Back to Journals » Drug Design, Development and Therapy » Volume 16
Design, Synthesis and Biological Evaluation of 1,4-Benzenesulfonamide Derivatives as Glyoxalase I Inhibitors
Authors Audat SA ![]() , Al-Balas QA
, Al-Balas QA ![]() , Al-Oudat BA, Athamneh MJ, Bryant-Friedrich A
, Al-Oudat BA, Athamneh MJ, Bryant-Friedrich A
Received 26 January 2022
Accepted for publication 10 March 2022
Published 28 March 2022 Volume 2022:16 Pages 873—885
DOI https://doi.org/10.2147/DDDT.S356621
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Suaad Abdallah Audat,1 Qosay Ali Al-Balas,2 Buthina Abdallah Al-Oudat,2 Mo’ad Jamil Athamneh,1 Amanda Bryant-Friedrich3
1Department of Chemistry, College of Science and Arts, Jordan University of Science and Technology, Irbid, 22110, Jordan; 2Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, 22110, Jordan; 3Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, Wayne State University, Detroit, MI, 48202, USA
Correspondence: Suaad Abdallah Audat, Department of Chemistry, College of Science and Arts, Jordan University of Science and Technology, P.O. Box 3030, Irbid, 22110, Jordan, Tel +962 2 720 1000, Fax +962 2 720 1071, Email [email protected]
Background: Glyoxalase system is one of the defense cellular mechanisms that protect cells against endogenous harmful metabolites, mainly methylglyoxal (MG), through conversion of cytotoxic methylglyoxal into the non-toxic lactic acid. Glyoxalase system comprises of two enzymes glyoxalase I, glyoxalase II, and a catalytic amount of reduced glutathione. Cancerous cells overexpress glyoxalase I, making it a target for cancer therapy. Many studies have been conducted to identify potent Glx-I inhibitors.
Methods: Aiming to discover and develop novel Glx-I inhibitors, a series of 1,4-benzenesulfonamide derivatives were designed, synthesized, and biologically evaluated in vitro against human Glx-I enzyme. Seventeen compounds were designed based on the hit compound that was obtained from searching the National Cancer Institute (NCI) database. The synthesis of the target compounds ( 13– 29) was accomplished utilizing an azo coupling reaction of aniline derivatives and activated substituted aromatic compounds. To understand the binding mode of the active compounds at the active site of Glx-I, docking studies were performed.
Results: Structure activity relationship (SAR) studies were accomplished which led to the identification of several compounds that showed potent inhibitory activity with IC50 values below 10 μM. Among the compounds tested, compounds (E)-2-hydroxy-5-((4-sulfamoylphenyl)diazenyl)benzoic acid ( 26) and (E)-4-((8-hydroxyquinolin-5-yl)diazenyl) benzenesulfonamide ( 28) displayed potent Glx-I inhibitory activity with IC50 values of 0.39 μM and 1.36 μM, respectively. Docking studies of compounds 26 and 28 were carried out to illustrate the binding mode of the molecules into the Glx-I active site.
Conclusion: Our results show that compounds 26 and 28 displayed potent Glx-I inhibitory activity and can bind the Glx-I well. These findings should lead us to discover new classes of compounds with better Glx-I inhibition.
Keywords: anticancer agents, glyoxalase I, structure activity relationship, SAR, molecular docking
Graphical Abstract:

Introduction
Advanced glycation end products (AGEs) are group of molecules produced from non-enzymatic glycation and oxidation of proteins and lipids.1–3 It has been established that AGEs are involved in many diseases including aging, neurological disorders, diabetic complications and cancer.4,5 α-Oxoaldehydes such as methylglyoxal (MG) are reactive dicarbonyl compounds contribute to the production of AGEs.6,7 MG is produced endogenously through different enzymatic and non-enzymatic pathways including metabolic glycolysis processes and fragmentation of triosephosphates glyceraldehyde-3-phosphate and dihydroxyacetone phosphate.7–12 Glyoxalase system is one of the cellular mechanisms that protects cells against AGEs production through conversion of MG into the non-toxic lactic acid13,14 and ultimately prevents the accumulation of reactive dicarbonyl compounds in cells (Figure 1).
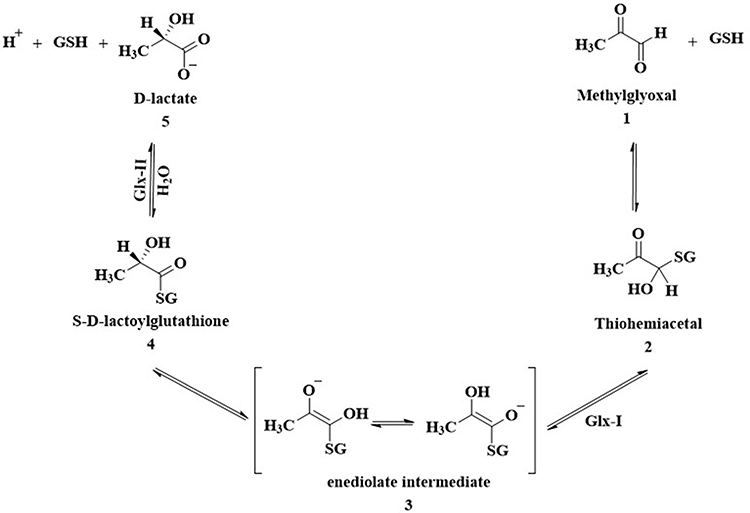 |
Figure 1 Glyoxalase enzyme system normal pathway. |
Glyoxalase system comprises glyoxalase I (Glx-I), glyoxalase II (Glx-II), and a catalytic amount of reduced glutathione.15 Under normal cellular conditions, thiohemiacetal 2 form from non-enzymatic condensation of glutathione (GSH) and MG 1. In the multistep pathway of glyoxalase system, Glx-I converts thiohemiacetal 2 into the S-D-lactoylglutathione 4 which is then hydrolyzed by Glx-II to produce D-lactate and glutathione.16–18
In cancerous cells, where inducing apoptosis to the cells is the ultimate goal, Glx-I enzyme is abnormally overexpressed.19–24 Inhibition of Glx-I enzyme should result in accumulation of MG25 at cytotoxic levels eventually leading to apoptosis.26 Therefore, inhibitors of Glx-I enzyme are considered effective anti-tumor agents.
Many studies have been conducted to identify potent Glx-I inhibitors.27,28 Different drug discovery methodologies have been employed to identify such inhibitors including glutathione derivatives,29–32 natural products and natural products derivatives,33–36 high-throughput screening,37,38 and in silico computer aided drug design.35,39–45 Recently, the active site of Glx-I was modeled using a structure-based pharmacophore approach that was utilized to construct a pharmacophore model, a tool that was used to search the NCI database to find new Glx-I inhibitors. Several promising hits were identified based on the search query, which were purchased and tested for their inhibitory potential, and showed good inhibitory activity.46 This study selected compound 6 as a lead compound toward the development of more potent Glx-I inhibitors. The selected compound has a benzenesulfonamide moiety and exhibited an IC50 value of 3.65 μM, as shown in Figure 2.
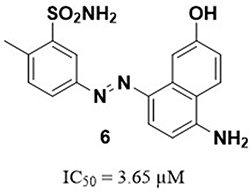 |
Figure 2 The structure of the lead compound 646. |
Recently, our research group has designed and synthesized 1,3-diazenylbenzenesulfonamide derivatives of the lead compound, that led to identification of promising Glx-I inhibitors.47 The present study, as a continuation of the previous research, aims to prepared 1,4-benzenesulfonamide derivatives as structural analogues of compound 6. The compounds were synthesized and biologically evaluated in vitro against human Glx-I enzyme. Structure-activity relationship (SAR) studies were performed after structural modifications of the synthesized compounds.
Materials and Methods
Chemistry
All organic reactions were performed with standard laboratory instruments with magnetic stirring. All chemicals or reagents and solvents used for syntheses were commercially available and were used as received without further purification. Analytical thin layer chromatography (TLC) was used to monitor all organic reactions in which the disappearance of the starting material and the formation of the product(s) indicated the completion of the reaction. TLC was performed on Merk silica gel 60 F254 aluminum backed plates. Merck silica gel 60 (0.2 mm) was used for column chromatography. Visualization of TLC was accomplished with UV light (254 nm). A Bruker AVANCE 400 MHz instrument was used to record 1H/13C NMR spectra at ambient temperature. The spectra were measured in ppm and calibrated with the residual proton peak of the solvent, DMSO-d6. The 1H NMR data are presented as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants, J (Hz), and integration. The 13C NMR analyses were reported in terms of chemical shift. 1H and 13C NMR spectra for all compounds are provided in the Supplementary Material (S1–S34). High resolution mass spectrometry (HRMS) was performed on an Impact II by Brucker using UHR QTOF MS (Ultra-High-Resolution Q-Time-Of-Flight) with full scan MS and broadband CID (bbCID) MS/MS methods located at the NABA HIKMA industrial and testing services, Fuhais, Jordan.
Synthesis of the Target Compounds
Utilizing modified published literature procedures, compounds (13–29) were synthesized using the following two steps:47–49
Step 1: General procedure for preparation of intermediates 10–12:
7.5 mL of 40% HCl was used to dissolve (0.50 g, 1.0 eq) of the aniline derivatives (7–9). After cooling down the solution to −5 °C, a sodium nitrite (1.2 eq, 2.3 M) was added dropwise and stirred for 15 min to generate the intermediate compounds 10–12.
Step 2: General procedure for preparation of compounds 13–29:
Procedure 1: To a solution of a R2 compound (1.0 equiv.) in a 2M sodium hydroxide (NaOH, 37.5 mL) at −5 °C was added dropwise the solution prepared in step 1. Immediately after mixing, the solution changed color, and a precipitate was formed. Stirring continued at the same temperature until the reaction was completed. The pH was then adjusted to 7.0 after the mixture was warmed to room temperature. After filtration, the collected solid was washed with deionized water, air dried, then purified utilizing flash column chromatography (5% MeOH/DCM) or by washing with hot ethanol to yield the final products.
Procedure 2: Similar procedure as in procedure 1 except in procedure 2 a saturated aqueous solution of NaOAc (7.5 mL) was used as a base instead of 2M NaOH.
Compound (13)
The title compound was synthesized as black powder using procedure 2. Yield (34%). 1H NMR (400 MHz, DMSO-d6) δ 8.63 (s, 1H), 8.34 (d, J = 8.8 Hz, 1H), 8.03 (s, 1H), 7.92–7.84 (m, 5H), 7.38 (s, 2H), 7.14 (d, J = 8.8 Hz, 1H), 7.06 (d, J = 10.4 Hz, 1H).13C NMR (DMSO-d6) δ 162.92, 162.03, 145.94, 139.43, 138.80, 132.16, 128.05, 127.36, 126.92, 118.98, 117.95, 116.01, 114.18, 107.95. HRMS (ESI, m/z): calculated for C16H15N4O3S [M+H]+ 343.0859; found 343.0900.
Compound (14)
The title compound was synthesized as black powder using procedure 2. Yield (43%). 1H NMR (400 MHz, DMSO-d6) δ 8.69 (d, J = 8.4 Hz, 1H), 8.12 (s, 1H), 7.94–7.88 (m, 5H), 7.55 (s, 1H), 7.41–7.34 (m, 3H), 6.96 (d, J = 8.0 Hz, 1H).13C NMR (DMSO-d6) δ 156.55, 155.62, 141.10, 134.95, 128.03, 127.26, 126.84, 125.37, 123.12, 122.30, 122.11, 118.54, 107.72, 106.98. HRMS (ESI, m/z): calculated for C16H15N4O3S [M+H]+ 343.0859; found 343.0905.
Compound (15)
The title compound was synthesized as black powder using procedure 2. Yield (75%). 1H NMR (400 MHz, DMSO-d6) δ 8.80 (d, J = 8.8 Hz, 1H), 8.42 (d, J = 8.4 Hz, 1H), 8.00–7.91 (m, 5H), 7.82 (t, J = 7.8 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.41 (s, 2H), 7.11 (d, J = 9.2 Hz, 1H).13C NMR (DMSO-d6) δ 161.05, 147.48, 140.58, 135.51, 133.30, 132.87, 127.67, 127.43, 126.44, 124.76, 123.72, 121.65, 117.64, 117.13. HRMS (ESI, m/z): calculated for C16H15N4O2S [M+H]+ 327.0910; found 327.0946.
Compound (16)
The title compound was synthesized as black powder using procedure 1. Yield (17%). 1H NMR (400 MHz, DMSO-d6) δ 15.96 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 9.6 Hz, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.35 (s, 2H), 7.23 (t, J = 8.0 Hz, 1H), 7.17 (s, 2H), 7.08 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 7.2 Hz, 1H), 6.61 (d, J = 9.6 Hz, 1H).13C NMR (DMSO-d6) δ 182.62, 146.06, 145.49, 144.55, 139.09, 134.76, 129.65, 127.84, 127.69, 125.61, 119.47, 118.86, 115.18, 112.04. HRMS (ESI, m/z): calculated for C16H15N4O3S [M+H]+ 343.0859; found 343.0875.
Compound (17)
The title compound was synthesized as an orange powder using method 1. Yield (25%). 1H NMR (400 MHz, DMSO-d6) δ 15.82 (s, 1H), 8.46 (d, J = 8.0 Hz, 1H), 7.96–7.90 (m, 5H), 7.73 (d, J = 7.6 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.49–7.45 (m, 3H), 6.78 (d, J = 9.6 Hz, 1H). 13C NMR (DMSO-d6) δ 175.58, 145.78, 142.23, 141.29, 132.67, 130.14, 129.52, 129.20, 128.09, 127.48, 126.83, 125.39, 121.81, 117.73. HRMS (ESI, m/z): calculated for C16H14N3O3S [M+H]+ 328.0750; found 328.0777.
Compound (18)
The title compound was synthesized as reddish-brown powder using procedure 1. Yield (76%). 1H NMR (400 MHz, DMSO-d6) δ 8.15–8.01 (m, 6H), 7.55 (s, 2H), 7.03 (s, 1H), 6.36 (d, J = 9.2 Hz, 1H).13C NMR (DMSO-d6) δ 175.32, 145.80, 142.10, 141.29, 132.63, 130.13, 129.44, 129.12, 128.04, 127.44, 126.75, 125.27, 121.75, 117.70. HRMS (ESI, m/z): calculated for C15H12N3O5S [M+H]+ 346.0492; found 346.0517.
Compound (19)
The title compound was synthesized as an orange powder using procedure 1. Yield (11%). 1H NMR (400 MHz, DMSO-d6) δ 15.81 (s, 1H), 8.46 (d, J = 8.0 Hz, 1H), 7.95–7.93 (m, 5H), 7.73 (d, J = 7.6 Hz, 1H), 7.60 (t, J = 7.8 Hz, 1H), 7.49–7.43 (m, 3H), 6.78 (d, J = 9.6 Hz, 1H). 13C NMR (DMSO-d6) δ175.46, 145.87, 142.27, 141.34, 132.71, 130.22, 129.59, 129.24, 128.14, 127.51, 126.89, 125.39, 121.85, 117.81.
Compound (20)
The title compound was synthesized as an orange powder using procedure 1. Yield (98%). 1H NMR (400 MHz, DMSO-d6) δ 15.82 (s, 1H), 8.48 (d, J = 8.0 Hz, 1H), 7.97–7.91 (m, 5H), 7.75 (d, J = 7.6 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.50–7.43 (m, 3H), 6.80 (d, J = 9.6 Hz, 1H). 13C NMR (DMSO-d6) δ 175.42, 145.78, 142.16, 141.29, 132.64, 130.13, 129.48, 129.16, 128.07, 127.45, 126.79, 125.33, 121.77, 117.72. HRMS (ESI, m/z): calculated for C17H17N4O5S [M+NH4]+ 389.0920; found 389.0958.
Compound (21)
The title compound was synthesized as reddish-orange powder using procedure 1. Yield (56%). 1H NMR (400 MHz, DMSO-d6) δ 15.88 (s, 1H), 8.45 (d, J = 8.0 Hz, 1H), 8.06–8.04 (m, 2H), 7.94 (d, J = 9.6 Hz, 1H), 7.88–7.85 (m, 2H), 7.74 (d, J = 7.6 Hz, 1H), 7.63–7.58 (m, 1H), 7.49–7.45 (m, 1H), 6.78 (d, J = 9.6 Hz, 1H). 13C NMR (DMSO-d6) δ175.86, 166.70, 146.56, 142.17, 132.67, 131.04, 130.13, 129.47, 129.14, 128.08, 128.04, 126.76, 125.45, 121.70, 117.29. HRMS (ESI, m/z): calculated for C18H11N2O5 [M-H]− 335.0673; found 335.0603.
Compound (22)
The title compound was synthesized as red powder using procedure 1. Yield (36%). 1H NMR (400 MHz, DMSO-d6) δ 15.76 (s, 1H), 8.55 (d, J = 8.4 Hz, 1H), 7.96 (d, J = 9.2 Hz, 1H), 7.87 (d, J = 8.0 Hz, 2H), 7.79 (d, J = 8.4 Hz, 1H), 7.64–7.60 (m, 1H), 7.55 (t, J = 7.8 Hz, 2H), 7.46 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 6.93 (d, J = 9.2 Hz, 1H). 13C NMR (DMSO-d6) δ 168.72, 145.09, 139.88, 132.72, 129.76, 129.18, 129.04, 128.84, 128.04, 127.80, 125.79, 123.85, 121.27, 118.92. HRMS (ESI, m/z): calculated for C17H13N2O3 [M+H]+ 293.0921; found 293.0961.
Compound (23)
The title compound was synthesized as brown powder using procedure 1. Yield (24%). 1H NMR (400 MHz, DMSO-d6) δ 8.92 (d, J = 8.4 Hz, 1H), 8.40 (d, J = 8.4 Hz, 1H), 8.31 (s, 1H), 8.17–8.15 (m, 2H), 8.05–8.02 (m, 2H), 7.92–7.88 (m, 1H), 7.76–7.72 (m, 1H), 7.55 (s, 2H).13C NMR (DMSO-d6) δ 172.53, 164.50, 153.63, 145.25, 138.42, 134.56, 130.92, 127.09, 126.92, 124.69, 123.61, 122.88, 122.78, 112.71, 106.86. HRMS (ESI, m/z): calculated for C17H14N3O5S [M+H]+ 372.0649; found 372.0703.
Compound (24)
The title compound was synthesized as dark violate powder using procedure 1. Yield (67%). 1H NMR (400 MHz, DMSO-d6) δ 8.90 (d, J = 8.4 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 8.30 (s, 1H), 8.16–8.14 (m, 2H), 8.08–8.06 (m, 2H), 7.91–7.87 (m, 1H), 7.75–7.71 (m, 1H). 13C NMR (DMSO-d6) δ172.45, 166.76, 164.62, 154.42, 138.33, 134.56, 132.01, 130.85, 130.59, 126.81, 124.72, 123.60, 122.81, 122.26, 112.77, 106.98. HRMS (ESI, m/z): calculated for C18H12N2O5Na [M+Na]+ 359.0644; found 359.0686.
Compound (25)
The title compound was synthesized as dark violate powder using procedure 1. Yield (15%). 1H NMR (400 MHz, DMSO-d6) δ8.91 (d, J = 8.4 Hz, 1H), 8.40 (d, J = 8.4 Hz, 1H), 8.22 (s, 1H), 8.03–7.99 (m, 2H), 7.91–7.87 (m, 1H), 7.75–7.71 (m, 1H), 7.64–7.54 (m, 4H). 13C NMR (DMSO-d6) δ172.76, 163.42, 152.40, 138.56, 134.43, 130.94, 130.56, 129.41, 126.66, 124.57, 123.48, 122.85, 122.55, 111.65, 106.32. HRMS (ESI, m/z): calculated for C17H13N2O3 [M+H]+ 293.0921; found 293.0962.
Compound (26)
The title compound was synthesized as brown powder using procedure 1. Yield (21%). 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.12 (d, J = 8.8 Hz, 1H), 8.02–7.99 (m, 4H), 7.53 (s, 2H), 7.18 (d, J = 9.2 Hz, 1H). 13C NMR (DMSO-d6) δ 171.22, 164.09, 153.37, 145.59, 144.47, 129.12, 127.04, 126.38, 122.76, 118.54, 113.85. HRMS (ESI, m/z): calculated for C13H12N3O5S [M+H]+ 322.0492; found 322.0535.
Compound (27)
The title compound was synthesized as brown powder using procedure 1. Yield (34%). 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.00–7.94 (m, 4H), 7.86 (d, J = 7.6 Hz, 2H), 7.50 (s, 2H), 6.97 (d, J = 7.6 Hz, 2H).13C NMR (DMSO-d6) δ 161.73, 153.69, 145.26, 145.01, 126.99, 125.38, 122.44, 116.11. HRMS (ESI, m/z): calculated for C12H12N3O3S [M+H]+ 278.0594; found 278.0628.
Compound (28)
The title compound was synthesized as dark red powder using procedure 1. Yield (99%). 1H NMR (400 MHz, DMSO-d6) δ 9.66 (d, J = 8.4 Hz, 1H), 9.12–9.11 (m, 1H), 8.20–8.15 (m, 3H), 8.04–7.98 (m, 3H), 7.57 (s, 2H), 7.46 (d, J = 8.8 Hz, 1H).13C NMR (DMSO-d6) δ 153.42, 146.67, 145.33, 139.01, 137.71, 132.41, 128.82, 127.33, 124.24, 122.87, 117.76, 115.50. HRMS (ESI, m/z): calculated for C15H13N4O3S [M+H]+ 329.0703; found 329.0754.
Compound (29)
The title compound was synthesized as a yellow powder using procedure 1. Yield (45%). 1H NMR (400 MHz, DMSO-d6) δ 9.30 (d, J = 8.4 Hz, 1H), 9.00–8.98 (m, 1H), 8.00–7.98 (m, 3H), 7.78–7.75 (m, 1H), 7.62–7.52 (m, 3H), 7.23 (d, J = 8.4 Hz, 1H).13C NMR (DMSO-d6) δ 157.86, 152.57, 149.09, 138.63, 138.04, 131.88, 130.81, 129.49, 127.62, 123.29, 122.51, 114.94, 111.84. HRMS (ESI, m/z): calculated for C15H12N3O [M+H]+ 250.0975; found 250.1003.
Biological Evaluation
The Glx-I inhibitory activity was conducted in vitro using established published procedure.35,42,43,47 The IC50 values of all compounds were calculated using GraphPad Prism 6 (2012). Myricetin was used as positive control with an IC50 equals to 3.38 ± 0.41 (µM).
Molecular Modeling Study
Using discovery studio software from Biovia®, the most active compounds were docked into the active site of Glx-I.50 The enzyme was prepared according to the default parameters using “Prepare protein” protocol and minimized while the necessary modifications of the zinc atom were conducted to agree with the previous work set by our research group. The parameters for the minimize protein was left as default and the zinc atom at the active site was assigned to be +2 charge with octahedral geometry. The docking protocols were the LibDock and Flexible Docking, which are validated build-in software supported by Discovery studio®. The LibDock parameters were set as default with the assignment of 10 A° as a docking sphere to enable the docking to reach all the active site three major areas (positively ionized entrance of the active site, zinc atom, deep hydrophobic pocket). Flexible docking was performed by allowing the amino acids within the active site to be flexible during docking. Parameters were left as default with 10 A° docking sphere.
Results and Discussion
Chemistry
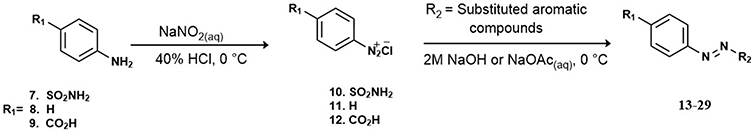
A series of structural analogues of the lead compound (6) were synthesized and biologically evaluated in vitro against human Glx-I enzyme in order to discover Glx-I inhibitors to be used as potential anticancer agents. The synthesis of the target compounds (13–29) was accomplished by diazo coupling reaction as illustrated in Scheme 1.
 |
Scheme 1 Synthesis of the target compounds (13–29). |
The synthesis of the target compounds (13–29) was accomplished by converting commercially available aniline derivatives (7–9) into the unstable intermediate diazonium chloride salts (10–12) via diazotization reaction. Next, coupling of the electrophilic diazonium ions with nucleophilic activated aromatic compounds (R2) through electrophilic aromatic substitution reaction led to the formation of the desired diazo compounds.
The synthesized diazo compounds (13–29) were chosen to have various R moieties (R1 and R2) containing polar, ionizable, heterocyclic, and hydrophobic moieties which are expected to bind to the active site of the enzyme via intermolecular forces that could enhance the inhibitory activity of the synthesized compounds. All compounds were analyzed, and their identities were validated by 1H-NMR, 13C-NMR and high-resolution mass spectrometry (HRMS), the results of these spectra indicated that the structures of these compounds are correct. All of the synthetic compounds gave satisfactory analytical and spectroscopic data, which were in full accordance with their depicted structures.
Structure-Activity Relationship (SAR) Studies
In this study compound 6 (IC50 = 3.65 µM) was used as a lead compound to identify more potent Glx-I inhibitors. From the perspective of compound structure, compound 6 possesses 5-diazenyl-2-methylbenzenesulfonamide moiety attached to position 8 of the 5-amino-2-naphthalenol moiety, in which the diazenyl moiety is located at the meta position relative to the benzenesulfonamide moiety.
A series of structural analogues of compound 6 were designed to locate diazenyl moiety at the para position relative to the benzenesulfonamide moiety. The analogues were synthesized and biologically evaluated in vitro against human Glx-I enzyme to discover potential Glx-I inhibitors. Structure activity relationship (SAR) studies were accomplished which led to identification of a series of analogues (13–29) that showed a broad range of inhibition (potent to moderate) as shown in Table 1. SAR studies started with the synthesis of (E)-4-((4-amino-7-hydroxynaphthalen-1-yl)diazenyl)benzenesulfonamide (13) that possesses 4-diazenylbenzenesulfonamide moiety attached to the position 8 of the 5-amino-2-naphthalenol moiety. Removing the methyl group from compound 6 and shifting the diazenyl moiety to the para position relative to the benzenesulfonamide moiety resulted in reduced inhibitory activity with IC50 39. 06 μM. Compounds 14 and 15 were synthesized to study the effect of naphthalene’s substituents on the activity of compound 13. Moving the hydroxyl group in compound 13 from position 2 to position 3 led to the formation of compounds 14 with enhanced inhibitory activity IC50 22.98 μM. Whereas removing the hydroxyl group from the naphthalene moiety (compound 15) significantly increased the inhibitory activity producing a potent compound with IC50 1.85 μM. These results suggest that enhancement of the activity possibly due to the naphthalene moiety forming much stronger interactions with the surrounding hydrophobic amino acids in the active site of the enzyme. In addition, the presence of the hydroxyl group at the naphthalene ring may cause unfavorable interactions in the hydrophobic pocket at the active site of the enzyme. Also, the removal of the OH reduced the desolvation penalty contributed to presence of polar functional groups in the structure.
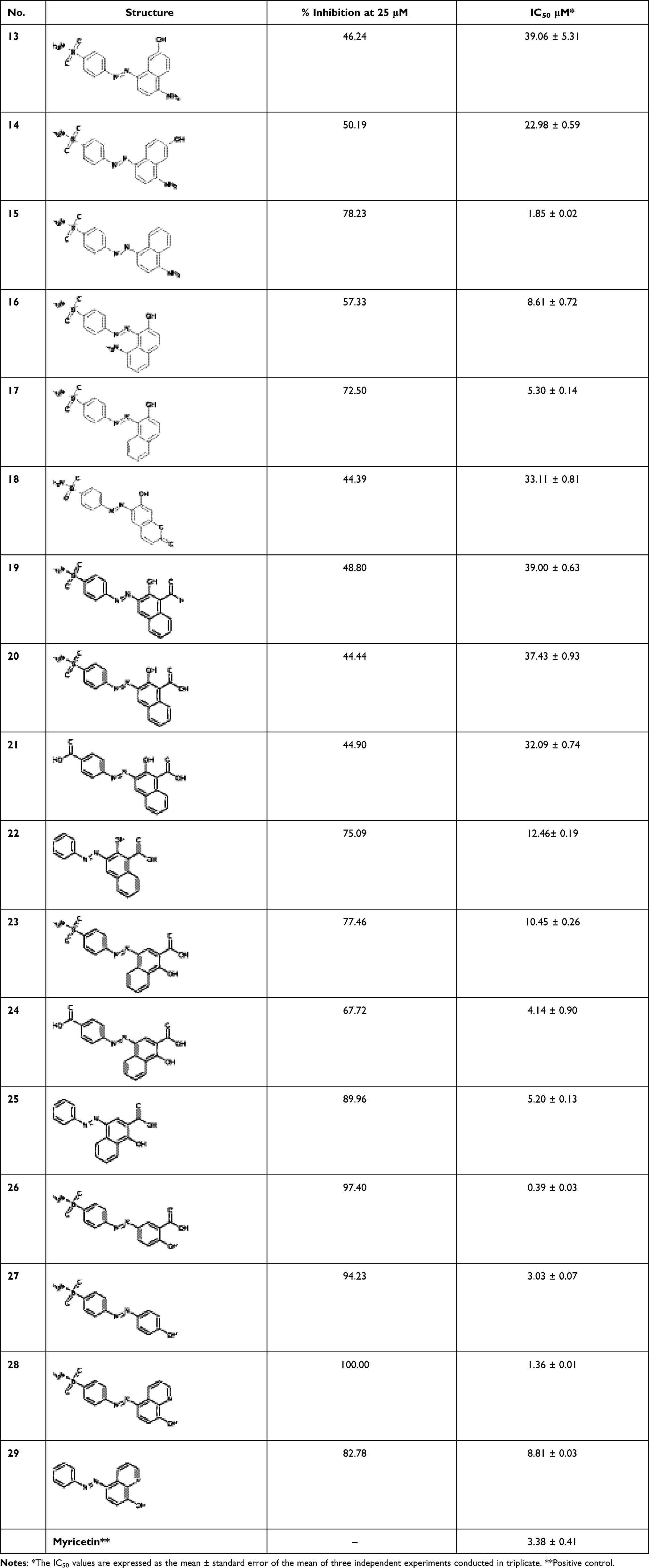 |
Table 1 Glx-I Inhibitory Activity of the Synthesized Compounds |
On the other hand, attaching the 4-diazenylbenzenesulfonamide moiety at position 1 of the 5-amino-2-naphthalenol moiety and moving the amino group from position 5 to position 8 produced compound 16 with good inhibitory activity (IC50 8.61 μM). Removing the amino group from the naphthalene moiety of compound 16 led to the formation of compound 17 with increasing inhibitory activity (IC50 5.30 μM). These results showed that the amino group at position 8 of the naphthalene ring reduced the inhibitory activity of compound 16, which may be due to the unfavorable interactions in the hydrophobic pocket at the active site of the enzyme.
Next, different analogues of compound 13 were synthesized in which the 5-amino-2-naphthalenol moiety was replaced with groups that possess the ability to interact with the zinc ion and the active site residues of the enzyme. First, replacement of the 5-amino-2-naphthalenol moiety with 7-hydroxyl-2H-chromen-2-one moiety led to moderately active compound (compound 18) with IC50 33.11 μM. Second, the replacement of 5-amino-2-naphthalenol moiety with 2-hydroxy-1-naphthaldehyde moiety led to the formation of compound 19 with similar inhibitory activities (IC50 39.00 μM), when compared to compound 13.
Furthermore, replacement of the formyl group in compound 19 with a carboxy group led to the formation of compound 20 with no noticeable improvement of the inhibitory activity. On the other hand, compounds 21–22 were synthesized to explore the SAR for compound 20. Isosteric replacement of the sulfonamide moiety with a carboxy group produced compound 21 (IC50 32.09 μM) with slight increase in activity, whereas, removing the sulfonamide moiety from compound 20 resulted in the formation of compound 22 with good inhibitory activity (IC50 12.46 μM). These results suggest that the presence of polar or ionizable groups at the para position of benzene ring reduces the inhibitory activity of the compounds, which may be attributed to an increase in the desolvation penalty. These groups have the potential to clasp water molecules tightly via a series of hydrogen bonds or via ion dipole interactions in the case of carboxylic acid. Most probably, the enthalpy gain from zinc chelation is not enough to compensate the desolvation penalty. Switching the position of the carboxyl and hydroxyl groups, led to compound 23 with significant enhancement in the inhibitory activity compared to compound 20 (IC50 10.45 μM). This suggests the importance of the position of the carboxyl and hydroxyl groups in bindings with the amino acid residues at the active site of the enzyme. An attempt to optimize compound 23, compounds 24–27 were synthesized. Isosteric replacement of the sulfonamide moiety in compound 23 with carboxy group produced potent compound with IC50 4.14 μM (compound 24). Removing the sulfonamide moiety from compound 23 resulting in the formation of compound 25 with IC50 5.20 μM. Further optimization of compound 23 was performed through replacing the 1-hydroxy-2-naphthoic acid moiety with 2-hydroxybenzoic acid moiety which gave compound 26 that was found to be the most active among the synthesized compounds with IC50 0.39 μM. This interesting result best explained due to the better positioning of the 2-hydroxybenzoic acid moiety within the active site with less steric clash caused by the other benzene ring within the naphthalene ring. To investigate the role of carboxy group in compound 26, compound 27 was synthesized and evaluated for their inhibitory activity. Removal of the carboxy group reduced the activity of compound 26, which indicate the importance of the carboxy group in binding to the amino acid residues of the active site. (Compound 27, IC50 = 3.03 μM).
Next, we synthesized compound 28 that contain 8-hydroxyquinoline moiety in the place of 5-amino-2-naphthalenol which provided a potent derivative with IC50 1.36 μM. Compounds 27 and 29 were synthesized to explore the SAR for compound 28. Investigating the importance of the pyridine moiety in compound 28 was accomplished by synthesizing compound 27 that lack the pyridine moiety. It was found to have less inhibitory activity when compared to compound 28. This result indicates that the pyridine moiety has a role in binding interactions within the active site of the enzyme. Compound 29 was synthesized to explore the importance of the sulfonamide moiety to the activity of compound 28. The result showed that removing the sulfonamide moiety causes significant decrease in the inhibitory activity of the compound (IC50 = 8.81 μM), possibly due to the loss of some binding interaction at the active site.
Binding Mode of 26 and 28 into the Active Site of Glx-I Enzyme
Molecular docking was performed using flexible docking and Libdock protocols. The poses of the most active compounds (26 and 28) were analyzed and studied to investigate their binding pattern within the active site of the enzyme and to explore the most important ligand-receptor interactions such as the role of zinc atom. Binding mode of the most active compound 26 into the catalytic site of Glx-I is shown in (Figure 3), which explains the good activity of the compound. The binding model showed that the ionizable carboxyl group chelates zinc atom cofactor in a bidentate pattern at the active site of the enzyme. Additionally, the sulfonamide group provided significant hydrogen bond interactions with Arg37, Asn103, and Arg122 that enhanced compound’s potency. Furthermore, the potency of compound 26 is also due to hydrophobic interactions between the aromatic benzene moieties of compound 26 and the hydrophobic amino acid residues (Phe162, Phe67, Phe160, and Met157) at the active site of the enzyme.
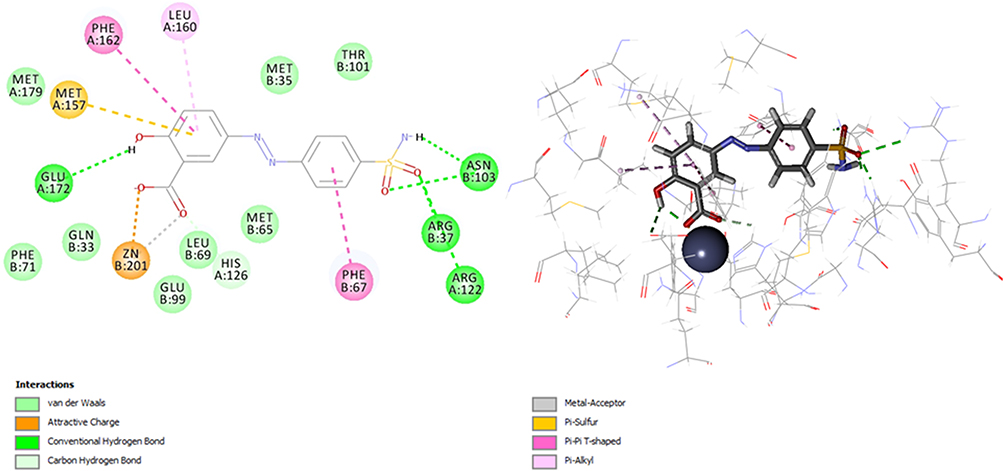 |
Figure 3 2D and 3D interaction map of the most active compound 26. |
On the other hand, binding model of compound 28 was generated by its docking into the active site of Glx-I. The probable binding mode is illustrated in (Figure 4). The binding model showed that the hydroxyl group chelates the zinc atom at the active site of the enzyme. Moreover, the sulfonamide group forms hydrogen bond interactions with Arg122, Arg37 and Asn103. Additionally, hydrophobic interactions occurred between the quinoline moiety and the hydrophobic amino acid residues (Phe162, Met157, Leu69, and Leu160) at the active site, whereas the benzene moiety forms hydrophobic interactions with the hydrophobic amino acid residue Phe67at the active site of the enzyme.
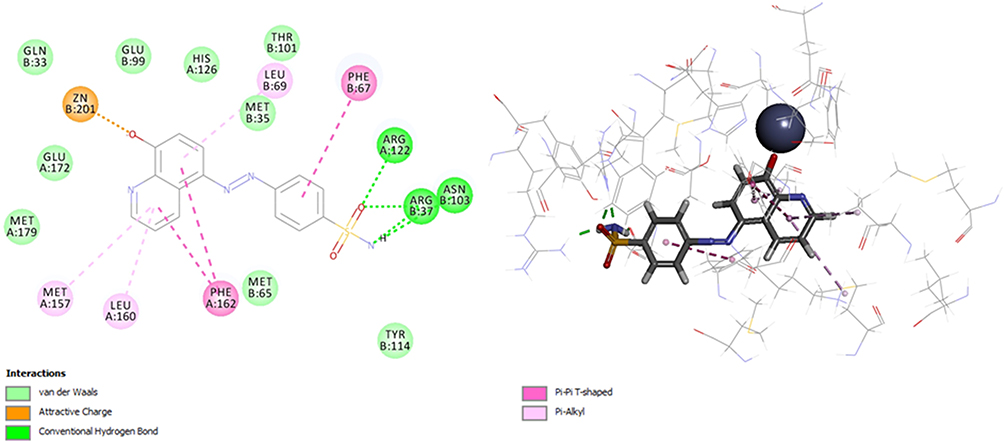 |
Figure 4 2D and 3D interaction map of the most active compound 28. |
Conclusion
To sum up, the previously reported Glx-I inhibitor (Compound 6, IC50 3.65 μM) was used as a lead compound to identify more potent Glx-I inhibitors. To achieve our goals, a series of structural analogues were synthesized and biologically evaluated in vitro against human Glx-I enzyme. Several analogues were found to exhibit potent activity with IC50 below 10 μM. Structural optimization of the lead compound (compound 6) produced potent compounds, compound 26 with IC50 0.39 μM and compound 28 with IC50 1.36 μM. SAR studies revealed that the carboxy moieties in compound 26 plays pivotal role in the inhibitory activity of the compound. Additionally, SAR evaluation of compound 28 signifies the importance of the sulfonamide and pyridine moieties for the inhibitory activity of the compound. Furthermore, molecular docking was performed to give the probable binding modes of compounds 26 and 28 into the catalytic site of Glx-I enzyme. These findings will open the doors for better discoveries of potent Glx I inhibitors.
Acknowledgments
The authors acknowledge the financial support given by the Deanship of Research at the Jordan University of Science and Technology (Grant No. 20200535).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ramasamy R, Vannucci SJ, Yan SSD, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15(7):16R–28R. doi:10.1093/glycob/cwi053
2. Jack M, Wright D. Role of advanced glycation endproducts and glyoxalase I in diabetic peripheral sensory neuropathy. Transl Res. 2012;159(5):355–365. doi:10.1016/j.trsl.2011.12.004
3. Zhang H, Tang L, Chen S, Yang Y, Chen M, Luo J. Effect of advanced glycation end products on the expression of hypoxia-inducible factor-1α and vascular endothelial growth factor proteins in RF/6A cells. Exp Ther Med. 2013;5(5):1519–1522. doi:10.3892/etm.2013.1015
4. Ahmed N, Argirov OK, Minhas HS, Cordeiro CA, Thornalley PJ. Assay of advanced glycation endproducts (AGEs): surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to N∈-carboxymethyl-lysine-and N∈-(1-carboxyethyl) lysine-modified albumin. Biochem J. 2002;364(1):1–14. doi:10.1042/bj3640001
5. Schröter D, Höhn A. Role of advanced glycation end products in carcinogenesis and their therapeutic implications. Curr Pharm Des. 2018;24(44):5245–5251. doi:10.2174/1381612825666190130145549
6. Degenhardt T, Thorpe S, Baynes J. Chemical modification of proteins by methylglyoxal. Cell Mol Biol. 1998;44(7):1139–1145.
7. Allaman I, Bélanger M, Magistretti PJ. Methylglyoxal, the dark side of glycolysis. Front Neurosci. 2015;9. doi:10.3389/fnins.2015.00023
8. Richard J. Mechanism for the Formation of Methylglyoxal from Triosephosphates. Portland Press Limited; 1993.
9. Ahmed MU, Thorpe SR, Baynes JW, Thorpe SR, Baynes JW. N ε-(Carboxyethyl) lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem J. 1997;324(2):565–570. doi:10.1042/bj3240565
10. Shinohara M, Thornalley P, Giardino I, et al. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. 1998;101(5):1142. doi:10.1172/JCI119885
11. Ray S, Ray M. Isolation of methylglyoxal synthase from goat liver. J Biol Chem. 1981;256(12):6230–6233. doi:10.1016/S0021-9258(19)69151-9
12. Chang T, Wu L. Methylglyoxal, oxidative stress, and hypertension. Can J Physiol Pharmacol. 2006;84(12):1229–1238. doi:10.1139/y06-077
13. Dakin H, Dudley H. An enzyme concerned with the formation of hydroxy acids from ketonic aldehydes. J Biol Chem. 1913;14(2):155–157. doi:10.1016/S0021-9258(18)88610-0
14. Dakin H, Dudley H. On glyoxalase. J Biol Chem. 1913;14(4):423–431. doi:10.1016/S0021-9258(18)88589-1
15. Thornalley PJ. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J. 1990;269(1):1. doi:10.1042/bj2690001
16. Neuberg C. The destruction of lactic aldehyde and methylglyoxal by animal organs. Biochem Z. 1913;49:502–506.
17. Racker E. Glyoxalases. Paper presented at: Federation Proceedings 1950.
18. Racker E. The mechanism of action of glyoxalase. J Biol Chem. 1951;190:685–696.
19. Chiavarina B, Nokin M-J, Durieux F, et al. Triple negative tumors accumulate significantly less methylglyoxal specific adducts than other human breast cancer subtypes. Oncotarget. 2014;5(14):5472. doi:10.18632/oncotarget.2121
20. Antognelli C, Talesa VN. Glyoxalases in urological malignancies. Int J Mol Sci. 2018;19(2):415. doi:10.3390/ijms19020415
21. Chiavarina B, Nokin M-J, Bellier J, et al. Methylglyoxal-mediated stress correlates with high metabolic activity and promotes tumor growth in colorectal cancer. Int J Mol Sci. 2017;18(1):213. doi:10.3390/ijms18010213
22. Antognelli C, Mandarano M, Prosperi E, Sidoni A, Talesa VN. Glyoxalase-1-dependent methylglyoxal depletion sustains PD-L1 expression in metastatic prostate cancer cells: a novel mechanism in cancer immunosurveillance escape and a potential novel target to overcome PD-L1 blockade resistance. Cancers. 2021;13(12):2965. doi:10.3390/cancers13122965
23. Antognelli C, Marinucci L, Frosini R, Macchioni L, Talesa VN. Metastatic prostate cancer cells secrete methylglyoxal-derived MG-H1 to reprogram human osteoblasts into a dedifferentiated, malignant-like phenotype: a possible novel player in prostate cancer bone metastases. Int J Mol Sci. 2021;22(19):10191. doi:10.3390/ijms221910191
24. Talesa VN, Ferri I, Bellezza G, Love HD, Sidoni A, Antognelli C. Glyoxalase 2 is involved in human prostate cancer progression as part of a mechanism driven by PTEN/PI3K/AKT/mTOR signaling with involvement of PKM2 and ERα. Prostate. 2017;77(2):196–210. doi:10.1002/pros.23261
25. Vince R, Daluge S. Glyoxalase inhibitors. A possible approach to anticancer agents. J Med Chem. 1971;14(1):35–37. doi:10.1021/jm00283a009
26. Holewinski RJ, Creighton DJ. Inhibition by active site directed covalent modification of human glyoxalase I. Bioorg Med Chem. 2014;22(13):3301–3308. doi:10.1016/j.bmc.2014.04.055
27. Jin T, Zhai J, Liu X, et al. Design, synthesis and biological evaluation of potent human glyoxalase I inhibitors. Chem Pharm Bull. 2017;65(5):455–460. doi:10.1248/cpb.c16-00800
28. Al-Balas QA, Hassan MA, Al-Shar’i NA, Al Jabal GA, Almaaytah AM. Recent advances in glyoxalase-I inhibition. Mini Rev Med Chem. 2019;19(4):281–291. doi:10.2174/1389557518666181009141231
29. Vince R, Wadd WB. Glyoxalase inhibitors as potential anticancer agents. Biochem Biophys Res Commun. 1969;35(5):593–598. doi:10.1016/0006-291X(69)90445-8
30. Vince R, Daluge S, Wadd WB. Inhibition of glyoxalase I by S-substituted glutathiones. J Med Chem. 1971;14(5):402–404. doi:10.1021/jm00287a006
31. Hamilton D, Creighton D. Inhibition of glyoxalase I by the enediol mimic S-(N-hydroxy-N-methylcarbamoyl) glutathione. The possible basis of a tumor-selective anticancer strategy. J Biol Chem. 1992;267(35):24933–24936. doi:10.1016/S0021-9258(19)73986-6
32. Murthy NS, Bakeris T, Kavarana MJ, Hamilton DS, Lan Y, Creighton DJ. S-(N-aryl-N-hydroxycarbamoyl) glutathione derivatives are tight-binding inhibitors of glyoxalase I and slow substrates for glyoxalase II. J Med Chem. 1994;37(14):2161–2166. doi:10.1021/jm00040a007
33. Liu M, Yuan M, Luo M, Bu X, Luo H-B, Hu X. Binding of curcumin with glyoxalase I: molecular docking, molecular dynamics simulations, and kinetics analysis. Biophys Chem. 2010;147(1–2):28–34. doi:10.1016/j.bpc.2009.12.007
34. Yuan M, Luo M, Song Y, et al. Identification of curcumin derivatives as human glyoxalase I inhibitors: a combination of biological evaluation, molecular docking, 3D-QSAR and molecular dynamics simulation studies. Bioorg Med Chem. 2011;19(3):1189–1196. doi:10.1016/j.bmc.2010.12.039
35. Al-Balas QA, Hassan MA, Al-Shar’i NA, et al. Novel glyoxalase-I inhibitors possessing a “zinc-binding feature” as potential anticancer agents. Drug Des Devel Ther. 2016;10:2623. doi:10.2147/DDDT.S110997
36. Zhang H, Zhai J, Zhang L, et al. In vitro inhibition of glyoxalase Ц# 134; by flavonoids: new insights from crystallographic analysis. Curr Top Med Chem. 2016;16(4):460–466. doi:10.2174/1568026615666150813150944
37. Yadav A, Kumar R, Sunkaria A, Singhal N, Kumar M, Sandhir R. Evaluation of potential flavonoid inhibitors of glyoxalase-I based on virtual screening and in vitro studies. J Biomol Struct Dyn. 2016;34(5):993–1007. doi:10.1080/07391102.2015.1064830
38. Chiba T, Ohwada J, Sakamoto H, et al. Design and evaluation of azaindole-substituted N-hydroxypyridones as glyoxalase I inhibitors. Bioorg Med Chem Lett. 2012;22(24):7486–7489. doi:10.1016/j.bmcl.2012.10.045
39. Takasawa R, Tao A, Saeki K, et al. Discovery of a new type inhibitor of human glyoxalase I by myricetin-based 4-point pharmacophore. Bioorg Med Chem Lett. 2011;21(14):4337–4342. doi:10.1016/j.bmcl.2011.05.046
40. More SS, Vince R. Design, synthesis, and binding studies of bidentate Zn-chelating peptidic inhibitors of glyoxalase-I. Bioorg Med Chem Lett. 2007;17(13):3793–3797. doi:10.1016/j.bmcl.2006.12.056
41. Perez C, Barkley-Levenson AM, Dick BL, et al. Metal-binding pharmacophore library yields the discovery of a glyoxalase 1 inhibitor. J Med Chem. 2019;62(3):1609–1625. doi:10.1021/acs.jmedchem.8b01868
42. Al-Balas QA, Hassan MA, Al Jabal GA, Al-Shar’i NA, Almaaytah AM, El-Elimat T. Novel thiazole carboxylic acid derivatives possessing a” zinc binding feature” as potential human glyoxalase-I inhibitors. Lett Drug Des Discov. 2017;14(11):1324–1334.
43. Al-Balas Q, Hassan M, Al-Oudat B, Alzoubi H, Mhaidat N, Almaaytah A. Generation of the first structure-based pharmacophore model containing a selective “zinc binding group” feature to identify potential glyoxalase-1 inhibitors. Molecules. 2012;17(12):13740–13758. doi:10.3390/molecules171213740
44. Al-Balas QA, Al-Smadi ML, Hassan MA, Al Jabal GA, Almaaytah AM, Alzoubi KH. Multi-armed 1, 2, 3-selenadiazole and 1, 2, 3-thiadiazole benzene derivatives as novel glyoxalase-I inhibitors. Molecules. 2019;24(18):3210. doi:10.3390/molecules24183210
45. Al‐Balas QA, Hassan MA, Al‐Shar’i NA, El‐Elimat T, Almaaytah AM. Computational and experimental exploration of the structure–activity relationships of flavonoids as potent glyoxalase‐I inhibitors. Drug Dev Res. 2018;79(2):58–69. doi:10.1002/ddr.21421
46. Al-Sha’er MA, Al-Balas QA, Hassan MA, Al Jabal GA, Almaaytah AM. Combination of pharmacophore modeling and 3D-QSAR analysis of potential glyoxalase-I inhibitors as anticancer agents. Comput Biol Chem. 2019;80:102–110. doi:10.1016/j.compbiolchem.2019.03.011
47. Al-Oudat BA, Hana’a MJ, Al‑Balas QA, Al-Shar’i NA, Bryant-Friedrich A, Bedi MF. Design, synthesis and biological evaluation of novel glyoxalase I inhibitors possessing diazenylbenzenesulfonamide moiety as potential anticancer agents. Bioorg Med Chem. 2020;28(16):115608. doi:10.1016/j.bmc.2020.115608
48. Carta F, Maresca A, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase inhibitors. Diazenylbenzenesulfonamides are potent and selective inhibitors of the tumor-associated isozymes IX and XII over the cytosolic isoforms I and II. Bioorg Med Chem. 2009;17(20):7093–7099. doi:10.1016/j.bmc.2009.09.003
49. Carta F, Pothen B, Maresca A, Tiwari M, Singh V, Supuran CT. Carbonic anhydrase inhibitors: inhibition of cytosolic carbonic anhydrase isozymes II and VII with simple aromatic sulfonamides and some azo dyes. Chem Biol Drug Des. 2009;74(2):196–202. doi:10.1111/j.1747-0285.2009.00842.x
50. Spassov VZ, Flook PK, Yan L. LOOPER: a molecular mechanics-based algorithm for protein loop prediction. Protein Eng Des Sel. 2008;21(2):91–100. doi:10.1093/protein/gzm083
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.