(15):6751–6762. | ||
Kaur G, Rana V, Jain S, Tiwary AK. Colon delivery of budesonide: evaluation of chitosan-chondroitin sulfate interpolymer complex. AAPS PharmSciTech. 2010;11(1):36–45. | ||
Bigucci F, Luppi B, Cerchiara T, et al. Chitosan/pectin polyelectrolyte complexes: selection of suitable preparative conditions for colon-specific delivery of vancomycin. Eur J Pharm Sci. 2008;35(5):435–441. | ||
Khan KA. The concept of dissolution efficiency. J Pharm Pharmacol. 1975;27(1):48–49. | ||
Song WH, Yeom DW, Lee DH, et al. In situ intestinal permeability and in vivo oral bioavailability of celecoxib in supersaturating self-emulsifying drug delivery system. Arch Pharm Res. 2014;37(5):626–635. | ||
Song SH, Chae BR, Sohn SI, et al. Formulation of controlled-release pelubiprofen tablet using Kollidon SR. Int J Pharm. 2016;511(2):864–875. | ||
Moustafine RI, Margulis EB, Sibgatullina LF, Kemenova VA, Van den Mooter G. Comparative evaluation of interpolyelectrolyte complexes of chitosan with Eudragit L100 and Eudragit L100-55 as potential carriers for oral controlled drug delivery. Eur J Pharm Biopharm. 2008;70(1):215–225. | ||
Davis SS, Stockwell AF, Taylor MJ, et al. The effect of density on the gastric emptying of single- and multiple-unit dosage forms. Pharm Res. 1986;3(4):208–213. | ||
Li J, Yang L, Ferguson SM, et al. In vitro evaluation of dissolution behavior for a colon-specific drug delivery system (CODES) in multi-pH media using United States Pharmacopeia apparatus II and III. AAPS PharmSciTech. 2002;3(4):59–67. | ||
Cummings JH, Macfarlane GT. The control and consequences of bacterial fermentation in the human colon. J Appl Bacteriol. 1991;70(6):443–459. | ||
Omwancha W, Kouba C, Yelamanchili S, Neau SH. Colon-specific drug delivery using ethylcellulose and chitosan in the coat of compression-coated tablets. Drug Dev Ind Pharm. 2011;37(8):945–953. | ||
Fukamizo T, Honda Y, Goto S, Boucher I, Brzezinski R. Reaction mechanism of chitosanase from Streptomyces sp. N174. Biochem J. 1995;311(2):377–383. | ||
Smoum R, Rubinstein A, Srebnik M. Chitosan-pentaglycine-phenylboronic acid conjugate: a potential colon-specific platform for calcitonin. Bioconjug Chem. 2006;17(4):1000–1007. | ||
Waterman KC, Sutton SC. A computational model for particle size influence on drug absorption during controlled-release colonic delivery. J Control Release. 2003;86(2–3):293–304. | ||
Iqbal Z, Babar A, Ashra M. Controlled-release naproxen using micronized ethyl cellulose by wet-granulation and solid-dispersion method. Drug Dev Ind Pharm. 2002;28(2):129–134. | ||
Desai J, Alexander K, Riga A. Characterization of polymeric dispersions of dimenhydrinate in ethyl cellulose for controlled release. Int J Pharm. 2006;308(1):115–123. | ||
Yeom DW, Song YS, Kim SR, et al. Development and optimization of a self-microemulsifying drug delivery system for atorvastatin calcium by using D-optimal mixture design. Int J Nanomedicine. 2015;10:3865–3878. | ||
Parhi R, Suresh P, Patnaik S. Formulation optimization of PVA/HPMC cryogel of Diltiazem HCl using 3-level factorial design and evaluation for ex vivo permeation. J Pharm Invest. 2015;45(3):319–327. | ||
Zu Y, Zhang Y, Zhao X, Zhang Q, Liu Y, Jiang R. Optimization of the preparation process of vinblastine sulfate (VBLS)-loaded folate-conjugated bovine serum albumin (BSA) nanoparticles for tumor-targeted drug delivery using response surface methodology (RSM). Int J Nanomedicine. 2009;4:321–333. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.