")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Design of oral agents for the management of multiple sclerosis: benefit and risk assessment for dimethyl fumarate
Authors Nicholas J, Boster A, Imitola J, O'Connell C, Racke M
Received 9 April 2014
Accepted for publication 6 June 2014
Published 7 July 2014 Volume 2014:8 Pages 897—908
DOI https://doi.org/10.2147/DDDT.S50962
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Jacqueline Ann Nicholas,1 Aaron Lee Boster,1 Jaime Imitola,1,2 Colleen O’Connell,1 Michael Karl Racke1,2
1Department of Neurology and Multiple Sclerosis Center, 2Department of Neuroscience, The Ohio State University, Columbus, OH, USA
Abstract: Dimethyl fumarate (DMF) is the most recent oral disease-modifying therapy approved by the US Food and Drug Administration and is indicated for the treatment of relapsing forms of multiple sclerosis (MS). Prior to approval for use in MS, DMF and its active metabolite, monomethyl fumarate, had been used for decades as two of the fumaric acid esters in Fumaderm®, a medication used in Europe for the treatment of psoriasis. The unique mechanism of action of DMF remains under evaluation; however, it has been shown to act through multiple pathways leading to shifts away from the Th1 proinflammatory response to the less inflammatory Th2 response. Preliminary data suggest that DMF may induce neuroprotective effects in central nervous system white matter, although further studies are needed to demonstrate these effects on inflammatory demyelination. The DMF Phase III clinical trials demonstrated its efficacy with regard to a reduction in the annualized relapse rate and reductions in new or enlarging T2 lesions and numbers of gadolinium-enhancing lesions on magnetic resonance imaging. DMF has a well-defined safety profile, given the experience with its use in the treatment of psoriasis, and more recently from the DMF clinical trials program and post-marketing era for treatment of MS. The safety profile and oral mode of administration of DMF place it as an attractive first-line therapy option for the treatment of relapsing forms of MS. Long-term observational studies will be needed to determine the effects of DMF on progression of disability in MS.
Keywords: multiple sclerosis, dimethyl fumarate, disease-modifying therapy, Nrf2 pathway, quality of life
Introduction
Approximately 350,000 people in the USA have physician-diagnosed multiple sclerosis (MS).1 It is the leading cause of nontraumatic neurologic disability in young adults in the USA, with most patients suffering from the effects of MS for most of their adult life. The cause of MS remains unknown, although because the disease involves perivascular mononuclear cell infiltrates and demyelination, features also characteristic of experimental autoimmune encephalomyelitis (EAE), an autoimmune process is hypothesized to be involved in the pathogenesis of the disease.2,3 Epidemiologic studies and studies examining the disease in identical twins suggest that both environment and genetics influence expression of the disease and play a role in pathogenesis of the disease.4 There are now ten disease-modifying therapies approved by the US Food and Drug Administration (FDA) for use in the treatment of MS in the USA; however, none of these agents are proven to be neuroprotective, so the need for better treatment strategies for MS remains.5–12 In addition, the unfortunate expression of progressive multifocal leukoencephalopathy (PML) in MS patients treated with natalizumab highlights the need for medications with a proven safety record.13–15
Dimethyl fumarate (DMF) offers a therapy with oral bioavailability and a record of effectiveness in human autoimmune diseases such as psoriasis and multiple sclerosis.12,16–18 However, little is known about how this small molecule exerts its protective effects in autoimmunity and whether it may have neuroprotective capability.
Relevance of EAE to MS
With the advent of transgenic and homologous recombination technology, it is increasingly clear that many powerful molecular tools are becoming available to study the immune response in pathologic processes such as EAE. While fumaric acid esters (FAE) have been used to treat psoriasis for over two decades, little was known about how it exerted its therapeutic effects in that disorder.16 In addition, there was no good animal model of psoriasis in order to study FAE and determine their effects on potentially pathogenic, autoreactive T-cells. We made use of the mouse transgenic model for the myelin basic protein Ac1-11-specific TCR to examine the mechanism of how manipulation of oxidative stress affects differentiation of antigen-specific T-cells.19,20 We had a great deal of experience with this transgenic mouse model, and importantly, because we were able to adoptively transfer disease into naïve recipients using TCR transgenic T-cells differentiated in vitro,21–23 we were able to examine the effects of FAE both in vitro and in vivo.19,20 For example, myelin-specific CD4+ T-cells activated in vitro in the presence of DMF had reduced ability to transfer disease into naïve recipients.19 Similarly, mice immunized with myelin proteins had reduced clinical signs of disease when fed DMF, and the myelin-specific T-cells obtained from these mice had less capacity to transfer disease.19 This suggested that FAE were able to reduce the pathogenic character of autoreactive, myelin-specific T-cells.
Phenotype of encephalitogenic T-cells
Although EAE was initially induced by immunization with myelin proteins emulsified in complete Freund’s adjuvant, it can also be induced by adoptive transfer of myelin-specific CD4+ Th1 and Th17 cells into naïve recipient mice.21–27 The observation that myelin-specific CD4+ Th1 cells were sufficient to induce EAE focused MS research on these interferon-gamma (IFNγ)-producing T-cells in MS patients. Although myelin-specific T-cells were found in both MS patients and healthy individuals, which raised questions as to the relevance of these cells in MS patients, it does appear that myelin-specific T-cells from MS patients are more likely to have a Th1 phenotype.27–31 Subsequently, several studies demonstrated that although healthy individuals had myelin-specific T-cells, these cells were naïve, whereas MS patients had activated and memory myelin-specific T-cells, indicating that these cells had been previously activated in MS patients.32–34 In addition, a clinical trial with an altered peptide ligand from myelin basic protein, which was intended to downregulate myelin-specific T-cells, actually exacerbated disease in several MS patients, and was associated with increased frequency of myelin basic protein-specific T-cells that produced IFNγ, suggesting that MS can be mediated by myelin-specific Th1 cells.35
As a result, several studies focused on IFNγ as the pathogenic molecule in EAE and MS. Surprisingly, IFNγ-deficient mice and mice given antibodies to neutralize IFNγ were still susceptible to EAE.32–34,36 The number of myelin-specific CD4+ T-cells was expanded in these mice, which may have occurred due to loss of regulatory cells that were dependent on IFNγ.37 However, several studies that specifically suppressed IFNγ in the myelin-specific T-cells prior to transfer into recipient mice demonstrated that altering the signaling pathway which results in IFNγ production in CD4+ T-cells decreased the encephalitogenic capacity of these cells.22,38,39 In addition, STAT4 and T-bet, transcription factors in the Th1 differentiation pathway, have been shown to be very important for induction of EAE.22,23,40–42 Together, these data suggest that the differentiation pathway that generates Th1 cells may be important in encephalitogenicity, but the downstream production of IFNγ by myelin-specific T-cells may not be critical. We examined how DMF affected the development of T-bet+, myelin-specific T-cells of both the Th1 and Th17 phenotype19,20 in the EAE model. Myelin-specific T-cells activated with antigen-presenting cells that had been exposed to FAE expressed lower levels of T-bet and RORgt, ie, transcription factors associated with the Th1 and Th17 lineages (Figure 1).19,20
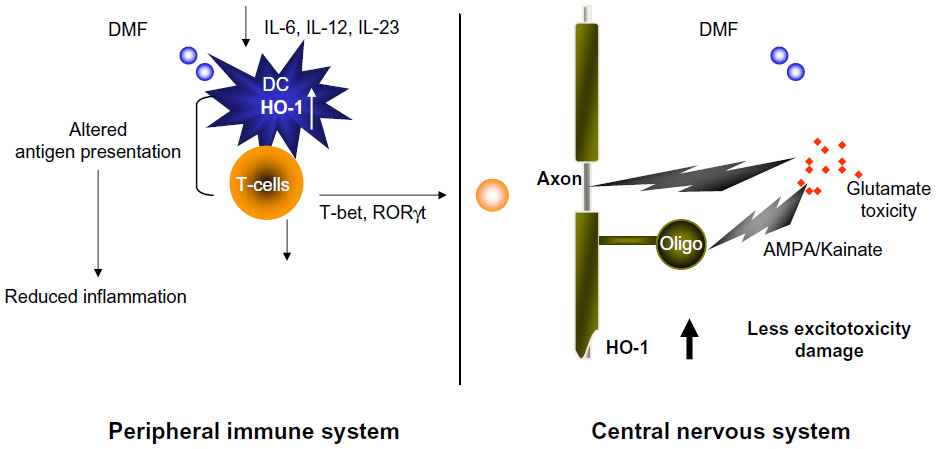 | Figure 1 Dimethyl fumarate (DMF) reduces peripheral inflammation and potentially enhances the cellular defense against glutamate toxicity in the central nervous system (CNS). |
The Th17 lineage, a new phenotype of CD4+ T-cells, was described in EAE and appeared to be pathogenic. Stimulation of myelin-specific T-cells from myelin-immunized mice with interleukin (IL)-23 promoted the expansion of T-cells expressing IL-17.43 Transfer of these myelin-specific IL-17-producing T-cells, called Th17 cells, into naïve mice induced EAE. In addition, microarray analysis of MS tissues indicates that IL-17 is present in MS lesions.44 We and others have shown that Th17 cells are found in the central nervous system of mice following adoptive transfer of myelin-specific Th1 cells; however, the Th17 cells are derived from the recipient and not the transferred Th1 cells.21,45 In addition, IL-17-deficient mice develop EAE, although the disease severity is reduced and the disease course is altered.46,47 These studies have raised the question as to whether the pathogenic T-cells in EAE and MS are actually Th1 or Th17 cells or whether both populations contribute to the disease. Our recent studies examining this issue suggest that both Th1 and Th17 cells can be pathogenic as long as they express the transcription factor T-bet.23 We subsequently examined the effects of DMF on the cytokine expression of dendritic cells and the subsequent effects on cytokine expression of both Th1 and Th17 cells.19,20 Exposure of dendritic cells to DMF prior to exposure to activating agents such as lipopolysaccharide resulted in decreased production of cytokines such as IL-12 and IL-23, which had been believed to be important in disease pathogenesis (Figure 1). This led to a change in the phenotype of the responding autoreactive T-cells, such that they were less likely to secrete proinflammatory cytokines such as IFNγ and IL-17 and more likely to secrete regulatory cytokines such as IL-4.19,20
Use of FAE in the treatment of autoimmune diseases
FAE were initially developed as a treatment for psoriasis, a putative Th1-mediated disorder.16 The compound utilized to treat patients in Europe was called Fumaderm®. Fumaderm was actually composed of a number of FAE, including DMF and monomethyl fumarate. After oral administration, it is known that DMF can become hydrolyzed to monomethyl fumarate, and this was thought to be the active ingredient for this compound.
Work with the drug initially supported the concept that FAE induced a shift in the immune response from Th1 to Th2. Increased production of Th2 cytokines such as IL-4, IL-5, and IL-10 was demonstrated.48,49 In addition, lipopolysaccharide-induced production of IL-12 and activation of NF-κB was also reported to be inhibited by FAE, although the mechanisms for these observations at a molecular level were unclear.50 Our own work supported the concept that FAE inhibited IL-12 production and NF-KB activation by antigen-presenting cells as well, making it less likely for responding T-cells to have a proinflammatory phenotype.20 Specifically, DMF inhibited MSK-1 expression, reducing p65 phosphorylation, and subsequent expression of inflammatory cytokines such as IL-6 and IL-12.
Studies conducted by Schilling et al using FAE in the EAE model indicated that there was a mild reduction in disease severity and that there was reduced infiltration of macrophages into the spinal cords of treated mice.51 In addition, there was an increase in IL-10 noted in the serum of FAE-treated mice. Additional studies by Linker et al showed that DMF upregulated the expression of Nrf-2 in neurons in the motor cortex and brainstem and in astrocytes and oligodendrocytes, suggesting a potential neuroprotective role.52 Another study by Chen et al has suggested that hydroxycarboxylic acid receptor 2 may be important for adhesion of neutrophils to endothelial cells in EAE.53 Since neutrophils are not prominent inflammatory cells in MS, the relevance of this finding to human disease remains unclear. In our preliminary studies, we utilized both monomethyl fumarate and DMF, and did not observe any significant differences between the two agents in terms of treatment effects. Both agents appeared to reduce the inflammatory infiltrates in the central nervous system and reduced the clinical signs of EAE.19 Because DMF has subsequently been utilized in the treatment of patients with MS and is better tolerated than the combination of FAE in Fumaderm, further studies have examined the effects of DMF.19,20
DMF was also studied in a rat model of myocarditis.54 Dark agouti rats were immunized with porcine cardiac myosin to induce autoimmune myocarditis. Rats were treated with 5 or 15 mg/kg of DMF on a daily basis. The higher dose of DMF reduced the severity of myocarditis and reduced the amount of tumor necrosis factor alpha (TNF-α) in the serum and supernatant of myosin-stimulated lymph node cells. Histopathology of the heart showed reduced mononuclear cell infiltration in the myocardium of the DMF-treated rats. Interestingly, no effect was shown on serum levels of IL-10. These results suggest that DMF is potentially effective in a number of autoimmune disorders.
Early MS clinical trials with FAE
Oral fumarates were initially utilized in a small pilot trial in ten patients with relapsing-remitting MS (RRMS). Fumaderm reduced the number and volume of gadolinium-enhancing lesions.18 These early results suggested that DMF may be a promising oral agent for the treatment of MS.
Oxidative stress and T-cell differentiation
Reactive oxygen species can initiate activation of NF-κB which can then induce expression of proinflammatory factors such as TNF-α and IL-12, molecules believed to be important in the pathogenesis of EAE and MS.55,56 Because excessive products of oxidation can exacerbate the inflammatory response, an important, efficient antioxidant defense system has evolved that is both enzymatic and nonenzymatic in nature.57,58 The enzymatic antioxidants include, but are not limited to, molecules such as superoxide dismutase, catalase, glutathione peroxidase, glutathione S-transferase, and thioredoxin. The nonenzymatic antioxidants include molecules such as glutathione, ascorbate (vitamin C), α-tocopherol (vitamin E), urate, bilirubin, and lipoic acid.59 Glutathione is the major intracellular thiol antioxidant that acts directly as a reactive oxygen species scavenger. The glutathione redox system is very important in regulating the intracellular redox balance and maintaining antioxidant function.60,61 We hypothesized that the ability of DMF to modulate cytokine production by antigen-presenting cells may have to do with its effects on reactive oxygen species. By manipulating intracellular glutathione levels in antigen-presenting cells, we could affect the subsequent cytokines produced by antigen-presenting cells when they were exposed to various activation stimuli.19
Accumulating evidence shows that heme oxygenase-1 (HO-1) is able to directly modulate the immune response. Under lipopolysaccharide stimulation, splenocytes from HO-1-deficient mice demonstrated elevated production of proinflammatory cytokines, including IL-6, IL-1, TNF-α, and IFN-γ, indicating that deficiency of HO-1 leads to an exaggerated inflammatory response.62 HO-1 has demonstrated an anti-inflammatory function in several autoimmune disease models. Systemic expression of HO-1 ameliorated type 1 diabetes and was associated with a decrease in the Th1 response.63 In models of arthritis and lupus, disease severity was reduced by induction of HO-1 and suppression of the inflammatory response.64,65 Clinical studies in humans have also supported an anti-inflammatory function of HO-1. A recent genetic study identified HO-1 expression as a marker for susceptibility to rheumatoid arthritis.66 This led us to examine whether DMF might lead to upregulation of HO-1 and be responsible for the observed effects on cytokine production in EAE and MS.19 Indeed, reactive oxygen species stress could upregulate the production of HO-1 in response to Toll-like receptor signaling, and this led to a number of effects that likely caused suppression of clinical signs of EAE, MS, and psoriasis in humans.19 We also showed that this HO-1 is subsequently cleaved and its N-terminal fragment interacts with AP-1 and NF-κB sites in the IL-23p19 promoter to inhibit IL-23p19 transcription.
HO-1 as a target in MS
During the pathogenesis of the MS lesion, monocyte-derived macrophages cause damage to the myelin sheath, oligodendrocytes, and axons. During the inflammatory process, macrophages/microglia produce a number of mediators, including cytokines, chemokines, nitric oxide, and reactive oxygen species. The reactive oxygen species likely contribute to evolution of the MS lesion.58 Expression of most antioxidant enzymes is regulated by the Nrf2/ARE pathway and is induced by oxidative stress. In the brains of MS patients, the presence of antioxidants regulated by the Nrf2/ARE pathway suggest that oxidative stress occurs in MS lesions.52 In EAE, the protective effects of exogenous antioxidants have been described.67 It does appear that ARE-driven genes are preferentially activated in astrocytes in the central nervous system, and this activation of astrocytes also prevented oxidative damage in neighboring neurons.68,69 Thus, antioxidant therapy may be a potential therapeutic strategy to protect both glia and neurons in diseases like MS.
Glutathione has been measured in the cerebrospinal fluid of patients with MS and found to be significantly lower than that in controls.70 Interestingly, plasma levels were increased in MS patients. Administration of oral L-acetylcarnitine to MS patients resulted in normalization of glutathione levels in cerebrospinal fluid. Treatment of the MS patients with L-acetylcarnitine also resulted in decreases in cerebrospinal fluid levels of nitric oxide reactive metabolites and protein nitration in addition to the higher glutathione levels. It is also of interest that acetylcarnitine was shown to have neuroprotective effects in rats exposed to global cerebral ischemia.71
Glutathione peroxidases may be more important than catalase in removing hydrogen peroxide from the central nervous system. Expression of glutathione peroxidase was also increased in active MS lesions.69 Studies by Guy et al suggested that treatment with glutathione peroxidases reduced blood–brain barrier breakdown in chronic EAE and indicated the potential for glutathione peroxidases as a therapy for neuroinflammatory diseases such as MS.72 Upregulation of HO-1 also reduced the clinical severity of EAE, while inhibiting HO-1 markedly exacerbated the disease.73 HO-1 expression has been shown to be increased in hypertrophic astrocytes in the MS spinal cord.74 Another study demonstrated that HO-1 was protective in the context of EAE.75 Mice deficient in HO-1 developed more severe EAE when compared with wild-type mice, while induction of HO-1 by administration of cobalt protoporphyrin after disease onset appeared to reverse signs of the disease. HO-1 also inhibited T-cell proliferation by inhibiting class II expression on antigen-presenting cells. These authors suggested that carbon monoxide might be a potential therapy for MS, although it is not clear how this would be achieved without its known toxicity. Another study suggested that DMF may mediate its actions through histone deacetylases. The inflammatory activation of astrocytes was inhibited by DMF, which was reversed by inhibition of HO-1.76 As a whole, all these studies suggest that manipulation of oxidative stress and targeting the HO-1 pathway may be a promising novel avenue for the treatment of MS. We have shown that administration of DMF results in increased production of HO-1 in both EAE and in patients with psoriasis (Figure 1).19 We were also able to show that increased HO-1 negatively regulated expression of the p19 subunit of IL-23. Further, we showed that nuclear HO-1 interfered with the transcriptional activity of AP-1 and NF-κB.19,20 We showed that HO-1 interfered with NF-kB binding at the promoter of p19 subunit of IL-23, suppressing its production by antigen-presenting cells.19 Thus, fumarates appear to induce type II dendritic cells that can ameliorate inflammatory autoimmune disorders such as MS and psoriasis.
Role of glutamate toxicity in EAE and MS
To delineate the neuroprotective effects of DMF, a model is desirable that replicates the pathomechanisms relevant to EAE and MS without involving inflammation, so that neuroprotective effects could be differentiated from anti-inflammatory effects. A study in which DMF was suggested to exert neuroprotective activity could have also produced reduced tissue damage in the central nervous system because of its anti-inflammatory activity.52 An obvious choice for such a pathomechanism is that of white matter excitotoxicity. Pitt et al and others have shown that glutamate excitotoxicity is a major effector mechanism in mouse EAE,77–79 where blockade of 2-(aminomethyl)phenylacetic acid/kainate receptors during acute disease significantly prevents oligodendroglial and axonal damage without altering the inflammatory response. Glutamate toxicity has also been implicated in MS by demonstrating that glutamate homeostasis is severely disrupted in white matter plaques.80,81
Preliminary data indicate that excitotoxic damage to axons is mediated by reactive oxygen species released by oligodendrocytes. It is therefore reasonable to speculate that in this paradigm, treatment aimed at inducing antioxidative enzymes may reduce axonal damage. Thus, DMF may be able to induce neuroprotective effects on white matter, although work is ongoing to prove that this mechanism is in play during DMF treatment of inflammatory demyelination.82
Benefit and risk assessment of DMF
An early open-label study of Fumaderm in ten patients with RRMS suggested that it was safe and efficacious by demonstrating significant reductions in MRI gadolinium-enhancing lesions along with stability in clinical measures of baseline Expanded Disability Status Scale, Ambulation Index, and the 9-Hole Peg Test.18 Subsequently, a dose-finding Phase IIb study of DMF compared with placebo demonstrated an overall 32% reduction in annualized relapse rate, significant reductions in gadolinium-enhancing lesions, and reductions in new or enlarging T2 lesions on magnetic resonance imaging (MRI) in the treatment arms as compared with placebo.17 Building on the evidence of efficacy in the early clinical studies, two 2-year, randomized, double-blind, placebo-controlled Phase III clinical trials confirmed DMF to be a safe and effective treatment for RRMS (Table 1).12,83
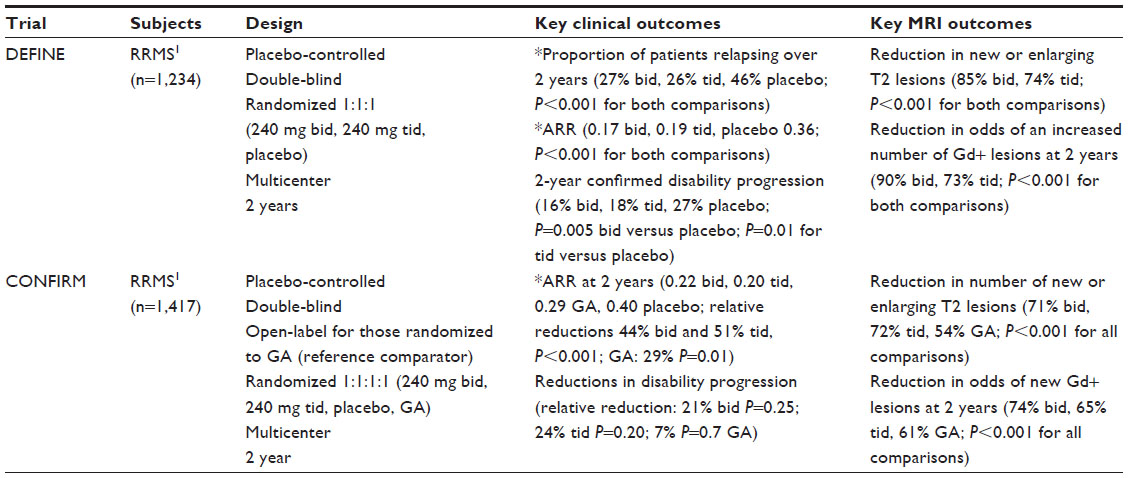 | Table 1 Efficacy data from pivotal Phase III clinical trials of dimethyl fumarate |
The DEFINE (Determination of the Efficacy and Safety of Oral Fumarate in RRMS) study compared two doses of DMF 240 mg twice daily and 240 mg three times daily against placebo.12 The primary endpoint of the study was met, with the proportion of patients who relapsed by the end of 2 years being 27% for DMF twice daily, 26% for DMF three times daily, and 46% for placebo (P<0.001 for both comparisons).12 Additionally, secondary endpoints of annualized relapse rate and time to disability progression were significant. The annualized relapse rate was 0.17 for DMF twice daily, 0.19 for DMF three times daily, and 0.36 for placebo (P<0.001 for both comparisons) consistent with a relative reduction of 48%–54% for the DMF arms,12 and confirmed disability progression occurring throughout the 2-year study was 16% for DMF twice daily, 18% for DMF three times daily and 27% for placebo (P=0.005 for DMF twice daily and P=0.01 for DMF three times daily).12 Further, MRI measures of new or enlarging T2 lesions and the number of gadolinium-enhancing lesions were significantly reduced in both DMF arms as compared with placebo (P<0.001 for both comparisons with placebo).12
CONFIRM (Comparator and an Oral Fumarate in RRMS; ClinicalTrials.gov identifier NCT00451451) compared DMF 240 mg twice daily and three times daily against placebo and included an open-label reference comparator of glatiramer acetate 20 mg subcutaneously once daily. The primary endpoint of annualized relapse rate, was significantly lower for both DMF arms, ie, 0.22 twice daily, 0.20 three times daily, with 0.29 for glatiramer acetate, and 0.4 for placebo, demonstrating a relative reduction of 44% for twice daily and 51% for three times daily (P<0.001 for both comparisons), and 29% for glatiramer acetate (P<0.01).83 There were also significant reductions in new or enlarging T2 lesions in all treatment arms as compared with placebo (P<0.001 for each comparison).83 In contrast with the DEFINE study, there was no significant difference in disability progression in the individual treatment arms when compared with placebo in CONFIRM.12,80 This discrepancy may be due to a higher percentage of patients with confirmed disability progression in DEFINE (27%) as compared with CONFIRM (17%).12,83
Common adverse events in the Phase III clinical trials included gastrointestinal (diarrhea, nausea, emesis, or abdominal pain) and flushing (erythema, warmth, pruritus, or burning sensation) events that were mild to moderate in the majority of patients.12,80 The incidence of side effects decreased with time on therapy. Reassuringly, DMF was not associated with an increased risk of malignancy, serious infections, or opportunistic infections.12,83 A decrease in mean white blood cell and lymphocyte counts and elevations in liver aminotransferases were identified in patients on DMF.12,80 In the first year on DMF, mean lymphocyte counts decreased by approximately 30% and then stabilized.84 In the placebo-controlled trials, 6% of patients on DMF and <1% on placebo had a decrease in lymphocyte counts <0.5×109/L (lower limit of normal 0.91×109/L). The incidence of infections was similar for patients on DMF and placebo (60% versus 58%) and also for serious infections (2% versus 2%).84 Elevations in hepatic aminotransferases in patients on DMF occurred primarily in the first 6 months on therapy, with the majority of these being less than three times the upper limit of normal.12,83 Elevations of three times or more the upper limit of normal were equal in patients on DMF and placebo.12,83,84 DMF is pregnancy category C and so not recommended for use in pregnant women.
In March 2013, DMF gained FDA approval in the USA as the third oral disease-modifying treatment for RRMS. The starting dose of DMF is 120 mg by mouth twice daily for 7 days, and then increased to 240 mg by mouth twice daily. A complete blood count is recommended prior to initiation of DMF, and should be repeated annually for safety monitoring.
Shortly after DMF gained FDA approval, four cases of PML were reported in patients treated with Fumaderm or compounded FAE for psoriasis, comprising three cases treated with Fumaderm and one treated with compounded FAE.85 One of these patients had a history of prior immunosuppressant use with methotrexate and developed severe lymphocytopenia one year after initiation of oral fumaric acid; however, it was not discontinued despite the manufacturer’s recommendation to discontinue therapy with severe lymphocytopenia.86 This patient developed PML 3 years after initiation of fumaric acid.86 A second patient, treated with compounded FAE (Psorinovo) for 6 years, with long-standing lymphopenia at 200 lymphocytes/mm3, developed PML.87 A third patient with a history of sarcoidosis treated with methotrexate and steroids developed PML one month after starting Fumaderm.85 A fourth patient on efalizumab for psoriasis was diagnosed with cancer, after which efalizumab was discontinued and Fumaderm was started for continued treatment of psoriasis. This patient eventually developed PML.85 Each of these patients had at least one or more known risk factors for PML, including lymphocytopenia, sarcoidosis, cancer, and efalizumab use.85 The risk of PML can be mitigated by routine lymphocyte monitoring. In the DMF clinical trials program, more than 2,600 MS patients were treated for up to 4 years with no reports of PML.12,83 With appropriate monitoring, DMF is a safe and efficacious disease-modifying treatment option for patients with RRMS.
Comparison of DMF with other disease-modifying therapies
Efficacy
There have been no head-to-head clinical trials comparing DMF with other disease-modifying treatments. Nevertheless, Hutchinson et al recently performed a meta-analysis using mixed treatment comparisons.88 Mixed treatment comparisons are typically used in the absence of sufficient direct head-to-head comparisons allowing for analysis across clinical trials. The investigators analyzed data from 27 randomized clinical trials of disease-modifying treatments using standard FDA-approved dosages and demonstrated that DMF 240 mg twice daily significantly reduces the annualized relapse rate as compared with placebo, IFN, glatiramer acetate, and teriflunomide. No significant difference was found when comparing DMF with fingolimod.88 Natalizumab was superior to DMF in reducing the annualized relapse rate in this meta-analysis.88
Safety and tolerability
Considering a disease-modifying treatment for a given MS patient requires consideration of the drug’s efficacy and safety profile. Unlike several other recently approved disease-modifying treatments, DMF does not require extensive screening tests prior to initiation (except for recent complete blood counts), or have known safety concerns regarding opportunistic infections, risk of malignancy, cardiac and ocular side effects, or teratogenicity.
Fingolimod and natalizumab have been associated with serious infections. There have been two reported cases of fingolimod-treated MS patients developing disseminated varicella zoster.89,90 Natalizumab is associated with a risk of PML.91 Fingolimod is associated with risk of first-dose bradycardia and atrioventricular block. It is also associated with a rare risk of macular edema.92 Teriflunomide is also pregnancy category X.93 In this respect, the side effect profile of DMF may be considered favorable. Fingolimod, natalizumab, teriflunomide, and DMF are all associated with a risk of elevated hepatic transaminases.9,12,92,94
Mode of administration and frequency of dosing for disease-modifying treatments is also a consideration in comparing these therapies. Injectable therapies may be less desirable than pills, given injection site reactions or associated flu-like symptoms with IFNs.95 Conversely, twice-daily dosing of DMF may be less favorable than once-daily administration of fingolimod or teriflunomide.
None of the FDA-approved disease-modifying treatments are recommended for use during pregnancy. Teriflunomide is pregnancy category X and carries significant risk during pregnancy as demonstrated by studies in animals.
Teriflunomide-treated MS patients must either wait up to 24 months or undergo rapid elimination with cholestyramine and activated charcoal before serum drug levels are low enough to allow safe conception.96 Natalizumab, fingolimod, IFNs, and DMF are pregnancy category C.93 Glatiramer acetate is pregnancy category B.93 Taken as a whole, the moderate efficacy, acceptable side effect profile, and oral route of administration of DMF make this therapy an attractive option for patients with relapsing forms of MS.
Effect of DMF on health-related quality of life in RRMS
Individuals living with MS report a lower health-related quality of life (HRQoL) as compared with healthy controls and patients with other chronic diseases.97,98 Moreover, HRQoL tends to decline in MS with disease progression.97 For these reasons, HRQoL may serve as an additional indicator of therapeutic efficacy along with traditional measures such as relapse rate, disability progression and MRI activity. Several measures of HRQoL in MS were assessed in the recently completed DEFINE and CONFIRM trials, which evaluated the efficacy and safety of DMF in RRMS.12,83 In this review, we will focus on the combined HRQoL analyses from both trials.99
HRQoL was assessed in both trials using three patient-reported outcome measures. The first was the Short Form 36 (SF-36), which was assessed in both trials at baseline, and at 6, 12, and 24 months. It is comprised of eight multi-item scales (0–100, higher is better), and patient-reported data is grouped into two summary scores, ie, the physical component summary (PCS) and the mental component summary (MCS). The second HRQoL measure used was the European Quality of Life-5 Dimensions Health Survey (EQ-5D), which was also administered in both trials at baseline, and at 6, 12, and 24 months. The EQ-5D scores are combined to provide a summary index score (higher is better). Lastly, both trials administered the patient global impression of well-being visual analog scale (GI-VAS) at baseline and every 3 months thereafter (0–100, higher is better).100,101 For both trials, the treatment effect of DMF on HRQoL was assessed in the intent to treat study population (combined placebo, n=711; DMF, twice daily, n=769; DMF three times daily, n=761) comparing mean changes at 2 years from baseline scores on all the above measures, adjusting for region. Post hoc analysis included calculating the proportion of patients manifesting a clinically relevant improvement from baseline (defined as a 5-point change) at 2 years on the PCS and MCS. Another post hoc analysis examined baseline SF-36 scores relative to baseline Expanded Disability Status Scale scores.
Consistent with prior investigations, the authors found baseline HRQoL scores were lower than in the general population.99 No significant between-group differences were found among baseline PCS and MCS scores. Baseline HRQoL was lower in patients with higher baseline Expanded Disability Status Scale scores.
There was a treatment effect favoring DMF over placebo in mean change from baseline for PCS and MCS scores in the integrated analysis. For PCS, this was statistically significant in the thrice-daily arm at 24, 48, and 96 weeks and in the twice-daily arm at 48 and 96 weeks. For the MCS score, differences from placebo were only statistically significant at 96 weeks for both treatment arms.99 When considering a clinically relevant change in HRQoL, there was a statistically significant 5-point change in both PCS and MCS scores at 2 years favoring DMF-treated patients over placebo (PCS, twice-daily arm odds ratio 1.26, P=0.02, and thrice-daily arm odds ratio 1.48, P=0.0001; MCS, twice-daily arm odds ratio 1.42, P=0.008, and thrice-daily arm odds ratio 1.63, P<0.0001). At 2 years, EQ-5D index summary scores were statistically improved in DMF-treated patients compared with the placebo arm. Similarly, the mean change from baseline to 2 years in the GI-VAS was significantly improved compared with placebo (−4.0) in both the twice-daily arms (−0.3, P<0.0001) and thrice-daily arms (0.1, P<0.0001). The authors conclude that in addition to significant treatment effects in traditional efficacy outcome measures, DMF-treated patients in the DEFINE and CONFIRM trials manifested improvements in their overall HRQoL.
Conclusion and place in therapy
In conclusion, DMF is the most recent FDA-approved oral disease-modifying treatment for the treatment of relapsing forms of MS. This therapy has demonstrated efficacy as a first-line therapy and should be considered as such in treatment selection for individuals with RRMS. Therefore, patients and clinicians may consider DMF over injectable therapies due to its demonstrated efficacy, ease of treatment initiation, and ease of administration. The Phase III clinical trials program demonstrated its success as an efficacious oral disease-modifying treatment with a favorable tolerability and safety profile. The early side effects of DMF, ie, gastrointestinal and flushing reactions, decrease with time on therapy and are generally self-limited. DMF used for the treatment of MS has not been associated with any serious or opportunistic infections. The data suggest that DMF positively affects quality of life in MS patients. Results from the Phase III clinical trials in MS and extensive clinical experience in psoriasis suggest that DMF has a robust long-term safety profile and supports its use in MS. Further studies will need to be done to completely delineate the neuroimmunologic effects of DMF and whether or not it is neuroprotective in inflammatory demyelination.
Acknowledgments
JAN is supported by a Sylvia Lawry Physician Award grant through the National Multiple Sclerosis Society. MKR is supported in part by a grant from the National Multiple Sclerosis Society. The authors would like to thank Haiyan Peng for designing Figure 1 for this manuscript. Dr Peng graduated from The Ohio State University Department of Neurology and Neurosciences Graduate Program in 2013 and is now in a post-doctoral research program at Biogen Idec.
Disclosure
JAN has received personal compensation for consulting from Novartis. She has received grant funding from the National Multiple Sclerosis Society via a Sylvia Lawry Physician Award grant and from Biogen Idec. ALB has received personal compensation from Merck Serono, Biogen, Novartis, Teva, Questcor, and Medtronic for consulting. He has also received financial support for research activities from Merck Serono, Biogen Idec, Novartis, Actilleon, Sun-Pharma, Roche, CNS Therapeutics, Jazz Pharmaceuticals, Accorda, the National MS Society and the National Institutes of Health. CO has nothing to disclose. JI has received research funding from the National Multiple Sclerosis Society but has nothing to disclose. MKR has received personal compensation for consulting from Revalesio, Inc., Mylan, and Novartis. He has received grant support from the National Multiple Sclerosis Society and the National Institutes of Health, and has received compensation as editor-in-chief of the Journal of Neuroimmunology and as a member of the editorial board of JAMA Neurology.
References
Anderson DW, Ellenberg JH, Leventhal CM, et al. Revised estimate of the prevalence of multiple sclerosis in the United States. Ann Neurol. 1992;31(3):333–336. | |
Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med. 2006;354(9):942–955. | |
Arnason BG. Relevance of experimental allergic encephalomyelitis to multiple sclerosis. Neurol Clin. 1983;1(3):765–782. | |
Sadovnick AD, Ebers GC. Epidemiology of multiple sclerosis: a critical overview. Can J Neurol Sci. 1993;20(1):17–29. | |
[No authors listed]. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. 1993;43(4):655–661. | |
Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol. 1996;39(3):285–294. | |
Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45(7):1268–1276. | |
Millefiorini E, Gasperini C, Pozzilli C, et al. Randomized placebo-controlled trial of mitoxantrone in relapsing-remitting multiple sclerosis: 24-month clinical and MRI outcome. J Neurol. 1997;244(3):153–159. | |
Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. | |
Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. | |
O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. | |
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | |
Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353(4):369–374. | |
Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353(4):375–381. | |
Van Assche G, Van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353(4):362–368. | |
Altmeyer PJ, Matthes U, Pawlak F, et al. Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J Am Acad Dermatol. 1994;30(6):977–981. | |
Kappos L, Gold R, Miller DH, Haghikia A. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2009;373(9672):1340–1340. | |
Schimrigk S, Brune N, Hellwig K, et al. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baseline-controlled pilot study. Eur J Neurol. 2006;13(6):604–610. | |
Ghoreschi K, Bruck J, Kellerer C, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208(11):2291–2303. | |
Peng H, Guerau-de-Arellano M, Mehta VB, et al. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor kappaB (NF-kappaB) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and mitogen stress-activated kinase 1 (MSK1) signaling. J Biol Chem. 2012;287(33):28017–28026. | |
Gocke AR, Cravens PD, Ben LH, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178(3):1341–1348. | |
Lovett-Racke AE, Rocchini AE, Choy J, et al. Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity. 2004;21(5):719–731. | |
Yang Y, Weiner J, Liu Y, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. 2009;206(7):1549–1564. | |
McDonald AH, Swanborg RH. Antigen-specific inhibition of immune interferon production by suppressor cells of autoimmune encephalomyelitis. J Immunol. 1988;140(4):1132–1138. | |
Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124(1):132–143. | |
Waldburger KE, Hastings RC, Schaub RG, Goldman SJ, Leonard JP. Adoptive transfer of experimental allergic encephalomyelitis after in vitro treatment with recombinant murine interleukin-12. Preferential expansion of interferon-gamma-producing cells and increased expression of macrophage-associated inducible nitric oxide synthase as immunomodulatory mechanisms. Am J Pathol. 1996;148(2):375–382. | |
Yura M, Takahashi I, Serada M, et al. Role of MOG-stimulated Th1 type “light up” (GFP+) CD4+ T cells for the development of experimental autoimmune encephalomyelitis (EAE). J Autoimmun. 2001;17(1):17–25. | |
Olsson T, Zhi WW, Hojeberg B, et al. Autoreactive T lymphocytes in multiple sclerosis determined by antigen-induced secretion of interferon-gamma. J Clin Invest. 1990;86(3):981–985. | |
Sun JB, Olsson T, Wang WZ, et al. Autoreactive T and B cells responding to myelin proteolipid protein in multiple sclerosis and controls. Eur J Immunol. 1991;21(6):1461–1468. | |
Voskuhl RR, Martin R, Bergman C, Dalal M, Ruddle NH, McFarland HF. T helper 1 (Th1) functional phenotype of human myelin basic protein-specific T lymphocytes. Autoimmunity. 1993;15(2):137–143. | |
Pelfrey CM, Rudick RA, Cotleur AC, Lee JC, Tary-Lehmann M, Lehmann PV. Quantification of self-recognition in multiple sclerosis by single-cell analysis of cytokine production. J Immunol. 2000;165(3):1641–1651. | |
Ferber IA, Brocke S, Taylor-Edwards C, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. 1996;156(1):5–7. | |
Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157(8):3223–3227. | |
Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-gamma. Eur J Immunol. 1996;26(10):2393–2398. | |
Bielekova B, Goodwin B, Richert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6(10):1167–1175. | |
Lublin FD, Knobler RL, Kalman B, et al. Monoclonal anti-gamma interferon antibodies enhance experimental allergic encephalomyelitis. Autoimmunity. 1993;16(4):267–274. | |
Chu CQ, Wittmer S, Dalton DK. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192(1):123–128. | |
Racke MK, Burnett D, Pak SH, et al. Retinoid treatment of experimental allergic encephalomyelitis. IL-4 production correlates with improved disease course. J Immunol. 1995;154(1):450–458. | |
Racke MK, Bonomo A, Scott DE, et al. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J Exp Med. 1994;180(5):1961–1966. | |
Chitnis T, Najafian N, Benou C, et al. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108(5):739–747. | |
Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200(1):79–87. | |
Nath N, Prasad R, Giri S, Singh AK, Singh I. T-bet is essential for the progression of experimental autoimmune encephalomyelitis. Immunology. 2006;118(3):384–391. | |
Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–240. | |
Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8(5):500–508. | |
Lees JR, Iwakura Y, Russell JH. Host T cells are the main producers of IL-17 within the central nervous system during initiation of experimental autoimmune encephalomyelitis induced by adoptive transfer of Th1 cell lines. J Immunol. 2008;180(12):8066–8072. | |
Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177(1):566–573. | |
Haak S, Croxford AL, Kreymborg K, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119(1):61–69. | |
de Jong R, Bezemer AC, Zomerdijk TP, van de Pouw-Kraan T, Ottenhoff TH, Nibbering PH. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur J Immunol. 1996;26(9):2067–2074. | |
Ghoreschi K, Mrowietz U, Rocken M. A molecule solves psoriasis? Systemic therapies for psoriasis inducing interleukin 4 and Th2 responses. J Mol Med (Berl). 2003;81(8):471–480. | |
Loewe R, Pillinger M, de Martin R, et al. Dimethylfumarate inhibits tumor-necrosis-factor-induced CD62E expression in an NF-kappa B-dependent manner. J Invest Dermatol. 2001;117(6):1363–1368. | |
Schilling S, Goelz S, Linker R, Luehder F, Gold R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol. 2006;145(1):101–107. | |
Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134 Pt 3:678–692. | |
Chen H, Assmann JC, Krenz A, et al. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE. J Clin Invest. 2014;124(5):2188–2192. | |
Milenkovic M, Arsenovic-Ranin N, Vucicevic D, Bufan B, Jancic I, Stojic-Vukanic Z. Beneficial effects of dimethyl fumarate on experimental autoimmune myocarditis. Arch Med Res. 2008;39(7):639–646. | |
Sean RD, Korner H, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD. Challenging cytokine redundancy: inflammatory cell movement and clinical course of experimental autoimmune encephalomyelitis are normal in lymphotoxin-deficient, but not tumor necrosis factor-deficient, mice. J Exp Med. 1998;187(9):1517–1528. | |
Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205(7):1535–1541. | |
Johnson JA, Johnson DA, Kraft AD, et al. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann N Y Acad Sci. 2008;1147:61–69. | |
Schreibelt G, van Horssen J, van Rossum S, Dijkstra CD, Drukarch B, de Vries HE. Therapeutic potential and biological role of endogenous antioxidant enzymes in multiple sclerosis pathology. Brain Res Rev. 2007;56(2):322–330. | |
van Muiswinkel FL, Kuiperij HB. The Nrf2-ARE signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord. 2005;4(3):267–281. | |
Franco R, Schoneveld OJ, Pappa A, Panayiotidis MI. The central role of glutathione in the pathophysiology of human diseases. Arch Physiol Biochem. 2007;113(4–5):234–258. | |
Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134(3):489–492. | |
Kapturczak MH, Wasserfall C, Brusko T, et al. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. 2004;165(3):1045–1053. | |
Hu CM, Lin HH, Chiang MT, Chang PF, Chau LY. Systemic expression of heme oxygenase-1 ameliorates type 1 diabetes in NOD mice. Diabetes. 2007;56(5):1240–1247. | |
Benallaoua M, Francois M, Batteux F, et al. Pharmacologic induction of heme oxygenase 1 reduces acute inflammatory arthritis in mice. Arthritis Rheum. 2007;56(8):2585–2594. | |
Takeda Y, Takeno M, Iwasaki M, et al. Chemical induction of HO-1 suppresses lupus nephritis by reducing local iNOS expression and synthesis of anti-dsDNA antibody. Clin Exp Immunol. 2004;138(2):237–244. | |
Rueda B, Oliver J, Robledo G, et al. HO-1 promoter polymorphism associated with rheumatoid arthritis. Arthritis Rheum. 2007;56(12):3953–3958. | |
Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. 2004;251(3):261–268. | |
Liu Y, Liu J, Tetzlaff W, Paty DW, Cynader MS. Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic Biol Med. 2006;40(6):960–967. | |
Tajouri L, Mellick AS, Ashton KJ, et al. Quantitative and qualitative changes in gene expression patterns characterize the activity of plaques in multiple sclerosis. Brain Res Mol Brain Res. 2003;119(2):170–183. | |
Calabrese V, Scapagnini G, Ravagna A, et al. Disruption of thiol homeostasis and nitrosative stress in the cerebrospinal fluid of patients with active multiple sclerosis: evidence for a protective role of acetylcarnitine. Neurochem Res. 2003;28(9):1321–1328. | |
Calvani M, Arrigoni-Martelli E. Attenuation by acetyl-L-carnitine of neurological damage and biochemical derangement following brain ischemia and reperfusion. Int J Tissue React. 1999;21(1):1–6. | |
Guy J, Ellis EA, Hope GM, Rao NA. Antioxidant enzymes reduce loss of blood-brain barrier integrity in experimental optic neuritis. Arch Ophthalmol. 1989;107(9):1359–1363. | |
Liu Y, Zhu B, Luo L, Li P, Paty DW, Cynader MS. Heme oxygenase-1 plays an important protective role in experimental autoimmune encephalomyelitis. Neuroreport. 2001;12(9):1841–1845. | |
Mehindate K, Sahlas DJ, Frankel D, et al. Proinflammatory cytokines promote glial heme oxygenase-1 expression and mitochondrial iron deposition: implications for multiple sclerosis. J Neurochem. 2001;77(5):1386–1395. | |
Chora AA, Fontoura P, Cunha A, et al. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest. 2007;117(2):438–447. | |
Kalinin S, Polak PE, Lin SX, et al. Dimethyl fumarate regulates histone deacetylase expression in astrocytes. J Neuroimmunol. 2013;263(1–2):13–19. | |
Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6(1):67–70. | |
Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6(1):62–66. | |
Kanwar JR, Kanwar RK, Krissansen GW. Simultaneous neuroprotection and blockade of inflammation reverses autoimmune encephalomyelitis. Brain. 2004;127 Pt 6:1313–1331. | |
Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50(2):169–180. | |
Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128 Pt 5:1016–1025. | |
Lukashev M, Zeng W, Goelz S, et al. Activation of Nrf2 and modulation of disease progression in EAE models by BG00012 (dimethyl fumarate) suggests a novel mechanism of action combining anti-inflammatory and neuroprotective modalities. Mult Scler. 2007;13 Suppl:S149–S149. | |
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. | |
Tecfidera® Full prescribing information. Dimethyl fumarate package insert. Research Triangle Park, NC, USA: Biogen Idec; 2013. Available from: http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=665d7e74-036c-5f68-5b67-ab84b9b49151. Accessed June 9, 2014. | |
Sweetser MT, Dawson KT, Bozic C. Manufacturer’s response to case reports of PML. N Engl J Med. 2013;368(17):1659–1661. | |
Ermis U, Weis J, Schulz JB. PML in a patient treated with fumaric acid. N Engl J Med. 2013;368(17):1657–1658. | |
van Oosten B, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med. 2013;368(17):1658–1659. | |
Hutchinson M, Fox RJ, Havrdova E, et al. Efficacy and safety of BG-12 (dimethyl fumarate) and other disease-modifying therapies for the treatment of relapsing-remitting multiple sclerosis: a systematic review and mixed treatment comparison. Curr Med Res Opin. 2014;30(4):613–627. | |
Ratchford JN, Costello K, Reich DS, Calabresi PA. Varicella-zoster virus encephalitis and vasculopathy in a patient treated with fingolimod. Neurology. 2012;79(19):2002–2004. | |
Uccelli A, Ginocchio F, Mancardi GL, Bassetti M. Primary varicella zoster infection associated with fingolimod treatment. Neurology. 2011;76(11):1023–1024. | |
Nicholas JA, Racke MK, Imitola J, Boster AL. First-line natalizumab in multiple sclerosis: rationale, patient selection, benefits and risks. Ther Adv Chronic Dis. 2014;5(2):62–68. | |
Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. | |
Coyle PK. Multiple sclerosis in pregnancy. Continuum (Minneap Minn). 2014;20(1 Neurology of Pregnancy):42–59. | |
O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. | |
Polman CH, Uitdehaag BM. New and emerging treatment options for multiple sclerosis. Lancet Neurol. 2003;2(9):563–566. | |
Aubagio® (teriflunomide) package insert. Cambridge, MA, USA: Genzyme Corporation; 2012. Available from: http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=4650d12c-b9c8-4525-b07f-a2d773eca155. Accessed June 9, 2014. | |
Miller A, Dishon S. Health-related quality of life in multiple sclerosis: the impact of disability, gender and employment status. Qual Life Res. 2006;15(2):259–271. | |
Rudick RA, Miller D, Clough JD, Gragg LA, Farmer RG. Quality of life in multiple sclerosis. Comparison with inflammatory bowel disease and rheumatoid arthritis. Arch Neurol. 1992;49(12):1237–1242. | |
Kita M, Fox RJ, Gold R, et al. Effect of BG-12(DMF) on quality of life in patients with MS. Presented at the Consortium of Multiple Sclerosis Centers and Americas Committee for Treatment and Research in Multiple Sclerosis 18th Annual Meeting, Orlando, FL, USA, May 29 to June 1, 2013. | |
Kappos L, Gold R, Arnold DL, et al. Quality of life outcomes with BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: the DEFINE study. Mult Scler. 2014;20(2):243–252. | |
Kita M, Fox RJ, Phillips JT, et al. Effects of BG-12 (dimethyl fumarate) on health-related quality of life in patients with relapsing-remitting multiple sclerosis: findings from the CONFIRM study. Mult Scler. 2014;20(2):253–257. | |
Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis:2005 revisions to the “McDonald Criteria.” Ann Neurol. 2005;58(6):840–846. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.