Back to Journals » ImmunoTargets and Therapy » Volume 14
Development and Characterization of 4A7: A High-Affinity Monoclonal Antibody Targeting Claudin18.2
Authors Wu Y, Tian J, Zhou Y, Zhang R, Gao X, Luo L
Received 22 September 2024
Accepted for publication 24 February 2025
Published 11 March 2025 Volume 2025:14 Pages 189—203
DOI https://doi.org/10.2147/ITT.S494696
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Jadwiga Jablonska
Yahui Wu,1,2,* Juan Tian,1,2,* Yangyihua Zhou,2,* Ran Zhang,1 Xiang Gao,1,2 Longlong Luo1,2
1Hunan Normal University Health Science Center, Changsha, Hunan Province, People’s Republic of China; 2State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing, 100850, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ran Zhang, Hunan Normal University Health Science Center, Changsha, Hunan Province, People's Republic of China, Email [email protected]; Longlong Luo, State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing, 100850, People’s Republic of China, Email [email protected]
Purpose: Claudin18.2 has emerged as a promising therapeutic target due to its high expression in gastric (GC) and pancreatic cancers (PC). However, patients with advanced, unresectable, or metastatic GC or PC face poor prognoses, highlighting the urgent need for more effective Claudin18.2-targeted therapies.
Methods and Results: We developed 4A7, a fully human monoclonal antibody with superior affinity and specificity for Claudin18.2, using a rigorous positive and negative screening strategy to eliminate cross-reactivity with Claudin18.1. In vitro, 4A7 demonstrated significantly enhanced binding activity, as well as robust antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), outperforming IMAB362, a clinical investigational antibody. In vivo, 4A7 exhibited remarkable tumor growth inhibition both as a monotherapy and in combination with anti-mPD-1, achieving superior efficacy compared to IMAB362. Additionally, 4A7 demonstrated a higher degree of humanization and comparable stability, supporting its translational potential.
Conclusion: 4A7 shows great promise as a next-generation therapeutic for Claudin18.2-positive cancers, offering improved efficacy and reduced immunogenicity. This study not only highlights 4A7’s potential to address unmet clinical needs but also provides a foundation for future innovations in monoclonal antibody-based cancer therapy.
Keywords: gastric cancer, Claudin18.2, monoclonal antibody, mAb, targeted therapy
Introduction
Gastric cancer (GC) is the sixth most prevalent cancer worldwide and the third leading cause of cancer-related deaths worldwide.1,2 In 2020, there were an estimated one million new gastric cancer diagnoses, resulting in 769,000 deaths. The asymptomatic nature of early-stage GC means that over 50% of patients are diagnosed at an advanced stage, resulting in a 5-year overall survival (OS) rate of less than 5% and a limited life expectancy of merely eight months.3–5 These statistics underline the urgent need for more effective systemic drug therapies, particularly those involving precision targeting and immunotherapy.6,7
Claudin18, a member of the Claudin family with four transmembrane domains, undergoes selective splicing to produce two isoforms: Claudin18.1, primarily expressed in lung tissue epithelial cells, and Claudin18.2, transiently expressed in gastric epithelial cells and abnormally overexpressed in various cancers, including gastric, pancreatic, esophageal, ovarian, and lung cancers.8,9 Both isoforms consist of 261 amino acids and possess four transmembrane domains and two extracellular loops (ECL1 and ECL2). The critical difference between Claudin18.1 and Claudin18.2 lies in only seven amino acid residues within the ECL1, which poses a significant challenge for designing monoclonal antibodies (mAbs) that selectively recognize Claudin18.2 without cross-reacting with Claudin18.1.10,11
IMAB362 (zolbetuximab) is a Claudin18.2-targeted chimeric IgG1 mAb in clinical development, showing potent antitumor activity.12,13 The FAST trial (NCT01630083) demonstrated that IMAB362 combined with EOX significantly improved progression-free survival (PFS) and OS in Claudin18.2-positive advanced or recurrent GC and gastroesophageal junction (GEJ) cancers.14,15 Additionally, Phase III trials, SPOTLIGHT (NCT03504397) and GLOW (NCT03653507), are currently evaluating IMAB362 in combination with mFOLFOX6 or CAPOX versus standard chemotherapy in Claudin18.2-positive, HER2 (Human epidermal growth factor receptor-2)-negative advanced GC/GEJ cancers.16,17 Despite these promising results, further research is necessary to enhance the affinity of mAbs for Claudin18.2, thereby improving immunotherapy efficacy and reducing the associated side effects.18,19
In this study, we present an effective strategy that combines negative and positive screening to develop antibodies with a high affinity and specificity for Claudin18.2. Among the selected candidates, 4A7 demonstrated superior binding characteristics, antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and peripheral blood mononuclear cells (PBMCs)-based cytotoxicity against Claudin18.2-positive tumor cells compared to IMAB362 in vitro. Additionally, in mouse xenograft models, both 4A7 alone and in combination with anti-mPD-1 showed significantly stronger anti-tumor efficacy than IMAB362 alone or in combination. Furthermore, 4A7 exhibited a higher level of humanization and similar stability compared with IMAB362. Collectively, 4A7 holds promising clinical development potential and offers differentiation advantages as a monoclonal antibody, bispecific antibody, antibody-drug conjugates (ADCs), and chimeric antigen receptor T-cell (CAR-T) therapy.
Materials and Methods
Reagents
IMAB362 and LXY-08 were obtained from our laboratory. APC-anti-human IgG Fc (Biolegend, Cat. No.: 410712), Anti-Human CD3 Functional mAb (Cat. No.: ks10H-3), and Anti-Human CD28 Functional mAb (Cat. No.: ks10H-28) were purchased from CoSinprotein Corporation. Poly-D-lysine solution (5 mg/mL, Cat. No.: C0312), Hoechst 33342 Staining Solution for Live Cells (100X, Cat. No.: C1027), and the Bio-Lumi™ II Firefly Luciferase Reporter Gene Assay Kit (Cat. No.: RG042M) were purchased from Beyotime Corporation. The cytotoxicity detection kit PLUS Lactate dehydrogenase(LDH) was purchased from Roche (Cat. No.: 04744934001). MEM (1x) medium (Gibco, Cat. No.: 2187311), fetal bovine serum (FBS) (MCE, Cat. No.: HY-T1000), Phosphate buffer saline (PBS, Servicebio, Cat. No.: G4200), and Phosphate buffered solution (PBST, 0.1% Tween 20 in PBS) were used in this study. Fluorescence-activated cell sorting (FACS) solution was prepared with 2% serum in PBS.
Cell Lines and Culture
The HEK-293, MC-38, NUGC4, KATOIII, NCI-N87 cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained according to the manufacturer’s instructions. HEK-293 (Claudin18.1), HEK-293 (Claudin18.2), MC-38 (Claudin18.2) and NCI-N87 (Claudin18.2) cell lines were genetically engineered to stably express human Claudin18.1 or Claudin18.2 and cultured in DMEM medium (Gibco) supplemented with 10% FBS. Specifically, the Claudin18.1 and Claudin18.2 gene sequences were cloned into the eukaryotic expression vector pcDNA-3.4, which harbors both a puromycin resistance gene and a green fluorescent protein (GFP) gene. The constructed pcDNA-3.4 plasmid was then transfected into host cells using the Sinofection Transfection Reagent (SinoBiological, Cat. No.: STF02). Stable transfectants expressing Claudin18.1 or Claudin18.2 were selected and enriched through flow sorting. To maintain these stable cell lines, puromycin was added to the DMEM medium. NUGC4 and KATOIII cells, which are human gastric cancer cell lines with exogenous Claudin18.2 expression, were cultured in RPMI-1640 medium (Gibco) supplemented with 10% FBS. Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors and cryopreserved in liquid nitrogen until use. All cell lines were maintained at 37°C in a 5% CO2 atmosphere.
Phage Display Selection
A human synthetic antibody phage library was employed to select Claudin18.2-targeted antibodies with high affinity and no binding to Claudin18.1. The library was amplified in E. coli TG1 and isolated using polyethylene glycol (PEG)/NaCl precipitation to induce phages. For negative selections, HEK-293 (Claudin18.1) cells were washed with ice-cold FACS buffer, blocked with 2% milk powder, and incubated with the phage library (approximately 5.7 × 10¹¹ plaque-forming units) for 1 hour at 30 rpm (the amplitude is Ф26mm) and 4°C in the incubator shaker model (Zhichu General Equipment, Cat. No.: ZQZY-88C). Bound phages were discarded by centrifugal sedimentation, and unbound phages in the supernatant were collected. For positive selections, HEK-293 (Claudin18.2) cells were washed with ice-cold FACS buffer and incubated with the amplified unbound phages (negative selection) for 1 hour at 30 rpm and 4°C. Unbound phages in the supernatant were discarded, and bound phages were eluted using 2.5% 3-cholamidopropyl dimethylammonio 1-propanesulfonate (CHAPS), then amplified in E. coli TG1 and isolated by PEG/NaCl precipitation. Negative and positive screenings were performed for three rounds of panning, and the input and output phages were quantified. Four clones were randomly picked from the third round, and sequencing analysis was performed.
Antibody Expression and Purification
The plasmids of the four clones were transfected into the CHO-S cells (ThermoFisher, Cat. No.: R80007). At 24 h post-transfection, the ExpiFectamine™ CHO Enhancer (ThermoFisher, Cat. No.: A29129) was added. Antibodies were secreted into the culture medium and collected after 8–9 days. The antibodies were purified using protein A-sepharose affinity chromatography. SDS-PAGE under reducing conditions and Coomassie Brilliant Blue G-250 staining were used to analyze the antibodies. Size exclusion chromatography HPLC (SEC-HPLC) was performed to assess purity, using a flow rate of 0.5 mL/min with a mobile phase of 0.15 M Na2HPO₄/NaH2PO₄, pH 7.4. The injection volume was 20 μL, and the column temperature was maintained at 35°C with detection at 280 nm.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted using the FastPure Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, Cat. No.: RC112-01), and cDNA was synthesized with the HiScript III All-in-one RT SuperMix Perfect for qRT-PCR (Vazyme, Cat. No.: R333-01) following the manufacturer’s instructions. Claudin18.1 and Claudin18.2 DNA sequences were amplified from the synthesized cDNA using the following primers:Claudin18.1: forward primer 18.1-F (5′-GGTTTATGGAGGAGCTGCGT-3′) and reverse primer 18.1-R (5′-CGCATAAACCGCTCACGATG-3′). Claudin18.2: forward primer 18.2-F (5′-ATCATCGCCGCCACTTGTAT-3′) and reverse primer 18.2-R (5′-CGCATAAACCGCTCACGATG-3′). qRT-PCR reactions were performed using the THUNDERBIRD® Next SYBR® qRT-PCR Mix (Toyobo, Cat. No.: QPX-201) on the QuantStudio 5 Real-Time PCR System (ThermoFisher). Relative expression levels of Claudin18.2 and Claudin18.1 were normalized to the reference gene GAPDH (Glyceraldehyde 3-phosphate dehydrogenase). The HEK-293 cell line was used as the control, and the ΔΔCT method was applied for data analysis.
Flow Cytometry
To test the binding of the four candidate antibodies to HEK-293 (Claudin18.1) and HEK-293 (Claudin18.2) cells, the antibodies were incubated with 3×105 cells at a concentration of 50 μg/mL for 30 minutes on ice. Cells were then washed with FACS buffer and incubated with APC-anti-human IgG Fc antibody at 4°C for 30 minutes, followed by additional washes with FACS buffer. Binding ability to Claudin18.2-positive cancer cell lines, including HEK-293 (Claudin18.2), MC-38 (Claudin18.2), NUGC4, and KATOIII, was analyzed by incubating cells with a threefold gradient dilution of antibodies starting from 60 µg/mL. Fluorescence measurements were acquired using a FACS Celesta Flow Cytometer (BD Biosciences) and analyzed with FlowJo software to determine mean fluorescence intensities (MFI).
Laser Scanning Confocal Microscopy and Live Cell Imaging
The antibodies were labeled with Percp-cy5.5. HEK-293 (Claudin18.1), HEK-293 (Claudin18.2), MC-38 (Claudin18.2), and NUGC4 cells were seeded in 15 mm glass-bottom culture dishes (Nest, China, 801002) pretreated with poly-D-lysine and cultured overnight. Percp-cy5.5-conjugated antibodies were added at 10 µg/mL and incubated at 37°C for 1 hour. The cells were washed twice with PBS and stained using standard protocols for Hoechst (Beytime, China, C1027). The images were deconvoluted and processed using NIS-Element (v5.21.03) and Fiji software (v2.1.0).
Cytotoxicity Assay
PBMCs were used as effector cells and MC-38 (Claudin18.2), NUGC4, and KATOIII cells were used as target cells in the PBMC-based assays. Briefly, 2.5 × 106 PBMCs were mixed with 2.5 × 105 target cells in MEM medium to achieve an effector-to-target (E:T) ratio of 10:1, with a final volume of 100 µL in 96-well white plates. Test antibodies were serially diluted in MEM medium and added to the assay system, followed by 4-hour incubation. Culture supernatants were collected to quantify cytotoxicity based on LDH activity (Roche).
ADCC and ADCP Assays
A reporter-based surrogate ADCC/ADCP bioassay was conducted using Jurkat cells (Pricella, Cat. No.: CL-0129) engineered to express FcγRIIIa or FcγRIIa and a nuclear factor of activated T cells (NFAT) response element driving firefly luciferase expression. Effector cells (2×104) were mixed with target cells (1.2×105), including MC-38 (Claudin18.2), NUGC4, and KATOIII, to achieve an E:T ratio of 1:6 in 50 µL of 96-well white plates. Test antibodies were serially diluted and added to the assay system, followed by a 6-hour incubation. Bright-Lumi™ II Firefly Luciferase Assay Kit (Beyotime, RG042M) was used to measure luminescence with a GloMax® Navigator Microplate Luminometer (Promega).
In vivo Treatment Efficacy Model
All mice were purchased from Vital River Laboratories (Beijing, China) and were maintained under specific pathogen free conditions. For monoclonal antibody treatment efficacy, 5×106 NCI-N87 (Claudin18.2) cells in 100 µL PBS were injected subcutaneously into the left flank of female NOD-SCID mice (6–8 weeks old). When tumor volumes reached approximately 100 mm³, mice were randomly divided into three groups and treated biweekly with 4A7 (15 mg/kg), IMAB362 (15 mg/kg), or PBS as a control, every 4 days for a total of 8 doses. Tumor volumes were monitored every 4 days and calculated using the formula (length × width²)/2. For combination treatment efficacy, female C57BL/6 mice (6–8 weeks old) were subcutaneously inoculated with MC-38 (Claudin18.2) cells (1×106 cells/mouse). Biweekly treatments were administered for 3 weeks with 4A7 (10 mg/kg), IMAB362 (10 mg/kg), anti-mPD-1 (3 mg/kg), or combinations of 4A7 or IMAB362 with anti-mPD-1 (10 mg/kg + 3 mg/kg). PBS was used as a control. Tumor volumes were monitored twice a week, and mice were sacrificed on day 24 post-tumor injection to record tumor weights.
Tox Studies
Five groups of BALB/C mice (6–8 weeks old, n = 3 per group) with similar body weights were used for the toxicity studies. Two groups received a single intraperitoneal (i.p). dose of 80 mg/kg or 10 mg/kg of the 4A7 antibody, while two additional groups received the same doses of the IMAB362 antibody. The fifth group, serving as the control, received an equivalent volume of PBS. Body weight was monitored daily for 14 days following administration and also recorded prior to dosing. At the end of the 14-day observation period, the mice were euthanized via carbon dioxide inhalation. The stomach, liver, and kidneys were collected and fixed in 4% formaldehyde for subsequent histopathological analysis.
Antibody Stability by UNCLE
Protein stability of the antibodies was evaluated using intrinsic fluorescence and static light scattering (SLS) analysis on the UNCLE instrument (Unchained Labs, Pleasanton, CA, USA). Thermal melting midpoint (Tm) was determined by intrinsic fluorescence (Blue-473 nm, filter 4), and thermal aggregation onset (Tagg) was measured by SLS (UV-226 nm, filter 3). Particle size and polydispersity were determined by dynamic light scattering, represented by the parameters Z-Ave.Dia, PDI, and Pk1 Mode Dia. Z-Ave.Dia reflects the average particle size, Pk1 Mode Dia represents the main peak value of particle size (usually approximately 10 nm for proteins), and PDI indicates the uniformity of the solution, with values below 0.2 indicating relatively uniform particle size. Data were concurrently collected and analyzed with UNCLE software, and values were directly exported.
Degree of Humanness of Antibody Sequence
The degree of humanness of the antibody sequences was calculated using abYsis (http://www.abysis.org/abysis/index.html) according to the method of described Abhinandan and Martin (2007). The “raw humanness” score was determined by scanning each human sequence against all other human sequences and calculating the mean sequence identity. This score was then standardized, and each sequence was assigned a final humanness score expressed as a number of standard deviations from the mean. Mouse sequences were compared against the human library similarly. The sequences of 4A7 and IMAB362 were also scanned against the human sequence library, and scores were calculated.
Statistical Analysis
All plots were created using the GraphPad Prism software (version 9.0), which was also used for statistical analysis. In vitro experiments were conducted in triplicate, and values were presented as mean ± SD (standard deviation). Individual or multiple group comparisons were performed using the 2-tailed unpaired Student’s t-test. Tumor volumes, blood values, and biochemical indices among the treatment groups were compared using two-way ANOVA. Statistically significant differences were defined as P < 0.05, with different levels of significance set as *P < 0.05, **P < 0.01, and ***P < 0.001.
Data Availability
The data generated in this study are available upon request from the corresponding author.
Results
Selection of Fully Human Antibodies Targeting Claudin18.2 Through Positive and Negative Screening
The close similarity between Claudin18.1 and Claudin18.2, differing by only seven amino acid residues in the ECL1 (Extracellular loop 1), presents a significant challenge in developing monoclonal antibodies (mAbs) that specifically recognize Claudin18.2 without cross-reacting with Claudin18.1. To address this, we employed a rigorous positive and negative screening strategy. Initially, a fully human phage display library was incubated with HEK-293 cells expressing Claudin18.1, and phages binding to Claudin18.1 were removed by centrifugation. The remaining phages were then incubated with HEK-293 cells expressing Claudin18.2 to enrich for phages specifically recognizing Claudin18.2 (Figure 1A). After three rounds of selection, there was a significant increase in the output-to-input ratio, indicating an enrichment of positive clones (Figure 1B).
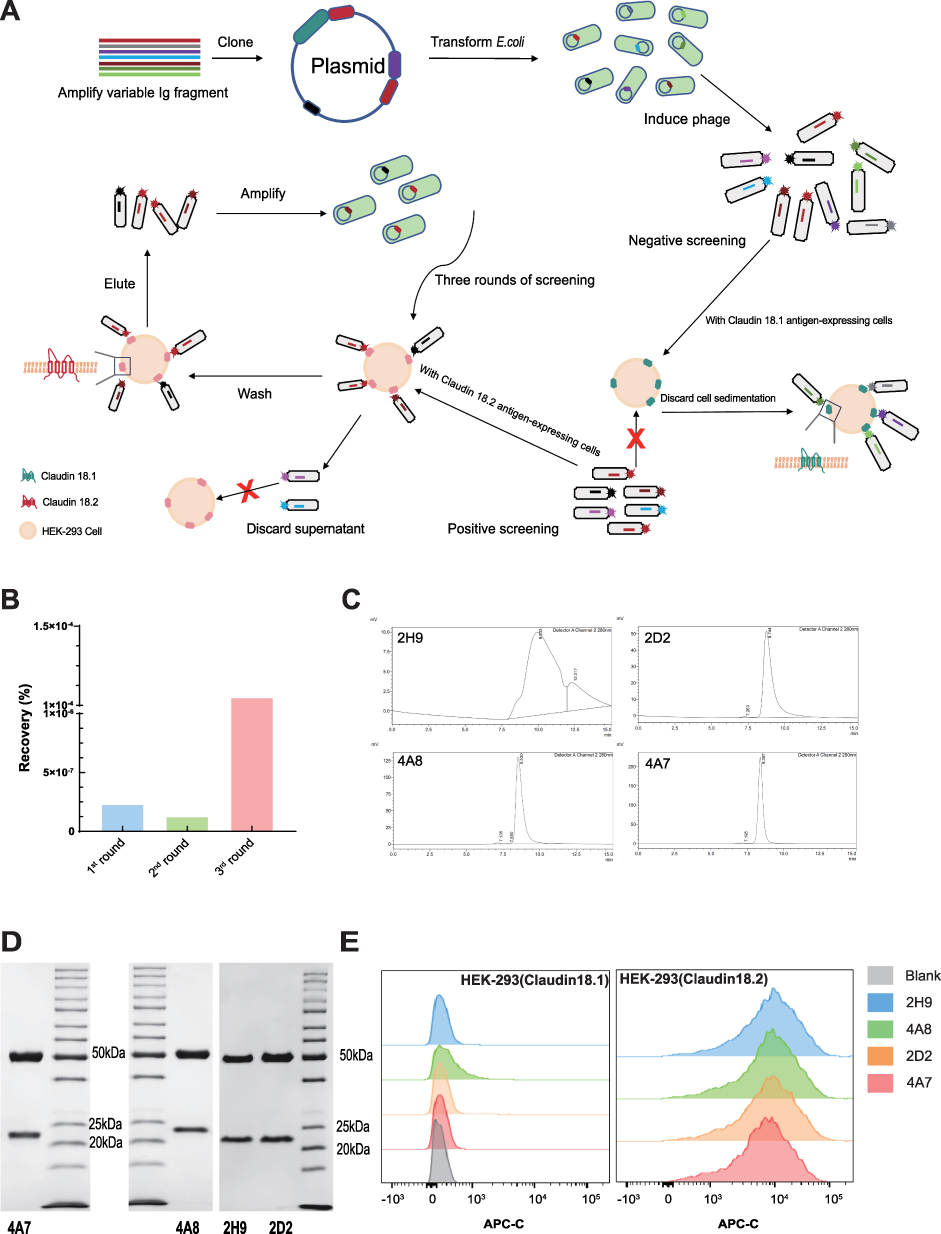 |
Figure 1 Antibody screening for specific targeting Claudin18.2. (A) Schematic diagram of negative/positive screening based on a fully human phage antibody library. (B) Comparison of the recovery ratios (output rate/input rate) in different rounds of phage bio-panning. (C) The full view of the SEC-HPLC UV chromatogram of the four clones. (D) SDS-PAGE analysis confirmed the four clones matching expected sizes under reducing condition. (E) Determination the specificity of the four candidate antibodies to Claudin18.2 by flow cytometric analysis. |
From the positive clones, four candidates (4A7, 4A8, 2D2, and 2H9) were selected. Upon expression and purification, all four clones exhibited a homogeneous single band at the expected size (approximately 25 kDa and 50 kDa under reducing conditions) as determined by Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Figure 1D). Except for clone 2H9, the other three clones (4A7, 4A8, and 2D2) demonstrated high purity (over 95%) as assessed by Size exclusion chromatography (SEC-HPLC) (Figure 1C).
The specificity of the four clones for Claudin18.2 versus Claudin18.1 was further evaluated using flow cytometry. Three clones (2H9, 4A7, and 2D2) exhibited strong binding activity to Claudin18.2 with no binding to Claudin18.1 (Figure 1E). Based on expression levels, purity, and cross-binding activity, 4A7 emerged as the most promising candidate for further studies.
4A7 Exhibits Superior Binding Activity to Claudin18.2 Compared to IMAB362
As depicted in Supplementary Figure 1A, neither 4A7 nor IMAB362 exhibited binding to HEK-293 cells overexpressing Claudin18.1, confirming their specificity. The binding activities of 4A7 and IMAB362 were subsequently evaluated using three Claudin18.2-overexpressing cell lines (HEK-293 (Claudin18.2), MKN-45-Claudin18.2, and MC-38 (Claudin18.2) and two Claudin18.2-positive gastric cancer cell lines (NUGC4 and KATOIII). The relative expression levels of Claudin18.1 or Claudin18.2 in each cell line were evaluated by qRT-PCR, and the results are shown in Supplementary Figure 1D. Figure 2A–E indicate that 4A7 significantly outperformed IMAB362 in binding to Claudin18.2 on the cell membrane surface. The half-maximal effective concentration (EC50) values for both antibodies were calculated across the five cell lines (Figure 2F). 4A7 demonstrated a significantly higher binding activity, with EC50 values ranging from 3- to 17-fold lower than those of IMAB362, underscoring its superior affinity for Claudin18.2.
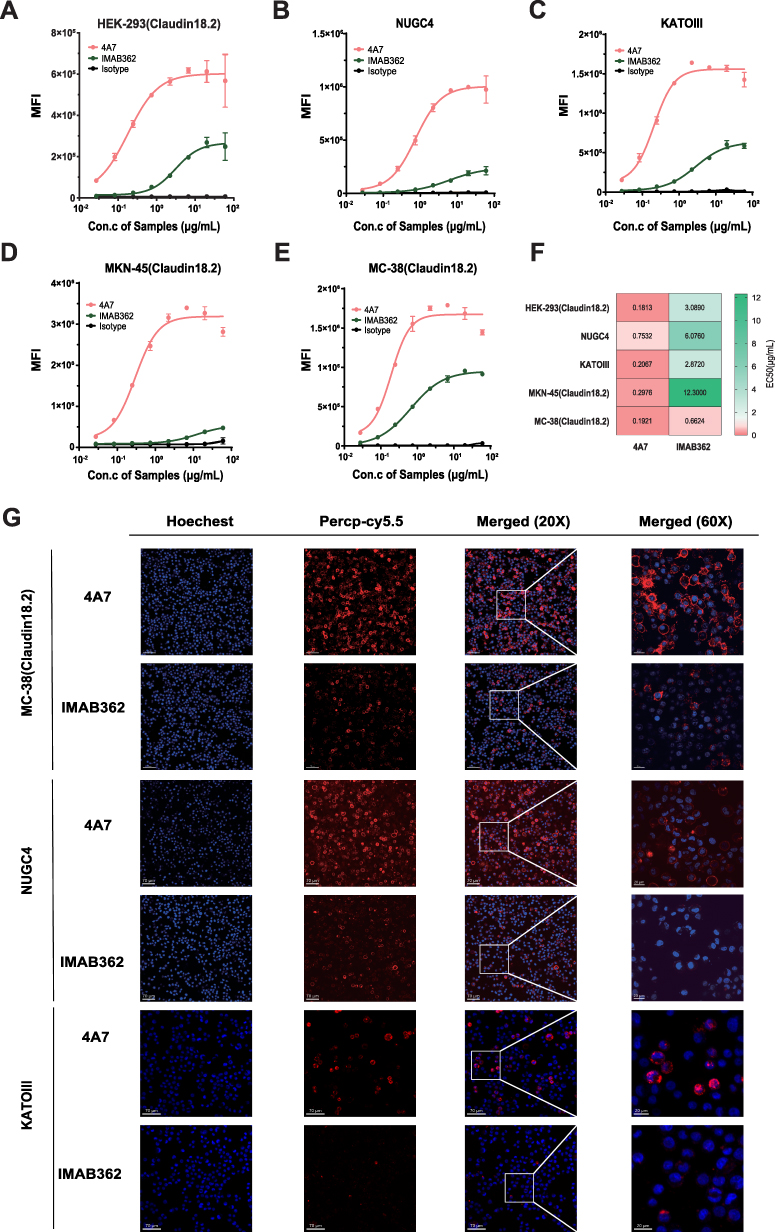 |
Figure 2 Binding activity of 4A7 and IMAB362 to Claudin18.2-positive cell lines. (A–E) Flow cytometry was utilized to assess the binding activity of 4A7 and IMAB362 to three human Claudin18.2-overexpressing cell lines—HEK-293 (Claudin18.2), MKN-45 (Claudin18.2), and MC-38 (Claudin18.2)—as well as to two Claudin18.2-positive gastric cancer cell lines, NUGC4 and KATOIII. Human IgG1 isotype served as the negative control. MFI denotes median fluorescence intensity. Values are presented as mean ± SD from one representative of two independent replicates. Con., concentration. (F) Heatmap of EC50 values for the binding activities of IMAB362 and 4A7 to the five cell lines. Colors range from red to green, the redder the color, the higher the antibody binding activity, and conversely, The darker the green color change, the worse the antibody binding activity. (G) Confocal immunofluorescence was employed to visualize the binding of 4A7 and IMAB362 to MC-38 (Claudin18.2), NUGC4 and KATOIII cells. Nuclei are stained with DAPI (blue); red indicates 4A7, IMAB362, or human IgG1 isotype conjugated with Percp-Cy5.5. Scale bars represent 70 µm and 20 µm. |
Further validation of the binding characteristics was performed using confocal immunofluorescence microscopy (Figure 2G). Cells stained with 4A7-Percp-cy5.5 exhibited strong red fluorescence signals on the membranes of MC-38 (Claudin18.2), NUGC4 and KATOIII cells, whereas IMAB362-Percp-cy5.5 produced notably weaker signals. Similar results were observed for HEK-293 (Claudin18.2) cells. No significant fluorescence was detected when the cells were stained with a negative control antibody or incubated with HEK-293 (Claudin18.1) cells (Supplementary Figure 1B and 1C). These findings confirm that 4A7 has a markedly higher binding activity to Claudin18.2 compared to IMAB362.
4A7 Demonstrates Superior Fc Receptor-Mediated Effector Functions Compared to IMAB362
The fragment of the antigen binding (Fab) region of an antibody achieves targeting by binding to the antigen, while the fragment crystallizable (Fc) region mediates immunological activity by recruiting innate immune cells via Fc gamma receptors (FcγRs), facilitating functions such as ADCC and ADCP.20,21 To compare the ADCC and ADCP activities of 4A7 and IMAB362, an engineered ADCC FcγRIIIa (158V)/FcγRIIa Jurkat Luciferase Reporter System was employed.
Both 4A7 and IMAB362 exhibited dose-dependent ADCC activity across the three target cell lines (NUGC4, KATOIII, and MC-38 (Claudin18.2). However, 4A7 demonstrated significantly higher ADCC activity, approximately 11- to 36-fold greater, with IC50 (half-maximal inhibition concentration) values of 0.06 μg/mL, 0.04 μg/mL, and 0.05 μg/mL, respectively, compared to IMAB362’s IC50 values of 2.15 μg/mL, 0.43 μg/mL, and 0.76 μg/mL (Figure 3A).
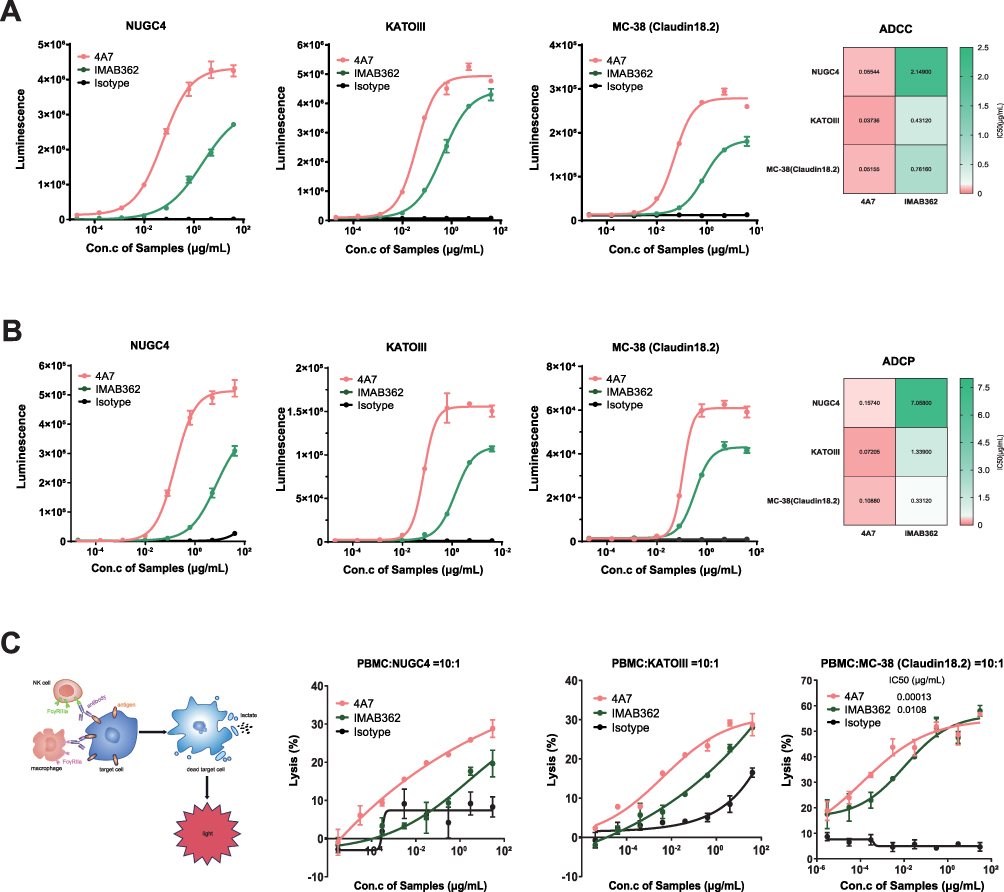 |
Figure 3 Fc receptor-mediated effector functions of 4A7 and IMAB362 to Claudin18.2-positive cell lines. (A) Dose-dependent ADCC activities of 4A7 and IMAB362 against NUGC4, KATOIII and MC-38 (Claudin18.2). Heatmap of IC50 values on the right for the ADCC activities of IMAB362 and 4A7 to the three cell lines. (B) Dose-dependent ADCP activities of 4A7 and IMAB362 against NUGC4, KATOIII and MC-38 (Claudin18.2). Heatmap of IC50 values on the right for the ADCP activities of IMAB362 and 4A7 to the three cell lines. (C) Dose-dependent cytotoxicity potential of PBMCs co-cultured with the cancer cells. The experimental principles diagram on the left. The results were measured LDH released from the target cells. All Data are plotted as mean ± SD of 2 technical replicates. Con., concentration. |
Similarly, in ADCP assays, 4A7 exhibited superior activity, approximately 3- to 44-fold higher, with IC50 values of 0.16 μg/mL, 0.07 μg/mL, and 0.11 μg/mL, compared to IMAB362’s IC50 values of 7.06 μg/mL, 1.34 μg/mL, and 0.33 μg/mL (Figure 3B).
To further evaluate the targeted killing activity of 4A7 and IMAB362, traditional cell-based assays and lactate dehydrogenase (LDH) release assays were conducted using peripheral blood mononuclear cells (PBMCs) co-incubated with tumor cells (Figure 3C). In these assays, 4A7 exhibited significantly higher cytotoxicity, with an IC50 of 0.00013 μg/mL, compared to IMAB362’s IC50 of 0.011 μg/mL against MC38-Claudin18.2 cells. Similar trends were observed in NUGC4 and KATOIII cells, with 4A7 showing statistically significantly higher cytotoxicity (****P < 0.0001) (Figure 3C).
Collectively, these results indicate that 4A7 exhibits markedly higher efficacy in mediating ADCC, ADCP, and PBMC-based cytotoxicity against Claudin18.2-positive tumor cell lines compared than IMAB362, establishing 4A7 as a superior candidate for therapeutic application.
4A7 Alone or in Combination with Anti-mPD-1 Exhibits Superior Anti-Tumor Activity Compared to IMAB362
To evaluate the therapeutic efficacy of 4A7, we first compared its effects as a monotherapy against IMAB362 in a Claudin18.2-overexpressing human gastric cancer cell line (NCI-N87) xenograft model in NOD-SCID mice (Figure 4A). The results demonstrated that mice treated with 4A7 exhibited significant tumor growth inhibition (TGI) compared with the PBS group (TGI: 42.34%, ***P < 0.001). In contrast, IMAB362 treatment did not result in a significant difference in tumor growth compared with the PBS group (TGI: 17.85%, P > 0.05) (Figure 4B). Notably, 4A7 treatment led to higher tumor suppression efficiency than IMAB362 (*P < 0.05).
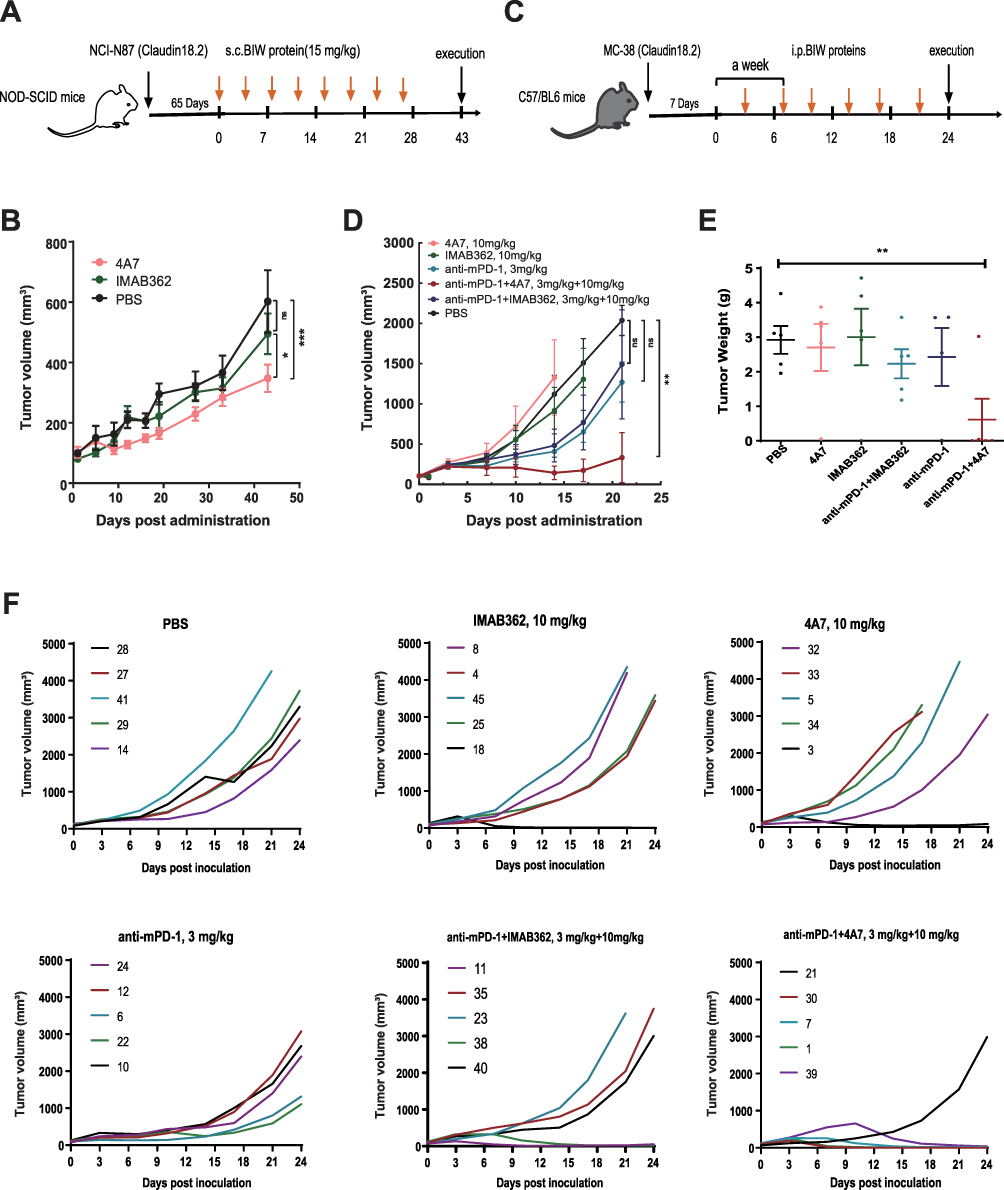 |
Figure 4 Anti-tumor activity of 4A7/IMAB362 alone and the combination with anti-mPD-1 in the mouse xenograft model. (A) Treatment schedule of 4A7 and IMAB362 at 15 mg/kg in NOD-SCID mice, using PBS as control group. Each group consisted of five mice (B) Tumor volumes of mice which were monitored every 4 days since the first treatment. (C) C57/BL6 mice were implanted with 1×106 MC-38 (Claudin18.2) cells to establish mouse xenograft models. Seven days after tumor inoculation, mice were intraperitoneally (i.p.) treated with 4A7, IMAB36, 4A7+ anti-mPD-1 or IMAB362 + anti-mPD-1 twice weekly. PBS were designated as control. Each group consisted of five mice. (D) Tumor volumes of mice which were monitored every 4 days since the first treatment. (E) Tumor weights of each mouse at the experiment’s end. (F) Individual tumor growth curves of mice in different treatment groups. Tumor volumes were calculated as (length × width2)/2. Mean tumor volumes ± SD are shown and compared between 2 groups as indicated by Two-way ANOVA test. ***P < 0.001; **P < 0.01 *P < 0.05. |
Given the limited clinical efficacy of monoclonal antibodies (mAbs) as monotherapies, combinations of chemotherapy or immunotherapy agents often enhance the therapeutic outcomes. We assessed the anti-tumor activity of 4A7 and IMAB362 in combination with anti-mPD-1 at equivalent doses. The treatment regimen included six groups: PBS, 4A7 (10 mg/kg), IMAB362 (10 mg/kg), anti-mPD-1 (3 mg/kg), anti-mPD-1 + 4A7 (3 mg/kg + 10 mg/kg), and anti-mPD-1 + IMAB362 (3 mg/kg + 10 mg/kg) (Figure 4C). Changes in tumor volume over time revealed that neither 4A7 nor IMAB362 monotherapy effectively suppressed tumor growth compared to the PBS group. However, the anti-mPD-1, anti-mPD-1 + IMAB362, and anti-mPD-1 + 4A7 groups all exhibited significant tumor suppression (TGI: 37.68%, 26.78%, and 83.68%, respectively; P > 0.05, P > 0.05, and **P < 0.01) (Figure 4C and Supplementary Figure 2A). Importantly, the combination of anti-mPD-1 + 4A7 demonstrated superior tumor growth inhibition compared to anti-mPD-1 alone (*P < 0.05), indicating a potent synergistic effect. At the end of the experiment, the tumor weights confirmed that only the anti-mPD-1 + 4A7 group had significantly smaller tumors than the PBS group (**P < 0.01), with no significant differences observed in the other groups (Figure 4E). Individual tumor growth curves showed that the anti-mPD-1 + 4A7 combination significantly impeded tumor proliferation, achieving complete regression in 80% of the mice (4 out of 5), compared to 40% (2 out of 5) in the anti-mPD-1 + IMAB362 group. In the 4A7 and IMAB362 monotherapy groups, complete regression was observed in only 20% of the mice (1 out of 5), and no complete regression occurred in the anti-mPD-1 monotherapy group (Figure 4F). Additionally, there were no significant differences in the body weight between the five groups suggesting that combination treatment is a safe and promising therapeutic strategy (Supplementary Figure 2B). Meanwhile, We further evaluated the potential toxicity of 4A7 and IMAB362 in the health mouse model, no significant weight loss was observed in the treated and untreated animals, and No changes in normal structure and infiltration of inflammatory cells were observed on the representative micrographs of the histopathological analyses of tissue sections of the stomach, kidneys and livers (Supplementary Figure 3).
These results collectively indicate that 4A7, whether as a monotherapy or in combination with anti-PD-1 antibodies, demonstrates superior in vivo anti-tumor activity compared to IMAB362, highlighting its potential as a more effective therapeutic agent.
4A7 Exhibits a Higher Degree of Humanness and Similar Stability Compared to IMAB362
To evaluate the degree of humanness of 4A7 and IMAB362, we calculated the Z-scores, a metric based on the percentage sequence identity between antibody sequences and human germline sequences. The results showed that both the heavy and light chains of the 4A7 had higher humanization scores compared to IMAB362, indicating a greater degree of humanness (Figure 5A).
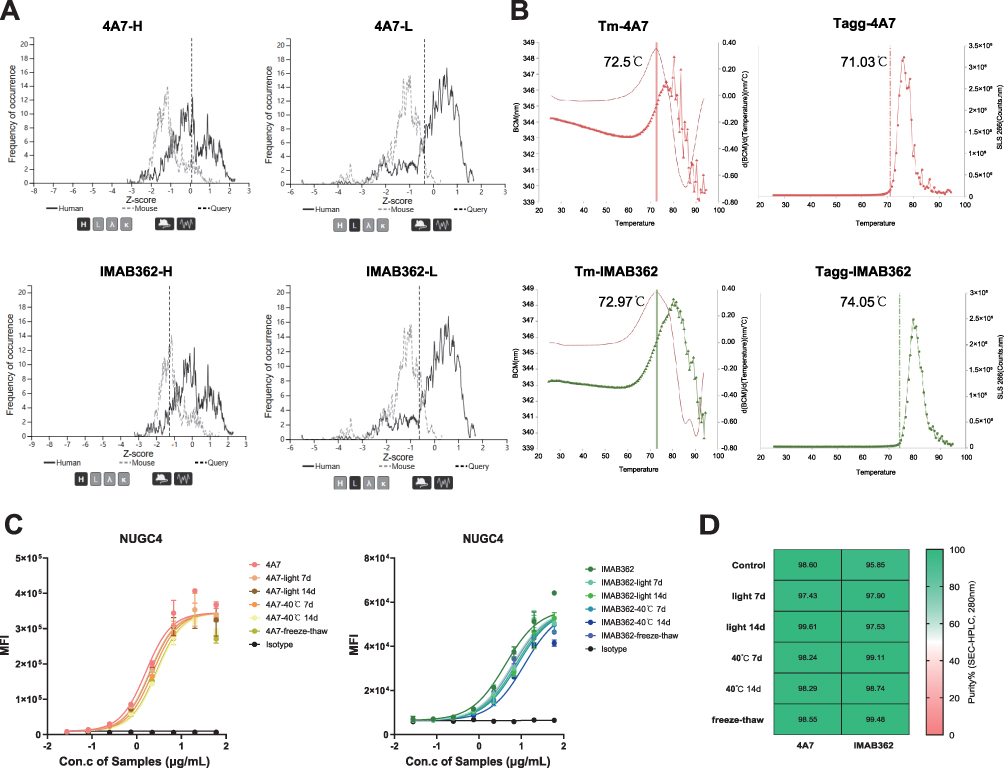 |
Figure 5 The humanized degree and the effect of accelerated storage conditions on the stability of 4A7 and IMAB362. (A) the Z-score analysis for 4A7 and IMAB362 indicated by vertical dashed lines overlaid on the distribution of humanness scores for (A and C) heavy chains and (B and D) lambda light chains. A distribution of scores for expressed human sequences and expressed mouse sequences were displayed by solid line and dotted line. (B) The fluorescence and scattered light spectra (SLS) represented the Tm and Tagg of 4A7 and IMAB362 respectively. (C and D) The binding affinity (C) and SEC-HPLC (D) of 4A7and IMAB362. The antibodies were applied to HPLC after ten freeze-thaw cycles and incubation at 40 °C or daylight for 7 and 14 days. |
Next, we assessed the thermal stability of 4A7 and IMAB362 using the UNCLE instrument (Unchained Labs, CA, USA). The midpoint transition temperatures (Tm) for both antibodies were approximately 70°C, indicating similar thermal stability (Figure 5B). Additionally, the aggregation temperature onset (Tagg), evaluated by static light scattering (SLS), showed comparable thermal stability for 4A7 and IMAB362, with Tagg values between 72–74°C (Figure 5B). Further analysis of particle size and polydispersity indicated that both 4A7 and IMAB362 had initial particle sizes of approximately 10 nm, with polydispersity indices (PDI) below 0.2, suggesting uniform particle size distribution (Supplementary Table 1).
To examine the robustness of these antibodies under accelerated conditions, we subjected them to ten freeze-thaw cycles and incubated them in light or at 40°C for 7 and 14 days. Subsequent binding affinity assays showed no significant changes in the binding activity of either antibody to target cells (Figure 5C). SEC-HPLC analysis confirmed no occurrence of protein aggregation under these conditions (Figure 5D and Supplementary Figure 3).
In summary, the 4A7 antibody not only exhibits a higher degree of humanness compared to IMAB362, but also demonstrates comparable thermal stability and robustness under stress conditions, making it a promising candidate for clinical application.
Discussion
Claudin18.2, a transmembrane protein, has emerged as a promising target for cancer therapy due to its selective and stable expression in specific tumor tissues, including gastric, pancreatic, esophageal, ovarian, and lung cancers.10,18,22 In normal healthy tissues, Claudin18.2 is predominantly localized within the tight junctions (TJ) of gastric mucosal cells. However, the epitopes of Claudin18.2 in the TJ supramolecular complex are largely inaccessible to monoclonal antibodies under normal conditions. During carcinogenesis, disrupted cell polarity exposes these previously hidden epitopes, allowing monoclonal antibodies to bind effectively.8–10 Consequently, for monoclonal antibodies targeting Claudin18.2, their distribution in patients is primarily confined to tumor tissues or subject to degradation. Despite the progress of IMAB362, a chimeric antibody (~70% human) approved in Japan for Claudin18.2-positive GC, its relatively low affinity for Claudin18.2 underscores the need for mAbs with higher degrees of humanization and targeting efficiency.6,16
In this study, a large-capacity, fully human phage synthetic antibody library was employed, followed by a rigorous negative and positive selection strategy to ensure high specificity for Claudin18.2 while avoiding cross-reactivity with Claudin18.1. This approach led to the development of 4A7, which demonstrated significantly stronger in vitro and in vivo activities compared to IMAB362. Specifically, 4A7 exhibited 3- to 17-fold stronger binding to Claudin18.2-positive cell lines and up to 40 times greater ADCC and ADCP activities. Additionally, in vivo experiments indicated superior TGI and reduced tumor volumes in a human tumor xenograft mouse model treated with 4A7.
Historically, the first-line treatment for advanced or metastatic GC/GEJ has been fluoropyrimidine and platinum-based chemotherapy.7,23 However, these treatments have limitations, and there is an unmet need for improved survival outcomes.19,24 Recent advancements in GC treatment have highlighted the potential of immunotherapy and biomarker-directed therapies.4,25 Anti-HER2 therapies combined with chemotherapy are now the standard treatment for advanced GC, although the heterogeneity of GC limits the number of patients who benefit from these treatments.19,26 Claudin18.2-targeted therapies offer a new approach, particularly for HER2-negative GC patients.11,27. Furthermore, the NCT01630083 study demonstrated that Claudin18.2 was expressed at ≥ 2+ levels in over 40% of tumor cells in patients with advanced or recurrent gastric cancer and gastroesophageal junction (GEJ) cancer. In these patients, Claudin18.2-targeted therapies combined with chemotherapy showed improved PFS and OS outcomes.15 Similarly, the MONO study, a multicenter phase IIa trial, reported significant therapeutic efficacy of Claudin18.2-targeted therapies in patients with recurrent or refractory advanced gastric adenocarcinoma.28 Additionally, immune checkpoint inhibitors (ICIs), such as nivolumab combined with chemotherapy, are increasingly being used as first-line treatments.19,25,29
Combining Claudin18.2-targeted therapy with PD-1 immune checkpoint inhibitors (ICIs) holds significant therapeutic potential. Preclinical studies have suggested that Claudin18.2-targeted therapies can enhance the immune response by promoting T-cell infiltration and antigen presentation.18,22 Early-stage clinical trials are currently investigating the combination of Claudin18.2-targeted antibodies with ICIs, which may offer a powerful new approach for treating Claudin18.2-positive cancers.11,27,30 Our study demonstrated that 4A7 combined with anti-mPD-1 had a good combined therapeutic effect, and the combined effect was significantly better than that of the IMAB362 and the anti-mPD-1 combined group. However, in the NOD-SCID mouse model of NCI-N87 (Claudin18.2) gastric cancer cell lines, 4A7 showed better tumor growth inhibition than IMAB362, while in the BALB/C mouse model of MC-38 (Claudin18.2) melanoma cell lines, the results were reversed. We speculated that this may be due to the differences in mouse tumor models, and the therapeutic effects of 4A7 and IMAB362 are different in different tumor cell lines and different immune states of mice (immune deficiency or healthy). Therefore, the animal experiments on the targeted therapy of MAB need to be further supplemented and improved. To explore as much as possible about the specific tumors in which 4A7 antibodies are effective.
Conclusion
This research underscores the superior targeted anti-tumor effects of 4A7 compared to IMAB362. Recent advancements in targeted therapies for GC have included monoclonal antibodies, CAR-T therapy, bispecific antibodies, and ADCs.31,32 ADCs and bispecific antibodies can overcome the limitations of monoclonal antibody therapy by redirecting immune cells or delivering cytotoxic payloads to tumors.20,33 The 4A7 antibody could potentially be further modified into a bispecific antibody or an ADC to enhance its efficacy.18,32 Additionally, understanding the immunological mechanisms underlying the enhanced efficacy of 4A7 could pave the way for novel therapeutic approaches.18,22
The development of 4A7 as a high-affinity monoclonal antibody targeting Claudin18.2 marks a significant advancement in targeted cancer therapy. Future research should explore the potential of 4A7 in the form of bispecific antibodies or ADCs to potentially overcome resistance mechanisms and enhance patient outcomes. Additionally, further studies are necessary to understand the synergistic effects of 4A7 when combined with other immunotherapeutic agents, such as ICIs, to optimize combination therapy strategies. A deeper understanding of the immunological mechanisms behind the enhanced efficacy of 4A7 could pave the way for novel therapeutic approaches and improved clinical management of Claudin18.2-positive cancers.
Abbreviations
GC, gastric cancers; PC, pancreatic cancers; ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; mAbs, monoclonal antibodies; OS, overall survival; EOX, epirubicin, oxaliplatin, and capecitabine; PFS, progression-free survival; GEJ, gastroesophageal junction; mFOLFOX6, 5-fluorouracil/leucovorin/oxaliplatin; CAPOX, capecitabine plus oxaliplatin; HER2, Human epidermal growth factor receptor-2; ADC, antibody-drug conjugate; CAR-T, chimeric antigen receptor T-cell; SDS-PAGE, Sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SEC-HPLC, Size exclusion chromatography-High performance liquid chromatography; PBS, Phosphate-Buffered Saline; EC50, half-maximal effective concentration; MFI, median fluorescence intensity; DAPI, 4′,6-diamidino-2-phenylindole; Fab, fragment of antigen binding; Fc, fragment crystallizable; LDH, lactate dehydrogenase; FcγRs, Fc gamma receptors; IC50, half-maximal inhibition concentration; PBMCs, peripheral blood mononuclear cells; TGI, significant tumor growth inhibition; Tm, midpoint transition temperatures; Tagg, aggregation temperature; SLS, static light scattering; PDI, polydispersity indices; ICIs, immune checkpoint inhibitors; CHO, Chinese Hamster Ovary.
Ethics Approval and Informed Consent
All animal experiments were conducted in accordance with the national regulations and ethical guidelines for experimental animal studies. The experimental protocol was approved by the Animal Ethics Committee of the Beijing Institute of Pharmacology and Toxicology (IACUC-DWZX-2024-P557).The PBMC experiments involved in this study have been formally approved by the Ethics Committee of the Beijing Institute of Pharmacology and Toxicology, with approval number AF/SC-08/02.366. This committee is a national-level independent ethics review body, and its review process complies with the Declaration of Helsinki and China’s Ethical Review Measures for Biomedical Research Involving Humans. All participants signed a written informed consent form prior to blood collection. Participants were healthy volunteers aged 25–35 years, screened to exclude infectious diseases and immune system abnormalities. The official verification documents of original consent forms were requested from the ethics committee.
Acknowledgments
Yahui Wu, Juan Tian, and Yangyihua Zhou are co-first authors for this study. The authors extend sincere gratitude to Ms. Ling Li for her exceptional contributions to this study. Her expertise was pivotal in the experimental design and execution, and she provided critical revisions that significantly improved the manuscript’s clarity and scientific rigor. We also acknowledge her generous support through the Natural Science Foundation of Hunan Province (Grant No. S202410542254).
Author Contributions
All authors contributed significantly to the work reported, including but not limited to the conception, study design, execution, data acquisition, analysis, and interpretation. They participated in drafting, revising, or critically reviewing the manuscript, provided final approval of the version to be published, agreed on the journal for submission, and accepted responsibility for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (82204262, 31970166), Beijing Nova Program (Z211100002121020, 20220484216) and Natural Science Foundation of Hunan Province (Grant. S202410542254).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Ferlay J, Colombet M, Soerjomataram I, et al. Cancer statistics for the year 2020: an overview. Int J Cancer. 2021;149:778–789. doi:10.1002/ijc.33588
3. Charalampakis N, Economopoulou P, Kotsantis I, et al. Medical management of gastric cancer: a 2017 update. Cancer Med. 2018;7(1):123–133. doi:10.1002/cam4.1274
4. Guan WL, He Y, Xu RH. Gastric cancer treatment: recent progress and future perspectives. J Hematol Oncol. 2023;16(1):57. doi:10.1186/s13045-023-01451-3
5. Li K, Zhang A, Li X, et al. Advances in clinical immunotherapy for gastric cancer. Biochim Biophys Acta Rev Cancer. 2021;1876(2):188615. doi:10.1016/j.bbcan.2021.188615
6. Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a Phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697. doi:10.1016/S0140-6736(10)61121-X
7. Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet. 2016;388(10060):2654–2664. doi:10.1016/S0140-6736(16)30354-3
8. Niimi T, Nagashima K, Ward JM, et al. claudin-18, a novel downstream target gene for the T/EBP/NKX2.1 homeodomain transcription factor, encodes lung- and stomach-specific isoforms through alternative splicing. mol Cell Biol. 2001;21(21):7380–7390. doi:10.1128/MCB.21.21.7380-7390.2001
9. Ohta H, Chiba S, Ebina M, et al. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell mol Physiol. 2012;302(2):L193–205. doi:10.1152/ajplung.00349.2010
10. Sahin U, Koslowski M, Dhaene K, et al. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin Cancer Res. 2008;14(23):7624–7634. doi:10.1158/1078-0432.CCR-08-1547
11. Cao W, Xing H, Li Y, et al. Claudin18.2 is a novel molecular biomarker for tumor-targeted immunotherapy. Biomark Res. 2022;10(1):38. doi:10.1186/s40364-022-00385-1
12. Sahin U, Schuler M, Richly H, et al. A Phase I dose-escalation study of IMAB362 (Zolbetuximab) in patients with advanced gastric and gastro-oesophageal junction cancer. Eur J Cancer. 2018;100:17–26. doi:10.1016/j.ejca.2018.05.007
13. Türeci Ӧ, Mitnacht-Kraus R, Wöll S, et al. Characterization of zolbetuximab in pancreatic cancer models. Oncoimmunology. 2019;8(1):e1523096. doi:10.1080/2162402X.2018.1523096
14. Sahin U, Türeci Ö, Manikhas G, et al. FAST: a randomised Phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann Oncol. 2021;32(5):609–619. doi:10.1016/j.annonc.2021.02.005
15. Rohde C, Yamaguchi R, Mukhina S, et al. Comparison of Claudin 18.2 expression in primary tumors and lymph node metastases in Japanese patients with gastric adenocarcinoma. Jpn J Clin Oncol. 2019;49(9):870–876. doi:10.1093/jjco/hyz068
16. Shitara K, Lordick F, Bang YJ, et al. Zolbetuximab plus mFOLFOX6 in patients with CLDN18.2-positive, HER2-negative, untreated, locally advanced unresectable or metastatic gastric or gastro-oesophageal junction adenocarcinoma (SPOTLIGHT): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2023;401(10389):1655–1668. doi:10.1016/S0140-6736(23)00620-7
17. Shah MA, Shitara K, Ajani JA, et al. Zolbetuximab plus CAPOX in CLDN18.2-positive gastric or gastroesophageal junction adenocarcinoma: the randomized, phase 3 GLOW trial. Nat Med. 2023;29(8):2133–2141. doi:10.1038/s41591-023-02465-7
18. Singh P, Toom S, Huang Y. Anti-claudin 18.2 antibody as new targeted therapy for advanced gastric cancer. J Hematol Oncol. 2017;10(1):105. doi:10.1186/s13045-017-0473-4
19. Janjigian YY, Shitara K, Moehler M, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398(10294):27–40. doi:10.1016/S0140-6736(21)00797-2
20. Liu R, Oldham RJ, Teal E, et al. Fc-engineering for modulated effector functions-improving antibodies for cancer treatment. Antibodies. 2020;9(4):64. doi:10.3390/antib9040064
21. Cao X, Chen J, Li B, et al. Promoting antibody-dependent cellular phagocytosis for effective macrophage-based cancer immunotherapy. Sci Adv. 2022;8(11):eabl9171. doi:10.1126/sciadv.abl9171
22. Osanai M, Takasawa A, Murata M, et al. Claudins in cancer: bench to bedside. Pflugers Arch. 2017;469(1):55–67. doi:10.1007/s00424-016-1877-7
23. Wang G, Huang Y, Zhou L, et al. Immunotherapy and targeted therapy as first-line treatment for advanced gastric cancer. Crit Rev Oncol Hematol. 2024;198:104197. doi:10.1016/j.critrevonc.2023.104197
24. Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018;4(5):e180013. doi:10.1001/jamaoncol.2018.0013
25. Forde PM, Spicer J, Lu S, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386(21):1973–1985. doi:10.1056/NEJMoa2202170
26. Körfer J, Lordick F, Hacker UT. Molecular targets for gastric cancer treatment and future perspectives from a clinical and translational point of view. Cancers. 2021;13(20):5216. doi:10.3390/cancers13205216
27. Qi C, Chong X, Zhou T, et al. Clinicopathological significance and immunotherapeutic outcome of claudin 18.2 expression in advanced gastric cancer: a retrospective study. Chin J Cancer Res. 2024;36(1):78–89. doi:10.21147/j.issn.1000-9604.2024.01.08
28. Dhaene K, Maurus D, Gold M, et al. A multicentre, phase IIa study of zolbetuximab as a single agent in patients with recurrent or refractory advanced adenocarcinoma of the stomach or lower oesophagus: the MONO study. Ann Oncol. 2019;30(9):1487–1495. doi:10.1093/annonc/mdz199
29. Sato H, Okonogi N, Nakano T. Rationale of combination of anti-PD-1/PD-L1 antibody therapy and radiotherapy for cancer treatment. Int J Clin Oncol. 2020;25(5):801–809. doi:10.1007/s10147-020-01666-1
30. Nakayama I, Qi C, Chen Y, et al. Claudin 18.2 as a novel therapeutic target. Nat Rev Clin Oncol. 2024;21(5):354–369. doi:10.1038/s41571-024-00874-2
31. Högner A, Moehler M. Immunotherapy in Gastric Cancer. Curr Oncol. 2022;29(3):1559–1574. doi:10.3390/curroncol29030131
32. Chen J, Xu Z, Hu C, et al. Targeting CLDN18.2 in cancers of the gastrointestinal tract: new drugs and new indications. Front Oncol. 2023;13:1132319. doi:10.3389/fonc.2023.1132319
33. Rader C. Bispecific antibodies in cancer immunotherapy. Curr Opin Biotechnol. 2020;65:9–16. doi:10.1016/j.copbio.2019.11.020
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.