Back to Journals » Drug Design, Development and Therapy » Volume 16
Designing a Dual GLP-1R/GIPR Agonist from Tirzepatide: Comparing Residues Between Tirzepatide, GLP-1, and GIP
Authors Wang L ![]()
Received 26 January 2022
Accepted for publication 18 May 2022
Published 25 May 2022 Volume 2022:16 Pages 1547—1559
DOI https://doi.org/10.2147/DDDT.S358989
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Video abstract presented by Lijing Wang.
Views: 1108
Lijing Wang
College of Life Sciences and Technology, China Pharmaceutical University, Nanjing, Jiangsu, People’s Republic of China
Correspondence: Lijing Wang, College of Life Science and Technology, China Pharmaceutical University, 639 Longmian Avenue, Jiangning District, Nanjing, Jiangsu, People’s Republic of China, Tel +86 182 860 69474, Email [email protected]; [email protected]
Abstract: Improving type 2 diabetes using incretin analogues is becoming increasingly plausible. Currently, tirzepatide is the most promising listed incretin analogue. Here, I briefly explain the evolution of drugs of this kind, analyze the residue discrepancies between tirzepatide and endogenous incretins, summarize some existing strategies for prolonging half-life, and present suggestions for future research, mainly involving biased functions. This review aims to present some useful information for designing a dual glucagon like peptide-1 receptor/glucose-dependent insulinotropic polypeptide receptor agonist.
Keywords: tirzepatide, GLP-1, GIP, Aib, structure–activity relationship, structure–function relationship
Introduction
From GLP-1 Receptor Agonists to Tirzepatide
Incretins are hormones that are secreted from the gastrointestinal tract into the circulatory system in response to nutrient ingestion, enhancing glucose-stimulated insulin secretion. Incretins are estimated to account for approximately 50–70% of the total insulin secretion after oral glucose administration, and this has been dubbed the “incretin effect”. To date, two incretins, glucagon like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), have been identified.1
GLP-1 receptor agonists are broadly applied in type 2 diabetes mellitus (T2DM) therapy. Exenatide is a non-human peptide analogue originally isolated from the saliva of the Gila monster (Heloderma suspectum). Compared to GLP-1, the second residue of exenatide is Gly rather than Ala; thus, it can circumvent the degradation of dipeptidyl peptidase-4 (DPP-4) and has a prolonged intravenous half-life of 30 min and a 2–3 h half-life after subcutaneous administration.2
Liraglutide is designed to bind to human albumin via a C16 fatty acid and a spacer covalently attached to Lys26. A residue substitution of Lys34 to Arg34 occurs to avoid the fatty acid being installed in a wrong place.3 Liraglutide has an intravenous half-life of 8–10 h and 13–15 h half-life after subcutaneous administration.2
Semaglutide converges these principles: the second residue is replaced by α-aminoisobutyric acid (Aib) to avoid DPP-4 degradation, Lys34 is replaced by Arg34, and a C18 fatty acid is linked to Lys26 via a γGlu-2xOEG spacer (Figure 1) providing higher affinity to albumin.2 The half-life of semaglutide after subcutaneous administration is up to 183 h.4
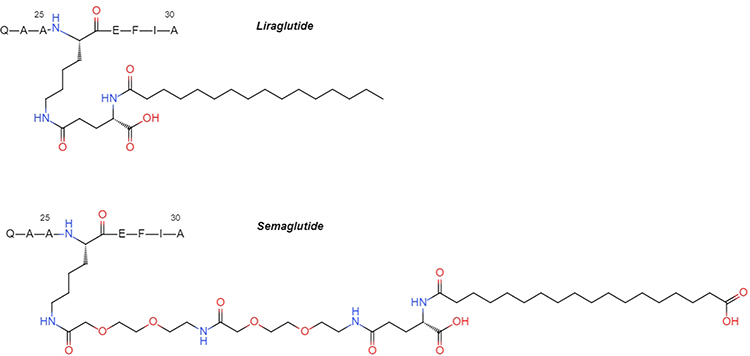 |
Figure 1 The structures of fatty acid side chains of liraglutide and semaglutide. |
GIP plays an important role in the incretin effect in healthy people. Unfortunately, its druggability is low. Infusions that achieve supraphysiological GIP concentrations fail to elicit a significant insulin secretory response in patients with T2DMs; thus, GIP infusions cannot rapidly normalize the blood glucose levels of patients with T2DMs.5 This blunted response is possibly caused by the downregulation of GIP receptors (GIPR) by the high level of circulating glucose. However, a substantial body of data suggests that GIP resistance can be largely overcome by agents that lower the circulating glucose level, paving the way for the consideration of GIP as an add-on to glucose-lowering therapies, like GLP-1.6 Moreover, GIPR signaling blocks emesis and attenuates other negative side effects of GLP-1 receptor (GLP-1R) activation.7
Tirzepatide is a unimolecular, bifunctional peptide invented by Eli Lilly and Company (Indianapolis, IN, USA), which simultaneously activates GLP-1R and GIPR. Tirzepatide is composed of 39 amino acids, is amidated at the C-terminal, conjugates a C20 fatty diacid moiety via a spacer connected to Lys20, and has a half-life of 116.7 h. Tirzepatide has a higher affinity with GLP-1R than GLP-1 does (pIC50: 8.90 vs 8.62), and its cAMP activation activity is significantly lower (1–2 decrease in pEC50). Tirzepatide has an obviously lower affinity with GIPR than GIP does (pIC50: 6.70 vs 7.87), and its cAMP activity is approximately equal to that of GIP.6,8
To date, tirzepatide has achieved an unprecedented effect on improving Hemoglobin A1c (HbA1c) and body weight in T2DM patients. The drugs used in T2DM were systematically compared by Alan Maloney et al9 (data updated on the website: https://www.comparediabetesdrugs.com): 15mg tirzepatide lowered HbA1c by 22.8 mmol/mol and weight by 10kg in patients, which were the best data ever. Another meta-analysis showed that HbA1c was reduced 2.1% and body weight 8.6kg with 15mg dose.10
To obtain dual activities, which seems to contribute the most in its illustrious clinical effects, tirzepatide not only fuses amino acid residue mainly from GLP-1 and GIP, but also uses some distinctive residues. In the next part, how tirzepatide is built is attempted to analyze based on existing reports.
Analysis of Tirzepatide Residues
Residues of tirzepatide are mainly from GLP-1, GIP and semaglutide and a few residues are unique (Figure 2). The contributions of each substitution are discussed in detail and mainly summarized in Table 1.
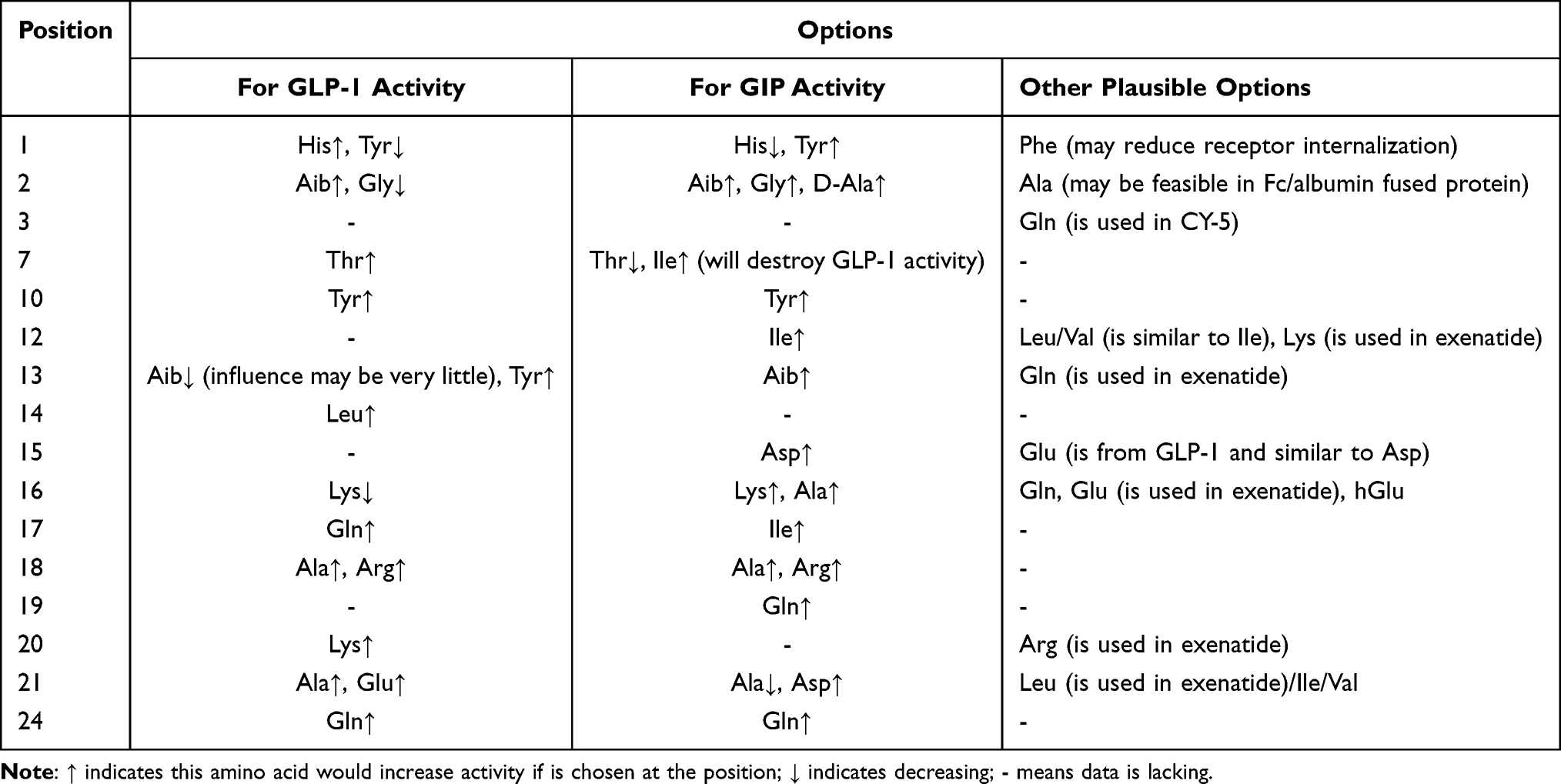 |
Table 1 Options of Residues in a GLP-1R/GIPR Dual Agonist |
 |
Figure 2 The structures of incretins and analogues. Different color indicates the derivation of each residue. Light blue: GLP-1; Yellow: GIP; dark blue: exenatide; Red: tirzepatide. |
Tyr1
GLP-1 (7–37) His7 is crucial to the activation of GLP-1R. In vivo, GLP-1 is a substrate for DPP-4 and is rapidly metabolized to GLP-1 (9–37) or GLP-1 (9–36) NH2 1. Among the series of analogues, including GLP-1 (1–37), GLP-1 (6–37), GLP-1 (7–37), GLP-1 (8–37), etc., only those which start with His7 retain their insulinotropic activity.11 GLP-1R Arg299 and Trp306 may interact with GLP-1 His7.12 Replacement of GLP-1 His7 with Phe7 does not influence either its receptor affinity or cAMP activity, indicating that the aromaticity of His is crucial to its activity. Replacement with Tyr7 lowers both the affinity (IC50: 0.26 nM→2.7 nM) and activity (EC50: 0.8 nM→5.4 nM). Replacement with Trp7 lowers the affinity (IC50: 0.26 nM→3.3 nM) and strongly lowers the activity (EC50: 0.8 nM→127 nM).13
The absence of Tyr1 from the N-terminal of GIP dramatically decreases its activity. GIPR Gln224, Arg300, and Phe357 have been described to interact with GIP Tyr1.14
The adoption of Tyr1 by tirzepatide may impair its GLP-1 activity while supporting its GIP activity. There were two other dual agonists whose sequence has been reported so far, Tyr1 has been used in MAR709 (designed by Finan et al in 2013, described by numerous other names: DA5-CH, DA-JC1, NNC0090-2746, RG7697, etc.) and His1 has been used in CY-5 (Figure 3).15,16 Additionally, Phe1 exenatide shows stronger long-term insulin release (which is dependent on β-arrestin recruitment reduction), faster agonist dissociation rates and lower receptor internalization than exenatide.17 Therefore, when designing a dual agonist, either His or Tyr can be chosen for position 1 by weighing the required GLP-1 and GIP activity, and Phe is also worth considering. Furthermore, several modifications of GIP Tyr1 that facilitate its insulinotropic activity18 may also be considered.
 |
Figure 3 The structures of other two GLP-1R/GIPR dual agonist. Residues in green means they are unique in MAR709 while in orange are unique in CY-5. |
Aib2
GLP-1 (7–37) Ala8 and GIP Ala2 are DPP-4 cleavage sites and many studies have attempted to reduce this degradation by substituting or modifying the first three residues. The findings suggest that substitution with Aib or Gly can fully prevent DPP-4 degradation. The affinity, cAMP activity, and insulinotropic activity in vitro all decline when Ala8 is substituted with Gly8 (IC50: 44.9 nM→220 nM;EC50: 0.15 nM→1.11 nM).19 Substitution with Aib8 hardly influences the cAMP activity and substitution with D-Ala8 induces little or no change in the affinity or cAMP activity.11
Considering GIP analogues, Gly2 GIP shows improved cAMP activity and insulinotropic activity in vitro,20 while the cAMP activity of D-Ala2 GIP matches that of GIP.21 The introduction of Aib2 into GIP has also been reported, and it seems that it does not obviously affect the cAMP activity.15,22
Comprehensively, using Aib2 is likely to only minimally influence the GLP-1 and GIP activities. Although Gly2 affects GLP-1 activity to some extent, overall, exenatide shows better activity than natural GLP-1. Thus, Gly2 may be feasible if a dual agonist is biosynthesized. Furthermore, since the instability of Ala2 is caused by degradation of DPP-4, Ala2 may not need to be changed if the peptide is fused with albumin or Fc domain (discussed later) for long-term activity, which creates a steric hindrance to prevent the enzyme from getting close.
Thr7
GLP-1 and GIP are partially homologous, in that GIP can partially activate GLP-1R in extreme doses and GLP-1 may also activate GIPR.15,23 Tyr1 and Ile7 are crucial for the selective activation of GIPR by GIP. Among the possible recombinant GIP analogues, the analogues that simultaneously conserve Tyr1 and Ile7 show similar GLP-1R-activating activity to natural GIP, and Ile7 is more important than Tyr1.23 Importing Ile7 into MAR709 caused a sharp decline in GLP-1 activity. The mechanism remains unclear; Moon et al suggested a hypothesis that GLP-1 Thr13 interacts with a binding pocket formed by Ile196, Leu232, and Met233 of GLP-1R because Thr7-containing chimeric peptides are highly sensitive for the Ile196 mutation;24 but it contradicts the model constructed by Zhang et al that Lys197 of GLP-1R is highly conserved and is hydrogen bonded to Thr13 of GLP-1.12 The substitution of GLP-1 Thr13 or GIP Ile7 with Ala obviously decreased their receptor-activating activities25,26 and the substitution of GIP Ile7 with Thr decreased its cAMP activity (pEC50: 9.76→9.58;Emax: 9.91→8.90).23 Incorporating Thr7 from GLP-1 into tirzepatide may lower its GIP activity slightly.
Tyr10
Positions 10, 12, 13, and 14 of exenatide (full GLP-1R agonist) are different from those of GLP-1, suggesting that these residues are unimportant to GLP-1. The mutation of GLP-1 positions 16, 17, 18, and 20 into Ala showed minor effects (IC50 from 0.27 nM to 1.7, 0.46, 0.68, and 1.7 nM; maximum EC50 varied from 2.6 to 7 nM).25 Ala substitutions in positions 10 and 11 of GIP had little negative effect on insulinotropic activity while position 12 and 14 mutations had a greater negative effect.27
Tyr10 and Ile12 from GIP are used in MAR709 and Ile12 plays a key role in the activation of GIPR 15. Tyr10 and Ile12 are also used in tirzepatide. In GLP-1 (7–37) NH2, both the affinity and activity are slightly promoted when Val16 is replaced with Tyr16.28 Thus, the Tyr10 in tirzepatide may have a positive influence on both GLP-1 and GIP activity.
Ile12
Using cryo-electron microscopy, Zhang et al found that a cluster of neighboring serines (Ser14, Ser17, and Ser18 of GLP-1) form polar interactions with Thr298, the backbone oxygen of Trp297, and the nitrogen of Arg299 in GLP-1R.12 However, Ala mutations at any of these positions hardly affected activity.25,29 In contrast, if GLP-1R Thr298 was mutated into Ala, it increased affinity to the ligand.30 Keliher et al ran a series of modifications of Lys12 in exenatide, indicating that this position is not important for interactions with GLP-1R and that it is, to some extent, tolerant of bigger groups.31
The mutation of GIP Ile12 to Ala caused a notable decrease in its insulinotropic effect.27 Hence, using Ile12 in tirzepatide seems not to weaken its GLP-1 activity and to strengthen its GIP activity. In GIP/GIPR binding models, Ile12, which is surrounded by hydrophilic components,32 might not interact with GIPR. However, the substitution of Ile12 with Ala remarkably decreased activity. Therefore, GIP Ile12 could function as a spatial obstruction, for example restricting GIPR Arg289 so that it interacts with GIP Asp15 rather than Asp9 (as a modeling in RCSB PDB: 7DTY, https://www.rcsb.org). Based on this assumption, Ile12 could be replaced with Leu or Val and retain the same effect. Even Phe could be considered as a potential replacement, and Lys from exenatide/CY-5 also seems feasible.
Aib13
GLP-1, GIP, GCG (glucagon), and GLP-2 share high homology so that it ought to be considered that GCGR and GLP-2R should sidestep the activation of the designed GLP-1/GIP dual agonist. In a CY-5 experiment, replacing position 13 with Aib rendered the agonist inactive toward GCGR and slightly altered GLP-1 and GIP activity.16 Substituting GLP-1 (7–37) Tyr19 with Ala notably decreased both the affinity and activity (IC50: 0.27 nM→3.5 nM;EC50: 2.6 nM→55 nM).25,29
In GIP, there is a small chance that activity can be influenced by replacing Ala13 with Aib. The Aib13 of tirzepatide seems to lower its GLP-1 activity without affecting its GIP activity, and to destroy its GCG activity. However, this hypothesis lacks supporting evidence. However, Aib13 is not necessary because other substitutions can fulfill the same role. For instance, in MAR709 Tyr13 from GLP-1 is retained, and yet it does not activate GCGR. GIP (1–14) is a partial agonist of GIPR. Substitution with Tyr13 scarcely affects the activity of GIP (1–14) analogues,33 implying that the use of Tyr13 in the designed dual agonist may not conspicuously affect GIP activity. Furthermore, considering Gln13 in exenatide, GLP-1 presumably interact with GLP-1R via a hydrogen bond. Tyr, Gln, Thr, Ser, and Lys are worth trying in this site.
Leu14
For certain functions, the Met14 of exenatide can be substituted with Leu14, because Met is easily oxidized and has little effect on activity.34
Asp15
The Glu21 of GLP-1 mutating into Ala or Gly negatively impacts activity.25,35 However, as Glu resembles Asp, using Asp15 from GIP in tirzepatide should not influence its GLP-1 activity.
Lys16
Replacing the Gly22 of GLP-1 with Ala or Aib slightly decreased both its affinity and activity (Ala IC50: 0.27 nM→0.57 nM;EC50: 2.6 nM→4 nM).25,36 In contrast, replacement with Glu increased both the affinity and activity to some degree.36 The attachment of long fatty acids relayed by a carboxylic linkage leaves the affinity and activity almost unaffected.2 A lactam bridge is reported to constrain the turning of the helix. A lactam bridge loaded at Glu22, either c[Lys18–Glu22] or c[Glu22–Lys26], shows lower affinity and activity than a simple substitution of Gly22 with Glu.28,36 Theoretically, the substitution of Gly22 with Ala would make the α-helix more stable, but increments in the affinity and activity were not found; substitution with Glu is likely to stabilize the α-helix as well; however, further increments in the affinity and activity were not found when a more stable lactam bridge was imported; instead, decrements are observed. Thus, the increments in the affinity and activity owing to Glu22 could be caused by a carboxyl group on Glu adding an extra binding site for the receptor. In this case, the affinity would weaken if Glu was replaced with a lactam bridge because the charge would be dispersed. Moreover, compared to c[Glu22–Lys26] Gly8 GLP-1, the affinity and activity of c[Asp22–Lys26] Gly8 GLP-1 are lower while those of c[hGlu22–Lys26] Gly8 GLP-1 are higher (hGlu: homoglutamic acid),36 indicating that the length of the amino acid side chain is an important factor.
Tirzepatide Lys16 may have a stacking interaction with GIPR Phe127.8 In CY-5 experiments, if Ser16 from GCG was changed to Lys from GIP, the GLP-1 activity decreased while the GIP activity increased.16 Surprisingly, replacing GIP Lys16 with Ala promotes the insulinotropic effect of GIP.27 The attachment of fatty acid chains to Lys16 via an amido bond does not conspicuously influence GIP activity.37 Thus, Gln seems feasible.
In summary, a carbon side chain of a certain length is favorable, or at least feasible, for interacting with both GLP-1 and GIPR. Apart from this hydrophobic region, it seems that GLP-1R prefers a carboxyl group at the end of the side chain while GIPR can accommodate, but does not favor, an alkaline group. Tirzepatide Lys16 may slightly decrease GLP-1 activity, but it enhances GIP activity. The following candidates are options: Ala, Gln, or Glu.
Ile17
MAR709 experiments have tested the concurrent substitution of Gln17, Ala18, and Ala19 from GLP-1 to Ile17, His18, and Gln19 from GIP, and found that GLP-1 activity decreased (EC50: 0.022 nM→0.140 nM) while that of GIP scarcely increased (EC50: 6.258 nM→5.530 nM).15 In terms of their overall effect, these changes seem not worthwhile.
Replacing GLP-1 Gln23 with Ala slightly lowered the affinity and activity (IC50: 0.27 nM→1.1 nM; EC50: 2.6 nM→5 nM).25 Changing GIP Ile17 to Ala massively lowered the insulinotropic effect.27 The Ile17 from GIP chosen in tirzepatide may be important to GIP activity but could cause a decline in GLP-1 activity.
Ala18
The mutation of GIP His18 to Ala reinforced its insulinotropic effect.27 In CY-5 experiments, replacing Arg18 from GCG with Ala simultaneously improved both the GLP-1 and GIP activity.16 Using the Ala18 from GLP-1 may support GIP activity by chance.
Gln19
GIP (1–30)-NH2, an active fragment of GIP with affinity and activity that align with those of integrated GIP, dramatically decreased somatostatinotropic activity in isolated perfused rat stomachs. Even when further broken down, GIP (1–14) and GIP (19–30) showed some activity. A study demonstrated that, among GIP (15–30), GIP (16–30), GIP (17–30), and GIP (19–30), GIP (19–30) was the only having activity although it is weak.38 GIP Gln19 may form hydrogen bonds with GIPR Gly29 and Thr31.32 The Gln19 adopted in tirzepatide seems to be crucial to GIP activity, but may still affect GLP-1 activity.
Lys20-X
Replacing GLP-1 Lys26 with Ala causes both the affinity and activity to decrease slightly.25 In semaglutide, a fatty acid chain is attached at this site and the affinity and activity are boosted.2 GLP-1 Lys26 may form a polar interaction with the receptor, which is presumably enhanced by Arg20 in exenatide, but creates no steric hindrance. GIP is similar, in that GIP Gln20 appears to form a hydrogen bond with GIPR Asn120 and expose unhindered into the extracellular matrix.32 A fatty acid chain attached to Lys20 in tirzepatide may boost the GLP-1 activity while the GIP activity remains unaffected.
Ala21
The mutation of GLP-1 Glu27 to Ala does not change the affinity but slightly increases activity.25 In CY-5 experiments, Asp21 from GIP was replaced by Ala; the GLP-1 activity changed little but slight decreases were observed in the GIP activity. Thus, the Glu27 of GLP-1 is not a critical point in the interaction with GLP-1R. GIP (1–42) Asp21 may form a hydrogen bond with GIPR Arg131 and the mutation of GIPR Arg131 to Ala decreases the affinity and activity. Surprisingly, this hydrogen bond between Asp21 and GIPR Arg131 vanishes in GIP (1–30).32 Since tirzepatide does not have a GIP (31–42) tail, chosen Ala21 was likely to increase the GLP-1 activity, despite it slightly decreasing the GIP activity. In humans, GLP-1 is not only damaged by DPP-4, but also by neutral endopeptidase 24.11 (NEP 24.11), and the main catalytic sites are Glu27–Phe28 and Trp31–Leu32.39 Using the Ala21 of tirzepatide can perhaps sidestep this NEP 24.11 degradation to a certain extent. Moreover, the Leu in position 21 of exenatide is unique, and its mutation seems to be unimportant. Thus, the spectrum to consider is expansive, including Glu from GLP-1, Asp similar to Glu from GIP, Ala, and even Leu/Ile/Val (forming a hydrophobic area along with Phe22).
Val23, Ile27
GLP-1 (7–37) Phe28, Trp31, and Leu32 are symmetrical with positions 22, 25, and 26 of GIP. GLP-1, GIP, and exenatide seem to have the same patterns here: Phe22 is centered to specifically interact with the receptor and the adjacent hydrophobic residues of positions 23, 25, and 26 are used to form a hydrophobic region. The residue of position 24 further away can interact polarly with the receptor and is probably beneficial in locating the hydrophobic region.
GLP-1R Trp78, Pro90, Trp91, and His212 could form a hydrophobic region with GLP-1 Phe28, Ile29, Trp31, Leu32, and Val33, and GIPR Leu35, Trp39, Tyr36, Met67, Tyr87, Trp90, and His115 could form a hydrophobic region with GIP Phe22, Val23, Trp25, Leu26, and Leu27.27 GLP-1 Ile29 and Val33 have similar properties as GIP Val23 and Leu27. The mutation of GLP-1 Phe28 to Ala causes it to lose nearly all of its affinity (IC50: 0.27 nM→357 nM), and Ile29 mutation decreases the affinity notably (IC50: 0.27 nM→25 nM), while Trp31, Leu32, and Val33 mutations have little influence, showing that they may be merely subsidiary.25 The Val23 and Ile27 used in tirzepatide are likely to have little impact on either GLP-1 or GIP activity because they are the same kind of residue and no specific interaction in these sites has been detected.
Gln24
Substituting GLP-1 Ala30 with Gln decreases the affinity (IC50: 0.27 nM→1.4 nM) but increases activity (EC50: 2.6 nM→0.5 nM).25 GIP Asn24 is similar to Gln; thus, the Gln24 adopted in tirzepatide could elevate GLP-1 activity and support GIP activity.
Ile27-
From positions 33–35 of GLP-1, each replacement with Ala increases the IC50 about five-fold but somewhat decreases the EC50.25 Although these replacements lower the affinity, ligands are made to ‘correct’ the construction. Linking position 30 and 34 with a lactam bridge simultaneously improves affinity and activity.40 Construction ‘correction’ could mean forming a more stable α-helix. GLP-1 Arg36 replacement with Ala obviously hinders affinity (IC50: 0.27 nM→4.6 nM) and activity (EC50: 2.6 nM→7 nM).25 Both affinity and activity reduce 10-fold if the Arg36 of GLP-1 is cut away,41 showing that it is crucial for interaction. Moreover, Gly35, Arg36, and Gly37 may contribute to selectivity of GLP-1.42
Compared to GLP-1, exenatide has nine extra residues in the C-terminal, which constitute a so-called ‘Trp-cage’ structure, folding and shielding the Trp25 indole ring from solvent exposure. Along with truncating the C-terminal, this exposes exenatide to NEP 24.11 degradation.43 Besides, attaching the GLP-1 C-terminal to exenatide (31–39) can prevent DPP-4 degradation.44
With C-terminal curtailing, the insulinotropic activity of exenatide gradually declines. Interestingly, when curtailed to exenatide (1–28), its insulinotropic activity recovers almost all integrity, but when it reaches exenatide (1–26), its insulinotropic activity suddenly disappears.19 Apparently, the Lys27 and Asn28 of exenatide play important roles in binding with the receptor. Most previous experiments were comparisons between exenatide (1–30) and GLP-1, thus exploring the function of the extra residues (31–39) in the exenatide C-terminal. However, in the above-mentioned experiments, the comparison should be between exenatide (1–28) and GLP-1, as exenatide (29–39) is truly the extra segment.
Cutting exenatide (29–39) away has a minimal influence on insulinotropic activity in vitro. Moreover, in MAR709 experiments, the attachment of exenatide (29–39) has a limited positive effect on GLP-1 activity (EC50: 0.022 nM→0.020 nM).15 Although it may decrease the degradation of exenatide in vivo, other strategies to lengthen the half-life can fulfill this effect. Hence, the attachment of an extra tail seems unnecessary.
In vivo, GIP can be scissored between GIP (1–30) and GIP (34–42) by prohormone convertases.32 Compared to GIP (1–42), GIP (1–30) has a symmetrical IC50, a slightly lower EC50, and a higher Emax.45 In mouse experiments, no variance has been found in the metabolic effects of GIP (1–30) and GIP (1–42).46 In a simulative binding model of GIP and the receptor, the fragment from position 31–42 neither interacted with the receptor, nor formed any specific structure, but it was able to cling to the receptor.32 Nevertheless, some aspects of the 31–42 segment of the GIP remain undiscovered, as GIP (3–30) is an antagonist while GIP (3–42) is a partial agonist of GIPR.14
GLP-1 (7–36) NH2, exenatide (1–28) and GIP (1–30), all have two polar residues starting from position 27: GLP-1 has Lys28 and Arg30, exenatide has Lys27 and Asn28, and GIP has Gln29 and Lys30. They seem to have these similar patterns to interact with the receptor. At the initial stage of ligand–receptor binding, the ligand clings to the receptor using its amorphous polar terminal. Then, gradually and thermodynamically, it moves to form the correct interface with the receptor. It further forms a stable helical conformation, with assistance from the receptor. Phe22(28) is inserted to match the hydrophobic region of the receptor, forming a relatively strong interaction. However, as the locations of the two polar residues are distinct, the receptor interaction sites are also different. The tirzepatide arrangements in GLP-1R and GIPR have an angular difference of 8.3°.8
An amorphous polar terminal could be implemented for practical applications, such as increasing the odds of contact between ligands and receptors under flowing conditions. In CY-5 experiments, substituting Lys27 with Ile27, or Lys27, Asn28 with Ile27, Ala28 decreased the EC50 of both GLP-1R and GIPR, but decreased hypoglycemic efficacies in mice.16
Tirzepatide Ile27 is akin to GLP-1 Val27 and GIP Leu27. Thus, their substitution seems not to affect activity. Using the Ala28 from GIP may decrease affinity with GLP-1R and increase the activities of the two receptors in vitro, but it is likely to decrease these activities in vivo. Moreover, tirzepatide elongated with exenatide (29–39) would most likely show no evident effect.
When designing a neo agonist, the length of the C-terminal may not be as important as previously thought. The crucial point could be that some polarity (such as alkalinity) is required. At least one alkaline Lys residue is contained in the C-terminals of GLP-1, exenatide, and GIP. The replacement of GLP-1 (7–37) Lys34 with Arg causes a two-fold reduction in the EC50.2 Whether the angular variance of tirzepatide binding to GLP-1R and GIP affects its activity has not yet been determined. If the number of polar residues increases, for example using the sequence of Ile27Lys28Gln29Lys30, making it possible to match two receptors, will this angular variance be eliminated?
Half-Life Extension Strategies
Apart from DPP-4 enzymolysis, another GLP-1/GIP degradation pathway, renal metabolism, can be overcome by augmenting the drug volume to prolong the half-life. Lipidation additionally induces concentration-independent liraglutide multimerization, resulting in a six- to eight-fold increase in size. However, it is unclear which of these, the size increase or HSA binding, are more pharmacologically relevant and account for the observed extension of the half-life.47 Another approach is PEGylating which may destroy the activity in vitro but retain the efficacy in vivo.15
Human serum albumin (HSA) and IgG have plasma half-lives of around 21 days, owing to HSA and the Fc domain of IgG interacting with the neonatal Fc receptor expressed in blood vessel endothelial cells, which rescues the proteins in the endosomes after pinocytosis, preventing them from entering the lysosomal pathway for destruction. By binding with HSA or IgG-Fc, the half-life of protein drugs could be extended. HSA and IgG-Fc can bind proteins not only through covalent attachments, but also through non-covalent labyrinthine interactions between endogenous albumin and long fatty acid chains, which have been added to GLP-1/GIP in some previous studies.47 The crucial elements of this strategy are the length of the fatty acid chain and the position of lipidation. Generally, the longer the fatty acid chain, the more readily it binds to HSA. However, excessive length may impede ligand to bind and activate receptor. The fatty acid side chain used in CY-5 is same as that of semaglutide, used in tirzepatide just having two more carbon as C20, used in MAR709 is a simple C16 chain without spacer located at Lys40.6,15,16 Taken together, the appropriate length is between C16 and C20 and most suitable lipidation position can be chosen from the following range of positions (starting from position 1): 10, 16, 20, 21, 28, and C-terminal.2,48 Apart from fatty acid chains, there are other molecules that could be used to connect to albumin and could be worth exploring.49
The semaglutide development process shows that the lipidation approach is decisive to the selectivity of the protein. Apparently, the intensity of the interaction between albumin and the fatty acid will not change if the length of the fatty acid remains unchanged. However, attaching the fatty acid chain to different sites can diversify the effects extensively. For example, in 2% albumin condition, the IC50 is 5.39 when lipidation occurs at position 27, yet at position 26 it reaches 357.2 It is possible that lipidation at certain sites can create steric hindrance that does not allow GLP-1Rs to capture ligands directly from the albumin, but only to trap free ligands released by the albumin. According to this hypothesis, shortening the fatty acid chain would decrease the affinity of the ligands to albumin while elongating it would impair their selectivity for binding albumin or the receptor, because they would be able to bind both simultaneously.
For biosynthesis, it seems the only strategy to prolong the half-life is a ligand fused with a terminal albumin or Fc domain, whose critical points are length and location. However, whether a protein of such size could infiltrate the blood–brain barrier to bind GLP-1R and GIPR in the brain, and whether receptors in the brain are important to the therapeutic effect, still need to be elucidated.
Plausible Orientations
The contribution of GIP part in dual agonist is still uncertain. Tirzepatide had finished its last Phase 3 clinical trial SURPASS-5 in February 2022,50 and it favors activating GIPR (for GIPR, native GIP EC50: 0.0334nM, tirzepatide EC50: 0.0224nM; for GLP-1R: native GLP-1 EC50: 0.0705nM, tirzepatide EC50: 0.934nM).6 MAR709 had finished its Phase 2a clinical trial in 2017,51 and it is a balanced dual agonist for GLP-1R and GIPR. ① Comparing the results of phase 2 clinical trials of these two drugs (baseline characteristics of populations were similar),51,52 tirzepatide seemed to be more efficacious in improving T2DM. After 12 weeks treatment, the reductions in HbA1c from baseline were −0.96% with 1.8mg MAR709, −0.9% with 1mg tirzepatide and −1.7% with 5mg tirzepatide. Other parameters were compared by Marie Bastin and Fabrizio Andreelli.53 The dose of 5mg MAR709 was determined to be not tolerated54 while a wide dose range of tirzepatide from 1mg to 15mg was established.52 The discrepancy of clinical efficacy of these two drugs is due at least partially to their difference of proportion of GLP-1 and GIP activity. In healthy people, postprandial GIP levels are approximately 4-fold higher than GLP-1 levels.55 The predicted quantity of GIPR occupied by tirzepatide is also more than that of GLP-1R (6.3-fold at 5mg, 4.6-fold at 10mg and 4.1-fold at 15mg).56 This may be the secret of the extraordinary efficacy of tirzepatide, but it is still possible that the best ratio of GLP-1 and GIP activity in a dual agonist has not been discovered.
Additionally, GIPR antagonists also exhibit improvement of metabolism on obese mice57 and GIPR agonists or antagonists both show therapeutical effects in combination with GLP-1R agonists.58 A GLP-1R agonist/GIPR antibody (antagonist) fused protein was reported promoting body weight loss.59 Nevertheless, whether a GLP-1R agonist/GIPR antagonist can improve T2DM needs more evidences. A GLP-1R agonist/GIPR antagonist peptide is theoretically possible but few full-length GIP-like antagonists were reported,60 therefore it requires an array of attempts, at least an alanine-scan experiment of GIP with measuring of affinity.
GLP-1R and GIPR belong to class B1 of the G protein-coupled receptors, and share high homology and a similar action pattern. G protein-coupled receptors include an N-terminal extracellular domain (ECD) and a C-terminal transmembrane domain (TMD), linked to each other through a soft sequence. In an inactive state, ECD favors a closed conformation covering the TMD. Subtle dynamics allows the C-terminal of the ligand to access the ECD binding site, and the binding of the ligand triggers further dissociation between the ECD and TMD, allowing the N-terminal to enter the orthosteric pocket in the TMD and activate the receptor. Small populations of open conformations exist as well and can smoothly accommodate the ligand.61
GLP-1R activation triggers Gαs-protein coupling, elevating cAMP, modulating calcium mobilization, and inducing β-arrestin recruitment.62 Different ligands lead to different interaction patterns with the receptor, generating various effects. For example, the P5 exenatide analogue is biased to reduce the recruitment of β-arrestin, resulting in better long-term hypoglycemic effects than those shown by exenatide, under the precondition that P5 has less cAMP activity.62 Tirzepatide is also biased toward cAMP activation and the activity of β-arrestin recruitment is lower than that of native GLP-1 for GLP-1R.8,56,63 After binding with a ligand for a certain period, the receptor will trigger the inward sinking of the membrane, forming an endosomal compartment, which is called agonist-induced receptor internalization.64 Some of these endosomal compartments return to the membrane, while others fuse with lysosomes.
For GLP-1R and GIPR, both tirzepatide and MAR709 showed strikingly decreased receptor internalization compared to mono agonist. GLP-1R internalization is mediated by Gαq signaling and influenced by β-arrestin 1/2; GIPR internalization is mainly influenced by β-arrestin 2.63 β-arrestin is involved in the desensitization of the receptor, and G-protein coupling will be blocked if the receptor is occupied with β-arrestin. Moreover, β-arrestin mediates receptor internalization and reduces the amount of receptors on the surface of the membrane.
The level of secreted incretin of diabetics commonly resembles that of healthy people, yet the number of incretin receptors is substantially lower. This may be relevant to internalization. Reducing the recruitment of β-arrestin and the residence time of the ligand towards the receptor, thus decreasing internalization, is presumably the key of incretin drugs. The mutation of GIPR Glu354 to Gln increases GIP affinity, enhances the agonist residence time by 25%, increases cAMP activity, and facilitates the rate of internalization by 2.1–2.3-fold. The throng with Gln354 GIPR has lower bone-mineral density, a > 50% increase in fracture risk, and a slightly increased plasma glucose.65 Does the paradox that either GIPR agonist or antagonist exhibits benefits in combination with GLP-1R agonist correlates with the likely reduction of internalization, following the decrement of β-arrestin recruitment or the acceleration of the dissociation rate (Koff) between the ligand and receptor, owing to competition between used agonists and antagonists in endogenous GIP?
Hence, the dynamic study of incretin drugs may be the center of future research. For the moment, a certain association rate (Kon) is required to ensure ligand binding to receptors in blood flow and to compete with endogenous incretins to decrease internalization. A rapid dissociation rate is also required to allow ligands to leave the receptor timeously after activation, to avoid internalization and ultimately maintain the quantity of the incretin receptors in the body.
Apart from the substitution of protein residues, an extra sequence connected to the N-terminal or C-terminal could change the biased interactions between the ligand and receptor. Using rapid selection, Wu et al discovered a surprising dual agonist composed of GIP (3–30) and an extra sequence of nine amino acids in the N-terminal.66 Compared to GIP (1–42), GIP (1–39) has a higher ability to modulate calcium and thus a stronger insulinotropic effect.67
Conclusion
GLP-1 analogs were utilized broadly in treatment of T2DM. Another incretin GIP is rather mystery and limited in clinical use, but scientists never stop exploring, tirzepatide is one of the rewards of these efforts.
Compared to the first-generation GLP-1R agonist, tirzepatide has three critical improvements: first, many residues in peptide backbone are changed to obtain GIPR-activating activity; second, C-terminal is prolonged with a sequence of C-terminal of exenatide; third, a fatty acid side chain is conjugated similar to semaglutide to prolong half-life.
By weighing different residues adopted in each position, the activity of a dual agonist could be customized. Initially, main concerns were receptor-activating activity while recently, downstream effects of GLP-1R and GIPR have been more and more revealed that not only receptor activation promotes cAMP and stimulates insulin releasing, but also other factors (β-arrestin recruitment, residence time, etc.) influence receptor internalization, which further elicits long-term effects in vivo. Most existed reports uncovered cAMP activity changes after residue substitutions and they were summarized in part 2. Future research may pay more attention to other activities like β-arrestin recruitment or ligand-receptor affinity.
All of three dual agonist peptides, tirzepatide, MAR709 and CY-5, have a C-terminal same as exenatide but no convincing reasons have been put forward and its use is more likely an “inertia” from MAR709.
Incretins are largely degraded by DPP-4 in vivo, which can be hindered by substitution of first three residues or increasing steric hindrance. The substitution of Aib at position 2 avoids DPP-4 acting and through a fatty acid side chain tirzepatide can adhere to albumin in vivo, whose native half-life is about three weeks, thus can weekly stay in blood circulation. PEGylating or fusing peptide with albumin/Fc domain are also feasible and even the DPP-4 would be resisted by steric hindrance if the latter method is taken.
Although GLP-1 kind has been deeply studied, many GIP aspect fundamental features should be more clarified, such as an alanine scanning, to support development of GLP-1R/GIPR bifunctional drugs. Recent researches suggested the internalization of receptors is the key point and thus more dynamic studies are required eagerly.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–2157. doi:10.1053/j.gastro.2007.03.054
2. Lau J, Bloch P, Schaffer L, et al. Discovery of the once-weekly Glucagon-Like Peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58(18):7370–7380. doi:10.1021/acs.jmedchem.5b00726
3. Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43(9):1664–1669. doi:10.1021/jm9909645
4. Christou GA, Katsiki N, Blundell J, Fruhbeck G, Kiortsis DN. Semaglutide as a promising antiobesity drug. Obes Rev. 2019;20(6):805–815. doi:10.1111/obr.12839
5. Pratley RE. GIP: an inconsequential incretin or not? Diabetes Care. 2010;33(7):1691–1692. doi:10.2337/dc10-0704
6. Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab. 2018;18:3–14. doi:10.1016/j.molmet.2018.09.009
7. Borner T, Geisler CE, Fortin SM, et al. GIP receptor agonism attenuates GLP-1 receptor agonist induced nausea and emesis in preclinical models. Diabetes. 2021;70:2545–2553. doi:10.2337/db21-0459
8. Zhao F, Zhou Q, Cong Z, et al. Structural basis for the therapeutic advantage of dual and triple agonists at the human GIP, GLP-1 or GCG receptors. bioRxiv. 2021. doi:10.1101/2021.03.15.435309
9. Maloney A, Rosenstock J, Fonseca V. A model-based meta-analysis of 24 antihyperglycemic drugs for type 2 diabetes: comparison of treatment effects at therapeutic doses. Clin Pharmacol Ther. 2019;105(5):1213–1223. doi:10.1002/cpt.1307
10. Bhagavathula AS, Vidyasagar K, Tesfaye W. Efficacy and safety of tirzepatide in patients with type 2 diabetes mellitus: a systematic review and meta-analysis of randomized phase II/III trials. Pharmaceuticals. 2021;14:10. doi:10.3390/ph14100991
11. Hui H, Zhao X, Perfetti R. Structure and function studies of glucagon-like peptide-1 (GLP-1): the designing of a novel pharmacological agent for the treatment of diabetes. Diabetes Metab Res Rev. 2005;21(4):313–331. doi:10.1002/dmrr.553
12. Zhang Y, Sun B, Feng D, et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature. 2017;546(7657):248–253. doi:10.1038/nature22394
13. Sarrauste de Menthiere C, Chavanieu A, Grassy G, et al. Structural requirements of the N-terminal region of GLP-1-[7-37]-NH2 for receptor interaction and cAMP production. Eur J Med Chem. 2004;39(6):473–480. doi:10.1016/j.ejmech.2004.02.002
14. Gabe MBN, van der Velden WJC, Smit FX, Gasbjerg LS, Rosenkilde MM. Molecular interactions of full-length and truncated GIP peptides with the GIP receptor - a comprehensive review. Peptides. 2020;125:170224. doi:10.1016/j.peptides.2019.170224
15. Finan B, Ma T, Ottaway N, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5(209):209ra151. doi:10.1126/scitranslmed.3007218
16. Liu C, Li C, Cai X, et al. Discovery of a novel GLP-1/GIP dual receptor agonist CY-5 as long-acting hypoglycemic, anti-obesity agent. Bioorg Chem. 2021;106:104492. doi:10.1016/j.bioorg.2020.104492
17. Jones B, Buenaventura T, Kanda N, et al. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat Commun. 2018;9(1):1602. doi:10.1038/s41467-018-03941-2
18. Gault VA, Flatt PR, O’Harte FP. Glucose-dependent insulinotropic polypeptide analogues and their therapeutic potential for the treatment of obesity-diabetes. Biochem Biophys Res Commun. 2003;308(2):207–213. doi:10.1016/S0006-291X(03)01361-5
19. Doyle ME, Theodorakis MJ, Holloway HW, Bernier M, Greig NH, Egan JM. The importance of the nine-amino acid C-terminal sequence of exendin-4 for binding to the GLP-1 receptor and for biological activity. Regul Pept. 2003;114(2–3):153–158. doi:10.1016/S0167-0115(03)00120-4
20. Gault VA, Flatt PR, Harriott P, Mooney MH, Bailey CJ, O’Harte FP. Improved biological activity of Gly2- and Ser2-substituted analogues of glucose-dependent insulinotrophic polypeptide. J Endocrinol. 2003;176(1):133–141. doi:10.1677/joe.0.1760133
21. Hinke SA, Gelling RW, Pederson RA, et al. Dipeptidyl peptidase IV-resistant [d-Ala2]Glucose-Dependent Insulinotropic Polypeptide (GIP) improves glucose tolerance in normal and obese diabetic rats. Diabetes. 2002;51(3):652–661. doi:10.2337/diabetes.51.3.652
22. Norregaard PK, Deryabina MA, Tofteng Shelton P, et al. A novel GIP analogue, ZP4165, enhances glucagon-like peptide-1-induced body weight loss and improves glycaemic control in rodents. Diabetes Obes Metab. 2018;20(1):60–68. doi:10.1111/dom.13034
23. Moon MJ, Kim HY, Kim SG, et al. Tyr1 and Ile7 of glucose-dependent insulinotropic polypeptide (GIP) confer differential ligand selectivity toward GIP and glucagon-like peptide-1 receptors. Mol Cells. 2010;30(2):149–154. doi:10.1007/s10059-010-0100-5
24. Moon MJ, Park S, Kim DK, et al. Structural and molecular conservation of glucagon-like Peptide-1 and its receptor confers selective ligand-receptor interaction. Front Endocrinol. 2012;3:141. doi:10.3389/fendo.2012.00141
25. Adelhorst K, Hedegaard BB, Knudsen LB, Kirk O. Structure-activity studies of glucagon-like peptide-1. J Biol Chem. 1994;269(9):6275–6278. doi:10.1016/S0021-9258(17)37366-0
26. Alana I, Parker JC, Gault VA, et al. NMR and alanine scan studies of glucose-dependent insulinotropic polypeptide in water. J Biol Chem. 2006;281(24):16370–16376. doi:10.1074/jbc.M510414200
27. Venneti KC, Malthouse JP, O’Harte FP, Hewage CM. Conformational, receptor interaction and alanine scan studies of glucose-dependent insulinotropic polypeptide. Biochim Biophys Acta. 2011;1814(7):882–888. doi:10.1016/j.bbapap.2011.04.002
28. Plisson F, Hill TA, Mitchell JM, et al. Helixconstraints and amino acid substitution in GLP-1 increase cAMP and insulin secretion but not beta-arrestin 2 signaling. Eur J Med Chem. 2017;127:703–714. doi:10.1016/j.ejmech.2016.10.044
29. Gallwitz B, Witt M, Paetzold G, et al. Structure/activity characterization of glucagon-like peptide-1. Eur J Biochem. 1994;225(3):1151–1156. doi:10.1111/j.1432-1033.1994.1151b.x
30. Wootten D, Reynolds CA, Smith KJ, et al. The extracellular surface of the GLP-1 receptor is a molecular trigger for biased agonism. Cell. 2016;165(7):1632–1643. doi:10.1016/j.cell.2016.05.023
31. Keliher EJ, Reiner T, Thurber GM, Upadhyay R, Weissleder R. Efficient (18)F-labeling of synthetic exendin-4 analogues for imaging beta cells. ChemistryOpen. 2012;1(4):177–183. doi:10.1002/open.201200014
32. Smit FX, van der Velden WJC, Kizilkaya HS, et al. Investigating GIPR (ant) agonism: a structural analysis of GIP and its receptor. Structure. 2021;29(7):679–693 e676. doi:10.1016/j.str.2021.04.001
33. Hinke SA, Manhart S, Speck M, Pederson RA, Demuth HU, McIntosh CH. In depth analysis of the N-terminal bioactive domain of gastric inhibitory polypeptide. Life Sci. 2004;75(15):1857–1870. doi:10.1016/j.lfs.2004.03.024
34. Hargrove DM, Kendall ES, Reynolds JM, et al. Biological activity of AC3174, a peptide analog of exendin-4. Regul Pept. 2007;141(1–3):113–119. doi:10.1016/j.regpep.2006.12.021
35. Watanabe Y, Kawai K, Ohashi S, Yokota C, Suzuki S, Yamashita K. Structure-activity relationships of glucagon-like peptide-1 (7-36) amide: insulinotropic activities in perfused rat pancreases, and receptor binding and cyclic AMP production in RINm5F cells. J Endocrinol. 1994;140(1):45–52. doi:10.1677/joe.0.1400045
36. Miranda LP, Winters KA, Gegg CV, et al. Design and synthesis of conformationally constrained glucagon-like peptide-1 derivatives with increased plasma stability and prolonged in vivo activity. J Med Chem. 2008;51(9):2758–2765. doi:10.1021/jm701522b
37. Irwin N, O’Harte FP, Gault VA, et al. GIP(Lys16PAL) and GIP(Lys37PAL): novel long-acting acylated analogues of glucose-dependent insulinotropic polypeptide with improved antidiabetic potential. J Med Chem. 2006;49(3):1047–1054. doi:10.1021/jm0509997
38. Hinke SA, Manhart S, Pamir N, et al. Identification of a bioactive domain in the amino-terminus of glucose-dependent insulinotropic polypeptide (GIP). Biochim Biophys Acta. 2001;1547(1):143–155. doi:10.1016/S0167-4838(01)00181-9
39. Hupe-Sodmann K, McGregor GP, Bridenbaugh R, et al. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1 (7-36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regul Pept. 1995;58(3):149–156. doi:10.1016/0167-0115(95)00063-H
40. Murage EN, Schroeder JC, Beinborn M, Ahn JM. Search for alpha-helical propensity in the receptor-bound conformation of glucagon-like peptide-1. Bioorg Med Chem. 2008;16(23):10106–10112. doi:10.1016/j.bmc.2008.10.006
41. Knudsen LB, Pridal L. Glucagon-like peptide-1-(9-36) amide is a major metabolite of glucagon-like peptide-1-(7-36) amide after in vivo administration to dogs, and it acts as an antagonist on the pancreatic receptor. Eur J Pharmacol. 1996;318(2–3):429–435. doi:10.1016/S0014-2999(96)00795-9
42. Finan B, Yang B, Ottaway N, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015;21(1):27–36. doi:10.1038/nm.3761
43. Lee JG, Ryu JH, Kim SM, et al. Replacement of the C-terminal Trp-cage of exendin-4 with a fatty acid improves therapeutic utility. Biochem Pharmacol. 2018;151:59–68. doi:10.1016/j.bcp.2018.03.004
44. Simonsen L, Holst JJ, Madsen K, Deacon CF. The C-terminal extension of exendin-4 provides additional metabolic stability when added to GLP-1, while there is minimal effect of truncating exendin-4 in anaesthetized pigs. Regul Pept. 2013;181:17–21. doi:10.1016/j.regpep.2012.12.012
45. Hansen LS, Sparre-Ulrich AH, Christensen M, et al. N-terminally and C-terminally truncated forms of glucose-dependent insulinotropic polypeptide are high-affinity competitive antagonists of the human GIP receptor. Br J Pharmacol. 2016;173(5):826–838. doi:10.1111/bph.13384
46. Gault VA, Porter DW, Irwin N, Flatt PR. Comparison of sub-chronic metabolic effects of stable forms of naturally occurring GIP (1-30) and GIP (1-42) in high-fat fed mice. J Endocrinol. 2011;208(3):265–271. doi:10.1530/JOE-10-0419
47. Bukrinski JT, Sonderby P, Antunes F, et al. Glucagon-like peptide 1 conjugated to recombinant human serum albumin variants with modified neonatal Fc receptor binding properties. impact on molecular structure and half-life. Biochemistry. 2017;56(36):4860–4870. doi:10.1021/acs.biochem.7b00492
48. Li Y, Wang Y, Wei Q, et al. Variant fatty acid-like molecules conjugation, novel approaches for extending the stability of therapeutic peptides. Sci Rep. 2015;5:18039. doi:10.1038/srep18039
49. Bech EM, Martos-Maldonado MC, Wismann P, et al. Peptide half-life extension: divalent, small-molecule albumin interactions direct the systemic properties of Glucagon-Like Peptide 1 (GLP-1) analogues. J Med Chem. 2017;60(17):7434–7446. doi:10.1021/acs.jmedchem.7b00787
50. Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534–545. doi:10.1001/jama.2022.0078
51. Frias JP, Bastyr EJ 3rd, Vignati L, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 2017;26(2):343–352 e342. doi:10.1016/j.cmet.2017.07.011
52. Frias JP, Nauck MA, Van J, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392(10160):2180–2193. doi:10.1016/S0140-6736(18)32260-8
53. Bastin M, Andreelli F. Dual GIP-GLP1-receptor agonists in the treatment of type 2 diabetes: a short review on emerging data and therapeutic potential. Diabetes Metab Syndr Obes. 2019;12:1973–1985. doi:10.2147/DMSO.S191438
54. Portron A, Jadidi S, Sarkar N, DiMarchi R, Schmitt C. Pharmacodynamics, pharmacokinetics, safety and tolerability of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 after single subcutaneous administration in healthy subjects. Diabetes Obes Metab. 2017;19(10):1446–1453. doi:10.1111/dom.13025
55. De Block CEM, Dirinck E, Verhaegen A, Van Gaal LF. Efficacy and safety of high-dose glucagon-like peptide-1, glucagon-like peptide-1/glucose-dependent insulinotropic peptide, and glucagon-like peptide-1/glucagon receptor agonists in type 2 diabetes. Diabetes Obes Metab. 2022;24(5):788–805. doi:10.1111/dom.14640
56. Willard FS, Douros JD, Gabe MB, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. 2020;5:17. doi:10.1172/jci.insight.140532
57. Irwin N, McClean PL, O’Harte FP, Gault VA, Harriott P, Flatt PR. Early administration of the glucose-dependent insulinotropic polypeptide receptor antagonist (Pro3) GIP prevents the development of diabetes and related metabolic abnormalities associated with genetically inherited obesity in ob/ob mice. Diabetologia. 2007;50(7):1532–1540. doi:10.1007/s00125-007-0692-2
58. Holst JJ, Rosenkilde MM. GIP as a therapeutic target in diabetes and obesity: insight from incretin co-agonists. J Clin Endocrinol Metab. 2020;105(8):e2710–e2716. doi:10.1210/clinem/dgaa327
59. Lu SC, Chen M, Atangan L, et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med. 2021;2(5):100263. doi:10.1016/j.xcrm.2021.100263
60. Gault VA, O’Harte FP, Harriott P, Flatt PR. Characterization of the cellular and metabolic effects of a novel enzyme-resistant antagonist of glucose-dependent insulinotropic polypeptide. Biochem Biophys Res Commun. 2002;290(5):1420–1426. doi:10.1006/bbrc.2002.6364
61. Wu F, Yang L, Hang K, et al. Full-length human GLP-1 receptor structure without orthosteric ligands. Nat Commun. 2020;11(1):1272. doi:10.1038/s41467-020-14934-5
62. Zhang H, Sturchler E, Zhu J, et al. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat Commun. 2015;6:8918. doi:10.1038/ncomms9918
63. Novikoff A, O’Brien SL, Bernecker M, et al. Spatiotemporal GLP-1 and GIP receptor signaling and trafficking/recycling dynamics induced by selected receptor mono- and dual-agonists. Mol Metab. 2021;49:101181. doi:10.1016/j.molmet.2021.101181
64. Widmann C, Dolci W, Thorens B. Agonist-induced internalization and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem J. 1995;310(Pt 1):203–214. doi:10.1042/bj3100203
65. Gabe MBN, van der Velden WJC, Gadgaard S, et al. Enhanced agonist residence time, internalization rate and signalling of the GIP receptor variant [E354Q] facilitate receptor desensitization and long-term impairment of the GIP system. Basic Clin Pharmacol Toxicol. 2020;126(Suppl 6):122–132. doi:10.1111/bcpt.13289
66. Wu Y, Ji T, Lv J, Wang Z. Rapid selection of a novel GLP-1/GIP dual receptor agonist with prolonged glycemic control and weight loss in rodent animals. Life Sci. 2020;257:118025. doi:10.1016/j.lfs.2020.118025
67. Xie L, Lu J, Ostenson CG, Xu T, Chen ZW. GIP1-39, a novel insulinotropic peptide form and aspects on its mechanism of action. Regul Pept. 2004;121(1–3):107–112. doi:10.1016/j.regpep.2004.04.013
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.