")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Design, Synthesis, and Biological Evaluation of 2-Anilino-4-Triazolpyrimidine Derivatives as CDK4/HDACs Inhibitors
Authors Wang S, Han S, Cheng W, Miao R, Li S , Tian X, Kan Q
Received 1 December 2021
Accepted for publication 29 March 2022
Published 11 April 2022 Volume 2022:16 Pages 1083—1097
DOI https://doi.org/10.2147/DDDT.S351049
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Georgios Panos
Suhua Wang,1,2 Siyuan Han,1,2 Weiyan Cheng,1,2 Ruoyang Miao,1,2 Shasha Li,1,2 Xin Tian,1,2 Quancheng Kan1,2
1Department of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, 450052, People’s Republic of China; 2Henan Key Laboratory of Precision Clinical Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, 450052, People’s Republic of China
Correspondence: Xin Tian; Quancheng Kan, Email [email protected]; [email protected]
Purpose: To enhance the cytotoxicities of our obtained CDK4 inhibitors and get CDK4/HDACs inhibitors with potent enzymatic inhibitory and anti-proliferative activities.
Methods: A series of novel CDK4/HDACs inhibitors were designed and synthesized by incorporating the HDAC pharmacophores (hydroxylamine or o-diaminoaniline) into the basic structure of our newly obtained 2-anilino-4-triazolpyrimidine based CDK4 inhibitors. The enzymatic inhibitory (HDAC1, CDK2, CDK4, and CDK6) activities and cytotoxicities of these compounds were evaluated. Moreover, HDAC isoforms inhibitory activity, cell cycle arrest assay, cell apoptosis analysis, cell migration, and cell colony formation assay were performed for the representative compound 11k.
Results: Most of these compounds showed excellent HDAC1 inhibitory activities (IC50s: 0.68∼ 244.5 nM) and anti-proliferative activities against cancer cell lines. Some compounds displayed potent CDK4 inhibitory activities and a certain selectivity towards CDK2 and CDK6. Compound 11k exhibited potent enzymatic (CDK4: IC50=23.59 nM; HDAC1: IC50=61.11 nM; HDAC2: IC50=80.62 nM; HDAC6: IC50=45.33 nM) and anti-proliferative activities against H460, MDA-MB-468, HCT116, and HepG2 cell lines with IC50 values 1.20, 1.34, 2.07, and 2.66 μM, respectively. Further mechanistic studies revealed that compound 11k could arrest the cell cycle in G0/G1 phase and induce apoptosis in HCT116 and MDA-MB-468 cells. In addition, compound 11k significantly inhibited the migration and cell colony formation of H460 and HCT116 cells.
Conclusion: This study suggested that the incorporation of the HDAC pharmacophore into CDK4 inhibitor scaffold to design CDK/HDAC inhibitors might be a tractable strategy to enhance the antitumor potency of compounds.
Keywords: CDK4, HDAC, inhibitor, 2-anilino-4-triazolpyrimidine, cancer
Introduction
The cyclin-dependent protein kinases (CDKs) are protein-serine/threonine kinases that display crucial effects in cell cycle and transcription regulation.1,2 At present, 21 members of the CDK family have been found, including CDK1-20.3 The over-expression of CDKs is closely related to the development of tumors. For example, the up-regulation of CDK2, CDK4, CDK6, CDK8, and CDK11 is positively correlated with the occurrence and deterioration of breast cancer,4 rectal cancer,5 esophageal cancer,6 and so on. CDKs have been considered as promising targets for the treatment of cancers. Many small molecule CDK inhibitors have been explored for the therapy of cancers,7 and Pfizer’s CDK4/6 inhibitor palbociclib (Figure 1)8 was the first CDK4/6 inhibitor approved in 2015 for the treatment of HR-positive and HER2-negative breast cancer. In 2017, Novartis’ ribociclib (Figure 1)9 and Lilly’s abemaciclib (Figure 1)10 were approved by the US Food and Drug Administration (FDA) for the therapy of certain breast cancers. In 2021, G1 Therapeutics’ trilaciclib (Figure 1)11 was approved, and this drug expanded the cancer treatment scope of the CDK4/6 inhibitor to small cell lung cancers. In addition to CDK4/6 inhibitors, many other CDK family inhibitors were advanced to clinic studies including the CDK2 inhibitor milciclib (Figure 1).12 Histone deacetylase (HDAC) is a catalytic enzyme in histone, which catalyzes the removal of acetyl from lysine.13,14 HDACs comprise a family of 18 genes in humans and are divided into four classes. Among them, the Class I (HDAC1, HDAC2, HDAC3, and HDAC8), II (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10), and IV (HDAC11) are considered “classical” HDACs which have a zinc-dependent active site.15 The over-expression of HDAC was observed in many tumors, so HDAC inhibition has been developed as one of the most efficient strategies for cancer treatment.16 Up to date, five HDAC inhibitors (vorinostat, romidepsin, belinostat, panobinostat, and chidamide, Figure 1) have been approved by FDA or China National Medical Products Administration (NMPA) for the therapy of cutaneous/peripheral T-cell lymphoma, multiple myeloma, and adult T-cell leukemia-lymphoma.17–21
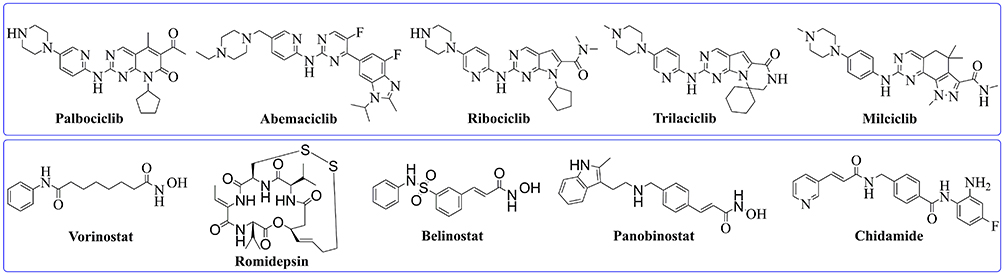 |
Figure 1 Representative structures of CDK inhibitors (above rectangle) and HDAC inhibitors (below rectangle). |
Dual (multiple)-target therapies have attracted much attention from medicinal chemistry researchers. The combinational suppression of two or more different pathways involved in disease progression often causes synergistic or additive effects and can reduce the potential of drug resistance development. In addition, dual (multiple)-target therapies show high efficacy and can reduce the therapeutic doses and side-effects when comparing with single target drug therapy.22 The introduction of HDAC inhibitory pharmacophores (hydroxylamine or o-diaminoaniline) into the structure of inhibitor for other target to develop HDAC-based dual or multiple inhibitors is a promising drug design strategy, and a large number of inhibitors are explored according to this way.14 Among these molecules, the HDAC/CDK dual (multiple) inhibitors have shown promising enzymatic inhibitory and anti-proliferative activities.23–25
In the initial exploration of CDK inhibitors, we designed and synthesized a series of 2-anilino-4-triazolpyrimidine derivatives with excellent CDK4 inhibitory activity yet poor anti-proliferative activity. In order to enhance the cytotoxicity, we introduce the HDAC inhibitory pharmacophores into the structures of these molecules to design a series of CDK4/HDACs inhibitors. The structure–activity relationship and the mechanism of action of these compounds were studied herein.
Materials and Methods
Chemistry
General Methods/Instruments
1H NMR were recorded on a BRUKER AVIII 500 MHz or AVII 400 MHz and 13C NMR were recorded on a BRUKER AVII 100 MHz spectrometer. Proton chemical shifts are expressed in parts per million (ppm) and coupling constants in Hz. Mass spectra were performed on a Finnigan LCQ DecaXP ion trap mass spectrometry or AB Sciex TripleTOF 6600 mass spectrometry. These data of related compounds are present in the Supporting Information (II. Chemistry).
General Synthetic Procedure of Trimethylsilylethynylpyrimidine Derivatives (2a, 2b)
To the solution of Pd (PPh3)2Cl2 (0.047 g, 0.067 mmol) and PPh3 (0.034g, 0.13 mmol) in a mixed solvent of THF (10 mL) and Et3N (15 mL), 2,4-dichloropyrimidine derivatives (1a or 1b, 13.4 mmol) were added. After replacing air with nitrogen by reducing pressure, CuI (0.026 g, 0.13 mmol) and trimethylsilylacetylene (1.579 g, 16.1 mmol) were added sequentially. The reaction mixture was heated at reflux temperature and cooled to room temperature (r.t.). The white precipitate (Et3N.HCl) was filtered off and the filtrate solution was concentrated and the residue was purified by silica gel column chromatography to get pure intermediate 2a or 2b.
General Synthetic Procedure of Ethynylpyrimidine Derivatives (3a, 3b)
To the solution of ethynylpyrimidine derivatives (2a or 2b, 4.7 mmol) in MeOH (10 mL), a solution of KOH (0.003 g, 0.05 mmol) in MeOH (5 mL) was added. After reacting for about 0.5 h at r.t., the mixture was concentrated and the residue was purified by silica gel column chromatography to get pure intermediate 3a or 3b.
General Synthetic Procedure of 2-Chloro-4-(1,2,3-Triazol-4-Yl) pyrimidine Derivatives (4a~4f)
To the solution of 2-chloro-4-ethynylpyrimidine derivative (3a or 3b, 1.28 mmol) and alkyl azide (1.53 mmol) in a mixed solvent of THF (5 mL), and H2O (5 mL), a solution of CuSO4.5H2O (65 mg, 0.26 mmol) and sodium ascorbate (103 mg, 0.52 mmol) in H2O (5 mL) was added. The reaction mixture was stirred at r.t. for about 1 hour. After completion, the reaction mixture was extracted with EtOAc (3×20 mL). The organic layer was dried over Na2SO4 and the excess solvent was removed under reduced pressure. Crude product was purified by column chromatography to get the pure compound 4a~4f.
General Synthetic Procedure of Compounds 6a~6d, 10a~10k, and 13a~13c
To the solution of halide substrate (0.30 mmol), aryl amine (0.33 mmol) in dioxane (10 mL), Pd(dba)2 (0.03 mmol), Xantphos (0.01 mmol), and Cs2CO3 (0.60 mmol) were added. The air in the reaction system is replaced with N2 for 3 times by decompression, and the reaction mixture was heated at 110°C for 2 hours and cooled to r.t. The solvent was removed under reduced pressure, and the residue was dissolved in CH2Cl2, washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum to afford crude compounds, which were purified by column chromatography to get the pure 6a~6d, 10a~10k, and 13a~13c.
General Synthetic Procedure of Compounds 8a and 8b
Cs2CO3 (1.89 g, 5.8 mmol) was added to the solution of nitrobenzene derivative (7a or 7b, 10.5 mmol) and methyl 7-bromoheptanoate (2.50 g, 11.2 mmol) in DMF (15 mL), the reaction mixture was stirred at 90°C for 3 hours and cooled to r.t. The solvent was removed under reduced pressure, and the residue was dissolved in CH2Cl2, washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum to afford crude compounds, which were purified by column chromatography to get the pure 8a or 8b.
General Synthetic Procedure of Compounds 9a and 9b
A solution of intermediate 8a or 8b (1.0 g) in MeOH (50 mL) was treated with H2 (1.0 bar, r.t.) in the presence of 10% palladium on charcoal (0.10 g). After completion (5 h), the catalyst was removed by filtration, and the solvent was evaporated to obtain the aniline 9a or 9b as yellow solid quantitatively.
General Synthetic Procedure of Target Compounds 11a~11k
To the solution of NH2OH·HCl (2.34 g, 34.3 mmol) in MeOH (12 mL), a solution of KOH (2.8 g, 50.9 mmol) in MeOH (7 mL) was dropwise added. After filtering the precipitate, a solution of NH2OK mixed with NH2OH was obtained. Compounds 10a~10k (0.5 mmol) were dissolved in the above solution (5 mL) and stirred overnight. After completion, the solvent was evaporated under vacuum, and the residue was acidified with 1 N HCl to a pH 3~4 and then extracted with CH2Cl2. The organic layer was washed with brine and dried over Na2SO4 and then evaporated, and the residue was purified by column chromatography to afford compounds 11a~11k.
General Synthetic Procedure of Compounds 14a~14c
Methyl benzoate derivative (13a, 13b, or 13c, 250 mg) was added to the solution of NaOH (50 mg) in MeOH (6 mL) and H2O (0.5 mL), the reaction mixture was stirred at 60°C for 5 hours and cooled to r.t. 2N HCl was added to adjust the pH value to 7.0 and then concentrated to dryness to afford crude compound, which was directly progressed to the next step without further purification.
General Synthetic Procedure of Compounds 15a~15c
2-(7-Azabenzotriazol-1-yl)-N, N, N’, N’-tetramethyluronium hexafluorophosphate (HATU, 322 mg, 0.85 mmol) and N, N-diisopropylethylamine (DIEA, 220 mg, 0.90 mmol) were added to a stirred solution of acid derivative (14a, 14b, or 14c, 0.77 mmol) and tert-butyl (2-aminophenyl) carbamate (161 mg, 0.77 mmol) in anhydrous DMF (5 mL). The reaction mixture was stirred overnight at r.t. After the reaction was completed, the reaction mixture had added DCM (20 mL) and was washed with saturated salt water (6×10 mL), the organic solvent was dried over sodium sulfate and concentrated, and the obtained crude product was further purified by column chromatography to get pure compound (15a, 15b, or 15c).
General Synthetic Procedure of Compounds 16a~16c
TFA (1 mL) was added dropwise to a stirred solution of tert-butyl carbamate derivative (15a, 15b, or 15c, 150 mg) in DCM (5 mL). The reaction mixture was stirred at r.t. for about 1 hour. After the reaction was completed, the reaction mixture was concentrated and the residue was purified by column chromatography to get the pure compound (16a, 16b, or 16c).
Cell Lines and Treatment
Human liver cancer cell line HepG2, breast cancer cell line MDA-MB-468, colorectal cancer cell HCT116, and lung cancer cell line H460 were all purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in high-glucose DMEM or RPMI-1640 medium (ThermoFisher, USA) supplemented with 10% fetal bovine serum (FBS, Invigentech, USA), 50 μg/mL streptomycin and 50 U/mL penicillin (Solarbio Science & Technology Co. Ltd, Beijing, China) at 37°C in a 5% CO2 humidified atmosphere.
Materials and Reagents
Palbociclib (S80974, purity 99%) was purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). CDK2/CycA2 (04–102), CDK4/CyclinD3 (04–105), and CDK6/CyclinD3 (04–107) were all purchased from Carna Biosciences, Inc. (Kobe, Japanese). Streptavidin-XL665 was obtained from Cisbio Bioassays (France). Annexin V-FITC/PI Apoptosis Kit was purchased from KeyGEN BioTECH (Nanjing, China). Cell cycle detection kit was obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Dimethyl sulfoxide (DMSO) was purchased from Beijing Solarbio Science & Technology Co. Ltd. (Beijing, China). Sulforhodamine B (SRB), Trichloroacetic acid (TCA), and Trizma® base were all obtained from Sigma-Aldrich (St. Louis, MO).
Evaluation of HDACs Inhibitory Activity
The in vitro HDACs inhibitory assay of all compounds was performed by Sundia MediTech Company, Ltd. (Shanghai, China). It was conducted by fluorescent assay. Briefly, all compounds were serially diluted to certain concentrations. Then, the HDAC1 enzymes (Active Motif, 31504), HDAC2 (BPS bioscience, 50002), HDAC5 (BPS bioscience, 50005), HDAC6 (BPS bioscience, 50006), HDAC8 (Active Motif, 31566), HDAC11 (BPS bioscience, 50011), and compounds solution were dissolved in 1×Assay buffer, which was then added into a 384-well microplate. It was mixed briefly with gentle centrifuging and the plate was incubated at r.t. Substrate peptide and Trypsin mixture were added to each well, shaken for 1 minute, and the 384-plate was placed in a BioTek Synergy plate reader for data collection.
CDKs Kinase Inhibitory Assay
CDKs kinase inhibitory assay were conducted using homogenous time resolved fluorescence (HTRF) assay. According to the assay protocol, various concentrations of tested compounds, CDKs kinase and their substrates were incubated in 1×Kinase buffer for 10 minutes. ATP was then added into the reaction mixture in a 384-well plate to start the enzymatic reaction. After incubated for 60–90 minutes at r.t., the reaction was blocked by addition of 1×Detection buffer with Streptavidin-XL665. Then the plate was placed in a PerkinElmer Envision plate reader for data collection, the IC50 values for inhibition were determined by GraphPad Prism.
SRB Assay for Cell Viability
The anti-proliferation assay was determined by SRB assay. Briefly, cells were seeded in 96-well plates and incubated overnight. Subsequently, cells were treated with the target compounds, palbociclib, vorinostat, and DMSO (1‰). After incubation at 37°C for 72 hours, 50 μL of ice-cold 50% TCA was added per well and fixed at 4°C for 1 hour, the plates were then washed with deionized water five times and air-dried. Next, fixed cells were stained with 0.4% SRB solution for 10 minutes at r.t., followed by washing with 1% acetic acid. Finally, bound SRB was dissolved with 10 mM Tris Base (150 μL/well), and the optical density at 515 nm was determined by a microplate reader. The IC50 values were calculated using GraphPad Prism 5.
Cell Cycle Analysis
HCT116 and MDA-MB-468 cells were inoculated in a 6-well plate and cultured for 12 hours. After being treated with 11k at different concentrations for 24 hours, cells were collected, fixed in 75% ethanol at −20°C for 1 hour, and then resuspended in RNase A for 30 minutes at 37°C, followed by being stained with propidium iodide (PI) for an additional 30 minutes. The stained cells were analyzed by flow cytometry (ACEA NovoCyte, Agilent Biosciences, Inc), and the percentage of cells in the G0/G1 and G2/M phases was assessed by ModFit software.
FITC Annexin V/PI Staining Assay for Cell Apoptosis
Analysis of cell apoptosis was conducted by flow cytometry. After treatment with 11k at different concentrations for 48 hours, MDA-MB-468 or HCT116 cells were harvested by trypsinization and resuspended in 500 μL 1×binding buffer, which was then added to 5 μL of PI and 5 μL of Annexin-V/FITC according to the manufacturer’s instructions. Cells were incubated for 15 minutes in the dark and analyzed within 1 hour by the ACEA NovoCyte flow cytometer (Agilent Biosciences, Inc). All experiments were performed at least three times.
Cell Migration Assay
MDA-MB-468 or HCT116 cells were plated into a 6-well plate and incubated overnight. After drawing a straight line with 10 μL pipette tips, cells were washed with PBS and exposed to 11k at the concentration of 2 μM and 5 μM, respectively. Vorinostat and palbociclib were used as positive control. Following incubation at 37°C, the cells were allowed to migrate. Images were taken at 24 hours and 48 hours, separately.
Colony Formation Assay
H460 or HCT116 cells were plated into a 6-well plate with 3,000 cells per well and incubated overnight. Subsequently, cells were treated with 11k, vorinostat and palbociclib. Following incubation for 7–10 days, cells were fixed and stained with 0.1% crystal violet.
Statistical Analysis
Data are presented as mean±standard deviation (SD). Statistical significance between multiple groups was analyzed by one-way analysis of variance with Dunnett’s test using GraphPad Prism 8.0 software. A value of p<0.05 was considered statistically significant.
Results and Discussion
Design of CDK4/HDACs Inhibitors
Many CDK inhibitors contained the scaffold of N-(pyridin-2-yl) pyrimidin-2-amine or N-phenylpyrimidin-2-amine (Figure 2).26 In order to get novel CDK inhibitors to overcome resistance and expand clinic indications, we introduced the substituted triazol groups to the 4-pyrimidine position of the N-phenylpyrimidin-2-amine to design a series of 2-anilino-4-triazolpyrimidine derivatives (6a~6d, Figure 2). In the in vitro activity assay, these compounds showed excellent CDK4 inhibitory activities (IC50s: 8.50~62.04 nM), while palbociclib had an IC50 of 11.03 nM. However, the anti-proliferative activities of these compounds were not ideal when testing against human lung carcinoma cell H460 and human breast cancer cell MDA-MB-468 (Table 1).
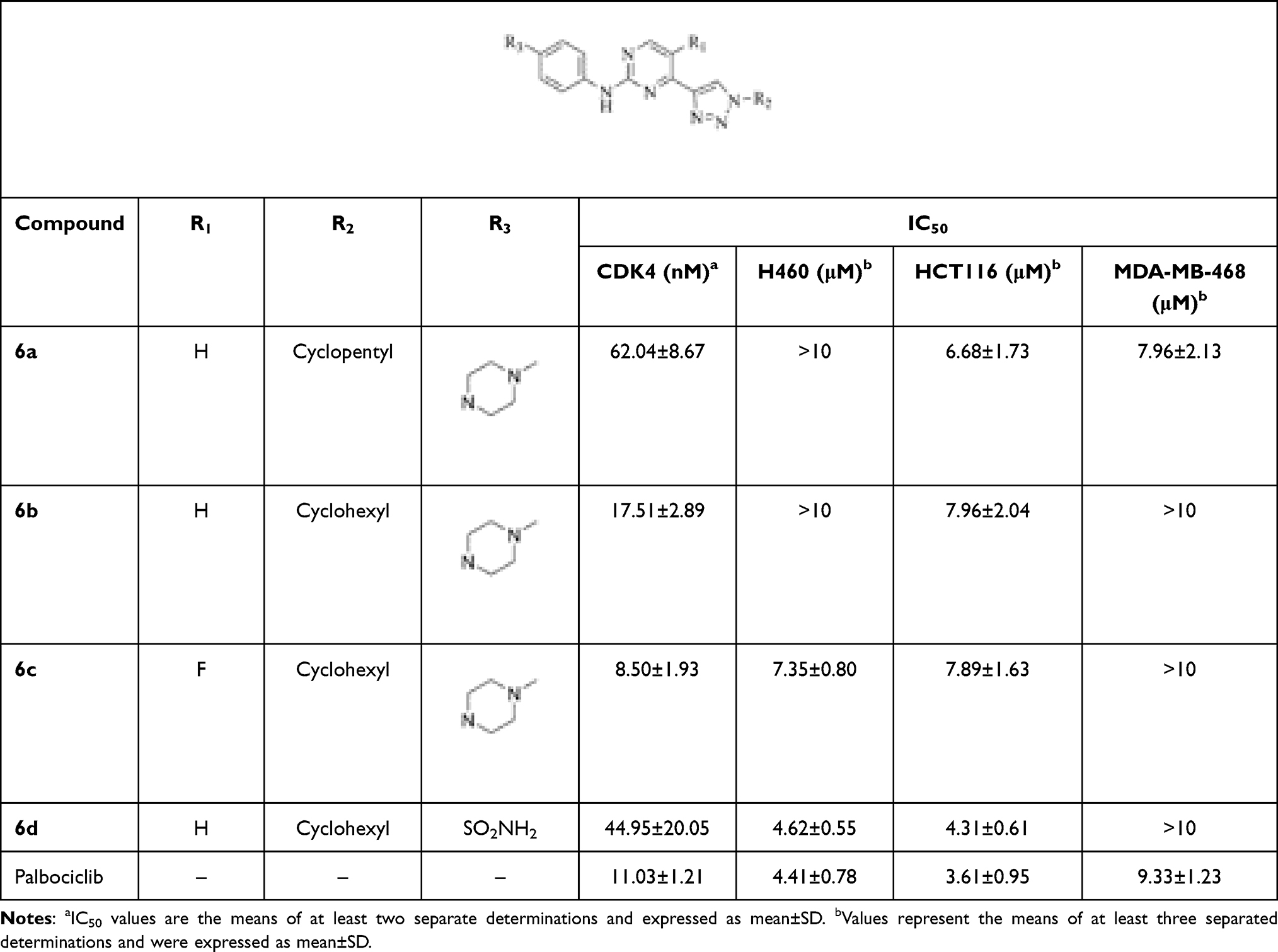 |
Table 1 CDK4 Inhibitory and Anti-Proliferative Activities of Compounds 6a~6d |
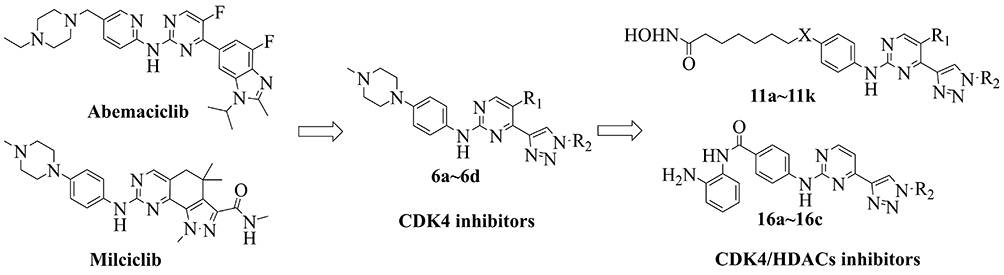 |
Figure 2 Design of CDK4 inhibitors and CDK4/HDACs inhibitors. |
It is reported that HDAC inhibitors showed potent cytotoxicities27 and the introduction of HDAC inhibitory pharmacophores effected minor to the inhibitors due to the low molecular weight.24 It has also been confirmed that the substitutes in the para position of aniline group for CDK inhibitors was injected in the solvent portion of targets. So we introduce the HDAC inhibitory pharmacophores (hydroxylamine or o-diaminoaniline) to the para-aniline position of the structures of 6a~6d to design a series of CDK4/HDACs inhibitors (Figure 2).
Chemistry
The synthetic routes to intermediates 4a~4f and target compounds 6a~6d are displayed in Scheme 1. Coupling of dichloropyrimidine derivative (1a or 1b) with ethynyltrimethylsilane gave intermediate 2a or 2b, subsequent removal of the TMS group led to the ethynylpyrimidine derivative 3a or 3b. The latter underwent click reaction with alkyl azides to afford the triazolpyrimidine derivatives 4a~4f. The target compounds 6a~6d were obtained through condensing 4a~4c with aniline 5a or 5b, respectively. The new compounds were characterized by the NMR and MS (or HRMS) spectrum, the purity of the target compounds was >95%.
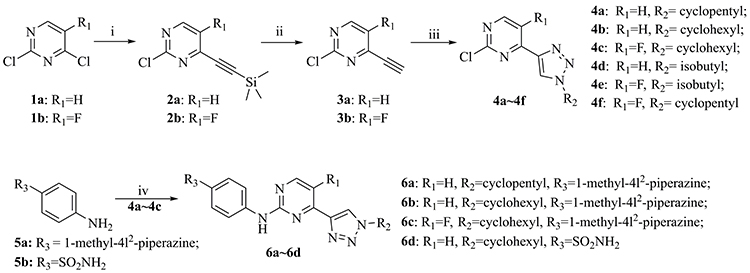 |
Scheme 1 Reagents and conditions: i) TMSA, PdCl2(PPh3)2, PPh3, CuI, THF, Et3N, reflux, 57%~62%; ii) KOH, MeOH, 90%~91%; iii) Alkyl azides, CuSO4.5H2O, sodium ascorbate, THF, H2O, r.t., 46%~81%; and iv) Pd2(dba)3, Cs2CO3, Xantphos, dioxane, reflux, 42%~67%. |
 |
Scheme 2 i) Methyl 7-bromoheptanoate, Cs2CO3, DMF, 90°C, 65%~76%; ii) H2, Pd/C, MeOH, r.t., 100%; iii) Pd2(dba)3, Cs2CO3, Xantphos, dioxane, reflux, 57%~73%; iv) NH2OH, KOH, MeOH, r.t., 74%~91%. |
The synthesis of the target compounds 11a~11k is shown in Scheme 2. Nucleophilic substitution reaction of nitrobenzene derivative 7a or 7b with methyl 7-bromoheptanoate afforded methyl heptanoate 8a or 8b, which was reduced by H2 to yield aniline 9a or 9b. Afterwards, the aniline (9a or 9b) was subjected to a Buchwald-Hartwig Cross Coupling Reaction with 4a~4f to furnish intermediate 10a~10k. Following reaction of 10a~10k with NH2OH got the target compounds 11a–11k. The new compounds were characterized by NMR and MS (or HRMS) spectrum, the purity of the target compounds was >95%.
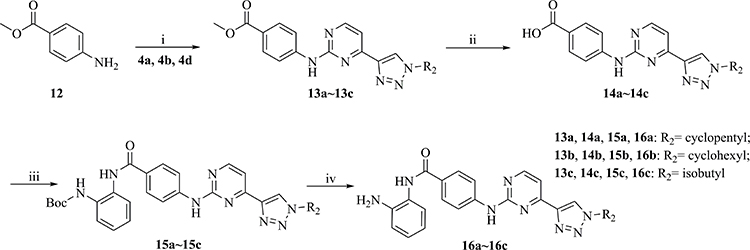 |
Scheme 3 i) Pd2(dba)3, Cs2CO3, Xantphos, dioxane, reflux, 80%~84%; ii) NaOH, MeOH, H2O, 60°C; iii) tert-Butyl (2-aminophenyl) carbamate, HATU, DIEA, DMF, r.t., 71%~74%; iv) TFA, DCM, r.t., 100%. |
The synthesis of the target compounds 16a~16c is shown in Scheme 3. Similar to 6a~6d, intermediates 13a~13c were synthesized by coupling of 4a, 4b, or 4d with aniline 12. Following hydrolytic acidification, the obtained 14a~14c condensed with tert-butyl (2-aminophenyl) carbamate to afford 15a~15c. Deprotection of the Boc groups in 15a~15c yielded compounds 16a~16c. The new compounds were characterized by NMR and MS (or HRMS) spectrum, the purity of the target compounds was >95%.
Enzymatic Inhibitory and Anti-Proliferative Activities of Compounds 11a~11k and 16a~16c
As HDAC1 based dual (multiple) inhibitors have been confirmed to possess synergistic or additive antitumor effects,25 the HDAC1 inhibitory activities of compounds 11a~11k and 16a~16c were first evaluated using a well-established fluorescent assay and vorinostat was employed as the positive control. As shown in Table 2, all the tested compounds exhibited potent HDAC1 inhibitory activities. Compounds with hydroxylamine groups (11a~11k, IC50s: 0.68~88.0 nM) showed more potent inhibitory activities than those with o-diaminoaniline groups (16a~16c, IC50s: 110.40~244.50 nM). The X group between the pharmacophores of HDAC1 and CDK impacted HDAC1 inhibition obviously, the O atom (11a~11f) was more beneficial than piperazin (11g~11k) to HDAC1 inhibitory activity.
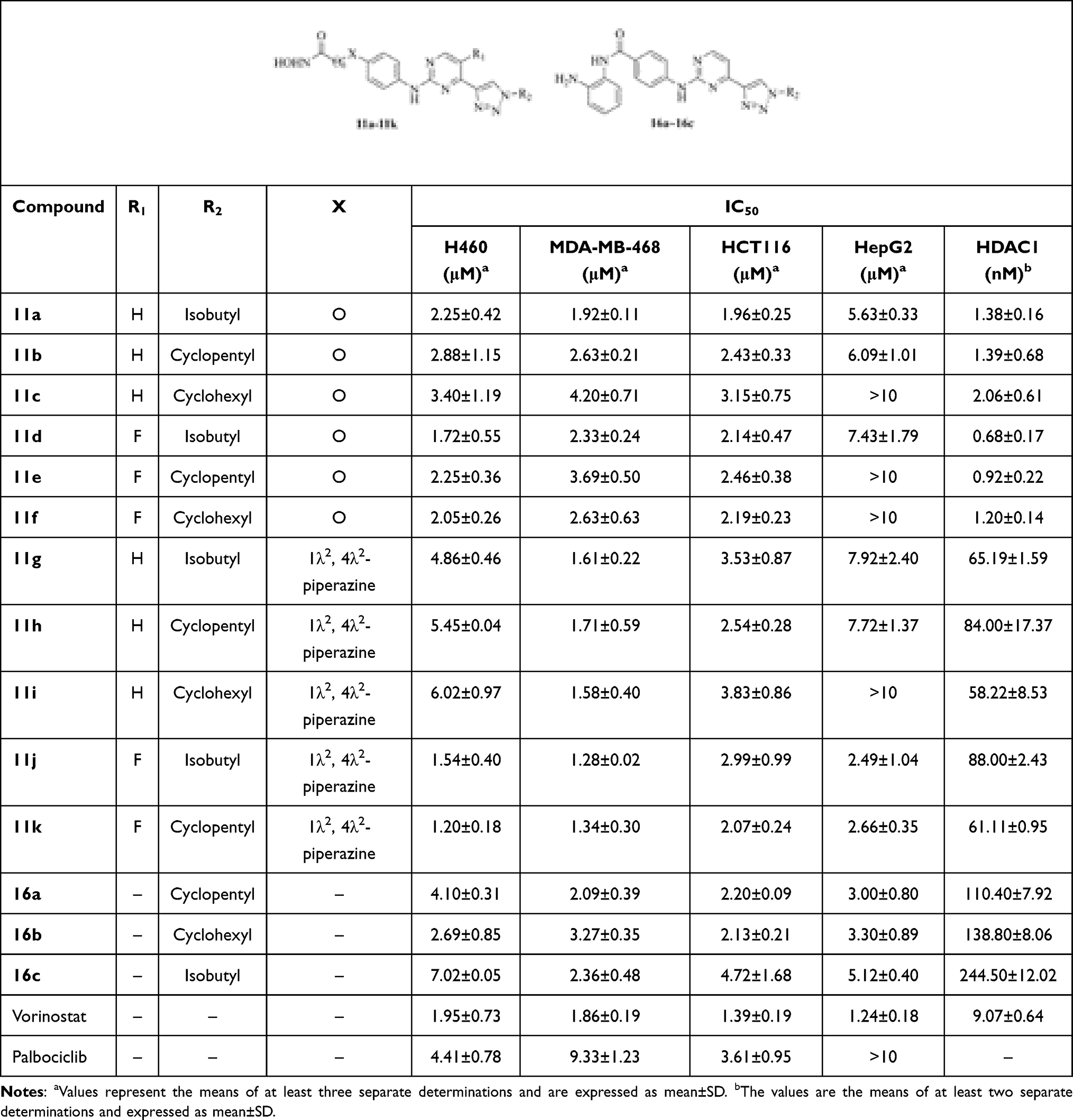 |
Table 2 The Anti-Proliferative and HDAC1 Inhibitory Activities of Compounds 11a~11k and 16a~16c |
Compared with compounds 6a~6d, the anti-proliferative activities of the new compounds were greatly enhanced. Most of these compounds exhibited IC50 values lower than 5.0 μM (Table 2) when testing against H460, MDA-MB-468, HCT116, and HepG2 cells. Especially, compound 11k showed IC50 values among 1.20~2.66 μM against these cell lines, which was better than the positive control palbociclib and comparable with vorinostat. These results suggested that the introduction of HDAC pharmacophores indeed enhanced the cytotoxicities of new compounds. In the primary stage of drug development, the main purpose is finding compounds with excellent anti-proliferative activities. Herein, the target compounds’ anti-proliferative activities are not so great, so their anti-proliferative activities against normal cell lines were not yet evaluated, and this will be investigated in subsequent studies when compounds with extraordinary cytotoxicities were found.
The CDK2, CDK4, and CDK6 inhibitory activities of these compounds were preliminarily evaluated in the concentrations of 5 μM and 50 nM, respectively (Table 3). In summary, the CDK4 inhibition of these compounds was better than CDK6, and the CDK2 inhibition was poor. Compounds 11a, 11d, and 11g~11k displayed 50% or higher inhibition for CDK2, CDK4, or CDK6 in the concentration of 5 μM. Especially, compounds 11g~11k showed 28%~73% inhibition for CDK4 in the concentration of 50 nM, which indicated that the presence of the piperazin group (X group) was in favor of CDK4 inhibition. This showed a certain degree of conflict with HDAC1 inhibition, which reminded us to balance the inhibition between HDAC1 and CDK4 for further structure modification. The IC50 values of compounds 11d and 11g~11k were evaluated (Table 4) as well. Among them, compounds 11g~11k showed the IC50 values ranging from 23.59 to 150.55 nM against CDK4, and from 128.60 to 526.95 nM against CDK6, respectively. Compound 11k showed the IC50 value of 23.59 nM against CDK4, which exhibited a >5-fold selectively for CDK6 (IC50: 128.60 nM). Compound 11k showed promising enzymatic and cellular inhibitory activities, and it was selected for further study.
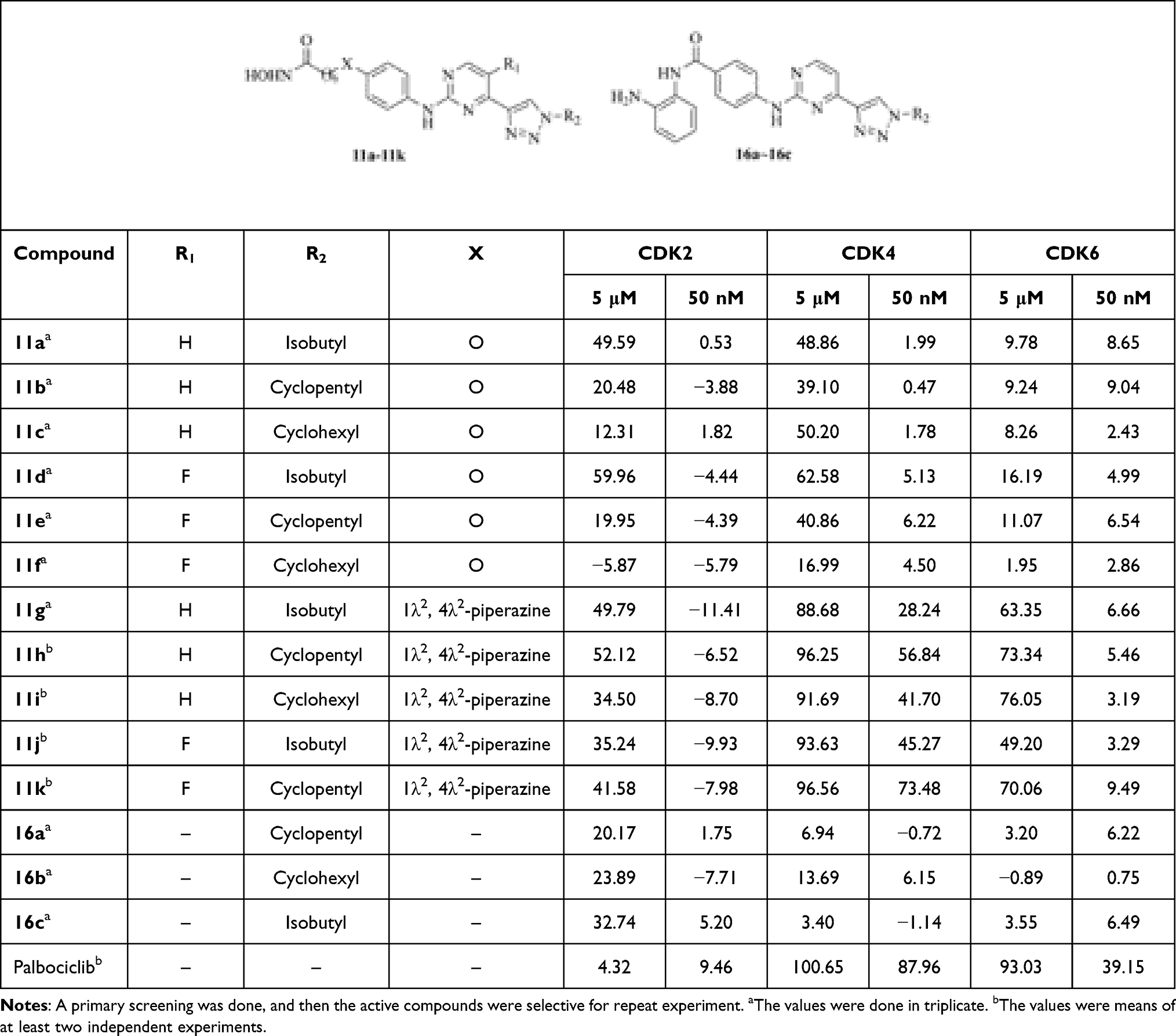 |
Table 3 Enzymatic Inhibitory Activities of Compounds 11a~11k and 16a~16c |
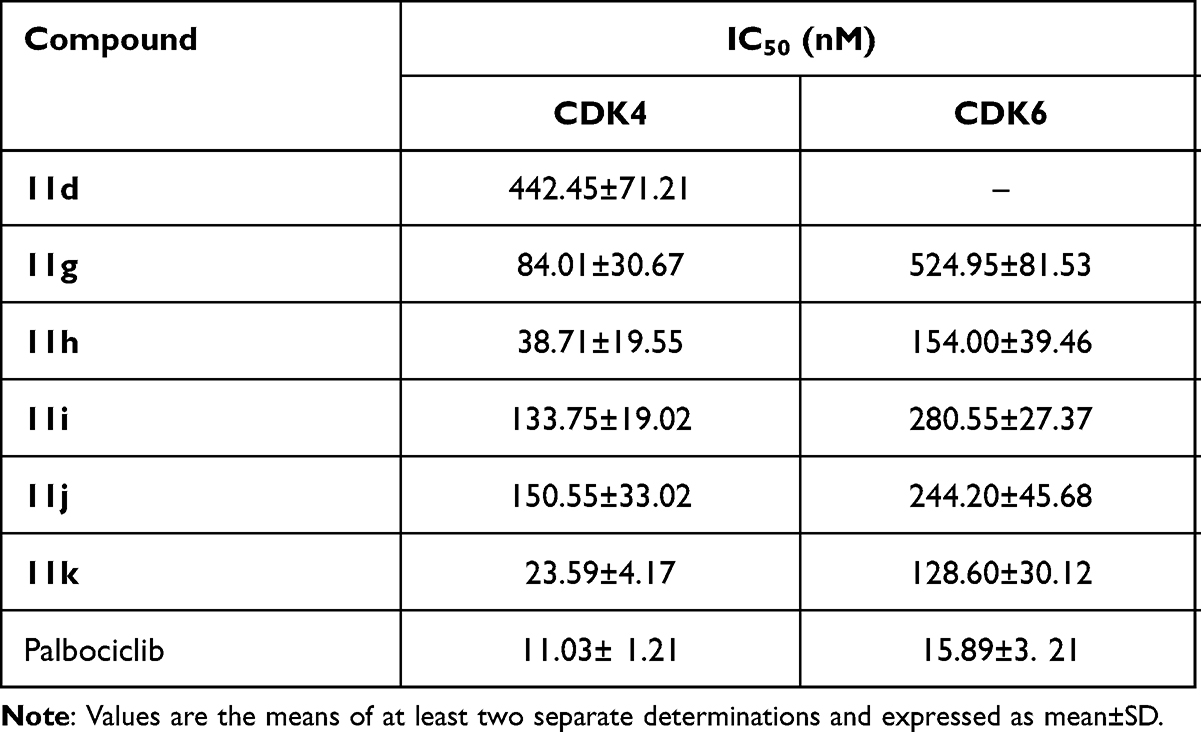 |
Table 4 CDK4/6 Inhibitory Activities of Representative Active Compounds |
HDAC Isoforms Inhibition Assay
To explore the inhibitory activity toward other HDAC isoforms, compound 11k was evaluated against HDAC1, HDAC2, HDAC5, HDAC6, HDAC8, and HDAC11 with vorinostat as the reference drug. As depicted in Table 5, besides HDAC1/2/6, compound 11k also displayed potent activity for HDAC8 with an IC50 value of 228.90 nM, showing a >5-fold selectivity in favor of HDAC8 versus HDAC6, while compound 11k displayed no significant HDAC5 and HDAC11 inhibition at 500 nM. With the exception of HDAC5 and HDAC11, compound 11k was less active than vorinostat against all other tested HDAC isoforms.
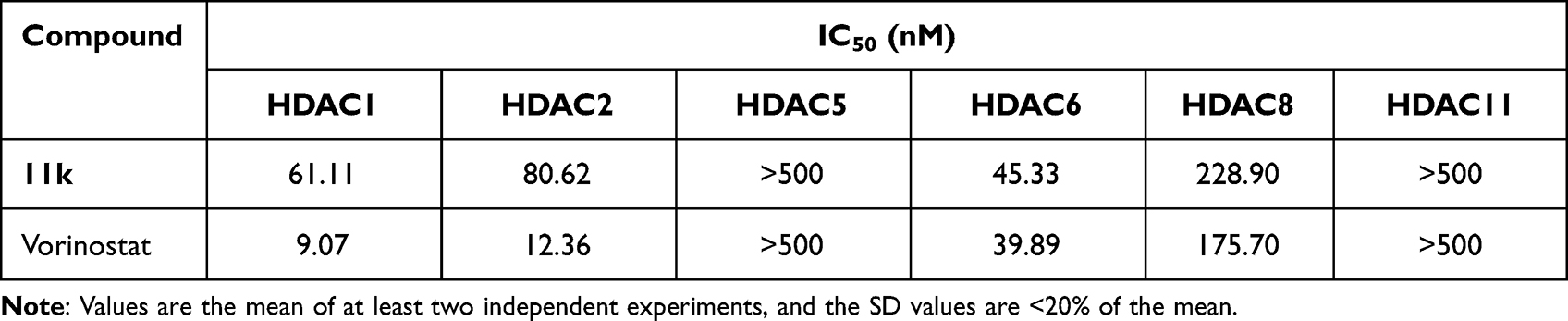 |
Table 5 Inhibition of HDAC Isoforms by Compound 11k |
Cell Cycle Arrest Assay of Compound 11k
11k was selected to test whether the kinase inhibitory effect of these compounds could partly cause cell cycle transition arrest. HCT116 and MDA-MB-468 cells were exposed to 11k at various concentrations, which were then stained with PI and analyzed by flow cytometry to determine the distribution of cells in phases of the cell cycle. Similar effects were observed in HCT116 and MDA-MB-468 cell lines (Figure 3). Cells with 11k-treatment demonstrated an increase of cells in G0/G1-phase and a large percentage of loss of cells in S-phase when compared with control (DMSO-treated cells). Similar to the 11k-treated group, palbociclib-treatment resulted in an accumulation of cells in G0/G1. 11k induced cell arrest in G0/G1 at low dose (0.5–2 μM), while at high dosage (5 μM), it induced an increase of cells in S-phase. CDK4 plays a key role in G1/S transition of cell cycle.28 It is expected that CDK4 inhibition blocks the cell cycle in G1 phase. Our results revealed that compound 11k showed the CDK4 inhibition activities and support the hypothesis of a blockage during G1 at lower doses. Thus, effects on G0/G1 arrest are consistent with the inhibition of CDK4. As a well-known HDACs inhibitor, vorinostat showed cells inhibition in G2-M phases.29 Compound 11k also induced cells arrest in G2/M phases at high concentration, which was similar to vorinostat. The results indicated that compound 11k has the inhibitory characteristic of CDK4 and HDACs and confirmed the anti-proliferative properties.
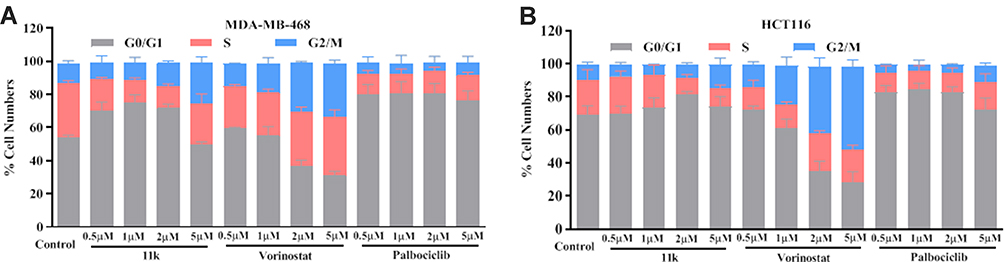 |
Figure 3 Cell cycle analysis of 11k on HCT116 and MDA-MB-468 cell lines. Notes: HCT116 (A) or MDA-MB-468 (B) cells were exposed to 11k at indicated concentrations (0, 0.5, 1, 2, and 5 μM) for 24 hours. Cells were stained by PI and analyzed by flow cytometry. Vorinostat and palbociclib were used as the positive control. Data represents the mean±standard deviation (SD) (n=3). |
Cell Apoptosis Analysis of Compound 11k
FITC-Annexin V/PI double staining was used to investigate whether compound 11k could inhibit cell growth through apoptosis. As illustrated in Figure 4A, compound 11k induced a dose-dependent increase in apoptosis of the MDA-MB-468 cell line, which was compared to vehicle-treated cells. Additionally, at the dosage of 5 μM, compound 11k induced more apoptosis in cells than the reference drug palbociclib. To further confirm the apoptosis induced by 11k, HCT116 and H460 were subjected to the apoptosis assay. Similar effects were observed in these two cell lines (Figure 4B and Supplementary Figure S1). However, the positive compound palbociclib did not produce a dose-dependent increase in apoptosis of H460. We speculated one possible reason the current experimental conditions were not so appropriate, increasing the dose or prolonging the induction time of palbociclib may get a better dose-dependent curve. Maybe there are other possible reasons here. As described in the article published by Li et al,25 at the dosage of 10 μM, vorinostat induces cell apoptosis of about 10% in H460 cells, while in our experiment, vorinostat caused about 20% percent apoptosis of H460 cells at 5 μM, but in HCT116 and MDA-MB-468 cell lines, the proportion of apoptosis is 64.72% and 49.19%, respectively. So it is speculated that H460 is not so sensitive to apoptosis inducer due to its own characteristics. All the above results are consistent with the effect on cell growth inhibition. Taken together, compound 11k could induce cell death via apoptosis.
 |
Figure 4 Effects of 11k, Vorinostat and palbociclib on the induction of cell apoptosis in MDA-MB-468 (A) and HCT116 (B) cell lines. Notes: Cells were treated to 11k, vorinostat and palbociclib for 48 hours and analyzed by flow cytometry with Annexin-V/FITC /PI staining. The percentage of apoptosis cells was defined as the sum of early- and late-stage apoptosis. The data represent at least three separate experiments. The Dunnett’s test was used for multi-group comparisons, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 vs the control group. |
Compound 11k Inhibited Cell Migration
To investigate the migration inhibitory property of compound 11k, HCT116 and MDA-MB-468 were subjected to cell scratch assay. The results are depicted in Figure 5. Compared to the control, 11k inhibited the migration of HCT116 and MDA-MB-468 cells in a dose-dependent manner, which was equivalent to the inhibitory activity of positive drugs vorinostat and palbociclib.
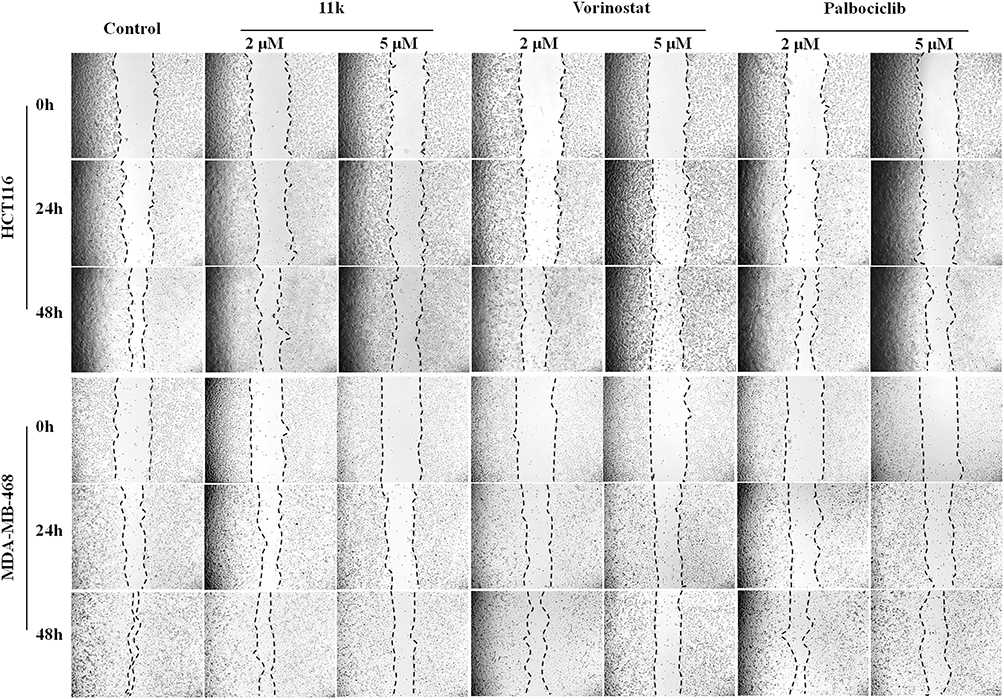 |
Figure 5 Effects of 11k on cell migration was determined by the scratch test. Notes: HCT116 and MDA-MB-468 cells were treated with 11k, vorinostat and palbociclib at 2 μM and 5 μM. Images were taken at 0, 24, and 48 hours, separately. One representative of two independent experiments was shown. Cells exposed to vehicle (DMSO) were used as controls. |
Cell Colony Formation Assay of Compound 11k
Cell colony formation assay was conducted to investigate the effect of compound 11k on cell clonogenicity. The results are provided in Figure 6. The colony formation capability of HCT116 and H460 cells was noticeably inhibited after compound 11k treatment compared to control group. Upon treatment of 11k at increased concentrations, the number of cell colony was gradually reduced. At the dosage of 2 μM, our compound 11k demonstrated a comparable inhibition of colony formation with reference drug palbociclib.
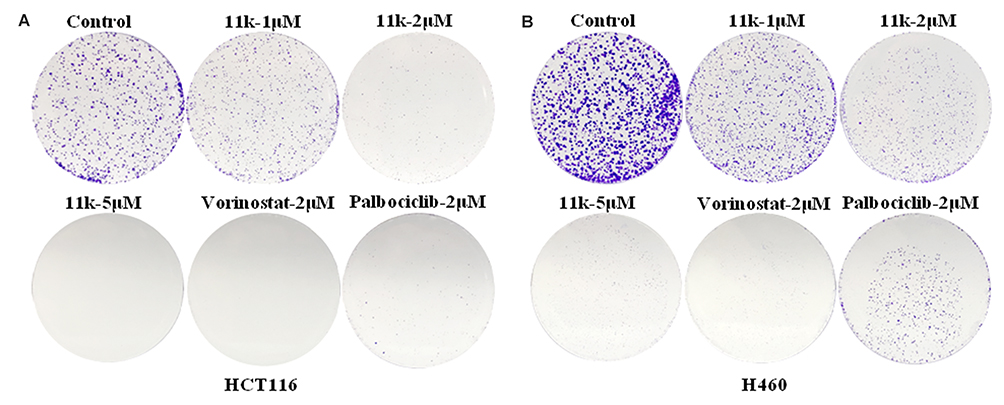 |
Figure 6 The effects of 11k on formation of clones in HCT116 (A) and H460 (B) cells. Note: At least two independent experiments were conducted. |
Conclusions
In order to get efficient anticancer agents, a series of novel CDK4/HDACs inhibitors were designed and synthesis based on our newly obtained 2-anilino-4-triazolpyrimidine scaffold of CDK4 inhibitors. Most of these compounds exhibited potent HDAC1 inhibitory activity and anti-proliferative activities. The CDKs inhibition evaluation confirmed that some compounds showed excellent CDK4 and inhibitory activities and certain selectivity toward CDK2 and CDK6. Compound 11k displayed significant anti-proliferative activities and enzymatic inhibition activities, the cell cycle assay, cell apoptosis analysis, cell migration, and cell colony formation assay further confirmed the mechanism action of 11k. Our results suggest that the incorporation of HDAC pharmacophore into CDK4 inhibitor scaffold might be a tractable strategy to enhance the antitumor potency of compounds.
Funding
We gratefully acknowledge the funding supports of grants from Major Science and Technology Project of Henan Province (161100311000), National Natural Science Foundation of China (31870809 and 32170594), and the Medical Science and Technology Project of Health Commission of Henan Province (2018020039).
Disclosure
The authors declare no potential conflicts of interest in this work.
References
1. Roskoski R
2. Zhang M, Zhang L, Hei R, et al. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res. 2021;11(5):1913–1935.
3. Cheng W, Yang Z, Wang S, et al. Recent development of CDK inhibitors: an overview of CDK/inhibitor co-crystal structures. Eur J Med Chem. 2019;164:615–639. doi:10.1016/j.ejmech.2019.01.003
4. Nie L, Wei Y, Zhang F, et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat Commun. 2019;10(1):5114. doi:10.1038/s41467-019-13105-5
5. Xu L, Cheng Z, Cui C, et al. Frequent genetic aberrations in the cell cycle related genes in mucosal melanoma indicate the potential for targeted therapy. J Transl Med. 2019;17(1):245. doi:10.1186/s12967-019-1987-z
6. Du Y, Yan D, Yuan Y, et al. CDK11(p110) plays a critical role in the tumorigenicity of esophageal squamous cell carcinoma cells and is a potential drug target. Cell Cycle. 2019;18(4):452–466. doi:10.1080/15384101.2019.1577665
7. Sanchez-Martinez C, Lallena MJ, Sanfeliciano SG, et al. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: recent advances (2015–2019). Bioorg Med Chem Lett. 2019;29(20):126637. doi:10.1016/j.bmcl.2019.126637
8. Braal CL, Jongbloed EM, Wilting SM, et al. Inhibiting CDK4/6 in breast cancer with palbociclib, ribociclib, and abemaciclib: similarities and differences. Drugs. 2021;81(3):317–331. doi:10.1007/s40265-020-01461-2
9. Syed YY. Ribociclib: first global approval. Drugs. 2017;77(7):799–807. doi:10.1007/s40265-017-0742-0
10. Johnston SRD, Harbeck N, Hegg R, et al. Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2-, node-positive, high-risk, early breast cancer (monarchE). J Clin Oncol. 2020;38(34):3987–3998. doi:10.1200/JCO.20.02514
11. Dhillon S. Trilaciclib: first approval. Drugs. 2021;81(7):867–874. doi:10.1007/s40265-021-01508-y
12. Aspeslagh S, Shailubhai K, Bahleda R, et al. Phase I dose-escalation study of milciclib in combination with gemcitabine in patients with refractory solid tumors. Cancer Chemother Pharmaco. 2017;79(6):1257–1265. doi:10.1007/s00280-017-3303-z
13. Ho TCS, Chan AHY, Ganesan A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J Med Chem. 2020;63(21):12460–12484. doi:10.1021/acs.jmedchem.0c00830
14. Duan YC, Zhang SJ, Shi XJ, et al. Research progress of dual inhibitors targeting crosstalk between histone epigenetic modulators for cancer therapy. Eur J Med Chem. 2021;222:113588. doi:10.1016/j.ejmech.2021.113588
15. Cai X, Zhai HX, Wang J, et al. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J Med Chem. 2010;53(5):2000–2009. doi:10.1021/jm901453q
16. McClure JJ, Li X, Chou CJ. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv Cancer Res. 2018;138:183–211.
17. Yang J, He J, Ismail M, et al. HDAC inhibition induces autophagy and mitochondrial biogenesis to maintain mitochondrial homeostasis during cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2019;130:36–48. doi:10.1016/j.yjmcc.2019.03.008
18. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30–39. doi:10.1172/JCI69738
19. Lee HZ, Kwitkowski VE, Del Valle PL, et al. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. 2015;21(12):2666–2670. doi:10.1158/1078-0432.CCR-14-3119
20. Laubach JP, Moreau P, San-Miguel JF, et al. Panobinostat for the treatment of multiple myeloma. Clin Cancer Res. 2015;21(21):4767–4773. doi:10.1158/1078-0432.CCR-15-0530
21. Lu X, Ning Z, Li Z, et al. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis Res. 2016;5(3):185–191. doi:10.5582/irdr.2016.01024
22. Liu T, Wan Y, Xiao Y, et al. Dual-target inhibitors based on HDACs: novel antitumor agents for cancer therapy. J Med Chem. 2020;63(17):8977–9002. doi:10.1021/acs.jmedchem.0c00491
23. Cheng C, Yun F, Ullah S, et al. Discovery of novel cyclin-dependent kinase (CDK) and histone deacetylase (HDAC) dual inhibitors with potent in vitro and in vivo anticancer activity. Eur J Med Chem. 2020;189:112073. doi:10.1016/j.ejmech.2020.112073
24. Cao Z, Yang F, Wang J, et al. Indirubin derivatives as dual inhibitors targeting cyclin-dependent kinase and histone deacetylase for treating cancer. J Med Chem. 2021;64(20):15280–15296. doi:10.1021/acs.jmedchem.1c01311
25. Li Y, Luo X, Guo Q, et al. Discovery of N1-(4-((7-cyclopentyl-6-(dimethylcarbamoyl)-7 H-pyrrolo[2,3-d]pyrimidin-2-yl)amino)phenyl)-N8-hydroxyoctanediamide as a novel inhibitor targeting cyclin-dependent kinase 4/9 (CDK4/9) and histone deacetlyase1 (HDAC1) against malignant cancer. J Med Chem. 2018;61(7):3166–3192. doi:10.1021/acs.jmedchem.8b00209
26. Goel B, Tripathi N, Bhardwaj N, et al. Small molecule CDK inhibitors for the therapeutic management of cancer. Curr Top Med Chem. 2020;20(17):1535–1563. doi:10.2174/1568026620666200516152756
27. Lin A, Giuliano CJ, Palladino A, et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med. 2019;11(509):eaaw8412. doi:10.1126/scitranslmed.aaw8412
28. Day PJ, Cleasby A, Tickle IJ, et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc Natl Acad Sci USA. 2009;106(11):4166–4170. doi:10.1073/pnas.0809645106
29. Munster PN, Troso-Sandoval T, Rosen N, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer. Cancer Res. 2001;61(23):8492–8497.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.