Back to Journals » Cancer Management and Research » Volume 17

Dermatotoxicity of Immune Checkpoint Inhibitors in Advanced Non-Small Cell Lung Cancer: Current Advances in Mechanistic Insights and Predictive Biomarker Identification
Authors Shao S, Zheng W, Li X, Qi A, Gu Y, Gong Y, Wang Q, Jiao L, Xu L
Received 16 March 2025
Accepted for publication 6 September 2025
Published 17 September 2025 Volume 2025:17 Pages 2035—2048
DOI https://doi.org/10.2147/CMAR.S528555
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Shiliang Shao,1 Wenjuan Zheng,2 Xuyang Li,1 Ao Qi,1 Yifeng Gu,1 Yabin Gong,1 Qin Wang,1 Lijing Jiao,1 Ling Xu1
1Department of Oncology, Yueyang Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 2Department of Oncology, Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan, People’s Republic of China
Correspondence: Ling Xu, Department of Oncology, Yueyang Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai University of Traditional Chinese Medicine, No. 110, Ganhe Road, Hongkou District, Shanghai, 200437, People’s Republic of China, Tel +8615901903361, Email [email protected] Lijing Jiao, Department of Oncology, Yueyang Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai University of Traditional Chinese Medicine, No. 110, Ganhe Road, Hongkou District, Shanghai, 200437, People’s Republic of China, Tel +8613524295060, Email [email protected]
Abstract: Immune checkpoint inhibitors (ICIs) have substantially improved clinical outcomes in patients with advanced non-small cell lung cancer (NSCLC). However, the cutaneous immune-related adverse events (cirAEs) they elicit—being the most frequent and earliest-emerging toxicities—not only compromise treatment adherence but also exhibit a distinct positive association with systemic immune activation and antitumor efficacy. Given these characteristics, elucidating the pathogenic mechanisms of cirAEs and identifying predictive biomarkers are critical for the early detection and intervention of cirAEs, as well as for forecasting the onset of other immune-related adverse events (irAEs) and assessing ICI therapeutic prognosis. This review systematically summarizes recent advances in the pathological mechanisms of cirAEs and predictive biomarkers. Mechanistically, cirAEs result from multifactorial interplay, including genetic predisposition, shared antigen-driven cross-reactivity, and breakdown of cutaneous immune tolerance. For predictive biomarkers, strategies span traditional predictors (eg, demographic and immunological features) and their clinical translation challenges to emerging methods leveraging multi-omics integration and radiomics. Finally, this review addresses future challenges and directions in cirAEs research: Specifically, the positive association between cirAEs and efficacy demands accurate differentiation of “manageable toxicities” from “high-risk toxicities”; furthermore, future studies must validate causal biomarkers via prospective multi-omics cohorts and develop AI-driven dynamic prediction models to enable toxicity-stratified management and optimization of personalized immunotherapy.
Keywords: non-small cell lung cancer, immune checkpoint inhibitors, cutaneous immune-related adverse events, immunotherapy, predictive biomarkers
Introduction
Lung cancer remains a leading cause of global cancer-related incidence and mortality.1 Over the past decade, immune checkpoint inhibitors (ICIs) have substantially reshaped the treatment paradigm for advanced and metastatic non-small cell lung cancer (NSCLC).2 These agents, notably monoclonal antibodies targeting programmed death-1/programmed death-ligand 1 (PD-1/PD-L1) and cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), counteract tumor cell-mediated suppression of T-cell activation (eg, via PD-L1 overexpression), consequently reinstating anti-tumor immunity.3–6 However, this therapeutic immune reactivation can concurrently trigger immune-related adverse events (irAEs) involving multiple organ systems.7 Among these toxicities, cutaneous immune-related adverse events (cirAEs) are the most frequent and earliest-emerging, posing significant clinical challenges. Importantly, grade ≥3 cirAEs (such as severe rash or bullous dermatitis) occur in a notable subset of patients, frequently necessitating treatment delays or cessation and consequently elevating the risk of disease progression.8–10
CirAEs represent the most prevalent and frequently earliest-onset irAEs during ICI therapy, occurring in 30–50% of patients.11 They generally emerge at ~4 weeks post-treatment initiation, which is markedly earlier than endocrine irAEs (~12 weeks) and gastrointestinal irAEs (~22.2 weeks).12–14 While most cirAEs are non-life-threatening, severe manifestations—including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), drug reaction with eosinophilia and systemic symptoms (DRESS), extensive/severe bullous pemphigoid, and grade 4 rash (>30% body surface area with severe symptoms or infection)—may occur and warrant treatment interruption or discontinuation.15 Furthermore, cirAEs demonstrate significant ICI class differences: CTLA-4 inhibitors typically induce a higher incidence and severity of cirAEs compared to PD-1/PD-L1 inhibitors, with an earlier onset.16–18 This incidence further exhibits variability across cancer types, histological subtypes, and disease stages, suggesting underlying mechanistic complexity.19
The onset of cirAEs demonstrates a significant positive correlation with subsequent multi-organ irAEs; specifically, in patients with multi-organ irAEs, cirAEs incidence is substantially higher than in those with isolated irAEs, indicating their potential role as sentinel biomarkers for predicting broader immune toxicities.20 Notably, patients experiencing cirAEs typically demonstrate superior objective response rates (ORR), prolonged progression-free survival (PFS) and overall survival (OS), along with reduced mortality risk compared to non-affected individuals.12,21–24 Furthermore, in patients with multi-organ irAEs, the number of irAEs positively correlates with both PFS and OS.25 Mechanistically, cirAEs fundamentally reflect direct cutaneous manifestations of systemic immune activation, offering unique insights into ICI mechanisms and toxicity profiles. As a visible manifestation window for localized immune events, skin pathologies (eg, vitiligo and dermatitis) exhibit consistent and significant correlations with systemic ICI-induced immune responses and clinical efficacy—making it the clinically best-established organ for such associations.25 Given their early emergence, high incidence, and unique associations with systemic immune activation and efficacy, elucidating the pathophysiology of cirAEs is therefore critical for deciphering ICI mechanisms of action and toxicity heterogeneity.
Consequently, elucidating the pathophysiology of cirAEs and identifying robust predictive biomarkers will not only enable early diagnosis and intervention of cirAEs but also inform risk prediction for other irAEs. While cirAEs represent a pan-cancer phenomenon in ICI therapy, NSCLC—as one of the earliest breakthrough and most extensively treated solid tumors with ICIs—has generated the richest clinical experience and research data, thus providing a robust foundation for mechanistic understanding of cirAEs and discovery of predictive biomarkers. Here, focusing specifically on NSCLC patients, this review synthesizes and critically evaluates current mechanistic insights into cirAEs within this disease context alongside predictive biomarkers, aiming to advance safer and more effective personalized immunotherapy.
Pathogenic Mechanisms of cirAEs
ICIs augment anti-tumor immunity through blockade of key inhibitory immune checkpoints (eg, PD-1/PD-L1 and CTLA-4). Notably, CTLA-4 and PD-1 demonstrate fundamentally distinct immunoregulatory mechanisms.26 Optimal T-cell activation necessitates two critical signals: 1) T-cell receptor (TCR) engagement with peptide-major histocompatibility complex (MHC) complexes on antigen-presenting cells (APCs); 2) CD28 molecule (CD28)-mediated co-stimulation via CD80/CD86 binding.27,28 CTLA-4 predominantly suppresses early-phase immune responses through three key mechanisms (Figure 1): 1) Initiating inhibitory signaling: CTLA-4-CD80/CD86 engagement recruits Src homology 2 domain-containing tyrosine phosphatase (SHP-2) and protein phosphatase 2A (PP2A), consequently inhibiting phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB) signaling;29–312) Promoting ligand transendocytosis: CTLA-4 internalizes CD80/CD86 from APCs via transendocytosis, reducing APC surface expression and impairing T-cell co-stimulation;32–34 3) Inducing immunosuppressive cytokines: CTLA-4 signaling can induce the production of immunosuppressive cytokines such as transforming growth factor-β (TGF-β) and interleukin-10 (IL-10). Conversely, PD-1 is widely expressed on activated T cells, B cells, natural killer (NK) cells, and other immune subsets. It engages two ligands: PD-L1 and PD-L2. PD-L1 exhibits broad expression across immune cells (T/B cells, dendritic cells (DCs), macrophages) and non-immune cells, including tumor cells. PD-L2 expression is more restricted, predominantly localized to APCs (eg, dendritic cells and macrophages).35,36 Critically, PD-1 engagement with its ligands elicits differential biological outcomes.37 Specifically, PD-1:PD-L2 binding favors T helper 2 (Th2)-polarized responses, while PD-1:PD-L1 engagement predominantly inhibits T-cell-mediated immunity.38,39 PD-1 functions predominantly during the effector and exhaustion phases, mediating broader immune regulation.40 Its core inhibitory mechanisms involve (Figure 1): 1) Attenuating TCR signaling: PD-1:PD-L1 binding phosphorylates intracellular immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) motifs (mainly via lymphocyte-specific protein tyrosine kinase (Lck)), recruiting SHP-2. Activated SHP-2 dephosphorylates proximal TCR components and suppresses PI3K/Akt and Ras/MEK/Erk cascades, inhibiting T-cell activation, proliferation, and effector functions;41–48 2) Disrupting CD28 co-stimulation: PD-1 directly impedes CD28 signaling, further constraining T-cell activity.41 Importantly, functional crosstalk exists between CTLA-4 and PD-1 pathways in regulating T-cell activation.49,50 Significantly, recent studies have identified PD-1 expression on select tumor cells themselves.51,52 Thus, PD-1 inhibitors can also restrain tumor growth by disrupting these tumor-intrinsic PD-1:ligand (eg, PD-L1) interactions.
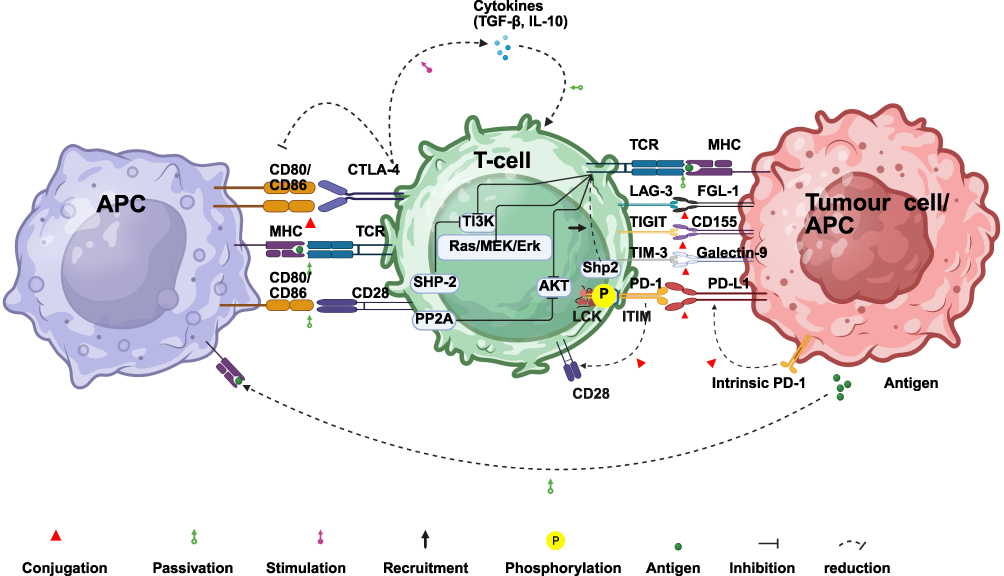 |
Figure 1 Mechanism of Immune Checkpoint Regulation in T-cell Responses. |
Beyond classical checkpoints, next-generation inhibitors targeting Lymphocyte Activation Gene-3 (LAG-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), and T cell immunoglobulin and mucin domain-containing protein-3 (TIM-3) are in clinical development, driven by growing interest in their distinct immunoregulatory mechanisms.53 LAG-3—expressed predominantly on activated/exhausted T cells and regulatory T cells (Tregs)—binds ligands (eg, major histocompatibility complex class II molecules, MHC-II) to attenuate TCR signaling, suppress CD8+ T-cell function/proliferation, and potentiate Treg-mediated suppression;54–57 TIGIT—expressed on T/NK cells—directly inhibits cytotoxic activity and cytokine production in effector T/NK cells via high-affinity binding to CD155 while concurrently inducing APCs such as DCs to secrete immunosuppressive cytokines and repress pro-inflammatory factors (eg, IL-12), thereby fostering an immunosuppressive milieu;58,59 TIM-3—expressed broadly on immune cells—induces T-cell exhaustion, apoptosis, or functional impairment upon engagement with ligands.60,61
Having outlined ICI immunomodulation, we now focus on cirAEs pathogenesis. Notably, cirAEs demonstrate heterogeneous clinical presentations, with underlying molecular mechanisms and drivers varying considerably among patients of divergent genetic/immunological profiles. Therefore, elucidating the complex mechanisms underlying NSCLC-associated cirAEs is critical for developing effective predictive and management strategies. The central mechanism for general irAEs involves ICI-induced disruption of peripheral tolerance mechanisms, which suppresses autoreactive T cells, thereby disrupting tissue homeostasis. However, the early emergence and high frequency of cirAEs strongly suggest unique contributing factors distinct from these generic irAEs mechanisms. Current evidence supports three principal mechanistic hypotheses specific to cirAEs: 1) genetic predisposition, 2) shared antigen-driven cross-reactivity, and 3) cutaneous immune tolerance breakdown.
Genetic Predisposition: Contributions of Human Leukocyte Antigen (HLA), Autoimmunity Genes and Deoxyribonucleic Acid (DNA) Methylation
Genetic predisposition significantly contributes to cirAEs development. Several cirAEs—including vitiligo, bullous pemphigoid, and psoriasiform eruptions—demonstrate clinical similarities to classical autoimmune disorders.62,63 Notably, psoriasiform eruptions—frequent cirAEs post PD-1/PD-L1 inhibition—exhibit autoimmune-mediated pathogenesis;64–67 this association is clinically significant, as approximately 86% of psoriasis patients develop psoriasiform eruptions following ICI therapy.68 Since autoimmune disorders generally exhibit strong genetic foundations, they offer a conceptual framework for deciphering ICI-triggered cirAEs. Supporting this genetic link, carriers of psoriasis-risk alleles (eg, HLA-Cw6, IL12B, IL23R, LCE3B (late cornified envelope 3B)/LCE3C) with parental psoriasis history demonstrate elevated susceptibility to psoriasiform eruptions.69,70 Similarly, genome-wide association studies (GWAS) have implicated genetic variants in CTLA-4 and PD-1/PD-L1 loci with autoimmune disease risks including Graves’ disease and systemic lupus erythematosus (SLE).63,71 Specific HLA alleles and subclinical autoimmunity markers (eg, elevated autoantibodies) may potentiate autoimmune responses.72,73 Beyond classical genetics, epigenetic regulation (especially DNA methylation)—bridging genetic predisposition and environmental triggers to modulate gene expression and immunity—has emerged as pivotal in oncogenesis and immune responses and is thus relevant to cirAE mechanisms. For instance, tumor suppressor promoter hypermethylation in NSCLC correlates with transcriptional silencing, adverse prognosis, and potential immunomodulatory effects via altered immune cell crosstalk in the tumor microenvironment (TME).74 Collectively, the above associations predominantly derive from parallels with autoimmune pathogenesis and phenotypic resemblances. It is important to note that direct evidence causally connecting specific genetic/epigenetic alterations to defined cirAEs risk in ICI-treated NSCLC patients is sparse. While such genetic/epigenetic perturbations may theoretically underpin ICI-triggered heterogeneous autoantibody/cytokine production,75,76 robust prospective cohort studies and functional validation demonstrating these variants as direct causal risk factors for ICI-associated cirAEs in NSCLC are lacking. Thus, prospective genetic/epigenetic association studies in ICI-treated NSCLC cohorts—integrated with functional validation—are imperative to establish causality, effect magnitudes, and predictive utility of these candidate factors for specific ICI-triggered cirAEs.
Shared Antigen-Driven Cross-Reactivity
Beyond genetic predisposition, numerous irAEs are mechanistically linked to shared antigen-driven cross-reactivity. ICIs augment immune recognition and targeting of tumor antigens. However, imperfect immune recognition specificity enables cross-reactivity against normal tissues expressing epitopes resembling tumor antigens during tumor targeting, precipitating irAEs.77–79 Critical evidence derives from melanoma studies: PD-1 blockade in melanoma patients induces tumor-targeting T cells to cross-react with melanocyte-shared antigens, driving vitiligo and other cirAEs. Crucially, shared T-cell receptor clonotypes detected in melanoma and autoimmune skin lesions directly demonstrate identical clones targeting both tumor and normal skin.80 Similarly, elevated baseline autoantibodies in subclinical autoimmunity correlate with irAEs development.81 Importantly, this paradigm extends to NSCLC. Clinical evidence supports the shared antigen hypothesis in NSCLC-associated cirAEs. NSCLC patients developing cirAEs demonstrate enhanced circulating TCR repertoire diversity.82 T-cell infiltration is consistently observed in post-ICI skin biopsies of NSCLC patients with cirAEs.80,83 While these findings offer strong clinical correlations, direct mechanistic validation (eg, demonstrating identical TCR clones targeting shared NSCLC tumor and skin antigens) is currently lacking. Thus, shared antigen-driven cross-reactivity provides a framework linking ICI efficacy and cirAEs in NSCLC: T-cell responses against epitopes shared between tumor and normal cutaneous tissues may simultaneously drive antitumor immunity and skin toxicity.84 This mechanism directly explains the association between cirAEs and enhanced systemic immune activation/improved clinical outcomes during ICI therapy.62,85–88 Notably, structural similarity between tumor antigens and tolerance-inducing self-antigens may impair APCs efficiency in priming tumor-specific T-cell responses. This APC dysfunction contributes to acquired ICI resistance and interpatient heterogeneity in therapeutic responses and irAEs profiles.89,90
Breakdown of Skin Immune Tolerance
Beyond genetic predisposition and the shared antigen mechanism, a pivotal mechanism underlying cirAEs is the breakdown of local immune tolerance within the skin. Importantly, genetic background may predetermine immune tolerance stability—modulating autoreactive T-cell repertoire size and activity—as noted in Section 2.1. Such genetic determinants may increase susceptibility to profound tolerance disruption and autoimmune responses during ICI therapy. Functionally, as the frontline barrier persistently encountering environmental antigens, skin exhibits a distinctive immune microenvironment designed to safeguard internal homeostasis against direct physical, chemical, and biological insults. To balance this constant surveillance with the need to avoid excessive inflammation, skin maintains a dense network of resident immune cells—including Langerhans cells (LCs), tissue-resident memory T cells (TRM), keratinocytes, and cytokine-secreting melanocytes—that actively surveil the microenvironment.91,92 Collectively, they constitute a hyper-vigilant “immune sentinel post” poised to mount rapid immune responses. However, given perpetual exposure to diverse (often benign) antigens (microbial/chemical/physical), skin must employ exquisite peripheral tolerance mechanisms to distinguish threats from innocuous signals, thereby preventing aberrant/autoimmune reactions. Central to the maintenance of this local tolerance is the persistent expression of immune checkpoint molecules on cutaneous immune cells. Notably, PD-1 is highly expressed on dermal DCs and LCs, serving as a crucial molecule for sustaining local immune tolerance.84 These molecules are essential for curbing nonessential inflammation and enforcing regional self-tolerance.93,94 Consequently, ICI-mediated blockade of these checkpoints prompts hyperactivation of resident/recruited immune cells, driving attacks against self-antigen-expressing cutaneous structures. This intrinsic reliance on checkpoint-dependent tolerogenic mechanisms explains skin’s heightened vulnerability as an early and frequent target of ICI-driven immune dysregulation.84,95–97 Although cutaneous tolerance breakdown offers a rational pathogenic framework, direct experimental evidence establishing causality in skin toxicity remains limited. Future studies must employ advanced approaches—such as single-cell sequencing of cutaneous infiltrates, spatial transcriptomics of key pathways, functional validation of checkpoint roles in tolerance—to establish direct causal evidence.
Predictive Biomarkers of cirAEs
The broad clinical adoption of ICIs underscores the critical need for accurate prediction and early recognition of cirAEs. Robust predictive biomarkers enable preemptive therapeutic planning for high-risk patients and comprehensive assessment of treatment benefit-risk profiles. Nevertheless, discovering biomarkers predictive of early therapeutic response and safety remains challenging. Current biomarkers for predicting ICI-associated cirAEs in NSCLC are broadly classified into multiple categories: traditional markers (eg, demographic/immunological features) and emerging approaches (eg, multi-omics integration and radiomics) (Figure 2). Therefore, critical evaluation of their predictive utility, limitations, and translational potential is essential for directing future research.
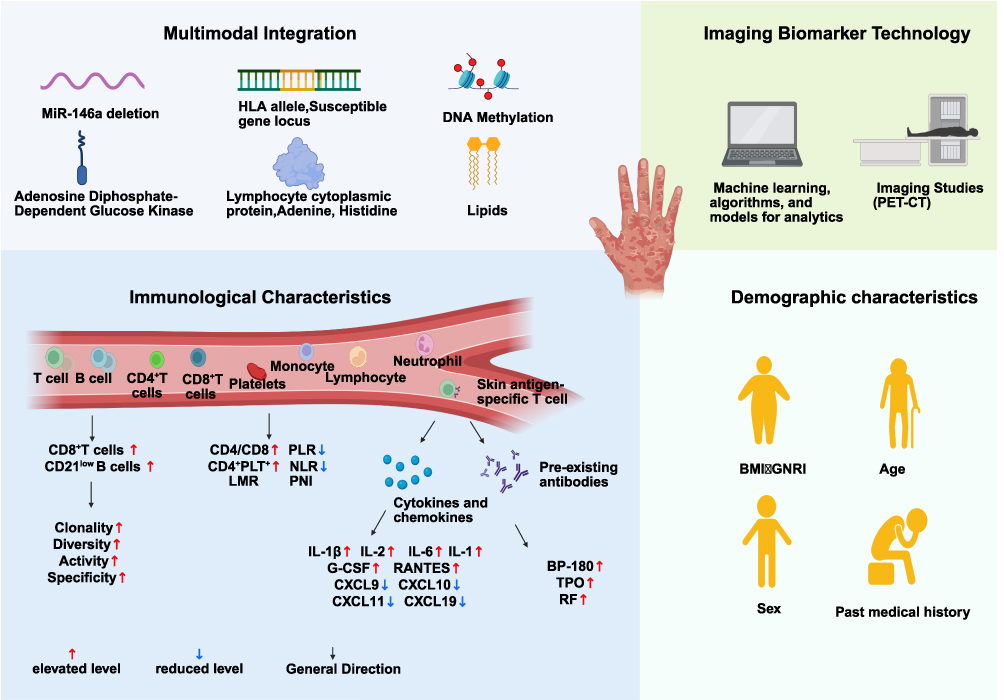 |
Figure 2 Predictive Biomarkers for cirAEs During ICI Treatment in NSCLC. |
Conventional Biomarkers: Demographic/Immunological Features and Translational Challenges
While traditional biomarkers—primarily demographic and immunological features—have been more extensively studied than emerging multi-omics/radiomics approaches, their clinical translation presents substantial challenges. Current investigations largely rely on cross-sectional, cohort, or case–control designs to examine associations between demographic factors and cirAEs.98–100 Reported associations include: Elevated body mass index (BMI) being implicated with heightened cirAE risk during pembrolizumab therapy in retrospective analyses;101,102 Advanced NSCLC patients aged ≥71 years exhibiting higher irAE incidence post-ICI than those aged 65–70 years;103–105 Elevated pretreatment geriatric nutritional risk index (GNRI) correlating with increased incidence of cirAEs (rash/pruritus).106 Sex and comorbidities have also been suggested to modulate cirAE risk.101,107 Despite these observed associations, these demographic factors typically function as risk modifiers rather than direct etiological agents, resulting in limited predictive accuracy and specificity. Consequently, their independent predictive utility and clinical generalizability warrant rigorous validation in large prospective cohorts.
Peripheral blood immune signatures represent valuable potential biomarkers for ICI-associated cirAE prediction due to their minimal invasiveness and serial sampling feasibility. Studies demonstrate that early CD8+ T-cell clonal expansion post-ICI correlates with severe irAEs;108 Combined anti-PD-1/CTLA-4 therapy induces early PD-L1-high clonal CD21low B-cell expansion correlating with severe irAEs.109 Regarding skin-specific toxicities, studies in NSCLC report that: Patients developing ICI-associated cirAEs exhibit early circulating TCR repertoire diversification and skin-antigen-specific T cells peripherally, indicating the predictive potential of these specific immune subsets.80,82 Additional proposed blood-based predictors include: Inflammatory indices: elevated neutrophil-to-lymphocyte ratio (NLR), reduced lymphocyte-to-monocyte ratio (LMR), increased platelet-to-lymphocyte ratio (PLR), and decreased prognostic nutritional index (PNI); Immune cell-based markers: CD4+/CD8+ ratios and circulating CD4+-platelet (PLT+) complexes.110,111
Cytokine and chemokine dynamics represent additional potential predictors for cirAEs. Pro-inflammatory cytokines (eg, IL-2, IFN-γ, TNF-α, IL-1) may correlate with both treatment response and irAE risk.112 Studies have identified several specific associations in NSCLC/lung cancer patients: Lower pretreatment serum levels of C-X-C motif chemokine ligand 9(CXCL9), CXCL10, CXCL11, and CXCL19 are linked to irAE development;113 Elevated post-treatment levels of granulocyte colony-stimulating factor (G-CSF) and regulated upon activation, normal T cell expressed and secreted (RANTES/CCL5) are associated with irAEs;114 Increased serum levels of IL-1β, IL-2, IL-6, and IL-10 during immunotherapy correlate with irAEs.115,116 Furthermore, baseline autoantibody levels constitute potential risk factors for post-ICI irAEs.117 For example: Elevated baseline anti-BP180 antibodies increase the risk of cirAEs in PD-1/PD-L1-blockade-treated NSCLC patients;118 Subclinical autoimmunity markers, such as anti-thyroid peroxidase antibody (anti-TPO), correlate with irAEs in advanced NSCLC;81 Elevated baseline rheumatoid factor (RF) independently predicts cutaneous reactions following ICI treatment.22
Despite extensive research on traditional biomarker-cirAE associations, their clinical predictive utility remains limited. Specifically, key limitations encompass: 1) Methodological constraints: Primarily retrospective designs with small cohorts; 2) Insufficient performance validation: Lack of rigorous evaluation of sensitivity/specificity (eg, ROC analysis, optimal cutoff identification); 3) Low mechanistic evidence: Predominantly observational associations without experimental validation of causality; 4) Heterogeneity and reproducibility issues: Significant inter-study variability with limited replicability across populations, hindering clinical translation.
Emerging Approaches: Multi-Omics Integration and Radiomics
Multi-omics integration (spanning genomics, transcriptomics, proteomics, and metabolomics) elucidates complex pathophysiological networks—from genetic alterations to immune-microbiome crosstalk—thereby surpassing traditional unidimensional approaches. This approach not only accelerates novel cirAE biomarker discovery but also addresses key limitations of conventional markers, such as sample heterogeneity and limited precision.
Multi-omics approaches have identified several promising biomarker candidates for irAEs and ICI response prediction. Through an integrated analysis of messenger RNA (mRNA), microRNA (miRNA), long non-coding RNA (lncRNA), proteins, and non-silent mutations across diverse cancer types, one study identified lymphocyte cytosolic protein 1 (LCP1) and ADP-dependent glucokinase (ADPGK)—both of which are intimately linked to T-cell activation—as candidate biomarkers for irAEs. A predictive model based on the combination of LCP1 and ADPGK showed superior predictive accuracy in linear regression models. In an initial validation cohort of 28 patients, LCP1/ADPGK expression was significantly higher in tissues from patients who experienced irAEs, with the cross-validated area under the curve (AUC) for the combined predictive metric achieving 0.80.119 Nevertheless, these findings warrant confirmation in large-scale, prospective cohorts and independent validation sets, with careful control of potential confounders (eg, limited sample size, single-center bias, and tumor heterogeneity), to further assess their potential for clinical translation. High-resolution HLA-I typing in 179 PD-1/PD-L1-blockade NSCLC patients showed HLA-I homozygosity associated with reduced pruritus/rash risk.120 Furthermore, HLA-DRB1*11:01 allele correlated with pruritus risk—aligning with its atopic dermatitis role—highlighting shared immunogenetics in cutaneous toxicity.121 16S rRNA sequencing implicates gut microbiome composition as a potential cirAE predictor.122,123 MiR-146a deficiency correlates with severe irAEs in preclinical/clinical studies, enabling circulating miRNA-based noninvasive prediction.124 Metabolomics identifies hypoxanthine, histidine,125 indoleamine 2,3-dioxygenase (IDO) activity,126 and very-long-chain fatty acid lipids127 as potential ICI response predictors. Proteomics reveals leukemia inhibitory factor as a novel ICI resistance biomarker.128 Functional proteomic analyses highlight the predictive value of detecting intact PD-1/PD-L1 complexes for ICI response in NSCLC.126,127,129 Importantly, multi-omics biomarkers initially predictive of ICI efficacy may also forecast irAEs, warranting specific validation for toxicity prediction.
Radiomics noninvasively characterizes tumors and their microenvironment through high-throughput extraction of quantitative features from computed tomography (CT), magnetic resonance imaging (MRI), and positron emission tomography-computed tomography (PET-CT) images. Current efforts leverage radiomics as imaging-based surrogates for the TME—particularly for quantifying tumor-infiltrating lymphocytes (TILs), which are established predictors of ICI response. Radiomics models derived from CT, PET-CT, or MRI features enable noninvasive TIL mapping, thereby indirectly informing efficacy and toxicity prediction.130 Furthermore, radiomics-based models (with or without clinical features) can predict tumor PD-L1 expression—a key correlate of both efficacy and toxicity.131–134 Though nascent in oncology, multi-omics-integrated radiomics represents a major frontier. Its noninvasive whole-lesion profiling captures spatial heterogeneity and enables longitudinal monitoring, offering powerful solutions for tumor heterogeneity assessment and dynamic monitoring. Robust radiomic models require validation in prospective trials—especially for organ-specific cirAEs—to achieve clinical utility.
Currently, most novel biomarkers are exploratory or derived from retrospective studies. Their predictive value and underlying causal mechanisms demand rigorous validation. Clinical translation faces challenges related to cost, throughput, and standardization. Future priorities include establishing prospective multi-omics cohorts analyzed using advanced bioinformatics. Such cohorts must control for confounders and address heterogeneity to develop quantifiable predictive models. Ultimately, validation in independent cohorts and interventional trials is essential to guide personalized risk management.135
Conclusions and Future Directions
While ICIs substantially improve outcomes in advanced NSCLC, treatment-induced cirAEs pose significant clinical challenges yet offer unique mechanistic insights. Although current mechanistic frameworks for cirAEs include genetic predisposition, shared antigen-driven cross-reactivity, and cutaneous immune tolerance breakdown, robust direct experimental validation of these hypotheses is still needed. Furthermore, their applicability to non-cutaneous irAEs (eg, pneumonitis, colitis, endocrinopathies) remains unconfirmed. Consequently, future studies should investigate connections between irAEs and classical immune dermatoses, and class-specific mechanisms underlying organ-specific irAEs.
Predictive biomarker development for cirAEs confronts substantial challenges. Despite numerous candidate biomarkers (particularly blood-based immune signatures), limited sensitivity/specificity and significant inter-study heterogeneity constrain their clinical utility. Critically, most candidates remain exploratory—derived primarily from small cohorts—and lack validation in large, independent prospective studies; these are major translational barriers.
A key unresolved clinical dilemma stems from the cirAEs-efficacy association: distinguishing “manageable toxicities” (reflecting beneficial systemic immune activation) from “high-risk toxicities” (potentially severe or life-threatening). Currently, no reliable biomarkers exist to early predict cirAE progression trajectories or identify cases where toxicity dissociates from clinical benefit. Indiscriminate immunosuppression (eg, corticosteroids) may control toxicity but risk compromising antitumor immunity and therapeutic efficacy. Therefore, future biomarker research should prioritize predicting cirAEs progression dynamics and efficacy-decoupling risk over binary occurrence prediction.
A central clinical dilemma arises from the positive association of cirAEs with improved ICI efficacy: how to differentiate “manageable toxicities” indicative of productive anti-tumor immunity from “deleterious toxicities” that might evolve into severe, life-threatening events or substantially compromise quality of life? For this review, “manageable toxicities” are defined as Grade 1–2 adverse events (AEs) (eg, localized maculopapular rash or mild pruritus) that exhibit a good response to topical corticosteroids or systemic immunosuppression (eg, ≤0.5 mg/kg/day prednisone equivalent) and do not require permanent cessation of ICI therapy. Conversely, “high-risk toxicities” denote Grade 3–4 AEs that may progress to life-threatening severe cutaneous adverse reactions (eg, SJS, TEN) despite intensive immunosuppression, or that necessitate permanent discontinuation of ICIs. This distinction highlights a current critical gap: the absence of robust biomarkers capable of precisely predicting, at an early stage, which cirAEs will progress to severity or, conversely, which are associated with a lack of anti-tumor benefit. Indiscriminate use of immunosuppressive agents such as glucocorticoids to quell all inflammatory activity, albeit effective for toxicity management, risks concurrently blunting anti-tumor immune responses, potentially compromising treatment efficacy. Consequently, a paramount goal for future biomarker discovery should be the development of tools that forecast the trajectory of cirAEs severity and the risk of their dissociation from anti-tumor response, moving beyond mere prediction of incidence. Future studies must transcend static correlative analyses and prioritize creating dynamic, biomarker-informed risk stratification models. Such models ought to integrate multi-omics data to identify the critical transition point where toxicity escalates from low to high risk. Promising investigative avenues include, but are not limited to: (1) Baseline genetic risk (eg, specific HLA alleles); (2) Dynamic early serum cytokine/chemokine profiles (eg, rate of change [slope] for IL-6, CXCL9/CXCL10 levels early in treatment); (3) Expansion kinetics and evolutionary patterns of specific T-cell clones in skin biopsies or peripheral blood (informed by the conceptual framework of Subudhi107 et al on the temporal dynamics of CD8+ T-cell clonal expansion and irAEs); (4) Evolving features on serial non-invasive skin imaging. The overarching objective of these efforts should be the identification of early warning signals predictive of toxicity escalation (eg, from Grade 2 to Grade 3), rather than simply forecasting the onset of any-grade toxicity.
Despite these challenges, integrating multi-omics data with AI in prospective studies will accelerate the discovery of robust predictive biomarkers. This will facilitate precision risk stratification, early intervention, and personalized management—preventing unnecessary treatment discontinuation—while advancing mechanistic understanding to optimize the safety–efficacy balance and survival outcomes in advanced NSCLC immunotherapy.
Abbreviations
ICIs, Immune checkpoint inhibitors; NSCLC, non-small cell lung cancer; cirAEs, cutaneous immune-related adverse events; irAEs, immune-related adverse events; PD-1/PD-L1, programmed death-1/programmed death-ligand 1; SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis; DRESS, drug reaction with eosinophilia and systemic symptoms; ORR, objective response rates; PFS, progression-free survival; OS, overall survival; TCR, T-cell receptor; MHC, major histocompatibility complex; APCs, antigen-presenting cells; SHP-2, Src homology 2 domain-containing tyrosine phosphatase; PP2A, protein phosphatase 2A; PI3K, phosphoinositide 3-kinase; AKT, protein kinase A; TGF-β, transforming growth factor-β; IL-10, interleukin-10; NK, natural killer; DCs, Dendritic cells; APCs, antigen-presenting cells; Th2, T helper 2; ITIM, tyrosine-based inhibitory motif; Lck, lymphocyte-specific protein tyrosine kinase; LAG-3, Lymphocyte Activation Gene-3; TIGIT, T cell immunoreceptor with Ig and ITIM domains; TIM-3, T cell immunoglobulin and mucin domain-containing protein-3; Tregs, regulatory T cells; MHC-II, major histocompatibility complex class II molecules; DCs, dendritic cells; HLA, Human Leukocyte Antigen; DNA, deoxyribonucleic acid; LCE3B, Late cornified envelope 3B; GWAS, genome-wide association studies; SLE, systemic lupus erythematosus; TME, tumor microenvironment; LCs, Langerhans cells; TRM, tissue-resident memory T cells; BMI, body mass index; GNRI, geriatric nutritional risk index; NLR, neutrophil-to-lymphocyte ratio; LMR, lymphocyte-to-monocyte ratio; PLR, platelet-to-lymphocyte ratio; PNI, prognostic nutritional index; PLT, Platelet; CXCL9, C-X-C motif chemokine ligand 9; G-CSF, granulocyte colony-stimulating factor; RANTES/CCL5, regulated upon activation, normal T cell expressed and secreted; anti-TPO, anti-thyroid peroxidase antibody; RF, rheumatoid factor; mRNA, messenger RNA; LCP1, lymphocyte cytosolic protein 1; ADPGK, ADP-dependent glucokinase; AUC, area under the curve; IDO, indoleamine 2, 3-dioxygenase; CT, computed tomography; MRI, magnetic resonance imaging; PET-CT, positron emission tomography-computed tomography; TILs, tumor-infiltrating lymphocytes.
Acknowledgments
We want to express our gratitude for the drawing materials provided by BioRender. The authors are grateful to Elsevier Language Editing Services for their help in revising the English in the final version of the paper.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82474484) and the National Key Research and Development Program of China [grant number: 2023YFC3503302].
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Yoneda K, Imanishi N, Ichiki Y, Tanaka F. Immune checkpoint inhibitors (ICIs) in non-small cell lung cancer (NSCLC). J UOEH. 2018;40(2):173–189. doi:10.7888/juoeh.40.173
3. Meissner TB, Schulze HS, Dale SM. Immune editing: overcoming immune barriers in stem cell transplantation. Curr Stem Cell Rep. 2022;8(4):206–218. doi:10.1007/s40778-022-00221-0
4. Starling S. MHC molecules: immune editing shapes the cancer landscape. Nat Rev Immunol. 2017;17(12):729. doi:10.1038/nri.2017.129
5. Sundar R, Huang -K-K, Kumar V, et al. Epigenetic promoter alterations in GI tumour immune-editing and resistance to immune checkpoint inhibition. Gut. 2022;71(7):1277–1288. doi:10.1136/gutjnl-2021-324420
6. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239
7. Vaddepally R, Doddamani R, Sodavarapu S, et al. Review of immune-related adverse events (irAEs) in non-small-cell lung cancer (NSCLC)—their incidence, management, multiorgan iraes, and rechallenge. Biomedicines. 2022;10(4):790. doi:10.3390/biomedicines10040790
8. Zhao H, Ning J, Gu Y, et al. Consecutive severe immune-related adverse events after PD-1 inhibitor induction and surgery in locally advanced non-small cell lung cancer: a case report. Transl Lung Cancer Res. 2021;10(8):3682–3688. doi:10.21037/tlcr-21-603
9. Leitinger M, Varosanec MV, Pikija S, et al. Fatal necrotizing encephalopathy after treatment with nivolumab for squamous non-small cell lung cancer: case report and review of the literature. Front Immunol. 2018;9:108. doi:10.3389/fimmu.2018.00108
10. Li D, He C, Xia Y, Du Y, Zhang J. Pembrolizumab combined with stereotactic body radiotherapy in a patient with human immunodeficiency virus and advanced non-small cell lung cancer: a case report. J Med Case Rep. 2018;12(1):104. doi:10.1186/s13256-018-1667-2
11. Keam SJ. Tremelimumab: first Approval. Drugs. 2023;83(1):93–102. doi:10.1007/s40265-022-01827-8
12. Cui S, Ge X, Li X. A real-world study on the incidence and outcome of immune-related adverse events in lung cancer patients. Zhongguo Fei Ai Za Zhi. 2023;26(4):257–264. doi:10.3779/j.issn.1009-3419.2023.101.08
13. Chang LS, Barroso-Sousa R, Tolaney SM, Hodi FS, Kaiser UB, Min L. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40(1):17–65. doi:10.1210/er.2018-00006
14. Miyahara K, Noda T, Ito Y, et al. an investigation of nine patients with gastrointestinal immune-related adverse events caused by immune checkpoint inhibitors. Digestion. 2020;101(1):60–65. doi:10.1159/000504647
15. Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83(5):1255–1268. doi:10.1016/j.jaad.2020.03.132
16. Puzanov I, Diab A, Abdallah K, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the society for immunotherapy of cancer (SITC) toxicity management working group. J Immunother Cancer. 2017;5(1):95. doi:10.1186/s40425-017-0300-z
17. Gu J, Shi L, Jiang X, et al. Severe immune-related adverse events of immune checkpoint inhibitors for advanced non-small cell lung cancer: a network meta-analysis of randomized clinical trials. Cancer Immunol Immunother. 2022;71(9):2239–2254. doi:10.1007/s00262-022-03140-5
18. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. 3303 efficacy and safety in key patient subgroups of nivolumab (NIVO) alone or combined with ipilimumab (IPI) versus IPI alone in treatment-naïve patients with advanced melanoma (MEL) (CheckMate 067). Euro J Cancer. 2015:51:S664–S665. doi:10.1016/s0959-8049(16)31822-6.
19. Nguyen N, Wan G, Ugwu-Dike P, et al. Influence of melanoma type on incidence and downstream implications of cutaneous immune-related adverse events in the setting of immune checkpoint inhibitor therapy. J Am Acad Dermatol. 2023;88(6):1308–1316. doi:10.1016/j.jaad.2023.02.014
20. Yamaguchi A, Saito Y, Narumi K, et al. Association between skin immune-related adverse events (irAEs) and multisystem irAEs during PD-1/PD-L1 inhibitor monotherapy. J Cancer Res Clin Oncol. 2023;149(4):1659–1666. doi:10.1007/s00432-022-04425-z
21. Li Y, Zhang Y, Jia X, et al. Effect of immune-related adverse events and pneumonitis on prognosis in advanced non-small cell lung cancer: a comprehensive systematic review and meta-analysis. Clin Lung Cancer. 2021;22(6):e889–e900. doi:10.1016/j.cllc.2021.05.004
22. Aso M, Toi Y, Sugisaka J, et al. Association between skin reaction and clinical benefit in patients treated with anti-programmed cell death 1 monotherapy for advanced non-small cell lung cancer. Oncologist. 2020;25(3):e536–e544. doi:10.1634/theoncologist.2019-0550
23. Cortellini A, Chiari R, Ricciuti B, et al. Correlations between the immune-related adverse events spectrum and efficacy of anti-PD1 immunotherapy in NSCLC patients. Clin Lung Cancer. 2019;20(4):237–247e1. doi:10.1016/j.cllc.2019.02.006
24. Medri M, Savoia F, Foca F, et al. A retrospective observational study on cutaneous adverse events induced by immune checkpoint inhibitors. Ital J Dermatol Venerol. 2023;158(6):437–444. doi:10.23736/S2784-8671.23.07542-4
25. Shankar B, Zhang J, Naqash AR, et al. Multisystem immune-related adverse events associated with immune checkpoint inhibitors for treatment of non-small cell lung cancer. JAMA Oncol. 2020;6(12):1952–1956. doi:10.1001/jamaoncol.2020.5012
26. Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. doi:10.1128/mcb.25.21.9543-9553.2005
27. Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303–310. doi:10.1084/jem.192.2.303
28. O’Neill RE, Cao X. Co-stimulatory and co-inhibitory pathways in cancer immunotherapy. Adv Cancer Res. 2019;143:145–194. doi:10.1016/bs.acr.2019.03.003
29. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39(1):98–106. doi:10.1097/COC.0000000000000239
30. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. 2018;131(1):58–67. doi:10.1182/blood-2017-06-741033
31. Marangoni F, Zhakyp A, Corsini M, et al. Expansion of tumor-associated treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell. 2021;184(15):3998–4015e19. doi:10.1016/j.cell.2021.05.027
32. Wang CJ, Kenefeck R, Wardzinski L, et al. Cutting edge: cell-extrinsic immune regulation by CTLA-4 expressed on conventional T cells. J Immunol. 2012;189(3):1118–1122. doi:10.4049/jimmunol.1200972
33. Kong KF, Fu G, Zhang Y, et al. Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat Immunol. 2014;15(5):465–472. doi:10.1038/ni.2866
34. Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. doi:10.1126/science.1202947
35. Marzec M, Zhang Q, Goradia A, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci USA. 2008;105(52):20852–20857. doi:10.1073/pnas.0810958105
36. Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. doi:10.1126/scitranslmed.3003689
37. Sheppard KA, Fitz LJ, Lee JM, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574(1–3):37–41. doi:10.1016/j.febslet.2004.07.083
38. Huber S, Hoffmann R, Muskens F, Voehringer D. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood. 2010;116(17):3311–3320. doi:10.1182/blood-2010-02-271981
39. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–122. doi:10.1016/j.immuni.2007.05.016
40. Akbari O, Stock P, Singh AK, et al. PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol. 2010;3(1):81–91. doi:10.1038/mi.2009.112
41. Hui E, Cheung J, Zhu J, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–1433. doi:10.1126/science.aaf1292
42. Kamphorst AO, Wieland A, Nasti T, et al. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science. 2017;355(6332):1423–1427. doi:10.1126/science.aaf0683
43. Xia L, Liu Y, Wang Y. PD-1/PD-L1 blockade therapy in advanced non-small-cell lung cancer: current status and future directions. Oncologist. 2019;24(Suppl S1):S31–S41. doi:10.1634/theoncologist.2019-IO-S1-s05
44. Liu Y, Wu L, Tong R, et al. PD-1/PD-L1 inhibitors in cervical cancer. Front Pharmacol. 2019;10:65. doi:10.3389/fphar.2019.00065
45. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA. 2001;98(24):13866–13871. doi:10.1073/pnas.231486598
46. Kuang D-M, Zhao Q, Peng C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206(6):418–424. doi:10.21873/invivo.13454
47. Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mole Cellular Biolog. 2023;25(21):9543–9553. doi:10.1128/mcb.25.21.9543-9553.2005
48. Appleman LJ, van Puijenbroek AAFL, Shu KM, Nadler LM, Boussiotis VA. CD28 costimulation mediates down-regulation of p27 kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J Immunol. 2002;168(6):2729–2736. doi:10.4049/jimmunol.168.6.2729
49. Schutz F, Stefanovic S, Mayer L, von Au A, Domschke C, Sohn C. PD-1/PD-L1 pathway in breast cancer. Oncol Res Treat. 2017;40(5):294–297. doi:10.1159/000464353
50. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26(1):677–704. doi:10.1146/annurev.immunol.26.021607.090331
51. Cao Z, Kon N, Liu Y, et al. An unexpected role for p53 in regulating cancer cell-intrinsic PD-1 by acetylation. Sci Adv. 2021;7(14). doi:10.1126/sciadv.abf4148
52. Yao H, Wang H, Li C, Fang J-Y, Xu J. Cancer cell-intrinsic PD-1 and implications in combinatorial immunotherapy. Front Immunol. 2018;9:1774. doi:10.3389/fimmu.2018.01774
53. Andrews LP, Marciscano AE, Drake CG, Vignali DA. A. LAG 3 (CD 223) as a cancer immunotherapy target. Immunol Rev. 2017;276(1):80–96. doi:10.1111/imr.12519
54. Johnson AM, Bullock BL, Neuwelt AJ, et al. Cancer Cell–intrinsic expression of MHC class II regulates the immune microenvironment and response to anti–PD-1 therapy in lung adenocarcinoma. J Immunol. 2020;204(8):2295–2307. doi:10.4049/jimmunol.1900778
55. Yan J, Yu Y, Wang N, et al. LFIRE-1/HFREP-1, a liver-specific gene, is frequently downregulated and has growth suppressor activity in hepatocellular carcinoma. Oncogene. 2004;23(10):1939–1949. doi:10.1038/sj.onc.1207306
56. Nagasaki J, Togashi Y, Sugawara T, et al. The critical role of CD4+ T cells in PD-1 blockade against MHC-II–expressing tumors such as classic Hodgkin lymphoma. Blood Adv. 2020;4(17):4069–4082. doi:10.1182/bloodadvances.2020002098
57. Hemon P, Jean-Louis F, Ramgolam K, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011;186(9):5173–5183. doi:10.4049/jimmunol.1002050
58. Yu X, Harden K, Gonzalez LC, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57. doi:10.1038/ni.1674
59. Stanietsky N, Simic H, Arapovic J, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA. 2009;106(42):17858–17863. doi:10.1073/pnas.0903474106
60. Gao X, Zhu Y, Li G, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7(2):e30676. doi:10.1371/journal.pone.0030676
61. Zhang Y, Cai P, Liang T, Wang L, Hu L. TIM-3 is a potential prognostic marker for patients with solid tumors: a systematic review and meta-analysis. Oncotarget. 2017;8(19):31705–31713. doi:10.18632/oncotarget.15954
62. Teulings H-E, Limpens J, Jansen SN, et al. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol. 2015;33(7):773–781. doi:10.1200/JCO.2014.57.4756
63. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. 2013;14(9):661–673. doi:10.1038/nrg3502
64. Ohtsuka M, Miura T, Mori T, Ishikawa M, Yamamoto T. Occurrence of psoriasiform eruption during nivolumab therapy for primary oral mucosal melanoma. JAMA Dermatolog. 2015;151(7):797–799. doi:10.1001/jamadermatol.2015.0249
65. Bonigen J, Raynaud‐Donzel C, Hureaux J, et al. Anti- PD 1-induced psoriasis: a study of 21 patients. J Eur Acad Dermatol Venereol. 2017;31(5):e254–e257. doi:10.1111/jdv.14011
66. Ayala-Fontanez N, Soler DC, McCormick TS. Current knowledge on psoriasis and autoimmune diseases. Psoriasis. 2016;6:7–32. doi:10.2147/PTT.S64950
67. Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5(9):699–711. doi:10.1038/nri1689
68. Hoa S, Laaouad L, Roberts J, et al. Preexisting autoimmune disease and immune-related adverse events associated with anti-PD-1 cancer immunotherapy: a national case series from the Canadian research group of rheumatology in immuno-oncology. Cancer Immunol Immunother. 2021;70(8):2197–2207. doi:10.1007/s00262-021-02851-5
69. Parisi R, Iskandar IYK, Kontopantelis E, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. 2020:
70. Ran D, Cai M, Zhang X. Genetics of psoriasis: a basis for precision medicine. Precis Clin Med. 2019;2(2):120–130. doi:10.1093/pcmedi/pbz011
71. Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015:518(7539):337–43. doi:10.1038/nature13835.
72. Allcock RJ. The major histocompatibility complex: a paradigm for studies of the human genome. Methods Mol Biol. 2012;882:1–7. doi:10.1007/978-1-61779-842-9_1
73. Hofstetter AR, Sullivan LC, Lukacher AE, Brooks AG. Diverse roles of non-diverse molecules: MHC class Ib molecules in host defense and control of autoimmunity. Curr Opin Immunol. 2011;23(1):104–110. doi:10.1016/j.coi.2010.09.009
74. Tran T-O, Lam LHT, Le NQK. Hyper-methylation of ABCG1 as an epigenetics biomarker in non-small cell lung cancer. Funct Integr Genomics. 2023;23(3):256. doi:10.1007/s10142-023-01185-y
75. Perricone C, Agmon-Levin N, Ceccarelli F, Valesini G, Anaya J-M, Shoenfeld Y. Genetics and autoantibodies. Immunol Res. 2013;56(2–3):206–219. doi:10.1007/s12026-013-8396-9
76. Ma W-T, Chang C, Gershwin ME, Lian Z-X. Development of autoantibodies precedes clinical manifestations of autoimmune diseases: a comprehensive review. J Autoimmun. 2017;83:95–112. doi:10.1016/j.jaut.2017.07.003
77. Esfahani K, Elkrief A, Calabrese C, et al. Moving towards personalized treatments of immune-related adverse events. Nat Rev Clin Oncol. 2020;17(8):504–515. doi:10.1038/s41571-020-0352-8
78. Berner F, Niederer R, Luimstra JJ, et al. Keratinocyte differentiation antigen-specific T cells in immune checkpoint inhibitor-treated NSCLC patients are associated with improved survival. Oncoimmunology. 2021;10(1):2006893. doi:10.1080/2162402X.2021.2006893
79. Berner F, Bomze D, Lichtensteiger C, et al. Autoreactive napsin A–specific T cells are enriched in lung tumors and inflammatory lung lesions during immune checkpoint blockade. Sci Immunol. 2022;7(75):eabn9644. doi:10.1126/sciimmunol.abn9644
80. Berner F, Bomze D, Diem S, et al. Association of checkpoint inhibitor-induced toxic effects with shared cancer and tissue antigens in non-small cell lung cancer. JAMA Oncol. 2019;5(7):1043–1047. doi:10.1001/jamaoncol.2019.0402
81. Toi Y, Sugawara S, Sugisaka J, et al. Profiling preexisting antibodies in patients treated with anti–PD-1 therapy for advanced non–small cell lung cancer. JAMA Oncol. 2019;5(3):376–383. doi:10.1001/jamaoncol.2018.5860
82. Oh DY, Cham J, Zhang L, et al. Immune toxicities elicted by CTLA-4 blockade in cancer patients are associated with early diversification of the T-cell repertoire. Cancer Res. 2017;77(6):1322–1330. doi:10.1158/0008-5472.CAN-16-2324
83. Hasan Ali O, Diem S, Markert E, et al. Characterization of nivolumab-associated skin reactions in patients with metastatic non-small cell lung cancer. Oncoimmunology. 2016;5(11):e1231292. doi:10.1080/2162402X.2016.1231292
84. Byrne EH, Fisher DE. Immune and molecular correlates in melanoma treated with immune checkpoint blockade. Cancer. 2017;123(S11):2143–2153. doi:10.1002/cncr.30444
85. Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatolog. 2016;152(1):45–51. doi:10.1001/jamadermatol.2015.2707
86. Downey SG, Klapper JA, Smith FO, et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade. Clin Cancer Res. 2007;13(22):6681–6688. doi:10.1158/1078-0432.CCR-07-0187
87. Haratani K, Hayashi H, Chiba Y, et al. Association of immune-related adverse events with nivolumab efficacy in non–small-cell lung cancer. JAMA Oncol. 2018;4(3):374–378. doi:10.1001/jamaoncol.2017.2925
88. Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatolog. 2015;151(11):1206–1212. doi:10.1001/jamadermatol.2015.1916
89. Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48(3):417–433. doi:10.1016/j.immuni.2018.03.007
90. Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348(6230):124–128. doi:10.1126/science.aaa1348
91. Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev. 2018;283(1):54–76. doi:10.21873/invivo.13454
92. Doebel T, Voisin B, Nagao K. Langerhans cells – the macrophage in dendritic cell clothing. Trends Immunol. 2017;38(11):817–828. doi:10.1038/ncomms10391
93. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Reviews Immunology. 2013;13(4):173–189. doi:10.7888/juoeh.40.173
94. Nam S, Lee A, Lim J, Lim J-S. Analysis of the expression and regulation of PD-1 protein on the surface of myeloid-derived suppressor cells (MDSCs). Biomol Ther. 2019;27(1):63–70. doi:10.4062/biomolther.2018.201
95. Esfahani K, Miller WH Jr. Reversal of autoimmune toxicity and loss of tumor response by interleukin-17 blockade. N Engl J Med. 2017;376(20):1989–1991. doi:10.1056/NEJMc1703047
96. Tarhini AA, Zahoor H, Lin Y, et al. Baseline circulating IL-17 predicts toxicity while TGF-β1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J Immunother Cancer. 2015;3(1):39. doi:10.1186/s40425-015-0081-1
97. Lo JA, Fisher DE, Flaherty KT. Prognostic significance of cutaneous adverse events associated with pembrolizumab therapy. JAMA Oncol. 2015;1(9):1340–1341. doi:10.1001/jamaoncol.2015.2274
98. Head L, Gorden N, Van Gulick R, et al. Biomarkers to predict immune-related adverse events with checkpoint inhibitors. J Clin Oncol. 2019;37(8_suppl):131. doi:10.1200/JCO.2019.37.8_suppl.131
99. Fujisawa Y, Yoshino K, Otsuka A, et al. Fluctuations in routine blood count might signal severe immune-related adverse events in melanoma patients treated with nivolumab. J Dermatol Sci. 2017;88(2):225–231. doi:10.1016/j.jdermsci.2017.07.007
100. Diehl A, Yarchoan M, Hopkins A, Jaffee E, Grossman SA. Relationships between lymphocyte counts and treatment-related toxicities and clinical responses in patients with solid tumors treated with PD-1 checkpoint inhibitors. Oncotarget. 2017;8(69):114268–114280. doi:10.18632/oncotarget.23217
101. Cortellini A, Bersanelli M, Santini D, et al. Another side of the association between body mass index (BMI) and clinical outcomes of cancer patients receiving programmed cell death protein-1 (PD-1)/ programmed cell death-ligand 1 (PD-L1) checkpoint inhibitors: a multicentre analysis of immune-related adverse events. Eur J Cancer. 2020;128:17–26. doi:10.1016/j.ejca.2019.12.031
102. Eun Y, Kim IY, Sun JM, et al. Risk factors for immune-related adverse events associated with anti-PD-1 pembrolizumab. Sci Rep. 2019;9(1):14039. doi:10.1038/s41598-019-50574-6
103. Gao J, Zhang P, Tang M, et al. Predictors of immune checkpoint inhibitor-related adverse events in older patients with lung cancer: a prospective real-world analysis. J Cancer Res Clin Oncol. 2023;149(11):8993–9006. doi:10.1007/s00432-023-04792-1
104. Mebarki S, Pamoukdjian F, Pierro M, et al. Safety and efficacy of immunotherapy according to the age threshold of 80 years. Bull Cancer. 2023;110(5):570–580. doi:10.1016/j.bulcan.2023.02.010
105. Storm BN, Abedian Kalkhoran H, Wilms EB, et al. Real-life safety of PD-1 and PD-L1 inhibitors in older patients with cancer: an observational study. J Geriatr Oncol. 2022;13(7):997–1002. doi:10.1016/j.jgo.2022.05.013
106. Shimizu A, Fukasawa M, Mitani K, et al. Association of geriatric nutritional risk index with immune checkpoint inhibitor treatment duration and adverse events in lung cancer. Vivo. 2024;38(1):418–424. doi:10.21873/invivo.13454
107. Kartolo A, Sattar J, Sahai V, Baetz T, Lakoff JM. Predictors of immunotherapy-induced immune-related adverse events. Curr Oncol. 2018;25(5):e403–e410. doi:10.3747/co.25.4047
108. Subudhi SK, Aparicio A, Gao J, et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc Natl Acad Sci USA. 2016;113(42):11919–11924. doi:10.1073/pnas.1611421113
109. Das R, Bar N, Ferreira M, et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J Clin Invest. 2018;128(2):715–720. doi:10.1172/JCI96798
110. Zamora C, Riudavets M, Anguera G, et al. Circulating leukocyte-platelet complexes as a predictive biomarker for the development of immune-related adverse events in advanced non-small cell lung cancer patients receiving anti-PD-(L)1 blocking agents. Cancer Immunol Immunother. 2021;70(6):1691–1704. doi:10.1007/s00262-020-02793-4
111. Zhang Z, Xie T, Qi C, Zhang X, Shen L, Peng Z. Peripheral blood biomarkers predictive of efficacy outcome and immune-related adverse events in advanced gastrointestinal cancers treated with checkpoint inhibitors. Cancers. 2022;14(15). doi:10.3390/cancers14153736
112. Duchemann B, Naigeon M, Auclin E, et al. CD8(+)PD-1(+) to CD4(+)PD-1(+) ratio (PERLS) is associated with prognosis of patients with advanced NSCLC treated with PD-(L)1 blockers. J Immunother Cancer. 2022;10(2). doi:10.1136/jitc-2021-004012
113. Khan S, Khan SA, Luo X, et al. Immune dysregulation in cancer patients developing immune-related adverse events. Br J Cancer. 2019;120(1):63–68. doi:10.1038/s41416-018-0155-1
114. Oyanagi J, Koh Y, Sato K, et al. Predictive value of serum protein levels in patients with advanced non-small cell lung cancer treated with nivolumab. Lung Cancer. 2019;132:107–113. doi:10.1016/j.lungcan.2019.03.020
115. Phillips GS, Wu J, Hellmann MD, et al. Treatment outcomes of immune-related cutaneous adverse events. J Clin Oncol. 2019;37(30):2746–2758. doi:10.1200/JCO.18.02141
116. Kaunitz GJ, Loss M, Rizvi H, et al. Cutaneous eruptions in patients receiving immune checkpoint blockade: clinicopathologic analysis of the nonlichenoid histologic pattern. Am J Surg Pathol. 2017;41(10):1381–1389. doi:10.1097/PAS.0000000000000900
117. Williams TJ, Benavides DR, Patrice KA, et al. Association of autoimmune encephalitis with combined immune checkpoint inhibitor treatment for metastatic cancer. JAMA Neurol. 2016;73(8):928–933. doi:10.1001/jamaneurol.2016.1399
118. Hasan Ali O, Bomze D, Ring SS, et al. BP180-specific IgG is associated with skin adverse events, therapy response, and overall survival in non-small cell lung cancer patients treated with checkpoint inhibitors. J Am Acad Dermatol. 2020;82(4):854–861. doi:10.1016/j.jaad.2019.08.045
119. Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466(7302):113–117. doi:10.1038/nature09114
120. Abed A, Law N, Calapre L, et al. Human leucocyte antigen genotype association with the development of immune-related adverse events in patients with non-small cell lung cancer treated with single agent immunotherapy. Eur J Cancer. 2022;172:98–106. doi:10.1016/j.ejca.2022.05.021
121. Horton R, Gibson R, Coggill P, et al. Variation analysis and gene annotation of eight MHC haplotypes: the MHC haplotype project. Immunogenetics. 2008;60(1):1–18. doi:10.1007/s00251-007-0262-2
122. Chaput N, Lepage P, Coutzac C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368–1379. doi:10.1093/annonc/mdx108
123. Dubin K, Callahan MK, Ren B, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016;7(1):10391. doi:10.1038/ncomms10391
124. Marschner D, Falk M, Javorniczky NR, et al. MicroRNA-146a regulates immune-related adverse events caused by immune checkpoint inhibitors. JCI Insight. 2020;5(6). doi:10.1172/jci.insight.132334
125. Nie X, Xia L, Gao F, et al. Serum metabolite biomarkers predictive of response to PD-1 blockade therapy in non-small cell lung cancer. Front Mol Biosci. 2021;8:678753. doi:10.3389/fmolb.2021.678753
126. Kocher F, Amann A, Zimmer K, et al. High indoleamine-2,3-dioxygenase 1 (IDO) activity is linked to primary resistance to immunotherapy in non-small cell lung cancer (NSCLC). Transl Lung Cancer Res. 2021;10(1):304–313. doi:10.21037/tlcr-20-380
127. Mock A, Zschabitz S, Kirsten R, et al. Serum very long-chain fatty acid-containing lipids predict response to immune checkpoint inhibitors in urological cancers. Cancer Immunol Immunother. 2019;68(12):2005–2014. doi:10.1007/s00262-019-02428-3
128. Loriot Y, Marabelle A, Guégan JP, et al. Plasma proteomics identifies leukemia inhibitory factor (LIF) as a novel predictive biomarker of immune-checkpoint blockade resistance. Ann Oncol. 2021;32(11):1381–1390. doi:10.1016/j.annonc.2021.08.1748
129. Sánchez-Magraner L, Gumuzio J, Miles J, et al. Functional engagement of the PD-1/PD-L1 complex but not PD-L1 expression is highly predictive of patient response to immunotherapy in non-small-cell lung cancer. J Clin Oncol. 2023;41(14):2561–2570. doi:10.1200/jco.22.01748
130. Boutros C, Tarhini A, Routier E, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13(8):473–486. doi:10.1038/nrclinonc.2016.58
131. Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19(3):345–361. doi:10.1007/s40257-017-0336-3
132. Quach HT, Johnson DB, LeBoeuf NR, Zwerner JP, Dewan AK. Cutaneous adverse events caused by immune checkpoint inhibitors. J Am Acad Dermatol. 2021;85(4):956–966. doi:10.1016/j.jaad.2020.09.054
133. Capdevila J, Wirth LJ, Ernst T, et al. PD-1 blockade in anaplastic thyroid carcinoma. J Clin Oncol. 2020;38(23):2620–2627. doi:10.1200/jco.19.02727
134. Ueno M, Ikeda M, Morizane C, et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: a non-randomised, multicentre, open-label, Phase 1 study. Lancet Gastroenterol Hepatol. 2019;4(8):611–621. doi:10.1016/s2468-1253(19)30086-x
135. Tran TO, Vo TH, Le NQK. Omics-based deep learning approaches for lung cancer decision-making and therapeutics development. Brief Funct Genomics. 2024;23(3):181–192. doi:10.1093/bfgp/elad031
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.