Back to Journals » Infection and Drug Resistance » Volume 12
Design and characterization of a new hybrid peptide from LL-37 and BMAP-27
Authors Al Tall Y ![]() , Abualhaijaa A, Alsaggar M, Almaaytah A, Masadeh M, Alzoubi KH
, Abualhaijaa A, Alsaggar M, Almaaytah A, Masadeh M, Alzoubi KH ![]()
Received 25 December 2018
Accepted for publication 13 March 2019
Published 30 April 2019 Volume 2019:12 Pages 1035—1045
DOI https://doi.org/10.2147/IDR.S199473
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Joachim Wink
Yara Al Tall,1 Ahmad Abualhaijaa,1 Mohammad Alsaggar,1 Ammar Almaaytah,1 Majed Masadeh,1 Karem H Alzoubi2
1Department of Pharmaceutical Technology; 2Department of Clinical Pharmacy, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan
Background and purpose: The world is heading to a post-antibiotic era where the treatment of bacterial infections will not be possible even with well-known last-line antibiotics. Unfortunately, the emergence of multidrug resistant bacterial strains is uncontrollable, and the humanity will face a life-threatening fate unless new antimicrobial agents with new bacterial target sites are promptly developed. Herein, we design a hybrid antimicrobial peptide (B1) from helical parts taken from the parent peptides: LL-37 and BMAP-27. The purpose of this design is to improve the potency and enhance the toxicity profile of the parent peptides.
Methods: Rational design was used to hybridize two antimicrobial peptides, in which two helical parts from the bovine analog BMAP-27, and the human cathelicidin LL-37 were used to generate a novel peptide (B1). The physicochemical properties were checked using in silico methods. The antimicrobial activities were tested against nine control and resistant strains of Gram-positive and Gram-negative bacteria. On the other hand, the antibiofilm activities were tested against four resistant strains. The cytotoxicity on mammalian cells was tested using HEK293, and the hemolysis activity was also investigated on human blood. Finally, synergistic studies were performed with four conventional antibiotics against four resistant strains of Gram-positive and Gram-negative bacteria.
Results: The new peptide B1 exhibited broad-spectrum activities against all tested strains. The concentration against planktonic cells ranged between 10 and 20 μM. However, 40–60 μM were needed to eradicate the biofilms. B1 showed reduced toxicity toward mammalian cells with minimal hemolysis risk. On the other hand, the synergistic studies showed improved activities for the combined conventional antibiotics with a huge reduction in their minimum inhibitory concentration values. The concentrations of B1 peptide combined with the tested antibiotics were also decreased markedly down to 0.5 μM in some cases.
Conclusion: B1 is a hybrid peptide from two cathelicidin peptides. It showed an improved activity compared to parent peptides. The hybridization was successful in this study. It generated a new potent broad-spectrum antimicrobial. The toxicity profile was improved, and the synergism with the convention antibiotics showed promising results.
Keywords: antimicrobial peptides, rational design, hybridization, antimicrobial resistance, antibiofilm activity, synergism
Introduction
The prevalence of antibiotic resistance is increasing worldwide, leading to serious challenges in public health, and sets forth extra pressure on the pharmaceutical industry and public health leaders to tackle the problem.1 This is coupled with the crisis in antimicrobial drug discovery progress, as evidenced by declined rates of development of new anti-infectious agents into the market.2,3 Therefore, it is urgently needed to explore different strategies to address antibiotic resistance and to develop new safer and more effective antimicrobial agents.
Antimicrobial peptides (AMPs) are ubiquitously expressed in various species in nature, including animals, insects, and plants,4 and have been shown to play essential roles in the immune system, as they represent host defense mechanism in expressing species. Investigating biological activity of these peptides has revealed promising antimicrobial activity that could be harnessed in clinical practice to treat wide variety of bacterial, parasitic, fungal, and viral infections.5 AMPs exert antimicrobial effects by different mechanisms; through disruption of bacterial membranes, inhibition of essential cellular processes like cell well, DNA and proteins synthesis, as well as enzymatic activity.6 Given the wide diversity in these peptides and their biological effects, increasing research is directed toward the development of novel AMPs, and/or further improvement of current AMPs for enhanced antimicrobial activities with minimal adverse events.
Cathelicidins encompass several families of AMPs expressed in epithelial and neutrophils to act as host defense peptides.7 While cathelicidins contain a conserved N-terminal domain (called cathelin domain) and a cationic C-terminal domain of variable lengths and sequences, the antimicrobial activity of cathelicidins resides in the C-terminal domain.8 Mammalian cathelicidins exhibit a wide variety of linear and circular structures that are distinct from amphibian and insect cathelicidins, with α-helix is the most common peptide conformation in environments of biological membranes.4,9 The primary features of cathelicidins that account for their antimicrobial activity are net cationic, as a result of the dominance of basic residues in their sequences, and overall amphipathic structural topology, together allowing these peptides to bind and be inserted into negatively charged macrobian membranes.10 The only cathelicidin that has been reported in human is hCAP18; a propeptide having a cathelin domain and the C-terminal domain of 37 residues called LL-37 peptide. The antimicrobial activity of hCAP18 is attributed to the C-terminal LL-37 region, which is characterized by α-helical conformation and amphipathic topology, with a net charge of +6 at physiological pH.11 LL-37 is a membrane-active agent with a broad-spectrum activity against Gram-positive (G+ve) and Gram-negative (G-ve) pathogens, and most importantly against common clinical isolates of uropathogens and wound pathogens, such as Escherichia coli HU734, Pseudomonas aeruginosa AK1, Klebsiella pneumoniae 3a, and Group A Streptococcus.12,13 BMAPs (bovine myeloid antimicrobial peptides) are bovine cathelicidins that share many structural and functional features with human cathelicidin.14 BMAP-27 is a 26 amino-acid peptide with amidated C-terminus. It has shown potent antimicrobial effects against various species of bacteria and parasites. However, it seems to have hemolytic effects on cultured blood cells, as well as cytotoxic effects to hematopoietic cells and activated human lymphocytes.15
In this study, we introduced a rationally designed peptide that combines essential features of LL-37 and BMAP-27 peptides to generate a hybrid peptide with enhanced antimicrobial activity along with minimized toxicity. In fact, our new peptide B1 demonstrated promising antimicrobial and antibiofilm activity against representative species of G+ve and G-ve bacteria, and displayed minimal toxicity compared to the parent peptides.
Material and methods
Bacterial strains
Methicillin-susceptible Staphylococcus aureus (MS S. aureus) ATCC 29213, methicillin-resistant Staphylococcus aureus (MR S. aureus) ATCC 33591, MR S. aureus ATCC 43300 (oxacillin-resistant), and MR S. aureus ATCC BAA-41, S. epidermidis ATCC12228, Escherichia coli (E. coli) ATCC 25922, E. coli ATCC 35218, Enterococcus faecium (E. faecium) BAA-2316 (vancomycin and teicoplanin-resistant), and multidrug-resistant (MDR) Pseudomonas aeruginosa (P. aeruginosa) ATCC BAA-2114 were acquired from the American Type Tissue Culture Collection (ATCC; Manassas, VA, USA).
Materials and chemicals
All antibiotics and chemical reagents were bought from Sigma Chemical Co. (St. Louis, MO, USA) and were dealt with according to the manufacturer’s recommendations.
Peptide design, molecular modeling, and in silico analyses
The HNN (Hierarchical Neural Network) software was used to calculate the helicity of the hybrid peptide,16 whereas ProtParam/ExPASy server17 and the antimicrobial peptide database (APD)18 were used to predict the physicochemical properties. The HHpred19 and MODELLER software20 from MPI Bioinformatics Toolkit server21 were used to run homology modeling, and the Ramachandran Plot22 were assessed using The RAMPAGE software. ProSA-web23 was also used to validate the model and finally, the I-TASSER24 was finally used to confirm the model reliability.
Peptide synthesis and purification
The peptide was purchased from GL Biochem (Shanghai) Ltd. Solid-phase peptide synthesis method was adopted to synthesize the new hybrid peptide (NH2- KFKKLFKKLSPVFKRIVQRIKDFLR-COOH). The purity of higher than 95% was then obtained and checked by reversed-phase high-performance liquid chromatography (RP-HPLC) and electrospray ionization mass spectrometry (ESI-MS).
Bacterial susceptibility assay
The minimum inhibitory concentrations (MICs) of the hybrid peptide were evaluated using the broth dilution method outlined by the Clinical and Laboratory Standards Institute guidelines.25
Fifty microliter of bacterial suspensions (1×106 CFU/mL) and 50 µL of the twofold serial dilutions of the peptide were placed in 96-well microtiter plates. The positive control consisted of 50 µL of Mueller-Hinton broth and 50 μL of bacterial suspension, whereas the negative control was 100 µL of sterilized Mueller-Hinton broth. The plates were then incubated for 18 hrs at 37°C and MICs were determined by an enzyme-linked immunosorbent assay (ELISA) microplate reader (EpochTM; BioTek, Winooski, VT, USA) at λ =600 nm. On the other hand, the minimum bactericidal concentration (MBC) was confirmed by plating 10 μL of the bacterial suspension from each well that showed no visible turbidity on Muller-Hinton Broth agar (MHB; Oxoid Ltd., Basingstoke, UK) at 37°C for 24 hrs or 48 hrs. The lowest concentration with less than 1% bacterial growth was considered the MBC.
Antibiofilm activity
Biofilms of S. aureus; ATCC 33591 and ATCC BAA-2114 were formed using the Calgary biofilm device (Innovotech, Innovotech Inc. Edmonton, Canada).26–29 Briefly, the biofilm formation was initiated by placing 150 μL of (1×106 CFU/mL) bacterial suspension of the tested microorganism inside each well of the 96-well base. The plate was covered by a lid consisting of 96 pegs and incubated in an orbital incubator at 37 °C for 16±2 hrs and 120 rpm. The lid with the pegs was then removed and washed three times with phosphate buffered saline (PBS) to remove unattached bacterial cells. The lid was then transferred to a 96-well challenge microtiter plate which contains 200 μL of different concentrations of the hybrid peptide in MHB medium and incubated for 4 hrs in the orbital shaker as previously. The peg-lids were then removed from the challenge plate, re-washed three times with PBS, and transferred into a 96-well recovery microtiter plate containing 150 μL PBS buffer for sonication. The sonication on high for 16 mins. The 96-pegs lid was removed, and a new lid covered the plate. The minimum biofilm eradication concentration (MBEC), which is the minimum concentration needed to inhibit the regrowth of biofilm after peptide exposure for 4 hrs was measured by determining the absorbance of the plate at OD=595 nm. On the other hand, the minimum bactericidal concentration (MBECb), which is the lowest concentration to kill 99.9% of the viable bacteria in a biofilm, was measured by colony count method; this was done by plating 10 μL on agar plates followed by incubation for overnight.30
Synergistic checkerboard assay
The synergistic effects were studied against four clinical bacterial isolates, and five antimicrobial agents; LVX, CHL, RIF, AMP, and ERY. The tests were performed on 96-well microplates according to previous studies.31,32 Each well contained 25 μL of one of the five antibiotics mentioned above with equal volume of the hybrid peptide, and 50 μL of 5×105 CFU/mL of a single bacterial strain suspension. The agents and the hybrid peptide were serially diluted along the microplates, which were then incubated for 18–24 hrs at 37°C. The MICs were then calculated using ELISA microplate reader (EpochTM, BioTeck, Winooski, VT, USA) at 600 nm.
Finally, the values of the fractional inhibitory concentrations (FIC) index were calculated by adding the FIC value of the peptide (in combination divided by its value alone) and the FIC value of the antibiotic used (in combination divided by its value alone). The reactions were considered synergistic when FICs are ≤0.5, additive when 0.5≤ FICs ≤1, indifferent when 1< FICs <2, and antagonistic when ≤2.
Hemolytic assay
This assay was performed using human blood purchased from Sigma Aldrich (St. Louis, MO, USA). Two milliliters of the blood was centrifuged at 2,000 rpm for 5 mins. The supernatant was aspirated and replaced by 50 mL PBS buffer at pH 7.4 and centrifuged as previously. The step was performed twice. The resulted supernatant was again discarded and replaced with 50 mL PBS to produce a suspension of 4% red blood cells (RBCs) (vortex). The final suspension of RBCs was divided into nine test tubes and tested under different concentrations of the hybrid peptide using equal volumes from each. The positive control contained 2 mL of RBCs suspension, 2 mL PBS, and 5 μL 10% Triton X-100 (Santa Cruz Biotechnology, Dallas, TX, USA), while the negative control only consisted of 2 mL of RBCs suspension and 2 mL PBS. Incubation was performed for all samples for 60 mins at 37°C followed by gentle vortex. One milliliter of each sample was then placed in pre-sterilized Eppendorf tubes and centrifuged as mentioned previously. Finally, the absorbance of each supernatant was measured using the ELISA microplate reader (450 nm). The percentage of hemolysis was measured by subtracting the absorbance value of the Triton X-100 treated suspension from the absorbance values of the tested solutions.
Mammalian cell cytotoxicity assay
Human Embryonic Kidney 293 cell line (HEK293), obtained from Sigma–Aldrich (St. Louis, MO, USA), was selected as a model to investigate the cell cytotoxicity and selectivity of B1. The cells were cultured in RPMI 1640, containing 10% fetal bovine serum, and 1% v/v concentration of penicillin and streptomycin. Cells were grown as monolayers in 5% CO2 and 95% air at 37°C.
The cells were seeded into a 96-well microtiter plate at a density of 5×103 cells/well. Cells were incubated for 18 hrs with different peptide concentrations. Subsequently, 20 μL of 5 mg/mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added and incubated for 6 hrs. Changing the color of MTT into purple indicates the formation of formazan by viable cells. The medium was replaced by 100 μL of dimethylsulfoxide and mixed to dissolve the formed formazan crystals. Absorbance was measured using an ELISA microplate reader at 550 nm, and the GraphPad Prism software was used for statistical analyses.
Results
Peptide design, molecular modeling, and in silico analyses
The B1 hybrid peptide has 25 amino acids. It consisted of two parts, the N-terminal side was taken from the sequence range of 9–20 from the parent peptide BMAP-27, and the C-terminal side was taken from the sequence range of 17–29 from human LL-37 peptide. The helicity percentages were predicted using HNN,16 which is considered as one of the most accurate softwares to predict helical structures.33
The prediction of the secondary structure of B1 showed 88% of the alpha helices and 12% of the random coils. These values were different from the parent peptides which adopted a lesser degree of helicity and more random coils as shown in Table 1.
 | Table 1 Predicted percentages of secondary structure for the parent peptides (LL-37 and BMAP-27) and B1 using HNN |
ProtParam software from the ExPASy server17 and the APD,18 were used to predict the physicochemical parameters of the parent peptides and B1 (Table 2). The molecular weight of B1 is 3163.98 g/mol, and the isoelectric point is 11.77. The instability index is 20.99; this value indicates the stability of the peptide in a test tube which should be less than 40. Furthermore, the aliphatic index value represents its thermostability. For B1, the value suggests that our peptide is thermostable, which is slightly higher in value compared to parent peptides. GRAVY value, on the other hand, is the Grand Average of Hydropathy.34 It is negative and close to zero for our peptide, which suggests the moderate hydrophilicity of the peptide. Finally, the last parameter calculated using the ProtParam tool was the charge. The total net charge for B1 is +9, which is higher than LL-37 charge but lower than BMAP-27 charge.
 | Table 2 The prediction of the physiochemical properties of LL-37, BMAP-27, and B1. |
For homology modeling, the HHpred,19 the MODELLER software20 then the RAMPAGE22 were used (Figure 1). HHpred suggested cathelicidin antimicrobial peptide LL-37 (pdb: 5NMN_A) as the best template with approximately 95% probability with an Expect value (E-value) of 0.011. The selected model was forwarded to MODELLER, which created a pdb file. The validation was performed using The RAMPAGE,22 and ProSA-web.23 ProSA calculated a z-score of −0.80 which suggests a model of good quality, and the RAMPAGE showed 100% of the residues in the favored region. The quality indicators; the C-score, the estimated TM score, and the estimated RMSD in I-TASSER24 confirmed a helical structure. The values obtained were as following: the C-score equal −0.11, the estimated TM score is 0.7±0.12, and the estimated RMSD is 1.5±1.4 Å.
 | Figure 1 Three-dimensional structure of B1. Note: The structure was generated by homology modeling using MODELLER20 and the figure was prepared using PyMol.35 |
Protein synthesis and purification
The solid-phase method was used for peptide synthesis)GL Biochem (Shanghai) Ltd.). (RP-HPLC) confirmed >95% purity, which is recommended for in vitro studies. Moreover, ESI-MS confirmed the identity of the hybrid peptide. The ESI-MS showed the following peaks: [M+3H]3+=1056.41 Da, [M+4H]4+ =792.52 Da, [M+5H]5+ =634.23 Da, [M+6H]6+ =528.67 Da.
Bacterial susceptibility assay
B1 showed similar potency against all tested strains as shown in Table 3. The MIC values were 20 μM against MS S. aureus ATCC 29213, S. epidermidis ATCC12228, MR S. aureus ATCC 33591, MR S. aureus ATCC 43300 (oxacillin-resistant), and MR S. aureus ATCC BAA-41 and E. faecium BAA-2316 (vancomycin and teicoplanin-resistant), E. coli ATCC 25922 and E. coli ATCC 35218. On the other hand, it showed enhanced potencies with MIC values of 10 μM against MDR P. aeruginosa ATCC BAA-2114. The MIC values were always equal to MBC values which indicated a cidal effect for B1 peptide.
 | Table 3 MICs of B1 against different bacterial strains |
Antibiofilm activity of B1
The antibiofilm activity of B1 was studied against MR S. aureus ATCC 33591 and MDR P. aeruginosa ATCC BAA-2114. The MBEC was defined as the minimum concentration of the peptide needed to inhibit the regrowth of the biofilms after an exposure of 4 hrs. The MBEC value for MR S. aureus ATCC 33591 was 40 μM, which is two-fold higher than the MIC value against the planktonic cells. The MBEC value was also 60 μM for MDR P. aeruginosa ATCC BAA-2114 which is sixfold higher than the MIC values. On the other hand, the viable count method showed complete eradication of biofilm cells (99.9% killing) (MBECb) at 140 μM for both resistant strains (Table 4). At MBEC concentrations, the percentage of viability in biofilm cells were reported to be 6.5% for MR S. aureus ATCC 33591% and 8.5% for MDR P. aeruginosa ATCC BAA-2114.
 | Table 4 The antibiofilm activity of B1 against MR S. aureus ATCC 33951 and MDR P. aeruginosa ATCC BAA-2114 |
Synergistic checkerboard assay
Synergistic studies were performed with five antibiotics; LVX, CHL, RIF, AMP, and ERY. The resistant bacterial strains used were MR S. aureus ATCC 33591, MR S. aureus ATCC 43300 (oxacillin-resistant), and MR S. aureus ATCC BAA-41 and MDR P. aeruginosa ATCC BAA-2114.
The FIC indices were calculated, and the values were interpreted according to EUCAST as synergistic, additive, indifferent, or antagonistic when the values are ≤0.5, 0.5< FIC index ≤1, 1< FICI <2 and ≥2, respectively.36 The results of the synergistic studies are presented in Table 5. The combination between B1 and LVX showed mixed effects against different strains; it was synergistic against MR S. aureus ATCC 33591 and MR S. aureus ATCC BAA-41 with 92.50% and 25% reduction in LVX MIC values, respectively. It was additive against MR S. aureus ATCC 43300 with a 60% reduction in LVX MIC but showed an indifferent effect against MDR P. aeruginosa ATCC BAA-2114, yet 58.33% reduction in LVX MIC.
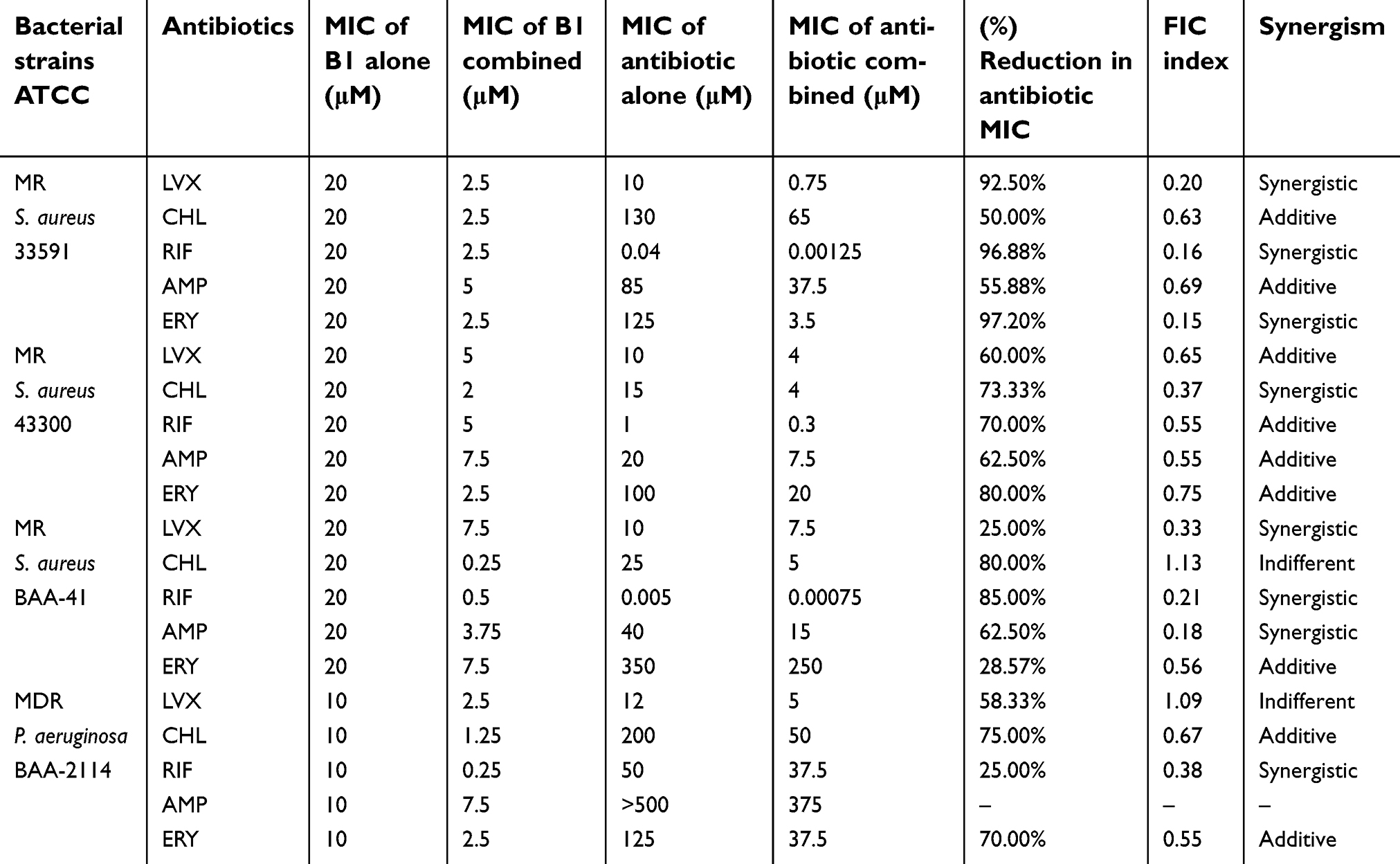 | Table 5 Correlation between FIC and the synergistic effect upon combination with antibacterial agents |
On the other hand, the combination between B1 and CHL was additive with a 50% and 75% reduction in MIC values against MR S. aureus ATCC 33591 and MDR P. aeruginosa ATCC BAA-2114, respectively; synergistic with 73.33% reduction in MIC value against MR S. aureus ATCC 43300, and indifferent against MR S. aureus ATCC BAA-41. Alternatively, RIF showed synergistic effects against three resistant strains: MR S. aureus ATCC 33591 with 96.88% reduction in the MIC value of the antibiotic, MR S. aureus BAA-41 with 85% MIC reduction and MDR P. aeruginosa BAA-2114 with 25% MIC reduction. However, B had an additive effect against MR S. aureus ATCC 43300 in the presence of the RIF peptide with a MIC reduction percentage of around 70%.
Moreover, AMP combination also showed additive effects against MR S. aureus ATCC 33591 and MR S. aureus ATCC 43300 with a reduction of over 50% in AMP MIC values; but showed a synergistic effect against MR S. aureus BAA-41 with over 60% reduction in its inhibitory concentration. Finally, the final combination with AMP against MDR P. aeruginosa BAA-2114 could not be tested since the MIC value of AMP alone was over 500 μM.
Meanwhile, the MICs of ERY changed markedly in this study. The combination showed a synergistic effect against MR S. aureus ATCC 33591 with a 97.20% reduction in ERY inhibitory concentration. On the other hand, the ERY combination with B1 against the other three resistant strains stated additive effects against MR S. aureus ATCC 43300 (80.00% MIC reduction), MR S. aureus ATCC BAA-41 (28.57% reduction) and MDR P. aeruginosa ATCC BAA-2114 (70.00% reduction).
Hemolytic assay
Different concentrations of B1 were tested against human erythrocytes. The concentration ranged from 200 μM to 10 μM. B1 showed a maximum concentration of 20 μM to exert its antimicrobial activity against different strains, and at this value, B1 caused slight hemolysis after 1-hr exposure time (Figure 2). Nonetheless, even with increasing the concentration of this hybrid peptide two times the MIC value, the hemolysis percentage is still less than 18%.
 | Figure 2 Hemolytic activity of B1 against human erythrocytes after an exposure time of 1 hr. |
Mammalian cell cytotoxicity assay
The average half maximal inhibitory concentration (IC50) of B1 against HEK293 mammalian cell line is 23.4 μM (Figure 3). This value is higher than the maximum concentration of B1 needed to inhibit the most resistant strains of bacteria, either be it alone or in combination. Therefore, B1 exhibits minimal cell cytotoxicity toward mammalian cells and can be safely used as an antimicrobial agent.
 | Figure 3 Cytotoxicity assay of B1 on HEK293 mammalian cell line using MTT assay. |
Discussion
The emergence of MDR bacterial strains is becoming a serious health concern.37 There is an urgent need to develop new antibiotics and to adopt new strategies to treat infections caused by MDR. AMPs have been proposed as one of the most promising antimicrobial agents, in particular, cationic antimicrobial peptides.38 Intensive studies have been done to discover and investigate new AMPs from different organisms.18 Trials have been made to synthesize new AMPs by a simple mutation in specific amino acids positions to natural and unnatural residues, by amidation of the C-terminal, and also by using computer-assisted methods.39
The mechanism of action of AMPs is complex, yet, it is well known that the cationic charge of any antimicrobial peptide and their hydrophobicity are critical for the mechanism of action. The cationic charge will facilitate the binding with the negatively charged bacterial cell membrane, which enables the permeabilizing of the amphiphilic peptide into the hydrophobic membrane, which can cause disruption of the membrane integrity or translocation of the peptide to affect intracellular targets.40,41 AMPs could also activate cell-wall lytic enzymes as suggested by Sahl et al,42 inhibit efflux pumps,43 protein synthesis and DNA replication,44–46 which make them an excellent antibiotic adjuvant.47–52
In this paper, we designed a hybrid peptide from two natural AMPs, checked it in silico and performed a homology modeling. The physicochemical properties were investigated by several online tools before proceeding with the in vitro experiments. The new hybrid, B1, consists of an N-terminal extracted from 9 to 20 of BMAP-27 and a C-terminal fragment obtained from 17 to 29 of LL-37. B1 consists of 25 amino acids in total with an improved helicity content of 88.0% (Table 1). It contains one negatively charged aspartic acid residue and ten positively charged residues including three arginine and seven lysine residues; this is created a net charge of +8 which is high enough to interact electrostatically with the negatively charged components in the cell membranes of the bacterial strains, such as the lipoteichoic acid of G+ve bacteria and the LPS moieties in G-ve bacteria.53 B1 also contains ten hydrophobic residues (ie, one isoleucine, two valine, three leucine, and four phenylalanine) with a total hydrophobic ratio of 45%, which facilitated the protein binding to the bacterial membranes and disrupted their integrity. The GRAVY value of B1 indicates the moderate hydrophilicity characteristic, and the stability and the aliphatic indices, calculated by the APD showed remarkable stability and thermostability (Table 2).18
B1 displayed a MIC value of 20 μM against all tested strains except MDR P. aeruginosa which showed an inhibitory concentration of 10 μM (Table 3). The resemblance in the MIC values against G+ve and G-ve despite the differences in their cell wall compositions was due to the amphiphilic nature of B1, since it contains a balanced ratio between the hydrophilic and the hydrophobic residues. Furthermore, the similarity in the MIC values and the MBC values indicates that B1 is bactericidal.
On the other hand, the MIC values of the parent peptide LL-37 were reported previously against all resistant strains of S. aureus to be more than 128 μM,54 and increased resistance to LL-37 was previously reported.55 Nevertheless, the MIC values of BMAP-27 were in the range of 4–19.5 μM.56
Antibiofilm activities were also investigated for B1. B1 showed excellent antibiofilm activities and eradicated the formation of biofilms. It was tested against MR S. aureus ATCC 33951 and MDR P. aeruginosa ATCC BAA-2114. These bacteria are known as biofilm formers.57,58 The MBEC values were two-fold and six-fold higher than the MIC values against the planktonic form of the aforementioned bacterial strains, respectively (Table 4).
The toxicity of B1 peptide toward mammalian cells using HEK293 cells was also examined. It is essential that the antimicrobial agent be active at a concentration that does not affect the mammalian cell viability. The (IC50) value of B1 was 23.4 μM, and this is higher than the MIC values (≤20 μM) needed to kill all the tested planktonic bacterial cells if used alone. Besides, the MIC values of B1 (20 μM) causes less than 8% hemolytic activity against human erythrocyte (Figure 2), which renders this hybrid safer to treat bacterial infections compared with the parent peptide BMAP-27, which showed a hemolytic activity at 6.2 μM,59 along with poor selectivity toward microbial organisms, which limited its potential therapeutic applications.14 However, using B1 at the concentrations needed to inhibit or kill tissues-associated biofilms inside the human body should be avoided, since these concentrations will affect the viability of the eukaryotic cells.
On the other hand, synergistic studies were also evaluated with five antibiotics against four resistant strains MR S. aureus ATCC 33591, MR S. aureus ATCC 43300, and MR S. aureus ATCC BAA-41 and MDR P. aeruginosa ATCC BAA-2114. The combination therapy or so-called synergistic studies are intensively studied during the application of peptides.47–52 Generally, peptides are pricey depending on the length of their sequence and the amino acid nature. And although bacterial resistance against these drugs has not emerged yet because of their unique mechanism of action, it could arise in the near future after extensive or abusive use of these agents. Therefore, combination therapy is a good strategy to avoid the possibility of the emergence of resistance, increase the efficacy of the agents, and finally, decrease the dose of the peptide; hence the cost.60
The antibiotics used for the synergistic studies were LVX, CHL, RIF, AMP, and ERY. LVX is a bactericidal agent that inhibits the DNA gyrase and topoisomerase IV enzymes. Whereas CHL is a bacteriostatic agent that inhibits protein synthesis.61 RIF, on the other hand, is bactericidal or bacteriostatic dependent on the type of the bacterial strains and the concentration of the drug.62 It suppresses the RNA synthesis by inhibiting the DNA-dependent ribonucleic acid (RNA) polymerase.63 AMP is a bactericidal cell wall inhibitor, whereas ERY is a protein synthesis inhibitor and it can be bacteriostatic or bactericidal for the same conditions mentioned for RIF.64 The emergence of resistance against these agents has been reported continuously in the past decades.65
The combinations of B1 with the five agents displayed synergistic and additive effects, except in two cases of “indifference” after using B1 with CHL against MR S. aureus ATCC BAA-41 and with LVX against MDR P. aeruginosa BAA-2114. B1 as an antimicrobial peptide is expected to disrupt the integrity of the bacterial membranes, and this could facilitate the entry of the combined agents. Also, B1 possibly inhibited the efflux pumps of the resistant strains as suggested by Rishi et al,43 and that could explain the synergistic effect on the activity of CHL, AMP, and ERY. However, the exact mechanisms of action of the combination therapy were not studied in this paper.
The results reported great enhancement in the activities of the five antibacterial agents with B1 against highly resistant strains with a massive reduction in the MIC values of the antibiotics and B1 as well (Table 5). The MIC values of B1 were measured as 20 μM if used alone against all tested resistant strains of S.aureus; ATCC 33591, ATCC 43300, and ATCC BAA-41. However, these MIC values were decreased markedly when combined with different antibiotics. The strongest synergism was seen with CHL and RIF (80%, and 85% reduction in MIC, respectively) against MR S. aureus ATCC BAA-41. The MIC values of B1 were decreased to 0.25 μM, and 0.5 μM when accompanied by CHL and RIF, respectively. In the other combinations against resistant strains of S. aureus, there was a range of concentration reduction for both the antibiotics and B1, however, the MIC value for B1 did not exceed 7.5 μM.
On the other hand, synergistic studies were also performed on MDR P. aeruginosa BAA-2144. The only synergism was found with RIF with a reduction percentage of 25% in RIF and the MIC value of B1 was reduced to 0.25 μM, which is 97.5% reduction in the MIC value. It is worthwhile to mention that these MIC values are extremely safe to use with less than 1% hemolysis risk.
Conclusion
Designing an antimicrobial peptide using computer-based methods is considered one of the fastest techniques in drug development. Herein, a new hybrid peptide, B1 was developed using the sequences of LL-37 and BMAP-27. B1 showed enhanced antimicrobial activity and excellent toxicity profile against planktonic G+ve and G-ve bacterial cells. B1 showed selectivity, reduced cytotoxicity with decreased hemolytic effect against mammalian erythrocytes. Also, the antimicrobial activities and the toxicity profiles were improved markedly upon combining with five different antibacterial agents.
On the other hand, B1 showed strong antibiofilm activities, but it can only be used to eradicate biofilms on abiotic surfaces since the concentrations needed to inhibit or kill the studied biofilms will risk the viability of mammalian cells.
Ethical approval
The study protocol was approved by the institutional review board of the Jordan University of Science and Technology.
Acknowledgments
This study was supported by the Deanship of Research at Jordan University of Science and Technology (grant number: 611-2017).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gottlieb T, Nimmo GR. Antibiotic resistance is an emerging threat to public health: an urgent call to action at the antimicrobial resistance summit 2011. Med J Aust. 2011;194(6):281–283.
2. Spellberg B. The future of antibiotics. Crit Care Lond Engl. 2014;18(3):228. doi:10.1186/cc13948
3. Spellberg B, Bartlett J, Wunderink R, Gilbert DN. Novel approaches are needed to develop tomorrow’s antibacterial therapies. Am J Respir Crit Care Med. 2015;191(2):135–140. doi:10.1164/rccm.201410-1894OE
4. Tossi A, Sandri L, Giangaspero A. Amphipathic, α-helical antimicrobial peptides. Biopolymers. 2000;55(1):4–30. doi:10.1002/1097-0282(2000)55:1<4::AID-BIP30>3.0.CO;2-M
5. Auvynet C, Rosenstein Y. Multifunctional host defense peptides: antimicrobial peptides, the small yet big players in innate and adaptive immunity. FEBS J. 2009;276(22):6497–6508. doi:10.1111/j.1742-4658.2009.07360.x
6. Gallo RL, Murakami M, Ohtake T, Zaiou M. Biology and clinical relevance of naturally occurring antimicrobial peptides. J Allergy Clin Immunol. 2002;110(6):823–831.
7. Bals R, Wilson JM. Cathelicidins – a family of multifunctional antimicrobial peptides. Cell Mol Life Sci CMLS. 2003;60(4):711–720.
8. Zanetti M, Gennaro R, Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995;374(1):1–5. doi:10.1016/0014-5793(95)01050-O
9. Zanetti M. The role of cathelicidins in the innate host defenses of mammals. Curr Issues Mol Biol. 2005;7(2):179–196.
10. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389–395. doi:10.1038/415389a
11. Gudmundsson GH, Agerberth B, Odeberg J, Bergman T, Olsson B, Salcedo R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur J Biochem. 1996;238(2):325–332.
12. Smeianov V, Scott K, Reid G. Activity of cecropin P1 and FA-LL-37 against urogenital microflora. Microbes Infect. 2000;2(7):773–777. doi:10.1016/S1286-4579(00)90359-9
13. Dorschner RA, Pestonjamasp VK, Tamakuwala S, et al. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol. 2001;117(1):91–97. doi:10.1046/j.1523-1747.2001.01340.x
14. Skerlavaj B, Gennaro R, Bagella L, Merluzzi L, Risso A, Zanetti M. Biological characterization of two novel cathelicidin-derived peptides and identification of structural requirements for their antimicrobial and cell lytic activities. J Biol Chem. 1996;271(45):28375–28381.
15. Risso A, Zanetti M, Gennaro R. Cytotoxicity and apoptosis mediated by two peptides of innate immunity. Cell Immunol. 1998;189(2):107–115. doi:10.1006/cimm.1998.1358
16. Guermeur Y. Combinaison de Classifieurs Statistiques, Application a La Prediction de La Structure Secondaire Des Proteines [PhD thesis]. Paris 6; 1997.
17. Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005:571–607. Available from:
18. Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44(D1):D1087–D1093. doi:10.1093/nar/gkv1278
19. Meier A, Söding J. Automatic Prediction of Protein 3D Structures by Probabilistic Multi-template Homology Modeling. PLOS Computational Biology: Methods. 2015:1004343. Available from:
20. Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2016;86(1):
21. Zimmermann L, Stephens A, Nam S-Z, et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018;430(15):2237–2243. doi:10.1016/j.jmb.2017.12.007
22. Lovell SC, Davis IW, Arendall III WB, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins Struct Funct Genet. 2003;50:437–450. doi:10.1002/prot.10286
23. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(suppl_2):W407–W410. doi:10.1093/nar/gkm290
24. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9(1):40. doi:10.1186/1471-2105-9-40
25.
26. Falciani C, Lozzi L, Pollini S, et al. isomerization of an antimicrobial peptide broadens antimicrobial spectrum to gram-positive bacterial pathogens. plos one. 2012;7(10):e46259. doi:10.1371/journal.pone.0046259
27. Luca V, Stringaro A, Colone M, Pini A, Mangoni ML. Esculentin(1-21), an amphibian skin membrane-active peptide with potent activity on both planktonic and biofilm cells of the bacterial pathogen Pseudomonas aeruginosa. Cell Mol Life Sci. 2013;70(15):2773–2786. doi:10.1007/s00018-013-1291-7
28. Feng X, Sambanthamoorthy K, Palys T, Paranavitana C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides. 2013;49:131–137. doi:10.1016/j.peptides.2013.09.007
29. Almaaytah A, Tarazi S, Alsheyab F, Al-Balas Q, Mukattash T. Antimicrobial and antibiofilm activity of mauriporin, a multifunctional scorpion venom peptide. Int J Pept Res Ther. 2014;20(4):397–408. doi:10.1007/s10989-014-9405-0
30. Parker AE, Walker DK, Goeres DM, Allan N, Olson ME, Omar A. Ruggedness and reproducibility of the MBEC biofilm disinfectant efficacy test. J Microbiol Methods. 2014;102:55–64. doi:10.1016/j.mimet.2014.04.013
31. Hsieh MH, Yu CM, Yu VL, Chow JW. Synergy assessed by checkerboard a critical analysis. Diagn Microbiol Infect Dis. 1993;16(4):343–349. doi:10.1016/0732-8893(93)90087-N
32. Dundar D, Otkun M. In-vitro efficacy of synergistic antibiotic combinations in multidrug resistant pseudomonas aeruginosa strains. Yonsei Med J. 2010;51(1):111. doi:10.3349/ymj.2010.51.1.111
33. Angamuthu K, Piramanayagam S. Evaluation of in silico protein secondary structure prediction methods by employing statistical techniques. Biomed Biotechnol Res J BBRJ. 2017;1(1):29. doi:10.4103/bbrj.bbrj_28_17
34. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132.
35.
36.
37. Oyston PCF, Fox MA, Richards SJ, Clark GC. Novel peptide therapeutics for treatment of infections. J Med Microbiol. 2009;58(Pt 8):977–987. doi:10.1099/jmm.0.011122-0
38. Jenssen H, Hamill P, Hancock REW. Peptide antimicrobial agents. Clin Microbiol Rev. 2006;19(3):491–511. doi:10.1128/CMR.00056-05
39. Bahar AA, Ren D. Antimicrobial peptides. Pharmaceuticals. 2013;6(12):1543–1575. doi:10.3390/ph6121543
40. Fjell CD, Hiss JA, Hancock REW, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. 2012;11(1):37–51. doi:10.1038/nrd3591
41. Hancock REW, Sahl H-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24(12):1551–1557. doi:10.1038/nbt1267
42. Sahl H-G, Pag U, Bonness S, Wagner S, Antcheva N, Tossi A. Mammalian defensins: structures and mechanism of antibiotic activity. J Leukoc Biol. 2005;77(4):466–475. doi:10.1189/jlb.0804452
43. Rishi P, Vij S, Maurya IK, Kaur UJ, Bharati S, Tewari R. Peptides as adjuvants for ampicillin and oxacillin against methicillin-resistant Staphylococcus aureus (MRSA). Microb Pathog. 2018;124:11–20. doi:10.1016/j.micpath.2018.08.023
44. Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3(3):238–250. doi:10.1038/nrmicro1098
45. Hsu C-H, Chen C, Jou M-L, et al. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005;33(13):4053–4064. doi:10.1093/nar/gki725
46. Marchand C, Krajewski K, Lee H-F, et al. Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. 2006;34(18):5157–5165. doi:10.1093/nar/gkl667
47. Cassone M, Otvos JL. Synergy among antibacterial peptides and between peptides and small-molecule antibiotics. Expert Rev Anti Infect Ther. 2010;8(6):703–716. doi:10.1586/eri.10.38
48. Reffuveille F, de la Fuente-Núñez C, Mansour S, Hancock REW. A broad-spectrum antibiofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrob Agents Chemother. 2014;58(9):5363–5371. doi:10.1128/AAC.03163-14
49. Simonetti O, Cirioni O, Ghiselli R, et al. In vitro activity and in vivo animal model efficacy of IB-367 alone and in combination with imipenem and colistin against Gram-negative bacteria. Peptides. 2014;55:17–22. doi:10.1016/j.peptides.2014.01.029
50. Ribeiro SM, de la Fuente-Núñez C, Baquir B, Faria-Junior C, Franco OL, Hancock REW. Antibiofilm peptides increase the susceptibility of carbapenemase-producing klebsiella pneumoniae clinical isolates to β-Lactam antibiotics. Antimicrob Agents Chemother. 2015;59(7):3906–3912. doi:10.1128/AAC.00092-15
51. de la Fuente-Núñez C, Cardoso MH, de Souza Cândido E, Franco OL, Hancock REW. Synthetic antibiofilm peptides. Biochim Biophys Acta BBA – Biomembr. 2016;1858(5):1061–1069. doi:10.1016/j.bbamem.2015.12.015
52. Lázár V, Martins A, Spohn R, et al. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat Microbiol. 2018;3(6):718–731. doi:10.1038/s41564-018-0164-0
53. Rao X-J, Yu X-Q. Lipoteichoic acid and lipopolysaccharide can activate antimicrobial peptide expression in the tobacco hornworm manduca sexta. Dev Comp Immunol. 2010;34(10):1119–1128. doi:10.1016/j.dci.2010.06.007
54. Mohamed MF, Abdelkhalek A, Seleem MN. Evaluation of short synthetic antimicrobial peptides for treatment of drug-resistant and intracellular Staphylococcus aureus. Sci Rep. 2016;6:29707. doi:10.1038/srep29707.
55. Ouhara K, Komatsuzawa H, Kawai T, et al. Increased resistance to cationic antimicrobial peptide LL-37 in methicillin-resistant strains of Staphylococcus aureus. J Antimicrob Chemother. 2008;61(6):1266–1269. doi:10.1093/jac/dkn106
56. Pompilio A, Crocetta V, Scocchi M, et al. Potential novel therapeutic strategies in cystic fibrosis: antimicrobial and anti-biofilm activity of natural and designed α-helical peptides against Staphylococcus aureus, Pseudomonas aeruginosa, and Stenotrophomonas maltophilia. BMC Microbiol. 2012;12(1):145. doi:10.1186/1471-2180-12-145
57. Dastranj M, Farahani A, Shoja S, Dinarvand G. State of globe: biofilm formation in Staphylococcus aureus isolates. J Glob Infect Dis. 2017;9(3):91–92. doi:10.4103/jgid.jgid_83_17
58. Murray J, Muruko T, Gill CIR, et al. Evaluation of bactericidal and anti-biofilm properties of a novel surface-active organosilane biocide against healthcare associated pathogens and Pseudomonas aeruginosa biolfilm. PLoS One. 2017;12(8):e0182624. doi:10.1371/journal.pone.0182624
59. Lee EK, Kim Y-C, Nan YH, Shin SY. Cell selectivity, mechanism of action and LPS-neutralizing activity of bovine myeloid antimicrobial peptide-18 (BMAP-18) and its analogs. Peptides. 2011;32(6):1123–1130. doi:10.1016/j.peptides.2011.03.024
60. Belanger CR, Mansour SC, Pletzer D, Hancock REW. Alternative strategies for the study and treatment of clinical bacterial biofilms. Emerg Top Life Sci. 2017;1(1):41–53. doi:10.1042/ETLS20160020
61. Hooper DC. Mechanisms of action and resistance of older and newer fluoroquinolones. Clin Infect Dis. 2000;31(Supplement_2):S24–S28. doi:10.1086/314056
62.
63. Wehrli W. Rifampin: mechanisms of action and resistance. Rev Infect Dis. 1983;5(Suppl 3):S407–S411.
64. Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol. 2010;8(6):423–435. doi:10.1038/nrmicro2333
65. Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev MMBR. 2010;74(3):417–433. doi:10.1128/MMBR.00016-10
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.