")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Design, synthesis, and biological evaluation of 3-vinyl-quinoxalin-2(1H)-one derivatives as novel antitumor inhibitors of FGFR1
Authors Liu Z, Yu S, Chen D, Shen G, Wang Y, Hou L, Lin D, Zhang J, Ye F
Received 14 May 2015
Accepted for publication 18 December 2015
Published 3 May 2016 Volume 2016:10 Pages 1489—1500
DOI https://doi.org/10.2147/DDDT.S88587
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Zhiguo Liu,1,* Shufang Yu,1,* Di Chen,1 Guoliang Shen,1 Yu Wang,1 Leping Hou,2 Dan Lin,1 Jinsan Zhang,1 Faqing Ye1
1School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, 2Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: FGFR1 is well known as a molecular target in anticancer drug design. TKI258 plays an important role in RTK inhibitors. Utilizing TKI258 as a lead compound that contains a quinazolinone nucleus, we synthesized four series of 3-vinyl-quinoxalin-2(1H)-one derivatives, a total of 27 compounds. We further evaluated these compounds for FGFR1 inhibition ability as well as cytotoxicity against four cancer cell lines (H460, B16-F10, Hela229, and Hct116) in vitro. Some compounds displayed good-to-excellent potency against the four tested cancer cell lines compared with TKI258. Structure–activity relationship analyses indicated that small substituents at the side chain of the 3-vinyl-quinoxalin-2(1H)-one were more effective than large substituents. Lastly, we used molecular docking to obtain further insight into the interactions between the compounds and FGFR1.
Keywords: FGFR1, synthesis, quinoxaline, antitumor activity, kinase inhibitor
Introduction
Fibroblast growth factor receptor (FGFR) has been extensively validated as a molecular target in anticancer drug discovery because of its regulatory function in multiple developmental processes including proliferation, motility, and differentiation.1–6 The FGFR family includes four highly conserved tyrosine kinase receptors: FGFR1–4.7,8 Among them, FGFR1 has been proposed as the most potent mutagen of the FGFR family,9,10 and FGFR1 is mutated in some of the deadliest human cancers (lung cancer,11 breast cancer,12 glioma, prostate cancer,13 and liver cancer). As such, several pharmaceutical inhibitors of FGFR1 have been developed to treat cancer.14 Current inhibitors of FGFR1 include SU5402,15 PD173074,16 and BGj39817 to name a few. There are only a few inhibitors used clinically. Due to limited number of FGFR1 inhibitors, as well as the side effects of presently available drugs, there is a pressing need for the development of novel FGFR1 inhibitors. In recent years, our group has engaged in the design and synthesis of FGFR1 inhibitors, and published a series of articles.18,19
Previous studies have shown that most of the known FGFR1 inhibitors target the ATP-binding site,7,10,20–22 as well as our early synthetic inhibitors.18,19 This study presents the development of quinoxaline FGFR1 inhibitors according to the lead compound – TKI258. TKI258 (Dovitinib), a novel, multitargeted RTK inhibitor plays an important role in myeloma,23,24 which is highly selective with an IC50 of 8 nM against FGFR1 tyrosine kinase.25 Additionally, several anticancer drugs containing a quinoxaline ring have been reported along with their pharmacological data, activities against solid tumors, and clinical trials.26,27 Our group studied modifications of quinoxaline mainly based on molecular docking between TKI258 and FGFR1. Molecular docking showed that three hydrogen bonds were formed between 1-N and Glu562, 3-N and Ala564, and 2-O and Ala564.9 Similar to the quinolinone of TKI258, the blue part depicted in Figure 1 is responsible for formation of hydrogen bonds with a kinase hinge region. As such, we chose quinoxaline because it has more hetero atoms as a nucleus, with the hope that it would form more hydrogen bonds with FGFR1 residues and significantly improve activity. At the same time, we introduced a vinyl at the 3-position. As is well known, a double bond has a rigidity effect. Therefore, with an introduction of vinyl, we hoped that the side chain would result in a deflection at a certain angle to form stronger hydrogen bonds when compounds are combined with FGFR1 kinase. In this way, it would be possible to improve the activity between kinase and inhibitors (rose-colored part in Figure 1).
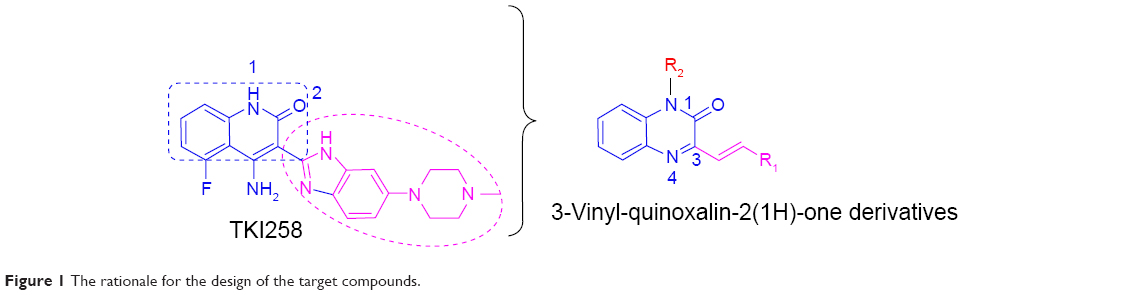 | Figure 1 The rationale for the design of the target compounds. |
Based on the above considerations and combined with our additional interest in the development of new FGFR1 inhibitors, we synthesized four series of 3-vinyl-quinoxalin-2(1H)-one analogs (Figure 2). Among them, we introduced methyl or benzyl at the 1-position, and analyzed through activity experiments whether the modification of 1-H had an impact on kinase inhibitory activity. Simultaneously, compound D was designed to verify 2-O and whether this had an impact on kinase inhibitory activities. Furthermore, the antitumor activities of all compounds were tested using methyl thiazolyl tetrazolium (MTT) assay and LANCE Ultra TR-FRET assay. Synthesis, a preliminary biological evaluation, and structure–activity relationship (SAR) were done on these derivatives.
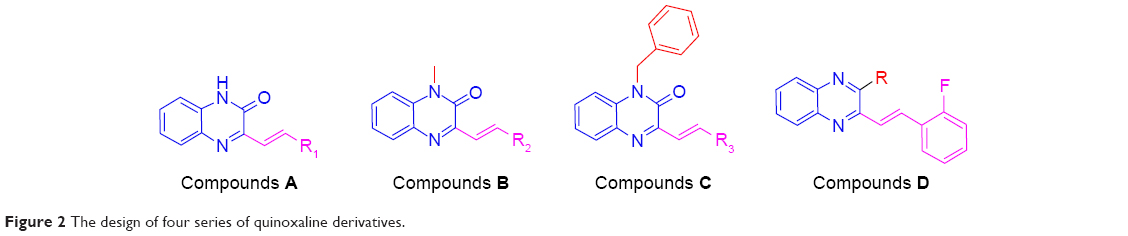 | Figure 2 The design of four series of quinoxaline derivatives. |
Materials and methods
Chemical experimental procedures
All chemical reagents and solvents were purchased from Aladdin (Beijing, People’s Republic of China). Silica gel (GF254) for thin-layer chromatography and column chromatography (100–200 mesh and 200–300 mesh) were obtained from Aladdin. Uncorrected melting points were determined using an XRC-1 micro-melting point apparatus. 1H nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Avance III spectrometer (600 MHz). Samples were dissolved in dimethyl sulfoxide (DMSO), while tetramethylsilane was used as an internal standard. Chemical shifts δ were recorded in ppm relative to tetramethylsilane, and J-values are expressed in Hz. Mass spectra were recorded using an Agilent-1200 LC mass spectrometer. Infrared spectroscopy was determined by PerkinElmer FT-IR 1605 spectrometer (Nicolet, USA). The detailed synthesis and spectral characterization of all compounds are described in the Supplementary material.
Cell line and reagents
All of the tested cancer cells (H460, B16-F10, Hela229, Hct116, and HL7702) were obtained from Wenzhou Medical University and were incubated with Dulbecco’s Modified Eagle’s Medium (Gibco®; Life Technologies, Carlsbad, CA, USA) and supplemented with 10% fetal bovine serum (Gibco®; Life Technologies), 100 U/mL of penicillin, and 100 mg/mL of streptomycin at 37°C with 5% CO2. TKI258, ATP, and MTT reagent were purchased from Sigma (St Louis, MO, USA). FGFR1 was obtained from Carna Bioscience Inc. (Kobe, Japan). All of the tissue culture reagents were obtained from Gino Biomedical Technology Co. (Shenzhen, People’s Republic of China). TKI258 and compounds A1–A14, B1–B5, C1–C5, and D1–D3 were dissolved in DMSO for the in vitro experiments.
LANCE Ultra TR-FRET assays
The ability of all target compounds to inhibit the activation of FGFR1 kinase domain was assessed using LANCE Ultra TR-FRET assays, and the inhibitor TKI258 was used as the control. The assays were carried out in a final volume of 50 μL per well in a white Packard OptiPlate-384 (PerkinElmer, Waltham, MA, USA). Each tested compound was diluted as 40, 4, and 0.4 μM in 1× kinase base buffer containing 50 mM (4-(2-hydroxyethyl)-1-piperazinethaneesulfonic acid) (pH 7.5), 10 mM MgCl2, 1 mM ethylenebis(oxyethylenenitrilo)tetraacetic acid, 2 mM DL-dithiothreitol, and 0.01% Tween-20. Then, 2.5 μL of the compounds was transferred to an assay plate. A standard enzymatic reaction, initiated by the addition of 5 μL of 2.5× peptide solution to 2.5 μL of 4× enzyme, contained 4 nM FGFR1 kinase, 11.9 μM ULight-JAK-1 (Tyr1023) peptide (PerkinElmer), 47 μM ATP, and 1× kinase base buffer. After 90-minute incubation at room temperature, the reaction was stopped by the addition of 5 μL termination buffer (40 mM ethylenediaminetetraacetic acid). Afterwards, 5 μL of 4× antibody (Eu-anti-phospho-tyrosine antibody [PT66] at a final concentration of 2 nM) was added to each well of the assay plate for 1 hour. The product and substrate in each independent reaction were separated using a 12-sipper microfluidic chip (Caliper Life Sciences) run on a Caliper LC3000 (Caliper Life Sciences). The resulting data were collected from the EnVision® Multilabel Reader (PerkinElmer). Then, the original values were converted into the inhibition ratio. All experiments were repeated independently three times.
In addition, some compounds were screened for their selective kinase inhibition. The inhibitory concentration or IC50 values on RTKs were determined by the same method.
MTT assay
An MTT assay was performed to evaluate cytotoxic and antitumor activities of all compounds. Four cell lines were seeded (2,000–20,000 cells per well) in 96-well plates, respectively. After incubation for 24 hours in serum-containing media, the cells were treated with inhibitors (50, 10, 2, and 0.4 μM), diluted with culture medium for 72 hours at 37°C under a 5% CO2 atmosphere. Thereafter, 20 μL of the MTT reagent (5 mg/mL) was added to each well, and the plates were incubated for 4 hours at 37°C. For the adherent cells, the media and MTT were carefully aspirated from each well, and formazan crystals were dissolved in 100 μL of DMSO. For the suspended cells, 50 μL of 20% acidified sodium dodecyl sulfate (w/v) was added to each well, and the cells were incubated overnight. Finally, absorbance at 490 nm was read using a Spectrophotometer (SMP500-13732-UJRK; MDC, Hayward, CA, USA). TKI258 was used as the positive control, and DMSO was used as a negative control. The resulting data were analyzed using GraphPad Prism™ software (GraphPad Software, Inc.). Furthermore, the viability of human liver HL7702 cells exposed to different doses of these inhibitors was determined using the same method. All experiments were performed in parallel, in triplicate. For data analysis, IC50 values were obtained by using GraphPad Prism version 5.0 (GraphPad Inc., San Diego, CA, USA).
Molecular docking analysis
The molecular modeling approach is widely used for the discovery, design, and prediction of the activity and mechanisms by which active compounds act. As such, in order to obtain further insight into the interactions mode between these 3-vinyl-quinoxalin-2(1H)-one derivatives and FGFR1, molecular docking analysis was done using Auto-Dock version 4.2.29,30 The protein structure (PDB:5AM6)31 of FGFR1 bound to the inhibitor TKI2589 was selected for the construction of the docking template. Subsequently, compounds from A5 were selected to dock with the validated template.
Statistical analysis
The results are presented as mean ± standard error of the mean. Student’s t-test was employed to analyze the differences between sets of data. Statistics were performed using GraphPad Prism 5.0. P-values <0.05 were considered indicative of significance. All experiments were repeated at least three times.
Results and discussion
Chemistry
Structural optimization was carried out by focusing on the C-3 and C-1 positions of the quinoxaline. There were four series of 3-vinyl-quinoxalin-2(1H)-one derivatives synthesized. The synthesis and structures of compounds A1–A14, B1–B5, C1–C5, and D1–D3 are shown in Figure 3. Comprising the intermediate products, a total of 27 target compounds were synthesized and contained six new compounds which have not been reported, including A14, B3, B5, C2, C3, and C4. Some compounds have been reported in other articles before.32,33 The structures of all compounds were characterized using 1H NMR, electrospray ionization mass spectroscopy, and infrared spectroscopy.
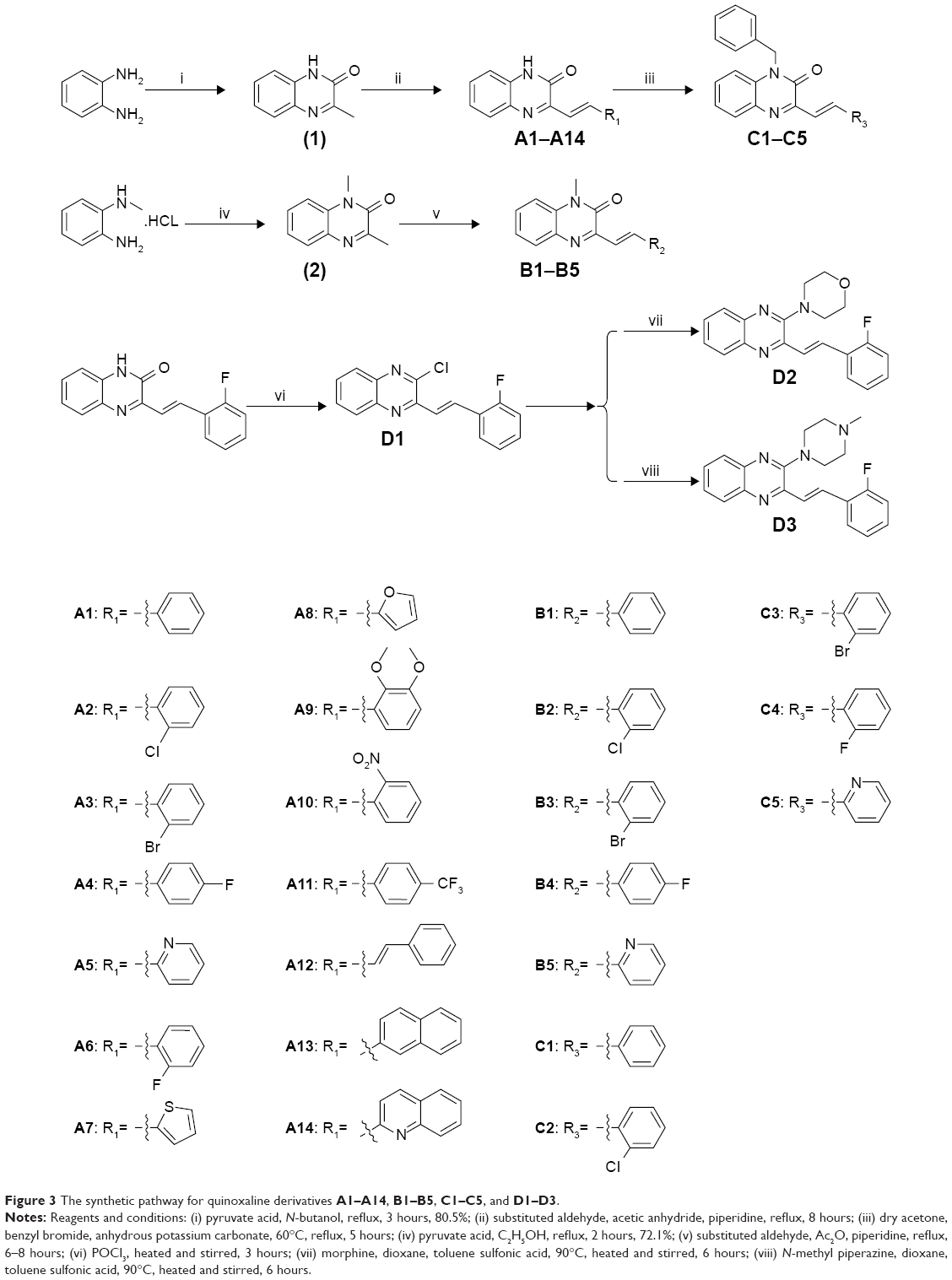 | Figure 3 The synthetic pathway for quinoxaline derivatives A1–A14, B1–B5, C1–C5, and D1–D3. |
Compounds that inhibited FGFR1 kinase
As shown in Figure 4, we were pleased to find that most of compounds had kinase inhibitory activities. Compounds A1, A2, A3, and A5 exhibited relatively good inhibitory activities of FGFR1 among all of the tested compounds, particularly at 10.0 μM. Therefore, the introduction of methyl groups or benzyl groups at the C-1 position of the quinoxaline scaffold seemed to have no significant positive effect on inhibition. While having the same R substitute, compounds A were more active than compounds B, and the inhibitory activity of compounds B was slightly better than compounds C. As such, compounds A had the potential to become a resource for developing FGFR1 inhibitors. Interestingly, substitution of the phenyl group onto the side chain (A1–A5) resulted in a higher level of kinase inhibition activity, compared to substitution of 4-trifluoromethylphenyl, 2-quinolyl, and 2-naphthyl groups (A11, A13, and A14). This may have been because the former group is relatively small. A5 was the most promising compound with an inhibition ratio of 79.33% at 10.0 μM. On the other hand, the concentration of TKI258, A1–A3, A5, B2, B5, and C2 resulting in IC50 is reported in Table 1. These studies demonstrated that A5 is a selective inhibitor with high potency against FGFR1 and has the potential to be a major resource for developing inhibitors against FGFR1.
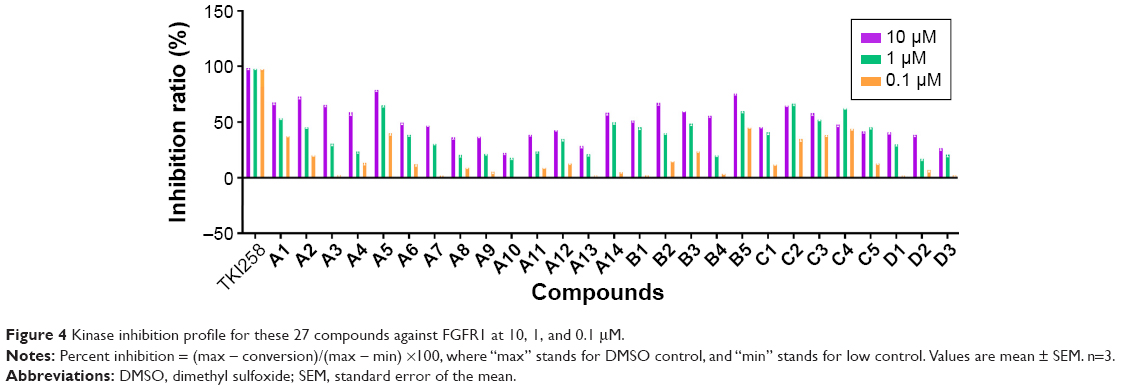 | Figure 4 Kinase inhibition profile for these 27 compounds against FGFR1 at 10, 1, and 0.1 μM. |
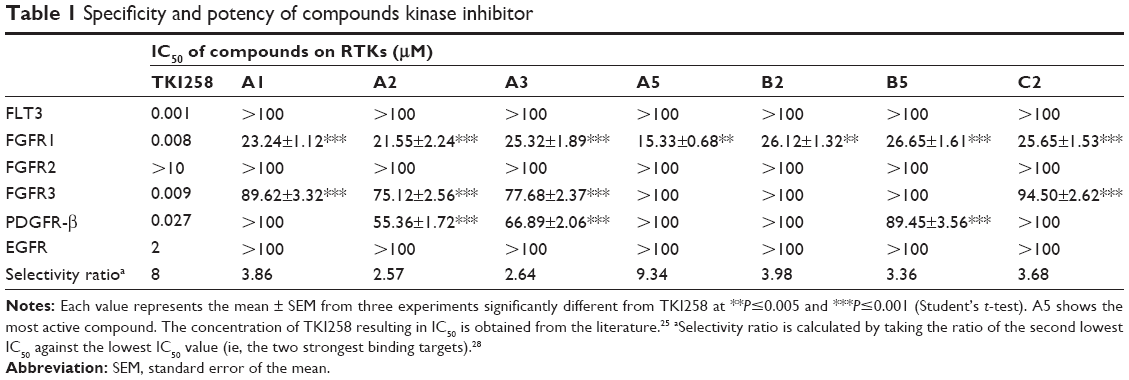 | Table 1 Specificity and potency of compounds kinase inhibitor |
Cytotoxic and antitumor activities of all compounds
The activities of 3-vinyl-quinoxalin-2(1H)-one derivatives were tested on cancer cell lines H460, Hct116, Hela229, and B16-F10, which are known to express high levels of FGFR1.34,35 The activity data regarding the anti-viability potency of compounds (A1–A14, B1–B5, C1–C5, and D1–D3) are summarized in Table 2. According to the results, most compounds showed antitumor activity on the four tumor cell lines, which appears to correspond to the inhibition of FGFR1 by the positive control drug (TKI258). Among them, compounds A5, A12, B2, B5, D2, and D3 exhibited remarkable improvement in activity against the four tumor cell lines. Additionally, A6, A8, A11, and A14 only affected B16-F10 cells. The cytotoxicity tests indicated that most of these 3-vinyl-quinoxalin-2(1H)-one derivatives had the advantage of lower toxicity against normal human liver HL7702 cells, compared with TKI258, a well-characterized anti-neoplastic agent (Table 2). In addition, as shown in Figure 5A and B, HL7702 cells had a higher survival rate by treatment with a relatively high concentration (2 and 10 μM) of A5 than with TKI258. Thus, the results were in line with our expectations.
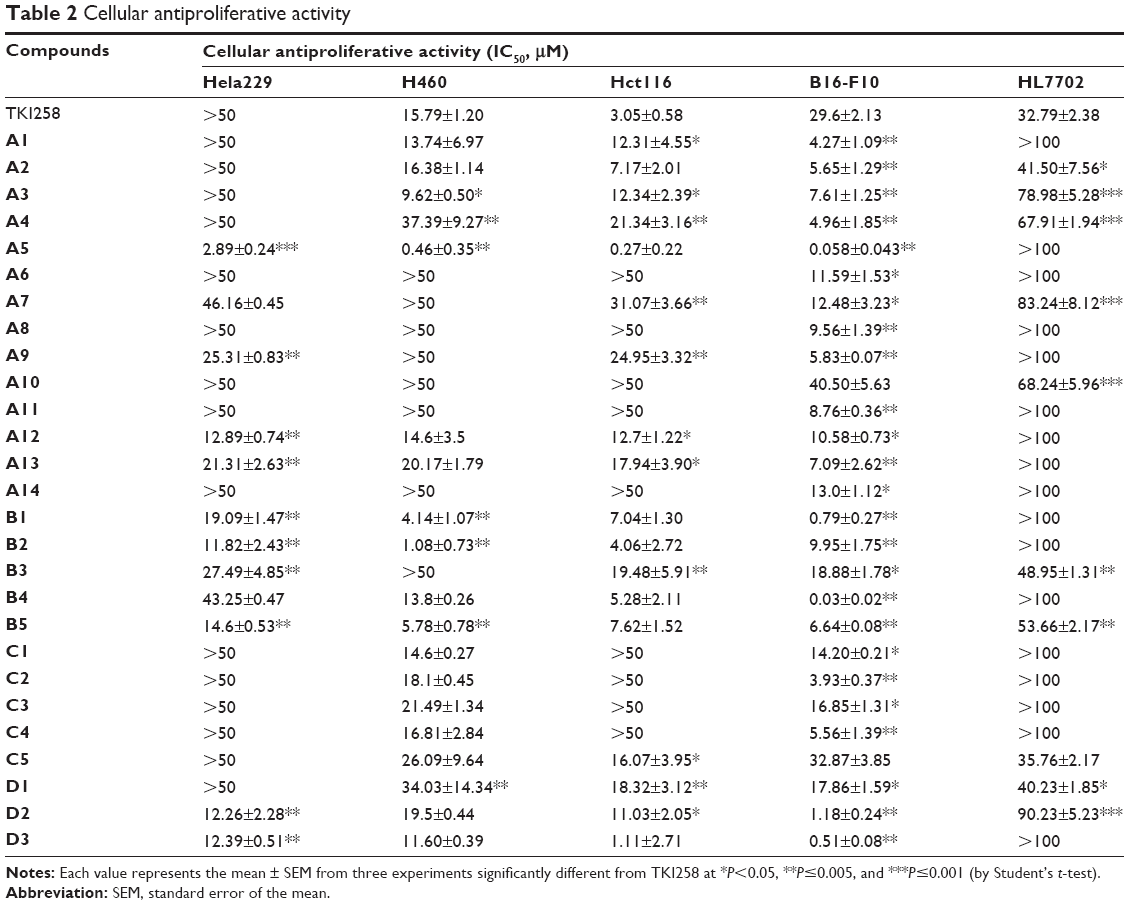 | Table 2 Cellular antiproliferative activity |
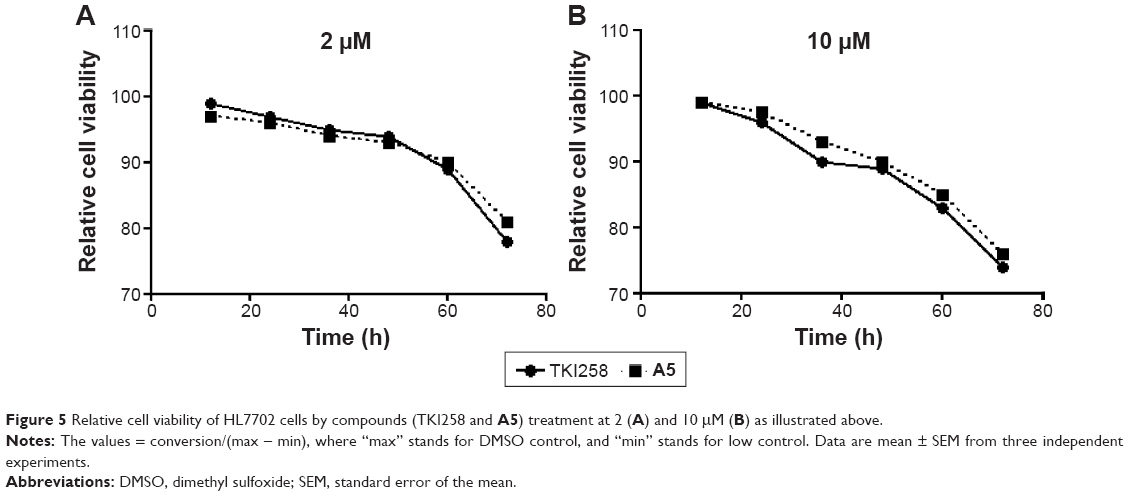 | Figure 5 Relative cell viability of HL7702 cells by compounds (TKI258 and A5) treatment at 2 (A) and 10 μM (B) as illustrated above. |
Molecular docking of compound A5 and FGFR1
Docking simulation of A5 to FGFR1 was carried out with the program Auto-Dock and MD/MM calculations. The binding profile of A5 to FGFR1 is −7.38 kcal/mol. Compared to TKI258, the docking results (Figure 6) showed that A5 exhibited preferable docking with FGFR1, and four hydrogen bonds were formed. This was a pleasant surprise, as this is different from TKI258. The oxygen atom of the carbonyl group on A5 forms two hydrogen bonds bound to Ala564 and Glu562 (distance cutoff: 2.8 and 3.4 Å). In addition, the 2-N of the pyridine ring is favorably oriented at ~1.8 Å from the Ala564. Probably because of the rigidity of the double bond, an interaction of the N–H and 2-O facilitated formation of hydrogen bonds. This may be the reason for its better inhibition activity. Additionally, the 1-N contributed another hydrogen bond that interacted with Glu562 in the active pocket with a distance cutoff of 2.2 Å. Therefore, the binding geometry provided potential explanations for the antitumor activity of A5 and was also in line with our initial design.
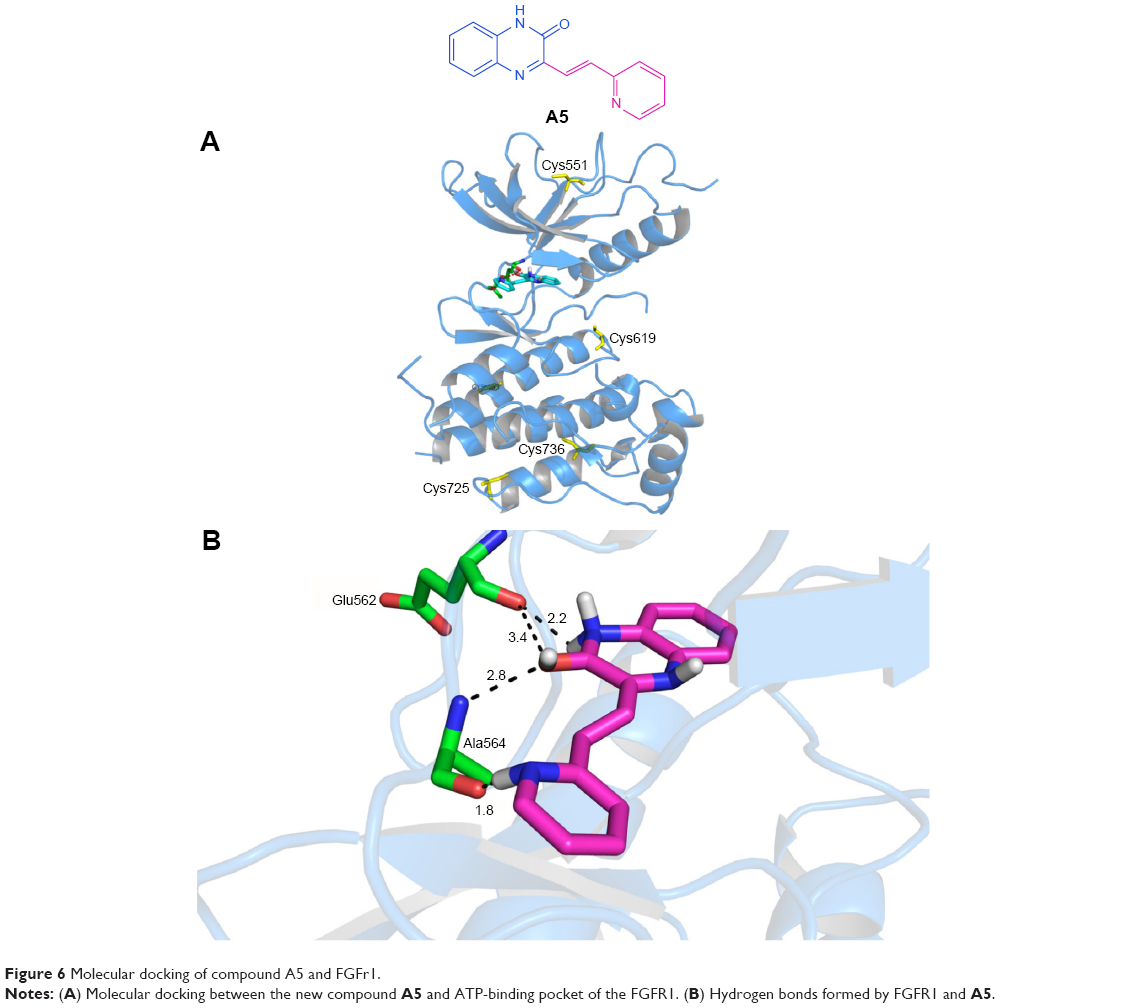 | Figure 6 Molecular docking of compound A5 and FGFr1. |
SAR of 3-vinyl-quinoxalin-2(1H)-one derivatives
The SAR obtained from two bioactivity assays potentially reports a combination of the inhibitory activity of the compounds against FGFR1 kinase and the ability of the compounds to inhibit proliferation of tumor cells. The SAR of these compounds is summarized as follows: 1) Comparing compound D with compounds A, B, and C, the compounds that contain a carbonyl group had better activity, probably due to the formation of two hydrogen bonds with FGFR1 kinase. 2) Comparing compound A with compounds B and C, when they had the same R substitute, and for compounds that had the non-N substitute, the kinase inhibitory activity was the largest, possibly due to an amide bond increase of its kinase affinity and more ready formation of hydrogen bonds with the hinge region; however, the substituted methyl or benzyl group did not integrate well into the adenine-binding site of FGFR1. 3) For compound A, with the introduction of different substitutions, in accordance with the pharmacophore model, the relatively small substitution may more easily fit into the active pocket, resulting in better inhibition.
Conclusion
In summary, we synthesized 27 (four series) 3-vinyl-quinoxalin-2(1H)-one derivatives as a new class of FGFR1 kinase inhibitors through a structure-based drug design and confirmed structures by 1H NMR and electrospray ionization mass spectroscopy. Meanwhile, we manifested common bioactivity for inhibition of FGFR1 and cellular toxicity against H460, Hct116, Hela229, and B16-F10 cell lines while being exempt of general toxicity to noncancerous cells. Different types of products exhibited their own activity against FGFR1; for example, the compounds substituted with small groups of side chains showed better selectivity toward FGFR1, such as A3 and A5. Moreover, different compounds showed their own selectively, as evidenced by compounds A6, A8, A11, and A14, which only affected B16-F10 cells. SAR studies centering on the C-1, C-2, and C-3 groups of the 3-vinyl-quinoxalin-2(1H)-one scaffold led to the discovery of some optimization methods. All of these results indicated that the 3-vinyl-quinoxalin-2(1H)-one scaffold had a preference for beneficial substitutions when combined with the ATP-binding site and may be utilized as candidates for inhibition of FGFR1. As such, detailed follow-up studies of the action mechanisms for these derivatives are underway and will be reported later.
Acknowledgments
Financial support was provided by the National Natural Science Foundation of China (81373262, 21202124, 21472142), Zhejiang Medical & Health Science and Technology Project (2013KYB168), Natural Foundation of Zhejiang Province (LY16B020010), and Xinmiao Talent Project of Zhejiang Province (ZHP).
Disclosure
The authors report no conflicts of interest in this work.
References
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nature Reviews Cancer. 2010;10(2):116–129. | ||
Haugsten EM, Wiedlocha A, Olsnes S, Wesche J. Roles of fibroblast growth factor receptors in carcinogenesis. Molecular Cancer Research. 2010;8(11):1439–1452. | ||
Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochemical Journal. 2011;437(2):199–213. | ||
Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clinical Cancer Research. 2012;18(7):1855–1862. | ||
Feng S, Zhou L, Nice EC, Huang C. Fibroblast growth factor receptors: multifactorial-contributors to tumor initiation and progression. Histology and Histopathology. 2015;30:13–31. | ||
Tiong KH, Mah LY, Leong CO. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis. 2013;18(12):1447–1468. | ||
Liang G, Chen G, Wei X, et al. Small molecule inhibition of fibroblast growth factor receptors in cancer. Cytokine & Growth Factor Reviews. 2013;24(5):467–475. | ||
Göke F, Bode M, Franzen A, et al. Fibroblast growth factor receptor 1 amplification is a common event in squamous cell carcinoma of the head and neck. Modern Pathology. 2013;26(10):1298–1306. | ||
Wu J, Wei T, Tang Q, Weng B, et al. Discovery and anti-cancer evaluation of two novel non-ATP-competitive FGFR1 inhibitors in non-small-cell lung cancer. BMC Cancer. 2015;15:276. | ||
Irschick R, Trost T, Karp G, et al. Sorting of the FGF receptor 1 in a human glioma cell line. Histochemistry and Cell Biology. 2013;139(1):135–148. | ||
Weiss J, Sos ML, Seidel D, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Science Translational Medicine. 2010;2(62):62ra93. | ||
Turner N, Pearson A, Sharpe R, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Research. 2010;70(5):2085–2094. | ||
Yang F, Zhang Y, Ressler SJ, et al. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Research. 2013;73(12):3716–3724. | ||
Liang G, Liu Z, Wu J, et al. Anticancer molecules targeting fibroblast growth factor receptors. Trends in Pharmacological Sciences. 2012;33(10):531–541. | ||
Chiang CC, Lin YH, Lin SF, et al. Discovery of pyrrole-indoline-2-ones as Aurora kinase inhibitors with a different inhibition profile. Journal of Medicinal Chemistry. 2010;53(16):5929–5941. | ||
Mohammadi M, McMahon G, Sun L, et al. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276(5314):955–960. | ||
GuagnanoV, Furet P, Spanka C, et al. Discovery of 3-(2, 6-dichloro-3, 5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. Journal of Medicinal Chemistry. 2011;54(20):7066–7083. | ||
Ye F, Chen L, Hu L, et al. Design, synthesis and preliminary biological evaluation of C-8 substituted guanine derivatives as small molecular inhibitors of FGFRs. Bioorganic & Medicinal Chemistry Letters. 2015;25(7):1556–1560. | ||
Ye F, Wang Y, Nian S, et al. Synthesis and evaluation of biological and antitumor activities of 5, 7-dimethyl-oxazolo [5, 4-d] pyrimidine-4, 6 (5 H, 7 H)-dione derivatives as novel inhibitors of FGFR1. Journal of Enzyme Inhibition and Medicinal Chemistry. 2015;30(6):961–966. | ||
Ravindranathan KP, Mandiyan V, Ekkati AR, et al. Discovery of novel fibroblast growth factor receptor 1 kinase inhibitors by structure-based virtual screening. Journal of Medicinal Chemistry. 2010;53(4):1662–1672. | ||
Fearon AE, Gould CR, Grose RP. FGFR signalling in women’s cancers. The International Journal of Biochemistry & Cell Biology. 2013;45(12):2832–2842. | ||
Wang Y, Cai Y, Ji J, et al. Discovery and identification of new non-ATP competitive FGFR1 inhibitors with therapeutic potential on non-small-cell lung cancer. Cancer Letters. 2014;344(1):82–89. | ||
Chan SL, Wong CH, Lau CPY, et al. Preclinical evaluation of combined TKI-258 and RAD001 in hepatocellular carcinoma. Cancer Chemotherapy and Pharmacology. 2013;71(6):1417–1425. | ||
Lopes de Menezes D, Hollenbach P, Tang Y, et al. TKI258 is an effective multitargeted receptor tyrosine kinase (RTK) inhibitor against prostate cancer models via potent inhibition of FGFR kinase. EJC Supplements. 2006;4(12):175–175. | ||
Trudel S, Li ZH, Wei E, et al. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105(7):2941–2948. | ||
Kim J, Lee G, Kim S, et al. Identification of a quinoxaline derivative that is a potent telomerase inhibitor leading to cellular senescence of human cancer cells. Biochemical Journal. 2003;373:523–529. | ||
Issa DAE, Habib NS, Wahab AEA. Design, synthesis and biological evaluation of novel 1, 2, 4-triazolo and 1, 2, 4-triazino [4, 3-a] quinoxalines as potential anticancer and antimicrobial agents. Medicinal Chemistry Communication. 2015;6(1):202–211. | ||
Ho HK, Yeo AHL, Kang TS, Chua BT. Current strategies for inhibiting FGFR activities in clinical applications: opportunities, challenges and toxicological considerations. Drug Discovery Today. 2014; 19(1):51–56. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. | ||
Wissner A, Fraser HL, Charles L, et al. Dual irreversible kinase inhibitors: quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2. Bioorganic & Medicinal Chemistry. 2007;15:3635–3648. | ||
Bunney TD, Wan S, Thiyagarajan N, et al. The effect of mutations on drug sensitivity and kinase activity of fibroblast growth factor receptors: a combined experimental and theoretical study. EBioMedicine. 2015;2(3):194–204. | ||
Badran MM, Moneer AA, Refaat HM, El-Malah AA. Synthesis and antimicrobial activity of novel quinoxaline derivatives. Journal of the Chinese Chemical Society. 2007;54:469–478. | ||
Noolvi MN, Patel HM, Bhardwaj V, Chauhan A. Synthesis and in vitro antitumor activity of substituted quinazoline and quinoxaline derivatives: search for anticancer agent. European Journal of Medicinal Chemistry. 2011;46:2327–2346. | ||
Yang J, Zhao H, Xin Y, et al. MicroRNA-198 inhibits proliferation and induces apoptosis of lung cancer cells via targeting FGFR1. Journal of Cellular Biochemistry. 2014;115(5):987–995. | ||
Wu X, Huang H, Wang C, et al. Identification of a novel peptide that blocks basic fibroblast growth factor-mediated cell proliferation. Oncotarget. 2013;4(10):1819. |
Supplementary material
The title compounds A1–A14, B1–B5, C1–C5, and D1–D3 were characterized as follows.
(E)-1,2-Dihydro-3-(2-phenyl ethenyl) quinoxalin-2-one (A1)
Yellow powder, 61.2% yield, mp 247.2°C–248.1°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.51 (br, 1H, NH), 8.07 (d, J=16.2 Hz, 1H, ethenyl H), 7.78 (d, J=7.8 Hz, 1H, ArH), 7.73 (d, J=7.2 Hz, 2H, ArH), 7.62 (d, J=16.2 Hz, 1H, ethenyl H), 7.48–7.51 (m, 1H, ArH), 7.43–7.45 (m, 2H, ArH), 7.37–7.40 (m, 1H, ArH), 7.30–7.32 (m, 2H, ArH). IR: 1,655.46 (C=O), 1,626.09 (C=N). ESI-MS m/z: 248.9 (M+H)+.
(E)-1,2-Dihydro-3-[2-(2-chlorophenyl) ethenyl]-quinoxalin-2-one (A2)
Yellow powder, 79.8% yield, mp 244.8°C–246.3°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.55 (s, 1H, NH), 8.44 (d, J=16.2 Hz, 1H, ethenyl H), 7.97–7.99 (m, 1H, ArH), 7.81 (d, J=7.2 Hz, 1H, ArH), 7.64 (d, J=16.2 Hz, 1H, ethenyl H), 7.51–7.56 (m, 2H, ArH), 7.39–7.44 (m, 2H, ArH), 7.31–7.33 (m, 2H, ArH). IR: 1,661.61 (C=O), 1,622.11 (C=N). ESI-MS m/z: 282.9 (M+H)+.
(E)-1,2-Dihydro-3-[2-(2-bromophenyl) ethenyl]-1,2-dihydro-quinoxalin-2-one (A3)
Yellow powder, 78.1% yield, mp 257.2°C–259.4°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.55 (s, 1H, NH), 8.40 (d, J=16.2 Hz, 1H, ethenyl H), 7.96 (d, J=7.8 Hz, 1H, ArH), 7.81 (d, J=7.8 Hz, 1H, ArH), 7.72 (d, J=7.8 Hz, 1H, ArH), 7.59 (d, J=16.2 Hz, 1H, ethenyl H), 7.51–7.54 (m, 1H, ArH), 7.45–7.48 (m, 1H, ArH), 7.33 (s, 3H). IR: 1,658.08 (C=O), 1,621.22 (C=N). ESI-MS m/z: 327.0 (M+H)+.
(E)-3-[2-(4-Fluorophenyl) ethenyl]-1,2-dihydro-quinoxalin-2-one (A4)
Yellow powder, 75.6% yield, mp 239.1°C–241.4°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.51 (s, 1H, NH), 8.06 (d, J=16.2 Hz, 1H ethenyl H), 7.79–7.82 (m, 2H, ArH), 7.77–7.78 (m, 1H, ArH), 7.57 (d, J=16.2 Hz, 1H ethenyl H), 7.48–7.51 (m, 1H, ArH), 7.28–7.33 (m, 2H, ArH), 7.25–7.28 (m, 2H, ArH). IR 1,664.02 (C=O), 1,626.11 (C=N). ESI-MS m/z: 266.9 (M+H)+.
(E)-3-[2-(Pyridine-2-yl) ethenyl]-1,2-dihydro-quinoxalin-2-one (A5)
Yellow powder, 71.2% yield, mp 208.6°C–210.5°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.53 (s, 1H, NH), 8.66 (d, J=3.6 Hz ,1H), 8.06 (s, 2H), 7.83–7.86 (m, 1H), 7.79–7.81 (m, 1H), 7.71 (d, J=7.8 Hz, 1H), 7.51–7.54 (m, 1H), 7.34–7.37 (m, 1H), 7.32 (d, J=7.8 Hz, 2H). IR: 1,655.43 (C=O), 1,629.59 (C=N). ESI-MS m/z: 250.1 (M+H)+.
(E)-3-[2-(2-Fluorophenyl) ethenyl]-1,2-dihydro-quinoxalin-2-one (A6)
Yellow powder, 84.1% yield, mp 235.5°C–237.6°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.54 (s, 1H, NH), 8.19 (d, J=16.2 Hz, 1H, ethenyl H), 7.88–7.90 (m, 1H, ArH), 7.79 (d, J=7.8, 1H, ArH), 7.69 (d, J=16.2 Hz, 1H, ethenyl H), 7.48–7.51 (m, 1H, ArH), 7.42–7.45 (m, 1H, ArH), 7.27–7.32 (m, 4H, ArH). IR: 1,655.61 (C=O), 1,621.51 (C=N). ESI-MS m/z: 267.1 (M+H)+.
(E)-3-[2-(Thiophene-2-yl) ethenyl]-1,2-dihydro-quinoxalin-2-one (A7)
Yellow powder, 62.1% yield, mp 241.8°C–243.4°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.49 (s, 1H, NH), 8.23 (d, J=15.6 Hz, 1H), 7.75 (d, J=7.8 Hz, 1H), 7.66 (d, J=5.4 Hz, 1H), 7.48–7.51 (m, 2H), 7.29–7.32 (m, 3H), 7.15–7.17 (m, 1H). IR: 1,656.14 (C=O), 1,614.25 (C=N). ESI-MS m/z: 254.9 (M+H)+.
(E)-3-[2-(Furan-2-yl) ethenyl]-1,2-dihydro-quinoxalin-2-one (A8)
Yellow powder, 59.1% yield, mp 249.4°C–252.1°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.47 (s, 1H, NH), 7.90 (d, J=16.2 Hz, 1H, ethenyl H), 7.84 (d, J=1.8 Hz, 1H), 7.74 (d, J=7.2 Hz, 1H), 7.46–7.49 (m, 1H), 7.36 (d, J=16.2 Hz, 1H, ethenyl H), 7.28–7.31 (m, 2H), 6.91 (d, J=3.6 Hz, 1H), 6.64–6.65 (m, 1H). IR: 1,661.86 (C=O), 1,624.25 (C=N). ESI-MS m/z: 238.9 (M+H)+.
(E)-1,2-Dihydro-3-[2-(2,3-dimethoxy phenyl) ethenyl]-quinoxalin-2-one (A9)
Yellow powder, 69.0% yield, mp 215.3°C–217.8°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.48 (s, 1H, NH), 8.30 (d, J=16.8 Hz, 1H), 7.80 (d, J=7.2 Hz, 1H, ArH), 7.62 (d, J=16.2 Hz, 1H), 7.46–7.51 (m, 1H, ArH), 8.38 (d, J=7.2 Hz, 1H, ArH), 7.30–7.32 (m, 2H, ArH), 7.08–7.15 (m, 2H, ArH), 3.82 (d, J=22.8 Hz, 6H, CH3×2). IR: 1,673.83 (C=O), 1,618.98 (C=N). ESI-MS m/z: 309.1 (M+H)+.
(E)-1,2-Dihydro-3-[2-(2-nitrophenyl) ethenyl]-quinoxalin-2-one (A10)
Yellow powder, 72.3% yield, mp 248.7°C–250.2°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.58 (s, 1H, NH), 8.36 (d, J=16.2 Hz, 1H, ethenyl H), 8.04–8.06 (m, 2H, ArH), 7.78–7.82 (m, 2H, ArH), 7.64–7.67 (m, 1H), 7.61 (d, J=16.2 Hz, 1H, ethenyl H), 7.52–7.55 (m, 1H, ArH), 7.32–7.34 (m, 2H, ArH). IR: 1,662.01 (C=O), 1,622.79 (C=N). ESI-MS m/z: 294.0 (M+H)+.
(E)-1,2-Dihydro-3-[2-(4-trifluoromethyl phenyl) ethenyl]-quinoxalin-2-one (A11)
Yellow powder, 78.2% yield, mp 206.1°C–208.7°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.57 (s, 1H, NH), 8.12 (d, J=16.2 Hz, 1H, ethenyl H), 7.96 (d, J=8.4 Hz, 2H, ArH), 7.76–7.80 (m, 3H, ArH), 7.74 (d, J=16.2 Hz, 1H, ethenyl H), 7.51–7.54 (t, 1H, ArH), 7.31–7.34 (t, 2H, ArH). IR 1,667.07 (C=O), 1,627.07 (C=N). ESI-MS m/z: 317.4 (M+H)+.
(E)-1,2-Dihydro-3-[(4-phenyl-1,3-butadienyl)-1-yl]-quinoxalin-2-one (A12)
Yellow powder, 65.3% yield, mp 252.1°C–254.8°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.43 (s, 1H, NH), 7.86–7.90 (m, 1H, ArH), 7.74 (d, J=7.8 Hz, 1H, ArH), 7.60 (d, J=7.2 Hz, 2H, ArH), 7.46–7.49 (m, 1H, ArH), 7.38–7.41 (t, 2H, ArH), 7.26–7.32 (m, 4H, ArH), 7.17 (d, J=15.6 Hz, 1H, ethenyl H), 7.06 (d, J=15.6 Hz, 1H, ethenyl H). IR: 1,661.65 (C=O), 1,612.51 (C=N). ESI-MS m/z: 275.1 (M+H)+.
(E)-1,2-Dihydro-3-[2-(naphth-2-yl) ethenyl]-quinoxalin-2-one (A13)
Yellow powder, 77.2% yield, mp 229.3°C–231.2°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.54 (br, 1H, NH), 8.24 (d, J=16.2 Hz ,2H), 7.93–8.00 (m, 4H), 7.81 (d, J=7.8 Hz, 1H), 7.76 (d, J=16.2 Hz, 1H), 7.54–7.57 (m, 2H), 7.50–7.52 (m, 1H), 7.31–7.34 (m, 2H). IR: 1,658.36 (C=O), 1,611.80 (C=N). ESI-MS m/z: 299.1 (M+H)+.
(E)-1-Dihydro-3-[2-(isoquinoline-3-yl) ethenyl]-quinoxalin-2-one (A14)
Yellow powder, 52.1% yield, mp 263.9°C–265.3°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 12.61 (s, 1H, NH), 8.42 (d, J=8.4 Hz, 1H), 8.26 (d, J=16.2 Hz, 1H, ethenyl H), 8.18 (d, J=16.2 Hz, 1H, ethenyl H), 8.06–8.09 (m, 1H), 7.96–8.00 (m, 2H), 7.84 (d, J=8.4 Hz, 1H), 7.78–7.81 (m, 1H), 7.61–7.63 (m, 1H), 7.54–7.56 (m, 1H), 7.33–7.36 (m, 2H). IR: 1,660.83 (C=O), 1,608.74 (C=N). ESI-MS m/z: 300.0 (M+H)+.
(E)-1,2-Dihydro-1-methyl-3-(2-phenyl ethenyl)-quinoxalin-2-one (B1)
Yellow powder, 54.4% yield, mp 125.3°C–127.6°C. 1H NMR (600 MHz, CDCl3-d1) δ (ppm): 8.13 (d, J=16.2 Hz, 1H, ethenyl H), 7.88–7.89 (m, 1H, ArH), 7.77 (d, J=16.2 Hz, 1H, ethenyl H), 7.74 (d, J=7.8 Hz, 2H, ArH), 7.52–7.54 (m, 1H, ArH), 7.38–7.41 (m, 2H, ArH), 7.26–7.38 (m, 3H, ArH), 3.76 (s, 3H, CH3). IR: 1,644.18 (C=O), 1,597.22 (C=N). ESI-MS m/z: 262.7 (M+H)+.
(E)-3-[2-(2-Chlorophenyl) ethenyl]-1,2-dihydro-1-methyl-quinoxalin-2-one (B2)
Yellow powder, 67.5% yield, mp 139.7°C–142.3°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.42 (d, J=16.2 Hz, 1H, ethenyl H), 7.97–7.99 (m, 1H, ArH), 7.85–7.87 (m, 1H, ArH), 8.68 (d, J=16.2 Hz, 1H, ethenyl H), 7.62–7.65 (m, 1H, ArH), 7.54–7.58 (m, 2H, ArH), 7.40–7.43 (m, 3H, ArH), 3.68 (s, 3H, CH3). IR: 1,635.59 (C=O), 1,595.02 (C=N). ESI-MS m/z: 297.1 (M+H)+.
(E)-3-[2-(2-Bromophenyl) ethenyl]-1,2-dihydro-1-methyl-quinoxalin-2-one (B3)
Yellow powder, 71.2% yield, mp 165.4°C–167.8°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.39 (d, J=15.6 Hz, 1H), 7.96 (d, J=6.6 Hz, 1H), 7.86 (d, J=8.4 Hz, 1H), 7.72 (d, J=6.6 Hz, 1H), 7.62–7.66 (m, 2H), 7.59 (d, J=7.8 Hz, 1H), 7.46–7.48 (m, 1H), 7.40–7.42 (m, 1H), 7.32–7.34 (m, 1H), 3.69 (s, 3H, CH3). IR: 1,636.16 (C=O), 1,595.07 (C=N). ESI-MS m/z: 340.9 (M+H)+.
(E)-1,2-Dihydro-3-[2-(4-fluorophenyl) ethenyl]-1-methyl-quinoxalin-2-one (B4)
Yellow powder, 77.2% yield, mp 162.1°C–164.2°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.03 (d, J=16.2 Hz, 1H), 7.80–7.84 (m, 3H), 7.60–7.64 (m, 2H), 7.56 (d, J=7.8 Hz, 1H), 7.39–7.41 (m, 1H), 7.25–7.28 (m, 2H), 3.68 (s, 3H, CH3). IR: 1,643.01 (C=O), 1,597.86 (C=N). ESI-MS m/z: 280.9 (M+H)+.
(E)-3-[2-(Pyridine-2-yl) ethenyl]-1,2-dihydro-1-methyl-quinoxalin-2-one (B5)
Yellow powder, 62.3% yield, mp 144.5°C–146.3°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.66 (d, J=3.6 Hz, 1H), 8.12 (d, J=16.2 Hz, 1H, ethenyl H), 8.02 (d, J=15.6 Hz, 1H, ethenyl H), 7.84–7.87 (m, 2H), 7.71 (d, J=7.8 Hz, 1H), 7.62–7.65 (m, 1H), 7.58 (d, J=7.2 Hz, 1H), 7.40–7.42 (m, 1H), 7.35–7.37 (m, 1H), 3.68 (s, 3H, CH3). IR: 1,649.70 (C=O), 1,600.62 (C=N). ESI-MS m/z: 264.0 (M+H)+.
(E)-1,2-Dihydro-3-(2-phenyl ethenyl)-1-phenylmethyl-quinoxalin-2-one (C1)
Chartreuse powder, 70.7% yield, mp 161.9°C–162.7°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.08 (d, J=16.2 Hz, 1H), 7.86 (d, J=8.4 Hz, 1H), 7.72–7.77 (m, 3H), 7.50–7.52 (m, 1H), 7.45–7.47 (m, 3H), 7.36–7.41 (m, 2H), 7.29–7.34 (m, 4H), 7.25–7.27 (m, 1H), 5.56 (s, 2H, CH2). IR: 1,654.75 (C=O), 1,598.64 (C=N). ESI-MS m/z: 339.3 (M+H)+.
(E)-3-[2-(2-Chlorophenyl) ethenyl]-1,2-dihydro-1-phenylmethyl-quinoxalin-2-one (C2)
Chartreuse powder, 72.2% yield, mp 108.2°C–110.9°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.46 (d, J=16.2 Hz, 1H, ethenyl H), 8.01–8.03 (m, 1H, ArH), 7.89 (d, J=6.6 Hz, 1H, ArH), 7.75 (d, J=16.2 Hz, 1H, ethenyl H), 7.52–7.57 (m, 2H, ArH), 7.41–7.46 (m, 3H, ArH), 7.37–7.39 (m, 1H, ArH), 7.29–7.34 (m, 4H, ArH), 7.25–7.27 (m, 1H, ArH), 5.57 (s, 2H, CH2). IR: 1,650.75 (C=O), 1,599.73 (C=N). ESI-MS m/z: 373.1 (M+H)+.
(E)-3-[2-(2-Bromophenyl) ethenyl]-1,2-dihydro-1-phenylmethyl-quinoxalin-2-one (C3)
Chartreuse powder, 69.9% yield, mp 139.3°C–141.6°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.43 (d, J=16.2 Hz, 1H, ethenyl H), 7.98–7.80 (m, 1H, ArH), 7.88–7.89 (m, 1H, ArH), 7.73 (d, J=6.6 Hz, 1H, ArH), 7.70 (d, J=16.2 Hz, 1H, ethenyl H), 7.52–7.55 (m, 1H, ArH), 7.45–7.49 (m, 2H, ArH), 7.37–7.39 (m, 1H, ArH), 7.29–7.35 (m, 5H, ArH), 7.25–7.27 (m, 1H, ArH), 5.57 (s, 2H, CH2). IR: 1,649.10 (C=O), 1,599.19 (C=N). ESI-MS m/z: 417.2 (M+H)+.
(E)-3-[2-(2-Fluorophenyl) ethenyl]-1,2-dihydro-1-phenylmethyl-quinoxalin-2-one (C4)
Chartreuse powder, 81.3% yield, mp 128.0°C–130.3°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.20 (d, J=16.2 Hz, 1H, ethenyl H), 7.88–7.93 (m, 2H, ArH), 7.81 (d, J=16.2 Hz, 1H, ethenyl H), 7.51–7.54 (m, 1H, ArH), 7.43–7.48 (m, 2H, ArH), 7.36–7.39 (m, 1H, ArH), 7.29–7.34 (m, 6H, ArH), 7.25–7.27 (m, 1H, ArH), 5.56 (s, 2H, CH2). IR: 1,644.32 (C=O), 1,601.77 (C=N). ESI-MS m/z: 357.2 (M+H)+.
(E)-3-[2-(Pyridine-2-yl) ethenyl]-1,2-dihydro-1-phenylmethyl-quinoxalin-2-one (C5)
Chartreuse powder, 70.11% yield, mp 148.3°C–151.1°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.67 (d, J=3.6 Hz, 1H), 8.17 (d, J=15.6 Hz, 1H, ethenyl H), 8.07 (d, J=15.6 Hz, 1H, ethenyl H), 7.85–7.89 (m, 2H), 7.74 (d, J=7.8 Hz, 1H), 7.52–7.55 (m, 1H), 7.45 (d, J=7.8 Hz, 1H), 7.36–7.39 (m, 2H), 7.29–7.34 (m, 4H), 7.25–7.27 (m, 1H), 5.57 (s, 2H, CH2). IR: 1,640.45 (C=O), 1,598.36 (C=N). ESI-MS m/z: 340.3 (M+H)+.
(E)-2-Chloro-3-[2-(2-fluorophenyl) ethenyl]-quinoxalin (D1)
Yellow powder, 76.4% yield, mp 154.0°C–155.4°C. 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.24 (d, J=16.2 Hz, 1H, ethenyl H), 8.16 (d, J=7.8 Hz, 1H), 7.97–7.99 (m, 1H), 7.85 (d, J=15.6 Hz, 1H, ethenyl H), 7.72–7.78 (m, 3H), 7.34–7.37 (m, 1H), 7.21–7.23 (m, 1H), 7.13–7.17 (m, 1H). IR: 1,623.68 (C=N). ESI-MS m/z: 285.1 (M+H)+.
(E)-2-(Morpholine-1-yl)-3-[2-(2-fluorophenyl) ethenyl]-quinoxalin (D2)
Yellow powder, 82.1% yield, mp 157.1°C–159.4°C. 1H NMR (600 MHz, CDCl3-d1) δ (ppm): 8.07 (d, J=16.2 Hz, 1H), 8.00 (d, J=7.8 Hz, 1H), 7.84 (d, J=8.4 Hz, 1H), 7.60–7.66 (m, 2H), 7.53–7.57 (m, 2H), 7.30–7.34 (m, 1H), 7.19–7.21 (m, 1H), 7.13–7.16 (m, 1H), 3.95–3.96 (m, 4H, CH2×2), 3.45–3.47 (m, 4H, CH2×2). IR: 1,628.07 (C=N). ESI-MS m/z: 336.4 (M+H)+.
(E)-2-(4-Methylpiperazin-1-yl)-3-[2-(2-fluorophenyl) ethenyl]-quinoxalin (D3)
Yellow powder, 62.3% yield, mp 196.3°C–198.5°C. 1H NMR (600 MHz, CDCl3-d1) δ (ppm): 8.06 (d, J=16.2 Hz, 1H), 7.93 (d, J=7.2 Hz, 1H), 7.82 (d, J=7.2 Hz, 1H), 7.65–7.67 (m, 1H), 7.58–7.61 (m, 1H), 7.51–7.55 (m, 2H), 7.30–7.33 (m, 1H), 7.19–7.21 (m, 1H), 7.12–7.15 (m, 1H), 3.51 (s, 4H, CH2×2), 2.70 (s, 4H, CH2×2), 2.43 (s, 3H, CH3). IR: 1,628.03 (C=N). ESI-MS m/z: 349.4 (M+H)+.
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.