Back to Journals » Drug Design, Development and Therapy » Volume 11
Detection approaches for multidrug resistance genes of leukemia
Received 12 February 2017
Accepted for publication 18 March 2017
Published 18 April 2017 Volume 2017:11 Pages 1255—1261
DOI https://doi.org/10.2147/DDDT.S134529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Ying Du, Baoan Chen
Department of Hematology and Oncology (Key Department of Jiangsu Medicine), School of Medicine, Zhongda Hospital, Southeast University, Nanjing, Jiangsu Province, People’s Republic of China
Abstract: Leukemia is a clonal malignant hematopoietic stem cell disease. It is the sixth most lethal cancer and accounts for 4% of all cancers. The main form of treatment for leukemia is chemotherapy. While some cancer types with a higher incidence than leukemia, such as lung and gastric cancer, have shown a sharp decline in mortality rates in recent years, leukemia has not followed this trend. Drug resistance is often regarded as the main clinical obstacle to effective chemotherapy in patients diagnosed with leukemia. Many resistance mechanisms have now been identified, and multidrug resistance (MDR) is considered the most important and prevalent mechanism involved in the failure of chemotherapy in leukemia. In order to reverse MDR and improve leukemia prognosis, effective detection methods are needed to identify drug resistance genes at initial diagnosis. This article provides a comprehensive overview of published approaches for the detection of MDR in leukemia. Identification of relevant MDR genes and methods for early detection of these genes will be needed in order to treat leukemia more effectively.
Keywords: leukemia, multidrug resistance, MDR, detection approaches
Introduction
The treatment of cancers which have developed multidrug resistance (MDR) is a major challenge, and rates of morbidity and mortality are extremely high.1 Also, in clinical practice we have found that the emergence of MDR has caused considerable levels of pain in many leukemia patients. MDR is defined as when cancer cells become resistant to a chemotherapeutic drug, after one or more treatment cycles, and this may lead to resistance to other chemotherapeutic drugs that have different structures and mechanisms.2
Numerous reports have focused on the different mechanisms of MDR in leukemia. These resistance mechanisms include: a) increased drug metabolism due to altered molecular targets; b) defective apoptotic machinery; c) over-expression of efflux pumps, such as P-glycoprotein (P-gp), multidrug resistance-associated proteins (MRP1, MRP2), lung resistance protein/major vault protein, and breast cancer resistance protein (BCRP); d) enzyme-mediated drug resistance mechanisms such as overexpression of glutathione S-transferase; e) microenvironmental resistance; and f) enhanced repair of drug-induced DNA damage.3,4 It is well known that the appearance of MDR has made effective treatment of leukemia very challenging. Therefore, the early, accurate, and sensitive detection of MDR genes is vital, and it is also beneficial to search for more effective chemotherapeutic approaches for use in the clinical setting. Notably, MDR efflux pumps are usually localized in epithelial cells, and their location is polarized. Moreover, they are often expressed in combination with other membrane proteins. All of these can serve as potential targets for convenient testing. MDR genes can usually be potentially detected by the RNA and protein levels by various approaches as described in this article.
Multidrug resistance
The occurrence of MDR is mainly due to the extracellular efflux of chemotherapy agents which involves mechanisms mediated by over expression of ATP-binding cassette (ABC), and MDR proteins.5 Both P-gp and MRP1 are thought to play pivotal roles in the MDR process.6 P-gp is a membrane-associated drug efflux pump which belongs to the ABC protein family and is encoded by the MDR1 gene (Figure 1). P-gp represents a 170 kDa glycosylated integral plasma membrane protein,7,8 which acts as a drug efflux pump to decrease intracellular drug concentrations.9 Multidrug resistance and chemotherapy failure are produced by intracellular anticancer drugs that increasingly flow from cells through the efflux pump.10,11 The protein includes two halves. Each part of the protein contains six hydrophobic trans-membrane domains, and one ATP binding domain.12 Powered by the hydrolysis of ATP, they efflux structurally diverse compounds. Hydrolysis of ATP is believed to reset the protein to the inward-facing form to begin a new cycle of drug binding and release.13 P-gp, which is located on chromosome 7q21, was the first MDR gene to be identified.14 Two isoforms of P-gp are expressed in humans, class I and III isoforms are drug transporters (MDR1/ABCB1), while the function of class II isoforms (MDR2/3/ABCB4) is to export phosphatidylcholine into bile.15 In addition to exporting chemotherapeutic agents, P-gp is able to transport a broad range of substrates, including amino acids, sugars, peptides, organic ions, metabolites, and numerous hydrophobic compounds.16,17 Several reports suggest that P-gp may regulate apoptosis, chloride channel activity, cholesterol metabolism, differentiation, proliferation, adhesion, and immune cell function.18 Significantly, elevated P-gp expression was found in chronic B-cell leukemia patients. Alterations in P-gp expression may also occur because of single nucleotide polymorphisms, which have been found in MDR1, and which affect drug-metabolism and result in altered pharmacokinetics of chemotherapeutics.19 Some investigations have proposed a model in which expression of ABC transporters prevent drug accumulation in resistant cells by increasing efflux, at the same time, the models also consider the other side of the equation in which drug influx is suppressed by the active reduction of endocytosis.20
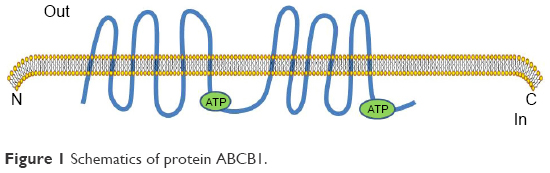 | Figure 1 Schematics of protein ABCB1. |
MRP1 was one of the first human ABC proteins identified and is encoded by the ABCC1 gene (Figure 2), located on chromosome 16p13.12 MRP1 is a glycosylated protein with molecular weight ~190 kDa and functions in the transport of sulfate, glutathione or glucuronate and anionic substances, in an ATP-dependent manner.16,21,22 Structurally, MRP1 has three membrane spanning domains (MSD0, MSD1, MSD2). MSD1 and MSD2 each has six transmembrane (TM) helixes, MSD0 has five TM segments with approximately 200 amino acids. MRP1 is also made up of two cytosolic nucleotide binding domains (NBDs). The cytosolic NBD is responsible for binding and hydrolysis of ATP to provide energy for substrate transport.16,23 A variety of anticancer drugs transported by MRP1, are bulky hydrophobic molecules that acquire entry to cells by simple diffusion across the lipid bilayer of the cell’s outer membrane.23,24 The mechanism of MRP1 involved multidrug resistance has not been exactly understood. GSH possibly plays a role in the occurrence of multidrug resistance.25 MRP1 also possibly serves a sequestration function to prevent drugs from reaching their intracellular targets, because it has been found in other subcellular organelles such as the endoplasmic reticulum and endocytic vesicles.26,27
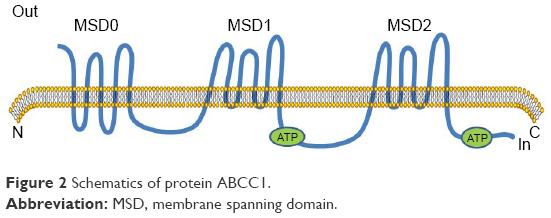 | Figure 2 Schematics of protein ABCC1. |
Detection approaches
The mechanisms underlying MDR are complex. Accurate and sensitive detection of these mechanisms is thought to be vital to improved treatment of leukemia. Since MDR1 gene amplification is almost non-existent in human tumors, detection of alterations in DNA copy number is not appropriate. Therefore, at present we usually measure MDR1 mRNA expression levels. Commonly used methods include polymerase chain reaction (PCR), in situ hybridization (ISH), and RNase protection assays (RPAs). Western blotting and immunohistochemistry (IHC) may also be used for protein detection.
Nucleic acid-based detection methods
PCR
PCR is a technique for the selective amplification of DNA or RNA segments of up to 2 kb or more in length.28 The three stages of PCR are exponential amplification, followed by leveling off, and the plateau stage. DNA polymerase and specific oligonucleotide primers are currently used for amplification of P-gp cDNAs.29 Specifically, PCR amplification relies on thermal cycling, consisting of cycles of repeated heating and cooling for DNA melting and enzymatic replication of the DNA.30 Primers for the PCR reactions are based on conserved sequences of the catalytic domain that are shared among all known drug efflux pumps protein tyrosine phosphatases.31 Primers could produce optimal amplicon size for quantitative PCR when amplifying regions for the partial P-gp sequences. For each reaction, for optimization, it was implemented to detect each primer pair and undertake amplification of a single PCR product only.29 Repeated cycles of three different reaction temperatures are needed: the first high temperature step for heat-denaturation of the DNA, the second for annealing, and the third for extension of the primers.32 As PCR is sensitive to contamination, care must be taken to include appropriate negative controls. In order to ensure that any contaminating material would not act as a template for PCR, buffers should be incubated with restriction enzymes that cut within the amplified fragment before amplification.33 This approach has been greatly simplified by automated procedures that use a thermostable enzyme DNA polymerase.34 This allows a single sample to be simultaneously amplified for several different markers, and has important economic implications.35 PCR is now routinely and universally applied in human genetics, basic molecular biology, and clinical investigations with the goal of monitoring the causes of disease.28 At the end of the 20th century, Vogelstein described a digital PCR method. Digital PCR is both a qualitative and quantitative method, and can sensitively and accurately detect genetic alterations.36
Fluorescence in situ hybridization (FISH)
FISH is a method to detect specific nucleic acids in fixed but otherwise intact cells, that has been shown to be both sensitive and specific.37,38 FISH for visualization of nucleic acids was developed as an alternative to older methods that used radiolabeled probes.39 The basic elements of the FISH procedure include selection of probe(s) for a sequence complementary to the target of interest, probe labeling, slide preparation, slide pretreatment, denaturation of probe and target, hybridization, washing, analysis, and interpretation.40 Probes are labeled either directly, by incorporation of fluorescent nucleotides, or indirectly, by incorporation of reporter molecules that are subsequently detected by fluorescent antibodies or other high affinity molecules (eg, streptavidin/biotin).41 Hybridization to the target loci is visualized by the detection of fluorescent signals on metaphase chromosomes or interphase nuclei.42 Many new technologies have been invented to overcome the shortcomings of FISH. For example, a high resolution multicolor banding method was developed to overcome the disadvantage of lack of detection of perientric and paracentric DNA inversions, and therefore to precisely identify chromosome breakpoints.43 Quantitative FISH can measure the length of telomere repeats in preparations of metaphase chromosomes.44 Many applications based on FISH have been proposed in different fields of investigation, including evolutionary studies, interphase nucleus architecture analyses, DNA sequence mapping, toxicology, microbial ecology, cancer diagnostics, and forensics.38,45 This technology provides a wide range of choices for us to target any desired tumor biomarkers or any of the chromosomes to be examined.46
RPA
The RPA is a sensitive means of quantitating mRNA transcripts initiated at a specific nucleotide, which was developed in the early 1980s.47 The procedure is based on the hybridization of the analyzed RNA to a radioactively labeled RNA probe, and the hybridization reactions are treated with ribonuclease to remove free probe. This leaves intact fragments of probe annealed to homologous sequences in the sample RNA.48,49 Notably, the position of radioactively labeled probe fragments can be detected by autoradiography. Alternatively, samples can be transferred to a membrane for secondary visualization if non-isotopically labeled probes are used.50 The hybridized RNA is then digested with ribonucleases, followed by precipitation and resuspension of the protected RNAs. Ultimately, electrophoresis and autoradiography of the protected RNAs is done on a denaturing polyacrylamide gel.51 The assay of mRNA by RPA is achievable because double-stranded RNA is not susceptible to degradation by ribonuclease.52 The advantage of the assay is that multiple mRNA species can be measured simultaneously in a single total RNA sample. Also, there are no amplification steps involved in ribonuclease protection, and therefore quantitative data are more reliable than reverse transcription (RT)-PCR. However, RNase protection is more time-consuming than RT-PCR and takes several days to complete, even by an experienced technician.53 For further technological development of this approach, microchip electrophoresis was considered in the analytical stages due to its characteristics: high speed, high throughput, low consumption of samples and reagents, miniaturization, and automation.54
Protein-based detection methods
Western blot
Western blotting is a technique that was first developed between 1977 and 1979 by Towbin and Staehelin, that has since become a common technique applied in research laboratories globally for the immune detection and quantitation of specific proteins.55,56 The principle of this method is based on specific antibody-antigen interaction. It could achieve the qualitative or semiquantitative identification of specific proteins. What is more important, the molecular weight of the proteins also be measured from a complex mixture. Gel electrophoresis of a protein sample, transfer of protein from a gel to a membrane support, and immune detection of a target antigen are the three necessary steps.57 Pelleting the cells by low speed centrifugation in order to drain the cell pellet well is usually performed when in the protein extraction. Also, it should be done at 4°C with protease inhibitors to prevent denaturing of the proteins, and incubated at room temperature for 5 min.58,59 Polyacrylamide gel electrophoresis is used to separate proteins in Western blotting.57 Some researchers indicated that the sensitivity of Western blotting was dependent on the quality of both the antigen and the antibody detection reagents.60 Similarly, increasing the volume of serum for low-titer specimens is critical.61 The critical steps in generating high-quality, quantitative Western blots are the following: 1) measuring protein concentrations; 2) sample preparation; 3) blocking the membrane; and 4) obtaining good quality and caliber of the primary antibody.62 A Western blotting minimal reporting standard and stain-free technology are recommended to improve the reproducibility of Western blot analysis.63 These are novel and unique quality control tools for data normalization in Western blotting workflows.55 The advantages of Western blotting are: simplicity, speed, and sensitivity. Furthermore, immunoblotting can distinguish different molecular forms of an antigen.64 Western blotting has been used to detect changes in MRP1 levels.65 Expression of MDR-1/P-gp levels in P388, P388/ADR, and HCT-15 cell lines have also been tested by Western blot analysis.7
IHC
IHC is a powerful method for detecting specific proteins in formalin-fixed, paraffin-embedded tissues based on antigen–antibody interactions. The process involves tissue processing, sectioning and epitope retrieval, antigen–antibody interaction for protein detection, and visualization through various approaches.66 Successful immunostaining depends on the source, specificity, and quality of the primary antibody. The condition of the specimen is also very important, as is the fixation procedure and antigen-detection strategy.67 Excellent morphology can be obtained if the cells are fixed while they are still in growth-culture medium dish/flask before removing them.68 IHC is a highly standardized, sensitive, and simple technique.66 In addition, it possesses the ability to delineate admixed normal cells bearing P-gp. All of these are beneficial when investigating the development and emergence of multidrug resistance in leukemia. Subjectivity is the primary disadvantage.69 Antigen localization, cell and tissue morphology can be observed simultaneously because IHC utilizes a light microscope for visualization.67 However, IHC is subject to qualitative and subjective assessment, and has been criticized due to a lack of stringency compared with PCR.70 As the enhanced diagnostic utility of IHC was realized, the demand for this technique has also increased. It will continue to be a rapidly evolving field with the ever-increasing numbers of new antibodies available.71 The development of automation has progressed to eliminate many of the manual steps in the process.72 The combination of mass spectroscopy with IHC has allowed multiplexed, direct quantitative imaging of tissue samples for basic and clinical research.73 Multiplexed IHC methods permit identification of at least three, and up to 30 discrete antigens.74 Some studies suggest that IHC may be sufficiently sensitive and specific to predict response to targeted therapy.75
Conclusion
The outcome of leukemia is usually fatal. Recent surveys indicate that leukemia is the most common cause of cancer-related death in men under the age of 40. Meanwhile, about half of new cancer-related deaths among females were attributed to leukemia. Equally alarming is the fact that leukemia is a major cause of death in females under the age of 20. For children under the age of 14, acute lymphocytic leukemia is the most common form of cancer.76 Chronic myeloid leukemia (CML) has an incidence of 1–1.5 per 100,000,77 with relapse occurring in 25% of patients being associated with a poor prognosis.78 As a global problem of increasing concern, MDR leads to inadequate treatment and poor prognosis in leukemia patients. In recent years, some progress has been made in understanding the mechanisms of MDR.79 Despite this poor prognosis, there has been some progress in the treatment of leukemia in recent years. Tyrosine kinase inhibitors (TKIs) have changed the clinical course of CML, improving the 5-year survival from 35% to more than 90% with imatinib mesylate.80 Some new therapies aimed at reducing drug resistance are being studied, such as targeting JAK/STAT signaling, and the combination of ATRA with TKIs.14,81 However, both P-gp and MRP1 have been shown to interact with imatinib.82 The challenges are sure to remain difficult in the future. Detection of MDR genes has been described using the methodology outlined in this article. For example, 56 cell membrane glycoproteins in leukemia samples could be successfully quantitated by antibody analysis of non-glycopeptides.83 Detection technology is constantly developing. At the end of the 20th century, DNA microarrays were starting to be developed, allowing an opportunity to test the expression of hundreds to thousands of genes in a single assay.84 Nowadays, high-throughput sequencing has risen to prominence and can also be used to detect MDR genes, diagnose cancer, and so on. The technology makes the direct application of RNA sequencing, while not through DNA synthesis, possible.85 DNA microarray and next generation sequencing are products of the implementation of high-throughput techniques. Microarrays have greater advantages in terms of parallelism, automation, and miniaturization. Without PCR-based signal amplification, next-generation DNA sequencing technologies also have the ability to read signals of a single fluorescent molecule.86 In comparison to traditional Sanger sequencing, next-generation sequencing allows multiplexing of samples and gene targets in one experimental setup.87 At the same time, it can be combined with microarrays, DNA co-immunoprecipitation, and so on. However, they still have some shortcomings and technical limitations. It is of critical importance to establish the most convenient and efficient method to detect MDR genes in the future. The ultimate goal is to detect resistance genes and reverse their activities so as to reduce the risk of relapse and improve patient prognosis.
Acknowledgments
This work was supported by the National Key Basic Research Program 973 of the People’s Republic of China (number 2010CB732404), the National Nature Science Foundation of the People’s Republic of China (numbers 81170492, 81370673), and the National High Technology Research and Development Program 863 Projects of the People’s Republic of China (number 2012AA022703).
Disclosure
The authors report no conflicts of interest in this work.
References
Kandi V, Kandi S. Antimicrobial properties of nanomolecules: potential candidates as antibiotics in the era of multi-drug resistance. Epidemiol Health. 2015;37:e2015020. | ||
Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347(2):159–166. | ||
Patel NR, Pattni BS, Abouzeid AH, Torchilin VP. Nanopreparations to overcome multidrug resistance in cancer. Adv Drug Deliv Rev. 2013;65(13–14):1748–1762. | ||
Ullah MF. Cancer multidrug resistance (MDR): a major impediment to effective chemotherapy. Asian Pac J Cancer Prev. 2008;9(1):1–6. | ||
Yu P, Du Y, Yang L, Fan S, Wu J, Zheng S. Significance of multidrug resistance gene-related proteins in the postoperative chemotherapy of gastric cancer. Int J Clin Exp Pathol. 2014;7(11):7945–7950. | ||
Ma H, Cheng L, Hao K, et al. Reversal effect of ST6GAL 1 on multidrug resistance in human leukemia by regulating the PI3K/Akt pathway and the expression of P-gp and MRP1. PLoS One. 2014;9(1):e85113. | ||
Ma P, Dong X, Swadley CL, et al. Development of idarubicin and doxorubicin solid lipid nanoparticles to overcome Pgp-mediated multiple drug resistance in leukemia. J Biomed Nanotechnol. 2009;5(2):151–161. | ||
Zhang L, Xiao R, Xiong J, et al. Activated ERM protein plays a critical role in drug resistance of MOLT4 cells induced by CCL25. PLoS One. 2013;8(1):e52384. | ||
Xu Y, Ohms SJ, Li Z, et al. Changes in the expression of miR-381 and miR-495 are inversely associated with the expression of the MDR1 gene and development of multi-drug resistance. PLoS One. 2013;8(11):e82062. | ||
Hu T, To KK, Wang L, et al. Reversal of P-glycoprotein (P-gp) mediated multidrug resistance in colon cancer cells by cryptotanshinone and dihydrotanshinone of Salvia miltiorrhiza. Phytomedicine. 2014;21(11):1264–1272. | ||
Jing X, Zhang H, Hu J, et al. β-arrestin 2 is associated with multidrug resistance in breast cancer cells through regulating MDR1 gene expression. Int J Clin Exp Pathol. 2015;8(2):1354–1363. | ||
Milojkovic M, Milacic N, Radovic J, Ljubisavljevic S. MDR1 gene polymorphisms and P-glycoprotein expression in respiratory diseases. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2015;159(3):341–346. | ||
Chufan EE, Sim HM, Ambudkar SV. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): recent biochemical and structural studies. Adv Cancer Res. 2015;125:71–96. | ||
Carmo CR, Lyons-Lewis J, Seckl MJ, Costa-Pereira AP. A novel requirement for janus kinases as mediators of drug resistance induced by fibroblast growth factor-2 in human cancer cells. PLoS One. 2011;6(5):e19861. | ||
Kapse-Mistry S, Govender T, Srivastava R, Yergeri M. Nanodrug delivery in reversing multidrug resistance in cancer cells. Front Pharmacol. 2014;5:159. | ||
de Moraes AC, Maranho CK, Rauber GS, Santos-Silva MC. Importance of detecting multidrug resistance proteins in acute leukemia prognosis and therapy. J Clin Lab Anal. 2013;27(1):62–71. | ||
Carrett-Dias M, Almeida LK, Pereira JL, et al. Cell differentiation and the multiple drug resistance phenotype in human erythroleukemic cells. Leuk Res. 2016;42:13–20. | ||
Cerezo D, Lencina M, Ruiz-Alcaraz AJ, et al. Acquisition of MDR phenotype by leukemic cells is associated with increased caspase-3 activity and a collateral sensitivity to cold stress. J Cell Biochem. 2012;113(4):1416–1425. | ||
Penna G, Allegra A, Alonci A, et al. MDR-1 polymorphisms (G2677T and C3435T) in B-chronic lymphocytic leukemia: an impact on susceptibility and prognosis. Med Oncol. 2011;28(4):1549–1554. | ||
Pisco AO, Jackson DA, Huang S. Reduced intracellular drug accumulation in drug-resistant leukemia cells is not only solely due to MDR-mediated efflux but also to decreased uptake. Front Oncol. 2014;4:306. | ||
Cole SP. Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Ann Rev Pharmacol Toxicol. 2014;54:95–117. | ||
Mahjoubi F, Golalipour M, Ghavamzadeh A, Alimoghaddam K. Expression of MRP1 gene in acute leukemia. Sao Paulo Med J. 2008;126(3):172–179. | ||
Yin J, Zhang J. Multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphism: from discovery to clinical application. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2011;36(10):927–938. | ||
Fazlina N, Maha A, Jamal R, et al. Expression of multidrug resistance (MDR) proteins and in vitro drug resistance in acute leukemias. Hematology. 2007;12(1):33–37. | ||
Akan I, Akan S, Akca H, Savas B, Ozben T. Multidrug resistance-associated protein 1 (MRP1) mediated vincristine resistance: effects of N-acetylcysteine and Buthionine sulfoximine. Cancer Cell Int. 2005;5(1):22. | ||
Cole SP. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J Biol Chem. 2014;289(45):30880–30888. | ||
Lu JF, Pokharel D, Bebawy M. MRP1 and its role in anticancer drug resistance. Drug Metab Rev. 2015;47(4):406–419. | ||
Vosberg HP. The polymerase chain reaction: an improved method for the analysis of nucleic acids. Hum Genet. 1989;83(1):1–15. | ||
Williamson SM, Wolstenholme AJ. P-glycoproteins of Haemonchus contortus: development of real-time PCR assays for gene expression studies. J Helminthol. 2012;86(2):202–208. | ||
Erlich HA. Polymerase chain reaction. J Clin Immunol. 1989;9(6):437–447. | ||
Yi T, Cleveland JL, Ihle JN. Identification of novel protein tyrosine phosphatases of hematopoietic cells by polymerase chain reaction amplification. Blood. 1991;78(9):2222–2228. | ||
Collasius M, Falk H, Ciesler C, Valet G. How to build an inexpensive cyclotherm instrument for automated polymerase chain reaction. Anal Biochem. 1989;181(1):163–166. | ||
Lawler M, Humphries P, McCann SR. Evaluation of mixed chimerism by in vitro amplification of dinucleotide repeat sequences using the polymerase chain reaction. Blood. 1991;77(11):2504–2514. | ||
Saito I, Servenius B, Compton T, Fox RI. Detection of Epstein-Barr virus DNA by polymerase chain reaction in blood and tissue biopsies from patients with Sjogren’s syndrome. J Exp Med. 1989;169(6):2191–2198. | ||
Bell J. The polymerase chain reaction. Immunol Today. 1989;10(10):351–355. | ||
Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96(16):9236–9241. | ||
Klinger KW. FISH: sensitivity and specificity on sorted and unsorted cells. Ann N Y Acad Sci. 1994;731:48–56. | ||
Vautrot V, Aigueperse C, Branlant C, Behm-Ansmant I. Fluorescence In situ hybridization of small non-coding RNAs. Methods Mol Biol. 2015;1296:73–83. | ||
Levsky JM, Singer RH. Fluorescence in situ hybridization: past, present and future. J Cell Sci. 2003;116(Pt 14):2833–2838. | ||
Tsuchiya KD. Fluorescence in situ hybridization. Clin Lab Med. 2011;31(4):525–542. | ||
Volpi EV, Bridger JM. FISH glossary: an overview of the fluorescence in situ hybridization technique. Biotechniques. 2008;45(4):385–386. | ||
Murthy SK, Demetrick DJ. New approaches to fluorescence in situ hybridization. Methods Mol Biol. 2006;319:237–259. | ||
Chudoba I, Plesch A, Lörch T, Lemke J, Claussen U, Senger G. High resolution multicolor-banding: a new technique for refined FISH analysis of human chromosomes. Cytogenet Cell Genet. 1999;84(3–4):156–160. | ||
Poon SS, Lansdorp PM. Quantitative fluorescence in situ hybridization (Q-FISH). Curr Protoc Cell Biol. 2001; Chapter 18: Unit 18.4. | ||
Nath J, Johnson KL. A Review of fluorescence in situ hybridization (FISH): current status and future prospects. Biotech Histochem. 2000;75(2):54–78. | ||
Lin PP. Integrated EpCAM-independent subtraction enrichment and iFISH strategies to detect and classify disseminated and circulating tumors cells. Clin Transl Med. 2015;4(1):38. | ||
Carey MF, Peterson CL, Smale ST. The RNase protection assay. Cold Spring Harb Protoc. 2013;2013(3). | ||
Mironov VN, Van Montagu M, Inzé D. High throughput RNase protection assay. Nucleic Acids Res. 1995;23(16):3359–3360. | ||
Gilman M. Ribonuclease protection assay. Curr Protoc Mol Biol. 2001; Chapter 4: Unit 7. | ||
Eyler E. Explanatory chapter: nuclease protection assays. Methods Enzymol. 2013;530:89–97. | ||
Young HA, Subleski JJ, Krebs SM. Multiprobe ribonuclease protection assay for simultaneous measurement of mRNA expression. Curr Protoc Immunol. 2003; Chapter 10: Unit 10.29. | ||
Mitchell A, Fidge N. Determination of apolipoprotein mRNA levels by ribonuclease protection assay. Methods Enzymol. 1996;263:351–363. | ||
Rottman JB. The ribonuclease protection assay: a powerful tool for the veterinary pathologist. Vet Pathol. 2002;39(1):2–9. | ||
Yamaguchi Y, Yatsushiro S, Yamamura S, et al. Ribonuclease protection assay on microchip electrophoresis. Analyst. 2011;136(11):2247–2251. | ||
Taylor SC, Posch A. The design of a quantitative western blot experiment. Biomed Res Int. 2014;2014:361590. | ||
Towbin H, Staehelin T. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76(9):4350–4354. | ||
Hnasko TS, Hnasko RM. The Western blot. Methods Mol Biol. 2015;1318:87–96. | ||
Eslami A, Lujan J. Western blotting: sample preparation to detection. J Vis Exp. 2010;(44). | ||
Mahmood T, Yang PC. Western blot: technique, theory, and trouble shooting. N Am J Med Sci. 2012;4(9):429–434. | ||
Gallo D, Diggs JL, Shell GR, Dailey PJ, Hoffman MN, Riggs JL. Comparison of detection of antibody to the acquired immune deficiency syndrome virus by enzyme immunoassay, immunofluorescence, and Western blot methods. J Clin Microbiol. 1986;23(6):1049–1051. | ||
Ashley RL, Militoni J, Lee F, Nahmias A, Corey L. Comparison of Western blot (immunoblot) and glycoprotein G-specific immunodot enzyme assay for detecting antibodies to herpes simplex virus types 1 and 2 in human sera. J Clin Microbiol. 1988;26(4):662–667. | ||
Silva JM, McMahon M. The fastest Western in town: a contemporary twist on the classic Western blot analysis. J Vis Exp. 2014;(84):e51149. | ||
Gilda JE, Ghosh R, Cheah JX, West TM, Bodine SC, Gomes AV. Western blotting inaccuracies with unverified antibodies: need for a Western blotting minimal reporting standard (WBMRS). PloS One. 2015;10(8):e0135392. | ||
Howe JG, Hershey JW. A sensitive immunoblotting method for measuring protein synthesis initiation factor levels in lysates of Escherichia coli. J Biol Chem. 1981;256(24):12836–12839. | ||
Ma SL, Hu YP, Wang F, et al. Lapatinib antagonizes multidrug resistance-associated protein 1-mediated multidrug resistance by inhibiting its transport function. Mol Med. 2014;20:390–399. | ||
Schacht V, Kern JS. Basics of immunohistochemistry. J Invest Dermatol. 2015;135(3):e30. | ||
Hofman FM, Taylor CR. Immunohistochemistry. Curr Protoc Immunol. 2013;103: Unit 21 4. | ||
Nuovo GJ. The Basics of Immunohistochemistry. In: In Situ Molecular Pathology and Co-Expression Analyses. London, UK: Academic Press; 2013:133–165. | ||
Beck WT, Grogan TM, Willman CL, et al. Methods to detect P-glycoprotein-associated multidrug resistance in patients’ tumors: consensus recommendations. Cancer Res. 1996;56(13):3010–3020. | ||
Elliott K, McQuaid S, Salto-Tellez M, Maxwell P. Immunohistochemistry should undergo robust validation equivalent to that of molecular diagnostics. J Clin Pathol. 2015;68(10):766–770. | ||
Ferringer T. Immunohistochemistry in dermatopathology. Arch Pathol Lab Med. 2015;138(1):83–105. | ||
Prichard JW. Overview of automated immunohistochemistry. Arch Pathol Lab Med. 2014;138(12):1578–1582. | ||
Rimm DL. Next-gen immunohistochemistry. Nat Methods. 2014;11(4):381–383. | ||
Dixon AR, Bathany C, Tsuei M, White J, Barald KF, Takayama S. Recent developments in multiplexing techniques for immunohistochemistry. Expert Rev Mol Diagn. 2015;15(9):1171–1186. | ||
Swanson PE. Immunohistochemistry as a surrogate for molecular testing: a review. Appl Immunohistochem Mol Morphol. 2015;23(2):81–96. | ||
Chen AH, Tsau YW, Lin CH. Novel methods to identify biologically relevant genes for leukemia and prostate cancer from gene expression profiles. BMC Genomics. 2010;11:274. | ||
Roundhill EA, Burchill SA. Detection and characterisation of multi-drug resistance protein 1 (MRP-1) in human mitochondria. Br J Cancer. 2012;106(6):1224–1233. | ||
Kourti M, Vavatsi N, Gombakis N, et al. Expression of multidrug resistance 1 (MDR1), multidrug resistance-related protein 1 (MRP1), lung resistance protein (LRP), and breast cancer resistance protein (BCRP) genes and clinical outcome in childhood acute lymphoblastic leukemia. Int J Hematol. 2007;86(2):166–173. | ||
Zhou H, Ma H, Wei W, et al. B4GALT family mediates the multidrug resistance of human leukemia cells by regulating the hedgehog pathway and the expression of p-glycoprotein and multidrug resistance-associated protein 1. Cell Death Dis. 2013;4:e654. | ||
Ichim CV. Kinase-independent mechanisms of resistance of leukemia stem cells to tyrosine kinase inhibitors. Stem Cells Transl Med. 2014;3(4):405–415. | ||
Wang Z, Liu Z, Wu X, et al. ATRA-induced cellular differentiation and CD38 expression inhibits acquisition of BCR-ABL mutations for CML acquired resistance. PLoS Genet. 2014;10(6):e1004414. | ||
Peng XX, Tiwari AK, Wu HC, Chen ZS. Overexpression of P-glycoprotein induces acquired resistance to imatinib in chronic myelogenous leukemia cells. Chin J Cancer. 2012;31(2):110–118. | ||
Li K, Sun Z, Zheng J, et al. In-depth research of multidrug resistance related cell surface glycoproteome in gastric cancer. J Proteomics. 2013;82:130–140. | ||
Martinez MA, Soto-Del Rio Mde L, Gutierrez RM, et al. DNA microarray for detection of gastrointestinal viruses. J Clin Microbiol. 2015;53(1):136–145. | ||
Park SJ, Saito-Adachi M, Komiyama Y, Nakai K. Advances, practice, and clinical perspectives in high-throughput sequencing. Oral Dis. 2016;22(5):353–364. | ||
Teng X, Xiao H. Perspectives of DNA microarray and next-generation DNA sequencing technologies. Sci China C Life Sci. 2009;52(1):7–16. | ||
Vollbrecht C, Mairinger FD, Koitzsch U, et al. Comprehensive analysis of disease-related genes in chronic lymphocytic leukemia by multiplex PCR-based next generation sequencing. PLoS One. 2015;10(6):e0129544. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.