")
Back to Journals » Drug Design, Development and Therapy » Volume 17
Design, Synthesis and Biological Evaluation of Glycosylated Derivatives of Silibinin as Potential Anti-Tumor Agents
Authors Xi JJ, Cao Y, He RY, Zhang JK , Zhao YM, Tong Q, Bao JF, Dong YC, Zhuang RX, Huang JS, Chen Y, Liu SR
Received 27 January 2023
Accepted for publication 30 June 2023
Published 11 July 2023 Volume 2023:17 Pages 2063—2076
DOI https://doi.org/10.2147/DDDT.S404036
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Frank Boeckler
Jian-Jun Xi,1,* Yu Cao,1,* Ruo-Yu He,1 Jian-Kang Zhang,2 Yan-Mei Zhao,1 Qiao Tong,1 Jian-Feng Bao,1 Yi-Chen Dong,3 Rang-Xiao Zhuang,1 Jin-Song Huang,1 Yongping Chen,4 Shou-Rong Liu1
1Department of Pharmacy, Hangzhou Xixi Hospital Affiliated to Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 2School of Medicine, Zhejiang University City College, Hangzhou, People’s Republic of China; 3Faculty of Chinese Medicine, Macau University of Science and Technology, Macau, People’s Republic of China; 4Department of Infectious Diseases, The First Affiliated Hospital of Wenzhou Medical University, Hepatology Institute of Wenzhou Medical University, Wenzhou Key Laboratory of Hepatology, Wenzou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongping Chen, Department of Infectious Diseases, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou Key Laboratory of Hepatology, Hepatology Institute of Wenzhou Medical University, Wenzhou, Zhejiang Province, 325025, People’s Republic of China, Email [email protected] Shou-Rong Liu, Department of Infectious Diseases, Hangzhou Xixi Hospital Affiliated to Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, 310023, People’s Republic of China, Email [email protected]
Objective: Silibinin, a natural product extracted from the seeds of the Silybum marianum, is versatile with various pharmacological effects. However, its clinical application was strongly hampered by its low bioavailability and poor water solubility. Herein, a series of glycosylated silibinin derivatives were identified as novel anti-tumor agents.
Materials and Methods: The cell viability was evaluated by CCK8 assay. Furthermore, cell apoptosis and cell cycle progression were tested by flow cytometry. In addition, the pharmacokinetic assessment of compound 15 and silibinin through intravenous administration (i.v., 2 mg/kg) to ICR mice were performed.
Results: The synthesized compounds showed better water solubilities than silibinin. Among them, compound 15 exhibited inhibitory activity against DU145 cells with IC50 value of 1.37 ± 0.140 μM. Moreover, it arrested cell cycle at G2/M phase and induced apoptosis in DU145 cells. Additionally, compound 15 also displayed longer half-life (T1/2 = 128.3 min) in liver microsomes than that of silibinin (T1/2 = 82.5 min) and appropriate pharmacokinetic parameters in mice.
Conclusion: Overall, glycosylation of silibinin would be a valid strategy for the development of silibinin derivatives as anti-tumor agents.
Keywords: glycosylation, silibinin derivatives, solubility, anti-proliferative activity
Graphical Abstract:
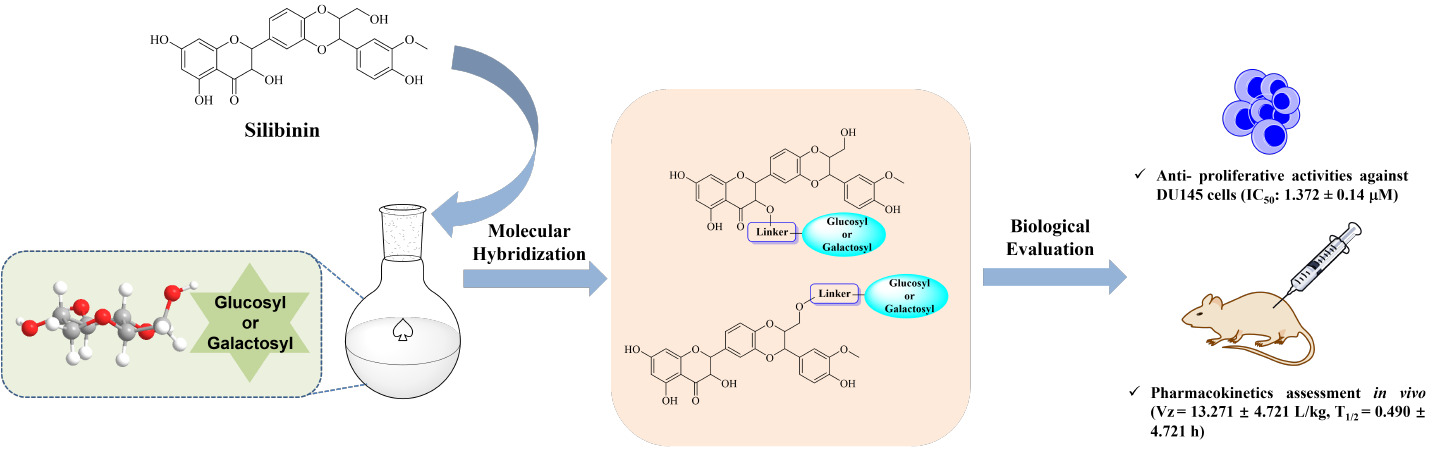
Introduction
Silymarin is a representative extract derived from the seeds of milk thistle (Silybum marianum), which consists of several flavonoid lignin-like compounds (silibinin, isosilybin and silychristin) (Figure 1).1–4 Silibinin, the most abundant active ingredient of silymarin, exhibits a variety of characteristics in terms of therapeutic activities, including antioxidant, free radicals scavenging, maintaining the stability of cell membrane, promoting hepatocyte proliferation and reducing blood lipids.5,6 Several animal studies in vivo have confirmed that silibinin possesses a significant inhibitory effect on different types of tumors including liver, colon, prostate, bladder and tongue through the suppression of receptor tyrosine kinase (RTK) represented by epidermal growth factor receptor (EGFR) and insulin-like growth factor 1 receptor (IGF-1R) and the activation of downstream signaling molecules.7–9 Additionally, enhanced efficacy of chemotherapy drugs can be achieved through combination with silibinin, avoiding the appearance of multidrug resistance (MDR).10
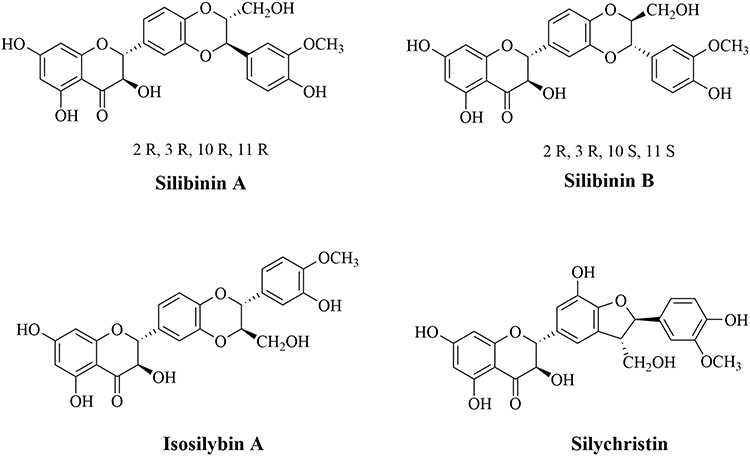 |
Figure 1 Chemical structures of respective components of silymarin. |
Despite a widespread use of silibinin for the treatment of various liver ailments for more than 3 decades, the pharmacological properties of silibinin are considerably hampered by its poor water solubility.11–14 Numerous approaches have been developed to overcome the low bioavailability of silibinin by means of the structural modification, delivery strategies and so on.15,16 On the one hand, silibinin derivatives can be obtained by the etherification, oxidation and esterification of hydroxyl groups of silibinin, formation of salts or the combination with functional groups using the splicing principle.17–19 On the other hand, silibinin can be modified via altering the cycle structures to expand the conjugated systems, adding unsaturated groups in the skeleton in order to improve the affinity on P-glycoprotein and the substitution of hydroxyl or alkoxy groups exerting antioxidant property.20,21 Apart from the above medicinal chemistry strategies, different delivery technologies have been employed to enhance the water solubility and biological activities, including solid dispersions, microspheres, nanoparticles and so forth.22–25 Nevertheless, the potency of some modified products is still weakened compared with original compounds. To improve drug-like properties and enhance antiproliferative activity of silibinin, further structural modifications are still needed.
It is reported that glycosylation can effectively improve the solubility of parent drug.26,27 The introduction of glycosyl moieties in the scaffolds generally enhances biological activities and targeting effects.28 In addition, cancer cells would consume more glucose during their rapid growth than normal cells through the effects on the glycolytic pathways, which probably due to the over-expression of membrane glucose transporter GLUT.29 Numerous studies have confirmed that the covalent binding of cancer drugs to carbohydrates contributes to targeting cancer cells.30 Hence, the glycosylated optimization could be a promising tool for improving bioactivities, reducing toxicity and enhancing targeting.
In general, the reactivity of hydroxyl groups in the 3 and 23 positions is higher than that of other positions which can be concluded from the structural characteristics of silibinin.15 Based on our previous studies with respect to the improvement of solution, selectivity and bioavailability, a pro-drug strategy has been used in drug design by introducing two glycosylated motifs in the 3 and 23 positions (Figure 2). Hence, a series of silibinin derivatives have been designed and synthesized to overcome the above problems.
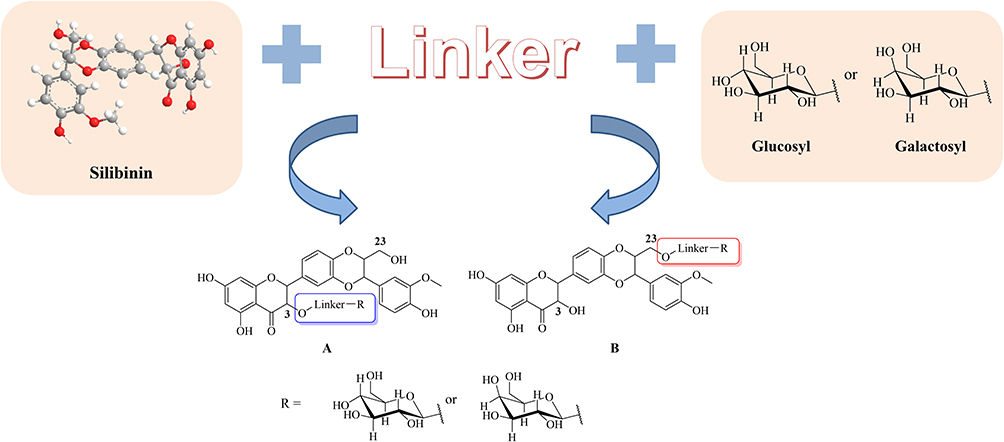 |
Figure 2 The design strategy of glycosylated derivatives of silibinin. |
Materials and Methods
Chemistry
The commercial reagents and solvents were used without further purification unless stated otherwise. Column chromatography was performed using silica gel (200–300 meshes). All yields are unoptimized and generally represent the result of a single experiment. The NMR spectra were recorded for 1H NMR at 400 MHz and for 13C NMR at 100 MHz. For 1H NMR, CDCl3 (δ = 7.26) and acetone (δ = 2.05) were served as internal standard and data were reported as follows: chemical shift, multiplicity, coupling constant in Hz and integration. For 13C NMR, CDCl3 (δ = 77.23) and acetone (δ = 206.68 and 29.92) were served as internal standard and spectra were obtained with complete proton decoupling. HRMS data were obtained on Agilent 1290 HPLC-6224 Time of Flight Mass Spectrometer. High-performance liquid chromatography (HPLC) analyses were performed on an Agilent 1100 system which was equipped with a photodiode array detector using an ChromCore 120C18 column (4.6 × 250 mm) and detected at 220 nm wavelength. The mobile phase A was 0.1% trifluoroacetic acid in 100% acetonitrile, and mobile phase B was 0.1% trifluoroacetic acid in water. A gradient of 12−100% A over 25 min was run at a flow rate of 1.0 mL/min.
General Procedure for the Synthesis of Fragments (3–4)
To a solution of compound 1–2 (0.20 mmol) in DCM (1 mL) was added DMAP (24.4 mg, 0.20 mmol) and tetrahydrofuran-2,5-dione (40.0 mg, 0.40 mmol). The mixture was stirred at 20 °C for 12 hr. TLC indicated compound 1–2 was consumed completely and one new spot formed. The reaction was clean according to TLC. The reaction mixture was quenched by addition 1 N HCl 5 mL at 20 °C, and then diluted with DCM 10 mL and extracted with DCM (10 mL × 2). The combined organic layers were washed with 1 N HCl 5 mL, dried over [Na2SO4], filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, DCM/MeOH = 19:1 to 4:1). Compound 3–4 was obtained as light-yellow oil.
4-Oxo-4-{[(2S,3R,4S,5R,6R)-3,4,5-tris(benzyloxy)-6-[(benzyloxy)methyl]tetrahydro-2H-pyran-2-yl]oxy}butanoic acid (3). Light yellow oil; Yield: 73.4%; HPLC purity: 95%; 1H NMR (400 MHz, CDCl3) δ = 7.31–6.99 (m, 20 H), 6.27 (br d, J=3.0 Hz, 0.7 H), 5.55 (d, J=8.0 Hz, 0.3 H), 4.90–4.80 (m, 1 H), 4.80–4.64 (m, 3 H), 4.63–4.46 (m, 3 H), 4.46–4.33 (m, 2 H), 3.89–3.74 (m, 1 H), 3.70–3.46 (m, 5 H), 2.66–2.43 (m, 4 H); HRMS (ESI, [M+H]+): 641.2746.
4-Oxo-4-{[(2S,3R,4S,5S,6R)-3,4,5-tris(benzyloxy)-6-[(benzyloxy)methyl]tetrahydro-2H-pyran-2-yl]oxy}butanoic acid (4). Light yellow oil; Yield: 88.2%; HPLC purity: 95%; 1H NMR (400 MHz, CDCl3) δ = 7.34–7.14 (m, 20 H), 6.32 (d, J=3.8 Hz, 0.5 H), 5.52 (d, J=8.0 Hz, 0.5 H), 4.87 (d, J=11.3 Hz, 1 H), 4.78–4.71 (m, 1 H), 4.69–4.59 (m, 3 H), 4.58–4.46 (m, 1 H), 4.43–4.28 (m, 2 H), 4.08 (dd, J=3.8, 10.0 Hz, 1 H), 3.96–3.86 (m, 2 H), 3.80 (dd, J=2.5, 10.0 Hz, 1 H), 3.64–3.42 (m, 3 H), 2.67–2.42 (m, 4 H); HRMS (ESI, [M+H]+): 641.2758.
The Procedure for the Synthesis of Intermediate 6
To a solution of compound 5 (964.9 mg, 2.0 mmol) in DMF (10 mL) was added K2CO3 (995.0 mg, 7.2 mmol) and BnBr (1.23 g, 7.2 mmol) at 0 °C. The mixture was stirred at 0–20 °C for 12 hr. TLC indicated that no compound 5 was remained, and one major new spot with lower polarity was detected. The reaction mixture was quenched by addition NH4Cl 20 mL at 20 °C, and then diluted with DCM 30 mL and extracted with DCM (30 mL × 2). The combined organic layers were washed with brine (30 mL × 5), dried over [Na2SO4], filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, DCM/Ethyl acetate = 100:1 to 9:1). Compound 6 (1.10 g, 1.46 mmol, 73.1% yield) was obtained as a light-yellow solid.
5,7-Bis(benzyloxy)-2-{3-[4-(benzyloxy)-3-methoxyphenyl]-2-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxin-6-yl}-3-hydroxychroman-4-one (6). Light yellow oil; Yield: 73.1%; 1H NMR (400 MHz, CDCl3) δ = 7.48 (d, J = 7.5 Hz, 2H), 7.42–7.21 (m, 14H), 7.14 (br d, J=14.6 Hz, 1H), 7.07–6.95 (m, 2H), 6.93–6.82 (m, 3H), 6.19 (s, 1H), 6.13 (dd, J=2.0, 6.3 Hz, 1H), 5.16–5.04 (m, 4H), 4.97 (d, J=2.5 Hz, 2H), 4.89 (br d, J = 9.5 Hz, 2H), 4.38 (dd, J = 6.4, 12.2 Hz, 1H), 4.03 (s, 1H), 3.97 (br d, J=6.5 Hz, 1H), 3.85 (s, 3H), 3.78–3.69 (m, 1H), 3.52–3.43 (m, 1H); HRMS (ESI, [M+H]+): 753.2706.
The Procedure for the Synthesis of Intermediate 7
To a solution of compound 6 (360.0 mg, 478.2 µmol) in DMF (5 mL) was added imidazole (97.7 mg, 1.43 mmol) and TBSCl (75.7 mg, 502.1 μmol). The mixture was stirred at 20 °C for 1 h. TLC indicated 5% Reactant 1 was remained and one new spot with lower polarity was detected. The reaction was clean according to TLC. The reaction mixture was quenched by addition saturated NH4Cl 10 mL at 20 °C, and then diluted with DCM 10 mL and extracted with DCM (10 mL × 2). The combined organic layers were washed with brine (20 mL × 5), dried over [Na2SO4], filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 9:1 to 4:1). Compound 7 (260.0 mg, 299.9 µmol, 62.7% yield) was obtained as a white solid.
5,7-Bis(benzyloxy)-2-{3-[4-(benzyloxy)-3-methoxyphenyl)-2-{[(tert-butyldimethylsilyl)oxy]methyl}-2,3-dihydrobenzo[b][1,4]dioxin-6-yl}-3-hydroxychroman-4-one (7). White solid; Yield: 62.7%; 1H NMR (400 MHz, CDCl3) δ = 7.50 (d, J = 7.3 Hz, 2H), 7.41–7.23 (m, 14H), 7.14 (dd, J=1.9, 13.7 Hz, 1H), 7.06–6.92 (m, 3H), 6.91–6.83 (m, 2H), 6.20 (s, 1H), 6.14 (dd, J = 2.1, 5.6 Hz, 1H), 5.16–5.07 (m, 4H), 5.01–4.93 (m, 3H), 4.90 (d, J = 12.0 Hz, 1H), 4.41 (ddd, J = 1.8, 5.2, 12.1 Hz, 1H), 4.04 (d, J = 1.3 Hz, 1H), 3.93–3.88 (m, 1H), 3.85 (s, 3H), 3.83–3.77 (m, 1H), 3.53–3.46 (m, 1H), 0.84 (s, 9H), 0.01 (d, J=6.3 Hz, 6H); HRMS (ESI, [M+H]+): 867.3556.
General Procedure for the Synthesis of Intermediates (8–9)
To a solution of compound 3–4 (171.7 µmol) in DCM (0.5 mL) was added EDCI (65.8 mg, 343.4 µmol), DMAP (21.0 mg, 171.7 µmol), DIPEA (88.8 mg, 686.7 µmol, 119.6 μL) and compound 7 (148.9 mg, 171.7 µmol). The mixture was stirred at 40 °C for 12 hr. TLC indicated that ~20% of compound 7 was remained, and one major new spot with larger polarity was detected. The reaction mixture was quenched by addition 1 N HCl 2 mL at 0 °C, and then diluted with DCM 5 mL and extracted with DCM (5 mL × 3). The combined organic layers were washed with brine 10 mL, dried over [Na2SO4], filtered and concentrated under reduced pressure to give a residue. The crude compound 8–9 (crude) as light-yellow oil was used into the next step without further purification.
General Procedure for the Synthesis of Intermediates (10–11)
To a solution of compound 8–9 (0.17 mmol) in THF (2 mL) was added TBAF (1 M, 340.0 μL) at 0 °C. The mixture was stirred at 20 °C for 2 hr. TLC indicated that compounds 8–9 were consumed completely and one new spot formed. The reaction was clean according to TLC. The reaction mixture was quenched by addition saturated NH4Cl 10 mL at 20 °C, and then diluted with EtOAc 10 mL and extracted with EtOAc (10 mL × 3). The combined organic layers were washed with brine 20 mL, dried over [Na2SO4], filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 2:1 to 1:1). Compound 10–11 were obtained as a white solid.
5,7-Bis(benzyloxy)-2-{3-[4-(benzyloxy)-3-methoxyphenyl]-2-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxin-6-yl}-4-oxochroman-3-yl [(2R,3S,4R,5S,6S)-3,4,5-tris(benzyloxy)-6-[(benzyloxy)methyl]tetrahydro-2H-pyran-2-yl] succinate (10). White solid; Yield: 68.4%; 1H NMR (400 MHz, CDCl3) δ = 7.44 (br d, J=7.5 Hz, 2H), 7.39–7.13 (m, 35H), 7.06 (br s, 3H), 6.95–6.79 (m, 5H), 6.31–6.19 (m, 1H), 6.14 (br d, J=17.3 Hz, 2H), 5.26–5.18 (m, 2H), 5.09 (br d, J=6.0 Hz, 4H), 4.99–4.82 (m, 4H), 4.74 (br t, J=11.4 Hz, 2H), 4.59–4.32 (m, 5H), 4.00–3.51 (m, 11H), 2.74–2.51 (m, 4H); HRMS (ESI, [M+H]+): 1375.5258.
General Procedure for the Synthesis of Intermediate (12–13)
To a solution of compound 6 (340.0 mg, 451.6 µmol) in DCM (5 mL) was added EDCI (173.2 mg, 903.3 µmol), HOBt (122.1 mg, 903.3 µmol), DIPEA (175.1 mg, 1.35 mmol, 236.0 μL) and compound 3–4 (542.0 µmol). The mixture was stirred at 20 °C for 12 hr. TLC indicated that ~20% of compound 6 was remained, and one major new spot with lower polarity was detected. The reaction mixture was quenched by addition 1 N HCl 2 mL at 20 °C, and then diluted with EtOAc 10 mL and extracted with EtOAc (10 mL × 3). The combined organic layers were washed with brine 20 mL, dried over [Na2SO4], filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate = 9:1 to 2:1). Compound 12–13 was obtained as colorless oil.
{3-[4-(benzyloxy)-3-methoxyphenyl]-6-[5,7-bis(benzyloxy)-3-hydroxy-4-oxochroman-2-yl]-2,3-dihydrobenzo[b][1,4]dioxin-2-yl}methyl [(2R,3S,4R,5S,6S)-3,4,5-tris(benzyloxy)-6-[(benzyloxy)methyl]tetrahydro-2H-pyran-2-yl] succinate (12). Colorless oil; Yield: 41.8%; 1H NMR (400 MHz, CDCl3) δ = 7.51–6.72 (m, 42H), 6.33–6.08 (m, 3H), 5.16–4.64 (m, 11H), 4.65–4.29 (m, 6H), 4.27–3.96 (m, 3H), 3.96–3.41 (m, 10H), 2.68–2.48 (m, 4H); HRMS (ESI, [M+H]+): 1375.5260.
General Procedure for the Synthesis of Target Compounds (14–17)
A mixture of compound 10–13 (116.3 µmol), Pd/C (160.0 mg, 10% purity) in MeOH (4 mL) was degassed and purged with N2 for 3 times, and then the mixture was stirred at 20 °C for 12 hr under H2 (15 PSI) atmosphere. LC-MS showed that no compound 10–13 was remained. Several new peaks were shown on LC-MS and trace desired compound was detected. The mixture was filtered through a pad of celite. The solvent was concentrated under reduced pressure to give a residue. The residue was purified by prep-HPLC (neutral condition). Compound 14–17 was obtained as a white solid.
5,7-Dihydroxy-2-[3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxin-6-yl]-4-oxochroman-3-yl [(2R,3S,4R,5R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl] succinate (14). White solid; Yield: 41.8%, HPLC purity: 88.43% (Supporting Information, S1); 1H NMR (400 MHz, Acetone-d6) δ = 11.61–11.48 (m, 1H), 10.04–9.86 (m, 1H), 7.81 (br d, J=4.3 Hz, 1H), 7.23–7.12 (m, 2H), 7.12–7.04 (m, 1H), 7.00 (dd, J=2.3, 8.3 Hz, 2H), 6.90 (d, J=8.0 Hz, 1H), 6.12 (d, J=3.8 Hz, 1H), 6.06–6.00 (m, 2H), 5.90 (dd, J=5.8, 11.8 Hz, 1H), 5.56–5.44 (m, 1H), 5.03 (dd, J=2.3, 8.0 Hz, 1H), 4.23–4.12 (m, 2H), 4.31 (br s, 1H), 3.89 (s, 3H), 3.84–3.42 (m, 9H), 3.07–3.07 (m, 1H), 2.76–2.52 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ = 191.12, 170.99, 170.85, 168.74, 163.72, 162.69, 148.10, 147.50, 144.53, 143.98, 128.81, 127.85, 121.05, 117.05, 116.77, 115.80, 112.17, 100.75, 97.14, 96.14, 95.05, 92.97, 89.49, 80.05, 78.64, 76.32, 76.27, 75.54, 75.04, 73.47, 72.70, 72.44, 71.11, 69.75, 60.85, 60.62, 56.18, 56.16, 29.09, 28.58 (see S2 for 1H and 13C NMR spectra of 14, Supporting Information); HRMS (ESI, [M+H]+-[H2O]): 727.1874.
5,7-Dihydroxy-2-[3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxin-6-yl]-4-oxochroman-3-yl [(2R,3S,4R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl] succinate (15). White solid; Yield: 30.4%, HPLC purity: 91.62% (Supporting Information, S1); 1H NMR (400 MHz, Acetone-d6) δ = 11.60–11.49 (m, 1H), 9.97 (br s, 1H), 7.81 (br s, 1H), 7.23–7.17 (m, 2H), 7.15 (s, 1H), 7.11–7.05 (m, 1H), 7.04–6.95 (m, 2H), 6.94–6.86 (m, 1H), 6.04 (t, J=2.5 Hz, 2H), 5.94–5.84 (m, 1H), 5.53–5.44 (m, 1H), 5.09–4.97 (m, 1H), 4.24–4.14 (m, 1H), 4.10–3.81 (m, 6H), 3.80–3.46 (m, 7H), 2.76–2.51 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ = 191.54, 171.00, 170.77, 167.80, 163.69, 162.78, 148.10, 147.50, 144.57, 143.99, 128.76, 127.86, 121.05, 117.07, 116.76, 115.80, 112.18, 101.07, 96.94, 95.90, 95.63, 93.44, 89.86, 80.11, 78.64, 76.66, 76.32, 74.17, 73.44, 72.83, 69.98, 68.88, 68.39, 60.61, 56.19, 29.11, 28.42 (see S2 for 1H and 13C NMR spectra of 15, Supporting Information); HRMS (ESI, [M+H]+-[H2O]): 727.1869.
[3-(4-hydroxy-3-methoxyphenyl)-6-(3,5,7-trihydroxy-4-oxochroman-2-yl)-2,3-dihydrobenzo[b][1,4]dioxin-2-yl]methyl [(2R,3S,4R,5R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl] succinate (16). White solid; Yield: 31.7%, HPLC purity: 90.03% (Supporting Information, S1); 1H NMR (400 MHz, Acetone-d6) δ = 11.74–11.64 (m, 1H), 9.89 (br s, 1H), 7.88 (br s, 1H), 7.22–7.09 (m, 3H), 7.08–6.95 (m, 2H), 6.91 (d, J=8.0 Hz, 1H), 6.15–5.95 (m, 3H), 5.13 (d, J=11.5 Hz, 1H), 5.07–4.97 (m, 1H), 4.68 (dd, J=2.5, 11.5 Hz, 1H), 4.53–4.44 (m, 1H), 4.36–4.26 (m, 1H), 4.12–4.03 (m, 1H), 3.90 (d, J=1.3 Hz, 3H), 3.78–3.62 (m, 4H), 3.59–3.41 (m, 3H), 2.81–2.59 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ = 172.07, 171.33, 162.83, 148.22, 147.76, 143.63, 143.46, 131.15, 127.09, 121.82, 121.06, 117.18, 116.91, 115.95, 112.18, 100.36, 94.73, 89.54, 82.83, 77.35, 76.19, 75.43, 75.03, 72.43, 71.92, 70.94, 70.46, 63.27, 61.35, 56.18, 29.14, 28.95 (see S2 for 1H and 13C NMR spectra of 16, Supporting Information); HRMS (ESI, [M-H]−): 743.1823.
[3-(4-hydroxy-3-methoxyphenyl)-6-(3,5,7-trihydroxy-4-oxochroman-2-yl)-2,3-dihydrobenzo[b][1,4]dioxin-2-yl]methyl [(2R,3S,4R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl] succinate (17). White solid; Yield: 28.8%, HPLC purity: 91.61% (Supporting Information, S1); 1H NMR (400 MHz, Acetone-d6) δ = 11.70 (s, 1H), 9.83 (br s, 1H), 7.87 (br s, 1H), 7.24–7.10 (m, 3H), 7.08–6.97 (m, 2H), 6.95–6.86 (m, 1H), 6.22–5.96 (m, 2H), 5.47 (dd, J=2.6, 7.9 Hz, 1H), 5.13 (d, J=11.5 Hz, 1H), 5.06–4.97 (m, 1H), 4.69 (br d, J=9.8 Hz, 1H), 4.47 (br s, 1H), 4.30 (br dd, J=2.6, 12.2 Hz, 1H), 4.12–4.01 (m, 2H), 3.99–3.58 (m, 9H), 2.76–2.60 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ = 197.88, 172.07, 163.80, 162.89, 148.22, 147.75, 143.65, 143.50, 131.06, 127.08, 121.82, 121.06, 117.21, 116.96, 115.95, 112.19, 100.72, 96.69, 95.67, 93.42, 82.91, 76.20, 75.49, 75.44, 73.48, 71.93, 71.85, 63.27, 60.67, 56.22, 28.96, 28.76 (see S2 for 1H and 13C NMR spectra of 17, Supporting Information); HRMS (ESI, [M-H]−): 743.1817.
Solubility Measurements
The synthesized compounds 14–17 were prepared in Milli-Q H2O as stock solution and successive dilutions of stock solution to obtain desired concentrations. The UV–vis spectrum of compounds presented a maximum which was centered at 286 nm. Molar absorption coefficients (ε) for each compound in water were determined from calibration curves. Then, the selected compounds of 5 mg were suspended in 10 mL of H2O under magnetic stirring in the dark for 10 min. After filtration, the absorption of compounds was measured using UV–vis spectrophotometer (UV-2550, SHIMADZU). The solubility of selected compounds in water was calculated using the Beer-Lambert law.
Evaluation of Cell Viability
For cytotoxicity assay, DU145 cells, Hep G2 cells, SNU-423 cells, Hep3B cells (ATCC, Rockville, MD, USA), LIXC-002 cells (yanhui, Shanghai, China), HL-7702 cells (Jihe, Shanghai, China), RWPE-1 cells (National Collection of Authenticated Cell Cultures, Shanghai, China) were seeded in 96-well plates and exposed to different concentrations of tested compounds for 72 h. Then, CCK8 (Beyotime, Shanghai, China) was added in cells for 4 h. The absorbance was measured at 450 nm by multifunction microplate reader (Berthold LB941). The growth inhibitory ratio was calculated as follows: Growth inhibitory ratio = (Acontrol − Asample)/Acontrol. IC50 values were obtained from a nonlinear regression model (curve fit) on the base of sigmoidal dose response curve (variable slope) and computed using GraphPad Prism version 5.0 (GraphPad Software).
Apoptosis Assays
DU145 cells were plated in a six-well plate and treated with vehicle, silibinin, or 15 for 24 h at 37 °C. After incubation, the cells were harvested and washed with ice-cold PBS. Then, an Annexin V-FITC Apoptosis Detection Kit (keygentec) was used for apoptosis analysis by flow cytometry.
Cell Cycle Assays
DU145 cells were seeded in 6 plates. All cells were treated with increasing concentrations of the indicated compounds 24 h post plating. Cells were harvested 24 h post-treatment, washed in phosphate buffered saline (PBS), and fixed in ice-cold 70% ethanol for at least 24 h. The fixed cells were then washed with room temperature PBS and stained with Propidium Iodide (50 μL) in the presence of RNase-A (3 μL) for 30 min at 37 °C. The stained cells were then analyzed using a Flow Cytometer, and the resulting data analyzed with cell cycle analysis software (FlowJo).
Metabolic Stability in Mouse Liver Microsomes
Add 2.0 μL of 150 μM control compound or test compound solutions to the incubation plates. Diclofenac was used as positive control in this study and the final concentration of test compound and diclofenac were 1 μM. The mixture was pre-warmed at 37 °C for 10 minutes. The reaction was started with the addition of 30 μL of the 10 mM NADPH. The final concentration of NADPH was 1 mM. Test compound sample without NADPH was used as a negative control. The incubation solution was incubated in water bath at 37 °C. Aliquots of 30 µL were taken from the reaction solution at 0, 5, 15, 30 and 60 minutes. The reaction was stopped by the addition of 200 μL of cold acetonitrile with IS (100 ng/mL Labetalol, 100 ng/mL tolbutamide). Samples were centrifuged at 1000 rpm for 10 minutes. Aliquot of 50 µL of the supernatant was mixed with 150 µL of ultra-pure H2O and then used for LC-MS/MS analysis.
Assessments of Pharmacokinetic Properties
Animals: ICR mice (male, 4–6 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology (Beijing, China). Animals ate food and water freely and were given a daily schedule of dark–light cycle. Before tests, they were housed for at least 12 hours.
The pharmacokinetic analysis of 15 was conducted in male ICR mice which were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd. The animals were administered a single dose of 2 mg/kg 15 by i.v. after fasting overnight. Blood was collected and centrifuged immediately to isolate plasma. The plasma concentrations were determined using high performance liquid chromatography with HPLC analysis on a Shimadzu Prominence-i LC-2030C 3D system. Pharmacokinetic parameters were estimated by non-compartmental model using WinNonlin 8.2.
Results and Discussion
Chemistry
The protocol for the synthesis of target compounds 14–15 was outlined in Scheme 1. Firstly, compounds 1 and 2 were transformed to corresponding acids (intermediate 3 and 4) in the presence of tetrahydrofuran-2,5-dione and DMAP. The challenge for the synthesis of glycosylated derivatives 14–15 lay in the competitive reactivity of phenolic hydroxy groups. The selective protection of C-5, C-7 and C-20 phenolic hydroxy group of silibinin has been achieved through compound 5 being treated with BnBr and K2CO3 at low temperature, with phenolic hydroxy groups being more reactive than alcoholic groups which are at C-3 and C-23. Compared with secondary alcoholic group at C-3, the primary alcoholic group at C-23 can be readily esterified. The primary alcoholic group at C-23 of intermediate 6 was protected with TBSCl for the better condensation of the secondary alcoholic group at C-3 with fragments 3–4 affording critical intermediates 8–9. Subsequently, the intermediates 8–9 were transferred to compounds 10–11 with TBAF. Ultimately, the debenzylation of 10–11 provided the target compounds 14–15 (Scheme 1).
Another synthetic route for target compounds 16–17 was depicted in Scheme 2. Condensation of the intermediate 6 with fragments 3–4 followed by debenzylation in the presence of Pd/C under H2 atmosphere to furnish the target compounds 16–17 (Scheme 2).
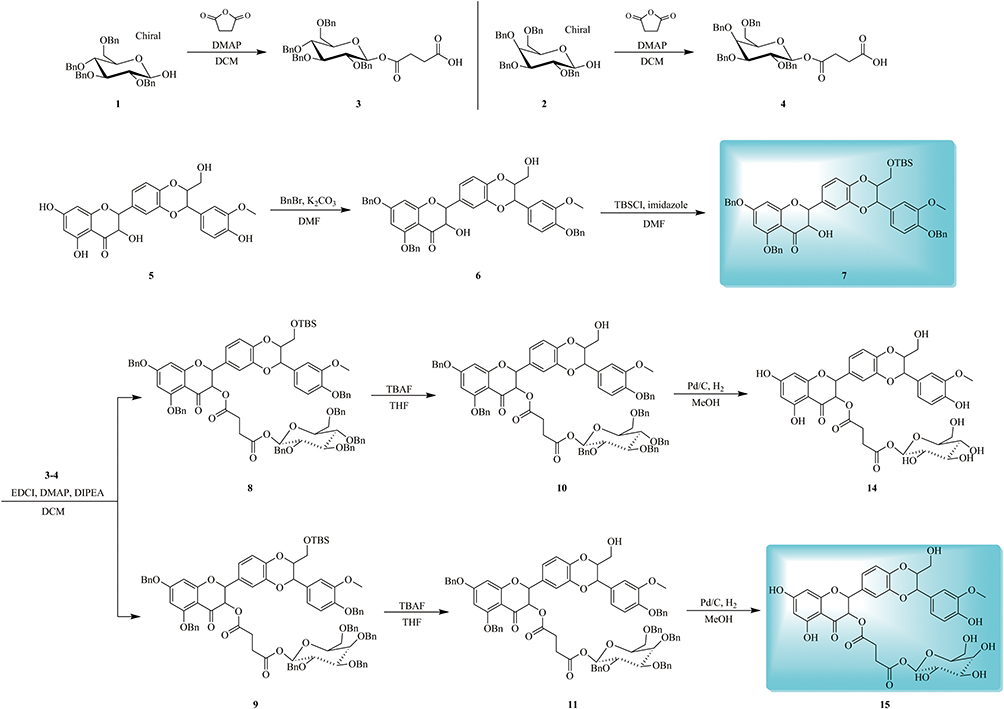 |
Scheme 1 Synthesis of silibinin derivatives 14–15. |
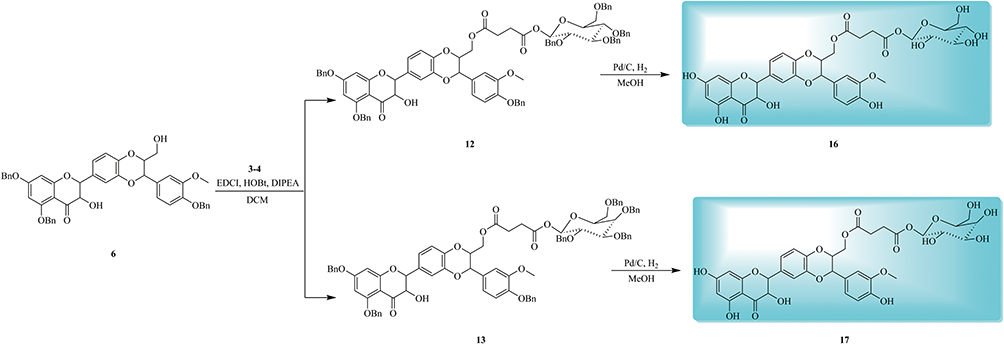 |
Scheme 2 Synthesis of silibinin derivatives 16–17. |
Water Solubility of the Synthesized Compounds
The measurement of solubility was carried out by dissolving the synthesized compounds in Milli-Q H2O for 10 min. By considering the relationship between UV maximum absorbance and compound concentration, calibration curve molar absorption coefficients (ε) for each compound in water were determined, with successive dilutions of a standard solution using the Beer-Lambert law.4 UV–Vis spectra are shown in Figure 3, and the linear response is at the maximum wavelength 286 nm (Figure 3). Approximately 0.5 mg of synthesized compounds was suspended in 10 mL H2O and the absorption was measured at 286 nm after filtration. Compared with the slight solubility of silibinin (0.4 mg·L−1), the synthesized compounds exhibited better solubility in water, ranging from 15.4 mg·L−1 to 33.0 mg·L−1, which demonstrated that the introduction of glycosyl can indeed improve the water solubility of molecules (Table 1).
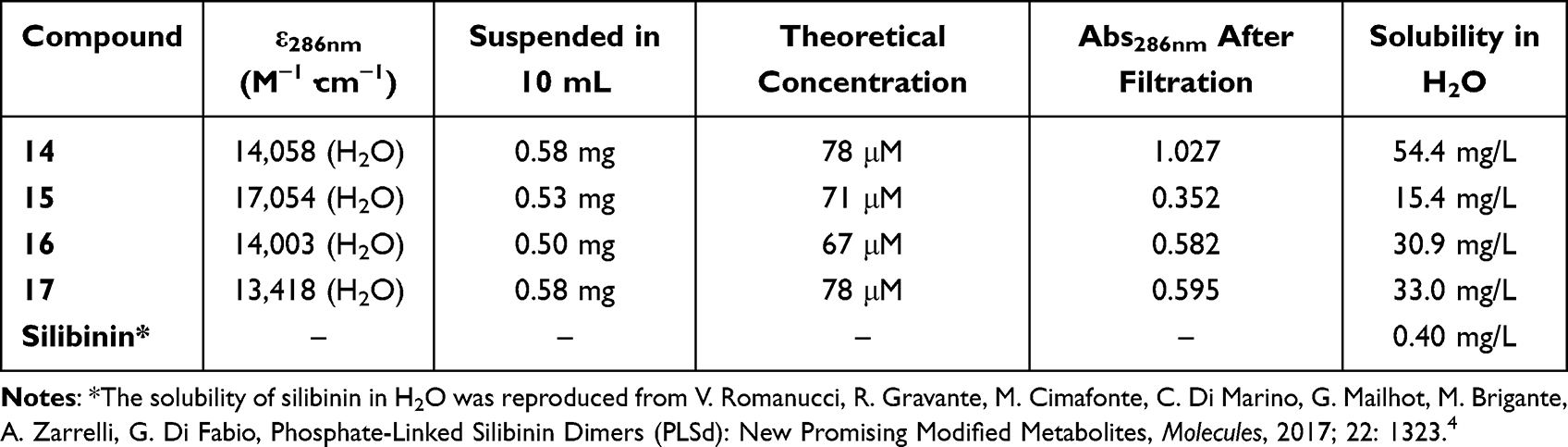 |
Table 1 Spectroscopic Characteristics and Solubility of the Synthesized Compounds (14–17) and Silibinin |
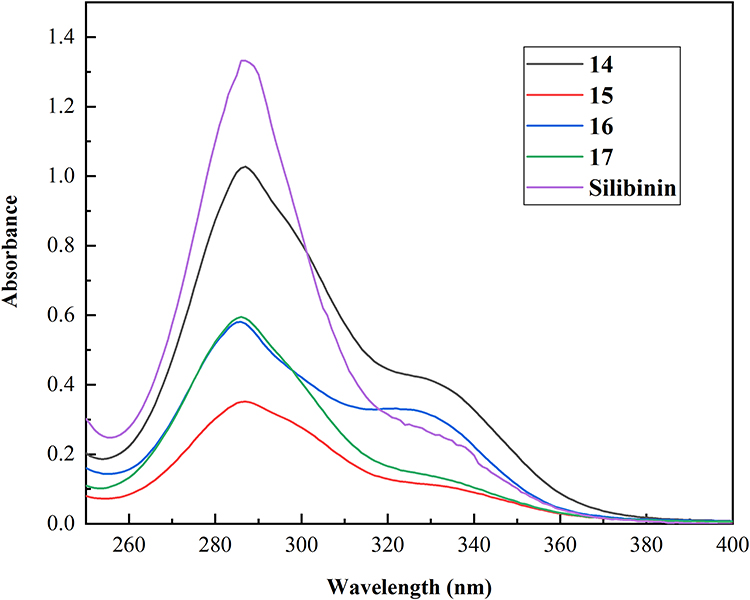 |
Figure 3 The UV–Vis spectra of the target compounds 14–17 and silibinin in H2O. |
In vitro Cytotoxicity Screening of the Synthesized Compounds
The anti-proliferative activities of the synthesized compounds 14–17 against five cancer cell lines including LIXC-002, Hep G2, SNU-423, Hep3B and DU145 were evaluated by CCK8 assay, with silibinin employed as control. The IC50 values of target compounds against tested cell lines are shown in Table 2. The majority of the target compounds displayed varying degrees of antitumor activities with IC50 values at low micromolar concentrations. Especially, several derivatives exhibited cytotoxic activity in the single-digit micromolar range, such as compounds 14 and 17 showed better anti-proliferative activities against SNU-423 cells with IC50 values of 7.82 ± 0.250 and 9.83 ± 0.890 μM. For DU145 cells, compounds 15 and 17 exerted strong cytotoxic activity with IC50 values of 1.37 ± 0.140 and 3.64 ± 0.240 μM. It was revealed that compound 15 exhibited inhibitory efficacy against LIXC-002 cells with IC50 values of 7.19 ± 0.940 μM and compound 14 displayed potent inhibitory activities against Hep3B cells with IC50 values of 7.55 ± 0.0600 μM, which was comparable to silibinin. Among these tested compounds, it seemed that the activity of derivative which was substituted with glucosyl group at 3 position was higher than that at 23 position (14 vs 16). Moreover, when glycosyl groups were anchored at 23 position, the analog 17 which was substituted with galactosyl group showed better inhibitory activity than that of compound 16 substituted with glucosyl group. Besides, LIXC-002 and DU145 were more sensitive to the compound 15 which displayed superior cytotoxicity with IC50 values of 7.19 ± 0.940 μM and 1.37 ± 0.140 μM compared with silibinin. Compounds 15 and 17 comprising galactosyl exhibited lower IC50 potency against DU145 cells compared with compounds 14 and 16 comprising glucosyl (Table 2). We also evaluated the toxicity of synthesized compounds against normal cells HL-7702 and RWPE-1. All of compounds exhibited low cytotoxic activity against normal cells with IC50 values higher than 300 μM.
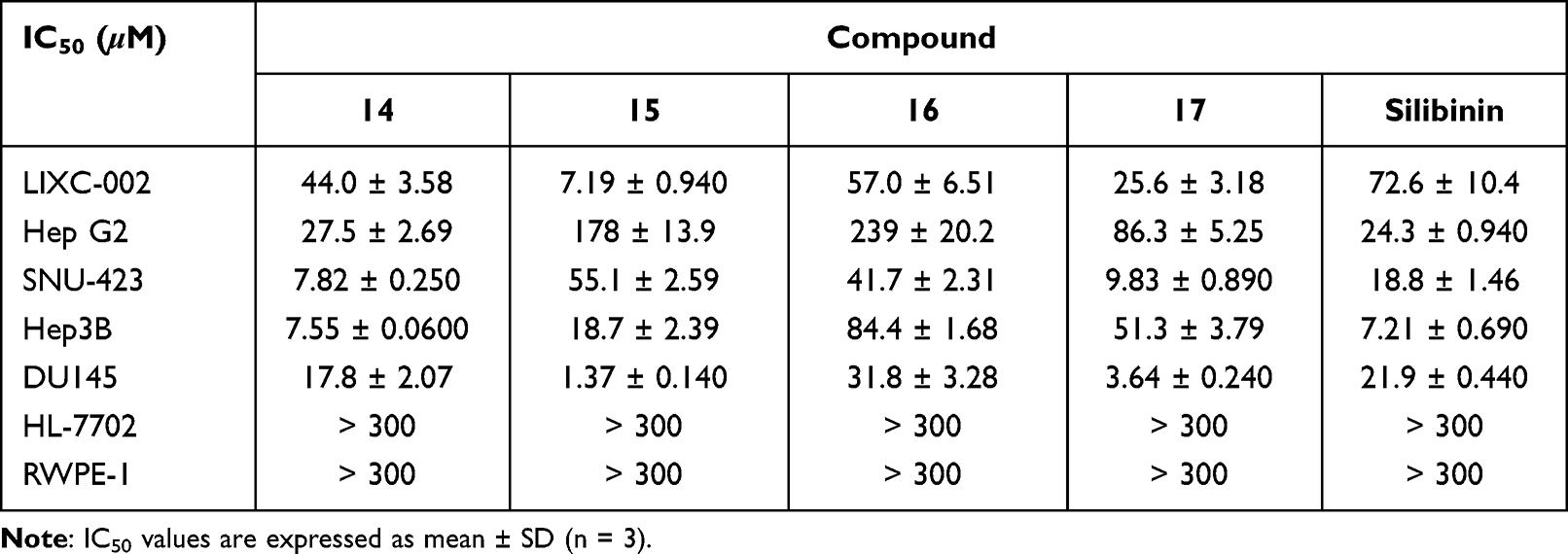 |
Table 2 In vitro Anti-Proliferative Activities of the Compounds Against Cancer Cell Lines and Normal Cell Lines |
Effects of Compound 15 on DU145 Cell Cycle Progression and Apoptosis
To investigate whether compound 15 induces apoptosis of DU145 cells, we evaluated compound 15-induced apoptosis of DU145 cells by flow cytometry using Annexin V-FITC and Propidium Iodide double staining. As shown in Figure 4, compound 15 markedly induced cell apoptosis in a dose-dependent manner. Treatment of DU145 cells with compound 15 (3, 10 and 30 μM) for 24 h resulted in Annexin V positive early-stage and late-stage apoptotic cells increasing compared with control groups. At the same concentration of 30 μM, the ability of 15 to promote DU145 cell apoptosis was approximately 2 times stronger than that of silibinin. To illustrate the mechanism of 15-induced cell inhibition, we analyzed the cell cycle in DU145 cells. Cells were treated with compound 15 in various concentrations and silibinin for 24 h according to IC50 values. As presented in Figure 4, cancer cells treated with compound 15 were demonstrated a significant increase in the percentage of cells in G2/M phase. It was evident that compound 15 blocked the tumor cells in G2/M -phase progression, which resulted in cell cycle inhibition on DU145 cell lines.
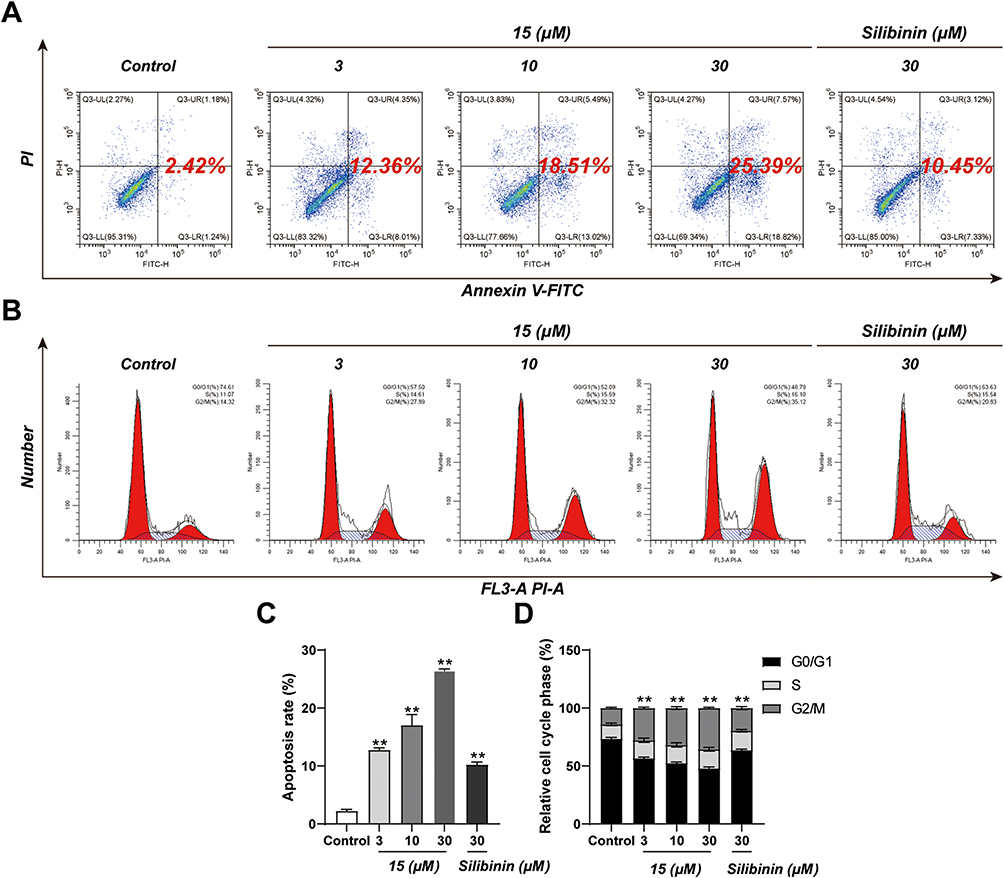 |
Figure 4 Apoptosis rate and cell cycle of DU145 cells treated with compound 15 or silibinin. DU145 cells were treated with compound 15 (3, 10, 30 μM) or silibinin (30 μM) for 24 hours. (A) Flow cytometry. (B) Cell cycle analysis by propidium iodide staining. (C) Quantitative analysis of apoptotic cells. (D) The percentage of cell cycle distribution. Results were mean ± SD for three individual experiments. **p < 0.01. |
The Metabolic Stability of 15 in Mouse Liver Microsomes
The in vitro liver microsomal metabolism of 15 and silibinin was tested. As illustrated in Table 3, compound 15 showed better metabolic stability with half-life of 128.3 ± 12.0 min in mouse liver microsomes and exhibited relatively low clearance rate (CLint: 42.8 mL/min/kg) compared with silibinin (Table 3).
 |
Table 3 The in vitro Metabolic Properties of 15 and Silibinin |
Pharmacokinetic Characteristics of Compound 15
The pharmacokinetic assessment of compound 15 and silibinin through intravenous administration (i.v., 2 mg/kg) to ICR mice were performed (Figure 5). Compound 15 presented high distribution volume (Vz) of 13.271 ± 4.721 L/kg and moderate half-life (T1/2) of 0.490 ± 4.721 h compared with silibinin. Besides, Compound 15 showed the maximum plasma concentration (Cmax) of 373.0 ± 75.4 ng/mL, AUC value of 111.0 ± 19.6 ng/mL·h, a clearance (CL) of 18.399 ± 2.961 L·kg−1·h−1 and mean residence time (MRT0−∞) was 0.1830 ± 0.0536 h (Table 4). These results revealed that compound 15 displayed favorable pharmacokinetics properties.
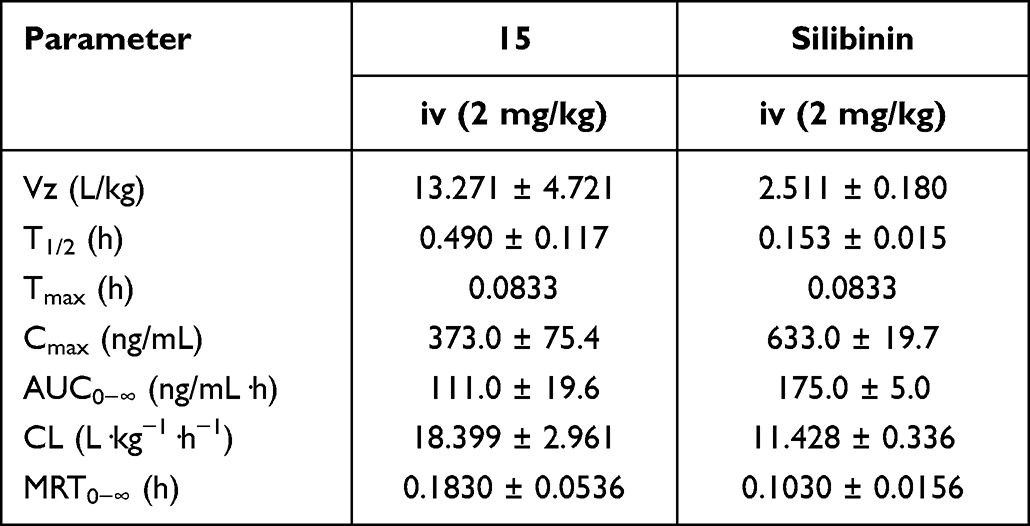 |
Table 4 Pharmacokinetic Characteristics of Compound 15 and Silibinin |
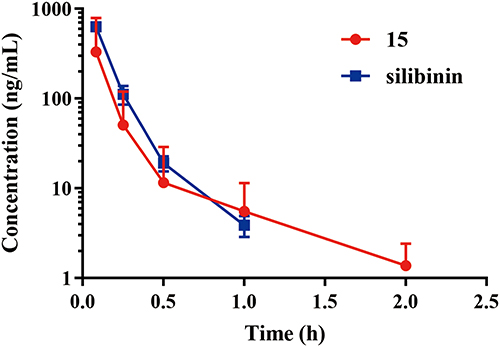 |
Figure 5 Plasma concentration vs time curves after iv (2 mg/kg) administration of compound 15 and silibinin to mice. Data are represented as the mean ± SD (n = 3). |
Conclusion
In summary, a series of glycosylated silibinin derivatives have been designed and synthesized as anti-tumor agents. Considering the strong limitations of silibinin, the water solubility of new derivatives has been evaluated. The glycosylated silibinin derivatives presented a good water solubility (more than 15 mg/L), while silibinin was poorly soluble (less than 0.4 mg/L). The biological evaluation highlighted that most of these new derivatives exhibited potent cellular antiproliferation activities. Moreover, the representative compound 15 stood out with potent inhibitory activity against LIXC-002 and DU145 cells (IC50 = 7.19 μM and 1.37 μM). Flow cytometric analysis indicated that compound 15 could cause G2/M phase arrest and induce apoptosis of DU145 cells. In addition, compound 15 showed better metabolic stability in liver microsomes (T1/2 = 128.3 min) compared with that of silibinin (T1/2 = 82.5 min) and good pharmacokinetics. All of results supported that the introduction of glycosyl in the 3-OH and 23-OH position of silibinin would be a feasible strategy for developing silibinin derivatives as anti-tumor agents.
Ethical Approval
The animal test subjects were entrusted to the contract research organization, Precedo Pharmaceuticals Co. Ltd. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Precedo Pharmaceuticals Co. Ltd. (IACUC-20220314). Animal welfare was performed according to the guidelines for ethical review of the welfare of laboratory animals (GB/T35892-2018).
Acknowledgment
The authors are grateful for the financial support from the Research Project on the Application of Public Welfare Technology supported by the Science and Technology Department of Zhejiang Province (LGF21H 300002), Zhejiang Provincial Key Laboratory for Accurate Diagnosis and Treatment of Chronic Liver Diseases (2020E10014), the Agricultural and Social Development Fund of the Hangzhou Science and Technology Committee (20201203B211), Medical health science and technology major project of Hangzhou Health Commission (Z20200022, Z20210004).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wei PS, Li X, Wang S, et al. Silibinin ameliorates formaldehyde-induced cognitive impairment by inhibiting oxidative stress. Oxid Med Cell Longev. 2022;2022:5981353. doi:10.1155/2022/5981353
2. Wu Q, Zeng J, Dong J. Synthesis and antitumor activity of novel silibinin and 2,3-dehydrosilybin derivatives with carbamate groups. Med Chem Res. 2022;31:533–544.
3. Gazak R, Walterova D, Kren V. Silybin and silymarin - new and emerging applications in medicine. Curr Med Chem. 2007;14(3):315–338. doi:10.2174/092986707779941159
4. Romanucci V, Gravante R, Cimafonte M, et al. Phosphate-Linked Silibinin Dimers (PLSd): new promising modified metabolites. Molecules. 2017;22:1323. doi:10.3390/molecules22081323
5. Park JW, Shin NR, Shin IS, et al. Silibinin inhibits neutrophilic inflammation and mucus secretion induced by cigarette smoke via suppression of ERK-SP1 pathway. Phytother Res. 2016;30:1926–1936. doi:10.1002/ptr.5686
6. Tong WW, Zhang C, Hong T, et al. Silibinin alleviates inflammation and induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes and has a therapeutic effect on arthritis in rats. Sci Rep. 2018;8:3241.
7. Mashhadi Akbar Boojar M, Mashhadi Akbar Boojar M, Golmohammad S. Overview of silibinin anti-tumor effects. J Herb Med. 2020;23:100375. doi:10.1016/j.hermed.2020.100375
8. Hou X, Du H, Quan X, et al. Silibinin inhibits NSCLC metastasis by targeting the EGFR/LOX pathway. Front Pharmacol. 2018;9:21.
9. Liu W, Otkur W, Li L, et al. Interference of silibinin with IGF-1R signalling pathways protects human epidermoid carcinoma A431 cells from UVB-induced apoptosis. Biochem Bioph Res Co. 2013;432:314–319.
10. Gu HR, Park SC, Choi SJ, et al. Combined treatment with silibinin and either sorafenib or gefitinib enhances their growth-inhibiting effects in hepatocellular carcinoma cells. Clin Mol Hepatol. 2015;21:49–59.
11. Punia R, Singh C, Singh RP, Singh M, Singh RP. Physicochemical properties of silibinin-phosphatidylcholine complex and its implications for drug formulations. Indian J Pharm Sci. 2022;84:979–987.
12. Bao V, Zhang Q, Chen Q-H, Wang G, Xiaojie P. Silibinin derivatives as anti-prostate cancer agents: synthesis and cell-based evaluations. Eur J Med Chem. 2016;109:36–46. doi:10.1016/j.ejmech.2015.12.041
13. Xu R, Qiu S, Zhang J, et al. Silibinin Schiff base derivatives counteract CCl(4)-induced acute liver injury by enhancing anti-inflammatory and antiapoptotic bioactivities. Drug Des Devel Ther. 2022;16:1441–1456. doi:10.2147/DDDT.S356847
14. Saller R, Melzer J, Reichling J, Brignoli R, Meier R. An updated systematic review of the pharmacology of silymarin. Forsch Komplementmed. 2007;14:70–80. doi:10.1159/000100581
15. Biedermann D, Vavříková E, Cvak L, Křen V. Chemistry of silybin. Nat Prod Rep. 2014;31:1138–1157. doi:10.1039/C3NP70122K
16. Chi C, Zhang CS, Liu Y, Nie HC, Zhou JP, Ding Y. Phytosome-nanosuspensions for silybin-phospholipid complex with increased bioavailability and hepatoprotection efficacy. Eur J Pharm Sci. 2020;144. doi:10.1016/j.ejps.2020.105212
17. Monti D, Gažák R, Marhol P, et al. Enzymatic kinetic resolution of silybin diastereoisomers. J Nat Prod. 2010;73:613–619. doi:10.1021/np900758d
18. Bosch-Barrera J, Queralt B, Menendez AJ. Targeting STAT3 with silibinin to improve cancer therapeutics. Cancer Treat Rev. 2017;58:61–69.
19. Si LL, Liu WW, Hayashi T, et al. Silibinin-induced apoptosis of breast cancer cells involves mitochondrial impairment. Arch Biochem Biophys. 2019;671:42–51. doi:10.1016/j.abb.2019.05.009
20. Schramm S, Huang G, Gunesch S, et al. Regioselective synthesis of 7-O-esters of the flavonolignan silibinin and SARs lead to compounds with overadditive neuroprotective effects. Eur J Med Chem. 2018;146:93–107. doi:10.1016/j.ejmech.2018.01.036
21. Vue B, Zhang X, Lee T, et al. 5- or/and 20-O-alkyl-2,3-dehydrosilybins: synthesis and biological profiles on prostate cancer cell models. Bioorg Med Chem. 2017;25:4845–4854.
22. Nawaz Q, Fuentes-Chandia M, Tharmalingam V, Rehman MAU, Leal-Egana A, Boccaccini AR. Silibinin releasing mesoporous bioactive glass nanoparticles with potential for breast cancer therapy. Ceram Int. 2020;46:29111–29119.
23. Perez-Sanchez A, Cuyas E, Ruiz-Torres V, et al. Intestinal permeability study of clinically relevant formulations of silibinin in Caco-2 cell monolayers. Int J Mol Sci. 2019;20(7):1606. doi:10.3390/ijms20071606
24. Dube D, Khatri K, Goyal AK, Mishra N, Vyas SP. Preparation and evaluation of galactosylated vesicular carrier for hepatic targeting of silibinin. Drug Dev Ind Pharm. 2010;36(5):547–555. doi:10.3109/03639040903325560
25. Zhang JQ, Liu J, Li YL, Jasti BR. Preparation and characterization of solid lipid nanoparticles containing silibinin. Drug Deliv. 2007;14:381–387. doi:10.1080/10717540701203034
26. Chen F, Huang GL. Application of glycosylation in targeted drug delivery. Eur J Med Chem. 2019;182:111612. doi:10.1016/j.ejmech.2019.111612
27. Cai LL, Gu ZP, Zhong J, et al. Advances in glycosylation-mediated cancer-targeted drug delivery. Drug Discov Today. 2018;23(5):1126–1138. doi:10.1016/j.drudis.2018.02.009
28. Huang G, Mei X. Synthetic glycosylated natural products have satisfactory activities. Curr Drug Targets. 2014;15(8):780–784. doi:10.2174/1389450115666140617153348
29. Huang G, Lv M, Hu J, Huang K, Xu H. Glycosylation and activities of natural products. Mini Rev Med Chem. 2016;16(12):1013–1016. doi:10.2174/138955751612160727164559
30. Zhang P, Ma J, Zhang QQ, et al. Monosaccharide analogues of anticancer peptide Rlycosin-I: role of monosaccharide conjugation in complexation and potential of lung cancer targeting and therapy. J Med Chem. 2019;62(17):7857–7873. doi:10.1021/acs.jmedchem.9b00634
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.