Back to Journals » ImmunoTargets and Therapy » Volume 3
Dendritic cell vaccination for glioblastoma multiforme: review with focus on predictive factors for treatment response
Authors Dejaegher J, Van Gool S, De Vleeschouwer S
Received 4 December 2013
Accepted for publication 22 January 2014
Published 13 March 2014 Volume 2014:3 Pages 55—66
DOI https://doi.org/10.2147/ITT.S40121
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Joost Dejaegher,1 Stefaan Van Gool,2 Steven De Vleeschouwer1
1Department of Neurosciences, 2Department of Microbiology and Immunology, KU Leuven, Leuven, Belgium
Abstract: Glioblastoma multiforme (GBM) is the most common and most aggressive type of primary brain cancer. Since median overall survival with multimodal standard therapy is only 15 months, there is a clear need for additional effective and long-lasting treatments. Dendritic cell (DC) vaccination is an experimental immunotherapy being tested in several Phase I and Phase II clinical trials. In these trials, safety and feasibility have been proven, and promising clinical results have been reported. On the other hand, it is becoming clear that not every GBM patient will benefit from this highly personalized treatment. Defining the subgroup of patients likely to respond to DC vaccination will position this option correctly amongst other new GBM treatment modalities, and pave the way to incorporation in standard therapy. This review provides an overview of GBM treatment options and focuses on the currently known prognostic and predictive factors for response to DC vaccination. In this way, it will provide the clinician with the theoretical background to refer patients who might benefit from this treatment.
Keywords: immunotherapy, personalized medicine, brain tumor, stratification
Introduction to glioblastoma multiforme
Glioblastoma multiforme (GBM), the World Health Organization (WHO) grade 4 malignant astrocytoma, is the most aggressive primary brain tumor.1 Clinically, radiologically and genetically, it can be subdivided into primary and secondary GBM. Primary GBM accounts for more than 90% of GBM cases and originates de novo, whereas secondary GBM arises after transformation from a prior low-grade glioma.2 The incidence of GBM is 2–3 per 100,000 per year for adults and 1.6–2 per million per year in children.3,4 It is the most common primary brain tumor, as it accounts for 16% of all central nervous system tumors.3,5 In spite of being infrequent, GBM is undoubtedly one of the most debilitating and lethal types of human cancer. This is illustrated by the fact that these central nervous system tumors have the highest average “years of life lost”, which is a measurement for the impact of cancer on the individual patient.6 Because of the fast and infiltrative growth pattern, the natural course of a GBM is unmerciful with progressive neurological deterioration resulting in death in the short term.7 To date, GBM remains incurable.
Current treatment options and outcomes
The current standard of care for newly diagnosed GBM in adult patients up to 70 years consists of maximal safe neurosurgical resection, followed by 6 weeks of radiotherapy with concomitant temozolomide and six adjuvant cycles of temozolomide. This so-called Stupp protocol was adapted from a landmark multicenter randomized Phase III trial.8 A significant improvement of median overall survival (OS) from 12.1 to 14.6 months, and of median progression-free survival (PFS) from 5.0 to 6.9 months was the result of adding temozolomide chemotherapy to surgery and radiotherapy. Strikingly, there was an important increase of long-term survivors, with 5-year survival rates of 9.8% in the radiotherapy and temozolomide group versus 1.9% in the radiotherapy alone group.9 Epigenetic silencing of the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) by promoter methylation accounts for the major part of the survival benefit of adding temozolomide, although MGMT promoter methylation was also found to be an independent prognostic factor.10
Despite multimodal treatment, relapse is universal and is associated with a very bad prognosis. No established standard of care exists for patients with a relapsed GBM, but a wide variety of treatment modalities and regimens is or has been the object of clinical trials. The intensive search for new treatments is being driven by a high clinical need and represents the more aggressive and less fatalistic attitude towards a relapsed GBM. We will only briefly discuss this topic to have a background for further reading, and refer to excellent and thorough reviews for more detailed information.11,12
At relapse, reoperation should always be considered. The exact benefit of re-resection, however, remains unknown and is further blurred by the inherent selection bias in case of repeat surgery.13 Moreover, reoperation is rarely performed without further postoperative adjuvant treatment. Re-irradiation with (hypofractionated) stereotactic radiotherapy or stereotactic radiosurgery for smaller recurrences can be options in selected patients, but their usefulness is still a matter of debate. More often, second-line or salvage chemotherapy is applied. Many chemotherapeutic agents and regimens have been studied. Rechallenge temozolomide and the use of nitrosoureas are the most often used types of chemotherapy in variable schedules and doses, but no regimen has proven to be superior. Overall, the median OS with chemotherapy in studies is reported to be around 5–13 months from start of retreatment. Combination regimens have not been demonstrated to be more effective. Bevacizumab, a monoclonal antibody against vascular endothelial growth factor (VEGF), has extensively been studied and has been approved for recurrent GBM in the US, but not in Europe. Median OS was 9.3 months in a recent meta-analysis.14 Other investigators studied the local implantation of chemotherapy wafers after resection, gene therapy, or electrical field therapy.15–17 Another emerging experimental therapy is dendritic cell (DC) vaccination, not only in the setting of relapsed GBM but also in the patients with newly diagnosed GBM. This will be the focus of the remainder of this review.
Introduction to immunotherapy with the focus on DC vaccination
Immunotherapy is theoretically appealing. If directed against tumor-specific antigens, immunotherapy can target all invasive tumor cells while sparing normal tissues with a high degree of specificity. Furthermore, active specific immunotherapy can induce an immunological memory leading to a long-lasting effect. Immunotherapy has been studied for many cancers, and has been approved by the Food and Drug Administration (FDA) of the US for prostate cancer and metastatic melanoma.18,19 As a treatment for GBM, it is still in the experimental stage. The rationale for immunotherapy and the different types, their mechanisms, and early clinical experiences have been extensively reviewed by our and other research groups during the last years.20–29 Briefly, one can distinguish four types of immunotherapy which have been studied in gliomas. Restorative immunotherapy consists of administration of cytokines, like interleukin (IL)-2, to enhance non-specific immunity. It has been largely abandoned because of toxicity. Adoptive immunotherapy consists of the administration of ex vivo-activated lymphocytes. These can be non-specific lymphokine activated killer (LAK) cells, studied from the late 1980s to the early 2000s, or specific cytotoxic T-lymphocytes (CTLs) activated against a specific antigen in later trials. In passive immunotherapy, monoclonal antibodies active against a tumor-associated antigen are infused. Bevacizumab belongs to this category. Finally, active specific immunotherapy aims for the induction and boosting of the patient’s own immune system specifically against the tumor. Only the latter can induce an immunological memory. This can be done by injecting peptide-based vaccines such as the EGFRvIII mutant vaccine,30 or by injecting antigen-presenting cells loaded with tumor antigens.26 For the latter application, DC are the most powerful antigen-presenting cells, and hence commonly used for that purpose.31 Different sources of antigens to load the DC have been tried in several clinical trials. Most commonly used are whole tumor lysates,32–48 autologous or synthetic tumor-associated peptides,49–56 or messenger RNA (mRNA) from autologous glioma cells or cancer stem cells.57,58 Other investigators have tried single-cell suspensions of tumor cells,59 interferon-γ (IFN-γ) immunologically enhanced and heat shock-treated cultured GBM cells,60 or heat shock-induced apoptotic GBM cells to load DC.61 Furthermore, Kikuchi et al used fusions of DC with autologous glioma cells62,63 and Okada et al used lysate-loaded DC together with IL-4 transfected fibroblasts.64 All of these clinical trials with DC vaccination were Phase I or II, focusing mainly on safety and feasibility. Late Phase II and Phase III trials are currently underway. In general, vaccination therapy was safe and well-tolerated, with only two serious adverse events reported. The first was an overwhelming inflammatory reaction in a patient with a large residual tumor,36 and the second was a cutaneous GBM after delayed-type hypersensitivity (DTH) testing, believed to rely on radioresistant tumor cells.38 It is difficult to extract other general conclusions from these trials, mainly due to the heterogeneity in the patient population, laboratory procedures used to produce the vaccines, origin of the antigens, administration route and timing, different monitoring assays used for immune monitoring, and different endpoints reported on. However, tumor-specific responses and prolonged survival have been observed in a subgroup of patients. A model to predict this subgroup is not yet available, and nor is a method to further increase the potential number of patients who might benefit from this type of immunotherapy. In general, it is believed that the incremental benefits obtained by tailoring the vaccine will be much smaller than the huge benefits that can be expected by exploring rational combinations of tumor vaccines and strong immunomodulators intervening with checkpoint molecules and key-hub pathways. This novel insight will be elaborated on in more detail in the following sections.
Who will benefit from DC vaccination?
Estimating which patients will benefit from DC vaccination is clinically important and relevant. Firstly, generating autologous tumor lysate DC vaccines implies the collection of tumor tissue obtained in an operation. Using alternative sources of antigens for vaccination other than tumor lysate will inherently restrict the approach to certain (highly selected) subgroups of patients. An operation or reoperation is also important to start the immunotherapy in a state of minimal residual disease (see below). In newly diagnosed patients, surgery is part of the standard therapy. In the setting of relapsed GBM however, a reoperation is not always clearly indicated. The decision to reoperate can depend on the objective to start postoperative vaccination therapy. Also, a leukapheresis has to be performed to harvest a large amount of peripheral blood monocytes as precursor cells for DCs. Thus, starting DC vaccination has to be considered as an invasive therapy. Secondly, the generation, storage, and administration of vaccines are all labor-intensive and expensive activities. In this section, we discuss the currently known prognostic and predictive factors for treatment response to DC vaccination.
Pretreatment patient characteristics
In GBM and other high-grade gliomas, pretreatment patient characteristics were stated to have more impact on outcome than any new treatment itself.65 Although provocative, it points out that every new treatment should stratify for these pretreatment variables. We discuss the most important in the setting of DC vaccination.
Age and recursive partitioning analysis (RPA) classification
Age is probably the most important predictor for outcome after DC vaccination.39 This can partially be attributed to the less aggressive biological behavior of GBM in younger patients.66 However, there is a decreased potency to elicit active immune responses in older patients, because of lower amounts of circulating naïve CD8+ T cells that can be primed against novel antigens.67 It has been shown that naïve recent thymic CD8+ emigrants accounted for the majority of tumor reactive T cells and their relationship with age was predictive for outcome after vaccination.33
RPA is a statistical model used to stratify patients based on several (pretreatment) variables. For newly diagnosed GBM, the first described six prognostic classes of the Radiation Therapy and Oncology Group were adapted and validated to the standard therapy from the European Organization for Research and Treatment of Cancer (EORTC) in 2006.68 The variables building up the class are age, WHO performance status, extent of resection, and Mini-Mental State Examination (MMSE) (Table 1). In our HGG-2006 Phase I/II trial, in which we integrated DC-vaccination in the standard of care for newly diagnosed GBM patients, we stratified 77 patients according to this RPA classification.45 There was a significant survival benefit (both PFS and OS) with lower RPA classification. We found a median OS of 39.7 months in class 3, which was the lowest class. The OS in class 4 was 18.3 months and dropped to 10.7 months in class 5.
 | Table 1 EORTC RPA classification for newly diagnosed GBM |
In relapsed high-grade glioma (HGG) patients, we have built and adapted a separate 4-class RPA classification based on age, pathology, Karnofsky Performance Score (KPS), and MMSE (Figure 1). There was a significant difference in OS and in the percentage of long-term survivors between the classes, with better outcome for lower classes.46 The extent of resection was not included, mainly because this parameter is not available in the pretreatment patient counseling but more part of the DC vaccination itself.
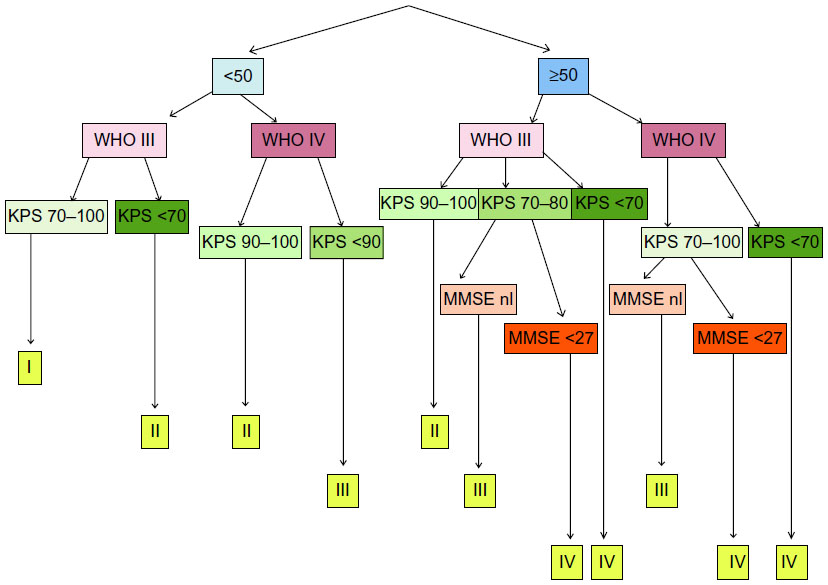 | Figure 1 HGG immuno RPA classification for relapsed HGG. |
To date, pretreatment patient stratification is only rarely reported in vaccination and other immunotherapy trials, probably because of limited numbers of included patients. However, due to heterogeneity of patients’ characteristics which influence outcome in general and probably also treatment response to DC vaccination, a stratification method is mandatory to more correctly compare the outcomes of different trials in this patient population.
Immune profile
In most clinical trials for DC vaccination, weaning from corticosteroids is considered an inclusion criterion. The immunosuppressive characteristics of chronically administered steroids are well-known and based on the inhibition of several immune cells and their function. In this context, an inhibitory effect on the activation of antigen-presenting cells, such as DCs, has been identified.69 On the other side of the spectrum, auto-immunity in the patient’s medical history is generally considered as an exclusion criterion.41 Of note, allergies and auto-immune diseases themselves reduce the lifetime risk for developing a glioma.70
Even without corticosteroids, glioma patients are to a certain extent systemically immunosuppressed, mainly by downregulated T cell and monocyte function.71 Also, the immunosuppressive myeloid-derived suppressor cells (MDSC) are more frequent in GBM patients, but their exact relationship to outcome after DC vaccination has not been clinically explored yet.72,73
As the major objective of DC vaccination is to activate lymphocytes against tumor antigens, the absolute lymphocyte count at the start of vaccination could theoretically be an easy measurable predictor. The prognostic influence of lymphocyte counts has been studied in various malignancies. In general, low pretreatment lymphocyte counts were found to be a negative prognostic factor, as was shown for hematologic malignancies such as follicular lymphomas and multiple myelomas74,75 and metastasis of solid tumors.76 After surgery, low lymphocyte counts were found to be negative prognostic factors in grade 4 melanomas and gall bladder carcinoma.77,78 More particular for immunotherapy, Schueneman et al reported that patients with lower postoperative lymphocyte counts after resection of pancreatic adenocarcinoma showed less response to immunotherapy with a granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting vaccine.79 For GBM, Grossman et al reported that lower CD4 counts at baseline and 2 months after the start of combined radio- and chemotherapy in newly diagnosed GBM patients, correlated significantly with shorter survival. The cause of death was early tumor progression and not opportunistic infection.80 This finding may be partially related to glucocorticoid administration, but the authors suggest that a more immunosuppressive state is related to poorer outcome. In this study, no immunotherapy was used. Interestingly, in a study on the use of an epidermal growth factor receptor class III variant (EGFRvIII) mutant peptide vaccine, Sampson et al found that there was an enhanced cellular and humoral immune response in patients with a more profound lymphopenia following a dose-intensified scheme of temozolomide.81 They stated that, in the phase of homeostatic lymphocyte proliferation, lymphocytes that encounter their antigen (in the form of a vaccine), have a proliferation advantage and become overrepresented. A survival benefit for those patients, when compared to patients with the standard temozolomide regimen, was not shown in this small study of 22 patients. Although the results of these studies seem to conflict, these data might suggest that patients with a more pronounced chemotherapy-induced lymphopenia, in general associated with poor outcome, can be good candidates for benefiting from DC vaccination, at least if bone marrow recovery is still possible. It also provides a rationale to start DC vaccination at the moment of (recovering) lymphopenia to maximally benefit from the phase of T cell homeostasis.82
Extent of resection
Gross total resection, defined by the neurosurgical report and the absence of residual contrast enhancement in an early (<72 hours) magnetic resonance imaging (MRI) after surgery, is a prognostic parameter for better outcome.83 The survival benefit of a complete resection as compared with an incomplete resection was calculated to be 4.9 months.84 When gross total resection is not possible, a more extensive, subtotal resection also improves outcome, in an incremental way correlated with the percentage of resection and the residual tumor volume.85 DC vaccination is generally only applied to patients with at least a subtotal resection, because of the fear for inflammatory peritumoral reactions in patients with large bulky residual tumors.36 Moreover, the state of minimal residual disease is believed to be essential to minimize the immunosuppressive effect of the GBM, and patients with low residual tumor volumes are less likely to need immunosuppressive corticosteroids.38,50 Liau et al showed in a study of 12 participants that patients with stable or minimal residual disease at the start of vaccination did develop systemic anti-tumor cytotoxicity, and at recurrence, intracranial T cell infiltration. Patients with tumor progression did not develop any of these.51 Another recent study dichotomized between more or less than 20 cm3 residual tumor volume at the start of DC vaccination, and showed a significant OS advantage after multivariate analysis in the group with lower residual tumor.47
Tumor characteristics
GBM, diagnosed on histopathological criteria1 is not a uniform pathological entity. Classical histopathology is increasingly being supplemented by genetic and epigenetic information, and subclassifications of GBM on the basis of genetic alterations or gene expression profiles are being developed.86 These additional data give more insight in the origin and characteristics of the disease itself, but they also provide new prognostic information and sometimes even therapeutic opportunities. We have to be careful when interpreting these data as the tumor itself can show marked intratumoral heterogeneity, so the results of the molecular analysis can depend on which part was examined.87 In the setting of autologous DC vaccination, the tissue available for advanced subclassification is usually small, as most of the tumor is used for the preparation of vaccines.
Mutations
The most well-known and clinically widely used mutation is IDH1, which discriminates between primary and secondary glioblastomas and which is associated with better survival.88–90 Other typical mutations, like NF-1, RB-1, and TP53 have been described with their prognostic relevance, and provide more insight in the pathogenesis of different types of GBM.2 The relevance for DC vaccination will certainly be elaborated during the next years when more extensive mutational analysis will be done on tumors of trial patients. Of particular interest, Parsa et al showed that loss of PTEN (phosphatase and tensin homolog) due to deletion or mutation leads to an increased expression of the immunosuppressive protein B7-H1 (or PD-L1). Glioma cells without PTEN expression were resistant to killing by tumor-specific T cells in culture.91 Tumors with PTEN loss showed fewer tumor infiltrating lymphocytes (TILs) and are associated with poor outcome.92,93 A nice example of the clinical application of mutational analysis to guide immunotherapy has been published by Sampson et al, who used a EGFRvIII-targeted peptide vaccine only in patients expressing this mutation, which occurs in approximately one-third of GBM. They reported good results, but also showed that upon recurrence, 82% of tumors had lost EGFRvIII, thereby proving immunologic escape after targeting a single antigen.30
Gene expression profiles and DNA methylation
There has been done extensive work done on developing molecular classification of GBM according to their preferential gene expression or transcriptional signature.93–95 Different experimental groups set up different subclassifications, which might trouble the final interpretation. The Phillips classification describes three (proneural, mesenchymal, and proliferative) subclasses and the Verhaak classification recognizes four (classical, neural, proneural, and mesenchymal) distinct gene expression profiles.93,95 Comparison led to the identification of robust proneural and mesenchymal subtypes.86 Recently, the work on subclassification of GBM has been further elaborated by a comprehensive study from Sturm et al, who classified GBM according to DNA-methylation patterns and hotspot mutations, and integrated this in gene expression profiles.96
Prognostic relevance has been attributed to certain profiles and genes. The proneural subtype is associated with IDH-1 mutations, tends to occur in younger patients, and has the best OS prognosis.96 Lee et al identified the proneural subtype as the main cause for the survival benefit in younger patients.66 Gene mutations and gene expression profiles are linked, as NF1 deletions and PTEN mutations predominantly occur in the mesenchymal subtype, and IDH1 and TP53 mutations in the proneural subtype. Secondary GBM were in fact mostly proneural. The mesenchymal gene profile is recognized by the overexpression of genes responsible for angiogenesis, invasion, and migration.93,94 The patients with this type of GBM have a poor prognosis. Interestingly and surprisingly, a recent Phase I study of Prins et al on DC vaccination showed a significant extended OS in nine patients with the mesenchymal gene expression profile, in comparison with a semi-matched group of 82 control patients with the same molecular profile.97 There was no survival benefit for tumors with the proneural profile. Also, they found a significantly higher number of CD3+ and CD8+ TILs in tumors with a mesenchymal signature when compared to tumors with the proneural profile. At recurrence, they noted an increase of CD3+ and CD8+ TILs in mesenchymal tumors but not in proneural tumors. Although these data have been generated in a small study group and although they seem to be in conflict with the PTEN immunosuppressive status in the mesenchymal subtype, defining a gene expression signature that predicts a good response to immunotherapy can be of major importance. Interestingly, in a recent paper, Naeini et al described MRI criteria to predict the mesenchymal profile.98 When validated, this finding could simplify the ongoing investigation of the relationship between DC vaccination and gene expression profiles.
Tumor microenvironment
Infiltrating lymphocytes
TILs are reported to be present in 28%–60% of gliomas. A relationship between the presence of TILs and enhanced survival has been reported in other cancers, including melanoma, ovarian carcinoma, and colorectal carcinoma.99 For GBM, several older studies have evaluated the effect of T cell infiltration on outcome. The results are inconsistent with studies describing a positive correlation between infiltrating lymphocytes and good outcome,100 no correlation,101 and even a negative correlation.102 These conflicting results are probably due to the lack of subclassification of TILs. Recent papers indeed showed a positive correlation between CD8+ effector T cells and longer survival.103,104 These findings provide some evidence for cellular immune responses that are involved in long-term tumor control. After DC vaccination, several groups have found CD4+ and CD8+ infiltrations (that were not present on initial histopathology) in patients undergoing re-resection or biopsy at relapse.32,33,50,59,60 This represents an immunological response to DC vaccination, but does not prove efficacy since this is only measured at time of relapse. Interestingly, recent studies showed that TILs were significantly associated with the mesenchymal transcriptional subclass, providing further evidence for the hypothesis that these transcriptional subgroups might be more immunogenic.92,97
The recognition of the importance of immunosuppressive regulatory T cells (Tregs) in the tumor microenvironment has changed the general view on TILs.105 This subpopulation of CD4+CD25+FoxP3+ or CD4+CD25+CD127dim cells was found to be present in about half of the GBM, whereas it is absent in normal brain tissue.106,107 There were clearly lower levels of Tregs in lower grade gliomas as compared to the high-grade gliomas. Also, it was shown that the proportion of Tregs in the total CD4+ population was not only increased in tumor tissue, but also in the peripheral blood of GBM patients, with clear immunosuppressive characteristics.108,109 The exact value of these Tregs is not well-established, illustrated by the fact that a significant relationship between Tregs infiltration and survival in humans could not be shown in three separate studies.99,107,110 In animal experiments, it has been shown that depletion of regulatory T cells in a murine glioma model was beneficial for survival, especially when combined with DC vaccination.111,112 In humans, many therapeutic agents to eliminate Tregs directly, or via its upstream regulator indoleamine-pyrrole 2,3-dioxygenase (IDO) are currently under investigation, but to date, no late phase clinical trials have been published.113,114 As Tregs are not present in all GBM samples, the benefits of these therapeutic actions may be limited to those tumors harboring high amounts of Tregs.
Other factors in the microenvironment
GBM are heavily infiltrated by microglia and macrophages, which are reported to make up to 30% of the tumoral mass.115,116 Although once thought to be an anti-tumor response, it has become clear that these cells, once recruited in the glioma, lose their anti-tumor efficacy. Even more strikingly, via multiple paracrine and synergistic interactions with the glioma cells, they adopt an immunosuppressive and pro-tumoral function. In this way, they promote tumor proliferation, angiogenesis, and invasion.117,118 Their important pro-tumoral function was shown in a recent publication by Pyonteck et al.119 They showed that blocking the CSF-1 receptor on macrophages led to significantly increased survival in a mouse proneural GBM model and in patient-derived glioma xenografts. Strikingly, they showed that the macrophages were not depleted in the tumor, but rather re-educated by downregulation of M2 macrophage-polarizing genes. Many other cell populations interact in the tumor microenvironment, including neural precursor cells, endothelial cells, astrocytes, and fibroblasts.115 The extensive and often reciprocal interactions between these cells and tumor cells are currently being unraveled. This will undoubtedly help to reveal the importance of the tumor microenvironment on the local immune interactions after systemic immunotherapy. Nevertheless, turning the immunosuppressive microenvironment into an anti-tumoral environment is a logical but challenging next step in brain cancer immunotherapy.
Monitoring during therapy
The immune-monitoring problem
Measuring the anti-tumor response as a surrogate marker to determine the efficacy of an immunologic treatment is a clinical problem that has not been solved completely. Misinterpreting the efficacy, however, can lead to unnecessary abortion of effective treatment, or, on the other hand, can lead to wasting time when one proceeds with an ineffective therapy.
To explore the immunologic response, most clinical trials have included some kind of immune monitoring, and have sometimes correlated this with clinical outcome. The variation in methods for immune monitoring and the sometimes conflicting results confuse the interpretation and comparison of outcomes. Furthermore, in the particular case of DC vaccination using DC pulsed with whole tumor lysates, the lack of knowledge of which (dominant) antigen the immunotherapy has been directed at impedes the measurement of specific immune responses. It is also possible that the peripheral immune response does not adequately reflect what is happening in the brain. Moreover, the sometimes extensive laboratory manipulations necessary for immune-monitoring procedures might influence the final results of the monitoring to a greater extent than the presumed immunization in the patient.
Selected examples of frequently used immune-monitoring tools
A positive DTH test has been reported to be associated with better clinical outcome in several reports,32,37,55 but other reports mention either a lack of positive DTH reactions,43 or a lacking correlation with outcome.39
IFN-γ production by peripheral blood mononuclear cells (PBMC), as a marker for tumor-specific Th1 T cell response against tumor antigens has frequently been studied. Wheeler et al observed post-vaccine, antigen-directed IFN-γ response using a quantitative polymerase chain reaction (qPCR) assay in 17/34 GBM patients, and found significantly longer PFS and OS in these vaccine responders.38 Yu et al also used qPCR and found IFN-γ mRNA accumulation after vaccination in response to tumor lysate, but did not correlate this with outcome.34 Measuring tumor-reactive CD8+ T cells after vaccination by IFN-γ ELISPOT, Yamanaka et al found a significantly better OS in 7 out of 16 patients, in whom they found tumor reactive effector T cells.37 Kikuchi et al also reported an increase in IFN-γ production in a culture of PBMCs and glioma cells, as measured by ELISA.62
Repeated fluorescence activated cell sorting (FACS) analysis of lymphocyte populations before and after vaccination is a conventional monitoring tool. However, to date, no robust and uniformly reproducible vaccination responses have been established. A recent publication found an increased natural killer (NK) cell ratio to be significantly correlated with prolonged OS and PFS on the one hand, and with decreased levels of the immunosuppressive TGF-beta on the other hand.47 A study by Yamanaka et al also reported an increase in NK cells,32 but other studies didn’t find significant changes in NK cells during or after vaccination.45,62,120 In an elaborated immune-monitoring study, Fong et al found predictive survival values for decreased ratios of Treg frequency and decreased CTLA-4 expression on CD4+ and CD8+ T cells after DC vaccination. They calculated cut-off points to provide a straightforward tool for immune monitoring and response prediction.120
We refer to other reviews for comparative summaries of the immune-monitoring results.21,23,25 In general, one can estimate that tumor-specific immune responses can be detected in approximately 50% of patients, but their relationships with clinical outcome remain mostly unclear.
Cluster analysis of multiple data
It is likely that the efficacy of treatment cannot be measured fully in one or two immune parameters, and a more complex interpretation of multiple parameters will be needed to cover the different physiological responses after immunotherapy. In this respect, some progress has been made during the last years by using cluster analyses.
Fadul et al performed hierarchical clustering of five post-vaccine lymphocyte functional parameters, which led to the identification of two distinct groups of immunological responders and non-responders.43 The group of responders had significant longer OS and a trend to longer PFS in this small study of ten patients. The single immune parameters themselves, however, were poor predictors of survival. In the discussion, they stated that by combining multiple assays, one can measure immune responses that can’t be detected by single assays. Ardon et al performed a cluster analysis on relative frequencies of several immune cell populations measured by FACS analysis on patients with newly diagnosed GBM.45 DC vaccination was started after concomitant radiochemotherapy and FACS was performed at the time of leukapheresis (LF) and the first vaccination (V1), thus before the start of vaccination. They explored clustering of several immune parameters, of which Treg and NK cell ratios (V1/LF) contributed most to cluster membership, which resulted in the determination of three clusters with a clear, although not significant, correlation with PFS. We indeed believe that combining multiple measurements can give a more accurate view of the subtle, reciprocal, and interactive changes in the immune system.
Radiology
In a neuro-oncological follow-up, MRI is performed at regular intervals, or in case of a clinical deterioration. As such, MRI results are one of the main non-invasive tools to define the response of a therapy. However, vaccine-induced immune responses often provoke a transient contrast enhancement, which might be difficult to distinguish from tumor relapse. To overcome this problem, advanced MRI techniques are being investigated. In a pilot study of eight patients with recurrent GBM treated with adjuvant DC vaccination as monotherapy, maximal lesional regional cerebral blood volume (rCBV) ratios was found to be higher in progressive compared to stable patients, but also higher in patients immediately before distinct progression was diagnosed based on conventional clinico-radiological characteristics.121 These results need of course to be validated in larger prospective trials, but show that modern MRI techniques have the possibility to monitor the effect of DC vaccination on gliomas to some extent. Also, MRI could theoretically be used to monitor the peripheral immune responses when using labeled DCs, as was shown in a study on mice by Ferguson et al.122 Besides or complementary to MRI, other imaging modalities such as positron emission tomography (PET) scans to evaluate the metabolic data can help,123 but have only anecdotally been explored for DC vaccination for GBM.35
Getting the whole picture: towards in silico oncology
The multiple factors discussed in this review interact in a complex manner with each other (Table 2), presumably to determine the resulting treatment response of a specific patient. Large, late Phase II and Phase III clinical trials will give us a large amount of information on multiple levels. To maximally integrate all data and get the whole picture, computational analysis may become a prerequisite. As the field of mathematical modeling and in silico oncology is progressing,124,125 assessing these multiscale data in a mathematical hypermodeling environment will give us more insight into the disease itself, and subsequently, in treatment response predictions.126,127 This, in particular, will be necessary to take the step from experimental to standard therapy.
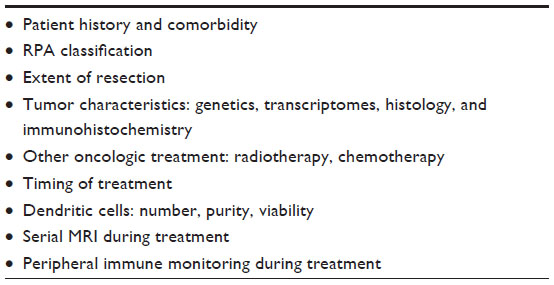 | Table 2 Multiscale parameters possibly predicting outcome after DC vaccination |
Disclosure
The research leading to these results has received funding from the European Union Seventh Framework Programme under grant agreement number 600841, Computational Horizons in Cancer (CHIC; http://www.chic-vph.eu). The vaccination program at KU Leuven is supported by the Olivia Hendrickx Research Fund, the Herman Memorial Research Fund, the James E Kearney Foundation, and the Leuven’s Association for Research against Cancer (ARCH). Further funding was obtained from the Fund for Scientific Research – Flanders (FWO-V) and the Agency for Innovation by Science and Technology (IWT), and charity funding from several private organizations. Stefaan Van Gool is senior clinical investigator at the FWO-V. The authors have no other conflicts of interest in this work.
References
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. | |
Dunn GP, Rinne ML, Wykosky J, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26(8):756–784. | |
Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol. 2012;14 Suppl 5:v1–v49. | |
Tamber MS, Rutka JT. Pediatric supratentorial high-grade gliomas. Neurosurg Focus. 2003;14(2):e1. | |
Fleury A, Menegoz F, Grosclaude P, et al. Descriptive epidemiology of cerebral gliomas in France. Cancer. 1997;79(6):1195–1202. | |
Burnet NG, Jefferies SJ, Benson RJ, Hunt DP, Treasure FP. Years of life lost (YLL) from cancer is an important measure of population burden – and should be considered when allocating research funds. Br J Cancer. 2005;92(2):241–245. | |
Claes A, Idema AJ, Wesseling P. Diffuse glioma growth: a guerilla war. Acta Neuropathol. 2007;114(5):443–458. | |
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | |
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | |
Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. | |
Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma – are we there yet? Neuro Oncol. 2013;15(1):4–27. | |
Anton K, Baehring JM, Mayer T. Glioblastoma multiforme: overview of current treatment and future perspectives. Hematol Oncol Clin North Am. 2012;26(4):825–853. | |
Chaichana KL, Zadnik P, Weingart JD, et al. Multiple resections for patients with glioblastoma: prolonging survival. J Neurosurg. 2013;118(4):812–820. | |
Wong E, Gautam S, Malchow C, Lun M, Pan E, Brem S. Bevacizumab for recurrent glioblastoma multiforme: a meta-analysis. J Natl Compr Cancer Netw. 2012;9(4):403–407. | |
Salmaggi A, Milanesi I, Silvani A, et al. Prospective study of carmustine wafers in combination with 6-month metronomic temozolomide and radiation therapy in newly diagnosed glioblastoma: preliminary results. J Neurosurg. 2013;118(4):821–829. | |
Chiocca EA, Aguilar LK, Bell SD, et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29(27):3611–3619. | |
Stupp R, Wong ET, Kanner AA, et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur J Cancer. 2012;48(14):2192–2202. | |
Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2012;363(5):411–422. | |
Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. | |
Marsh JC, Goldfarb J, Shafman TD, Diaz AZ. Current status of immunotherapy and gene therapy for high-grade gliomas. Cancer Control. 2013;20(1):43–48. | |
Xu X, Stockhammer F, Schmitt M. Cellular-based immunotherapies for patients with glioblastoma multiforme. Clin Dev Immunol. 2012;2012:764213. | |
Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol. 2011;13(1):3–13. | |
Vauleon E, Avril T, Collet B, Mosser J, Quillien V. Overview of cellular immunotherapy for patients with glioblastoma. Clin Dev Immunol. 2010;2010. pii: 689171. | |
Van Gool S, Maes W, Ardon H, Verschuere T, Van Cauter S, De Vleeschouwer S. Dendritic cell therapy of high-grade gliomas. Brain Pathol. 2009;19(4):694–712. | |
De Vleeschouwer S, Rapp M, Sorg RV, et al. Dendritic cell vaccination in patients with malignant gliomas: current status and future directions. Neurosurgery. 2006;59(5):988–999. | |
De Vleeschouwer S, Van Gool SW, Van Calenbergh F. Immunotherapy for malignant gliomas: emphasis on strategies of active specific immunotherapy using autologous dendritic cells. Childs Nerv Syst. 2005;21(1):7–18. | |
Aguilar LK, Arvizu M, Aguilar-Cordova E, Chiocca EA. The spectrum of vaccine therapies for patients with glioblastoma multiforme. Curr Treat Options Oncol. 2012;13(4):437–450. | |
Thomas AA, Ernstoff MS, Fadul CE. Immunotherapy for the treatment of glioblastoma. Cancer J. 2013;18(1):59–68. | |
Kim W, Liau LM. Dendritic cell vaccines for brain tumors. Neurosurg Clin N Am. 2011;21(1):139–157. | |
Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–4729. | |
Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. | |
Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89(7):1172–1179. | |
Wheeler CJ, Black KL, Liu G, et al. Thymic CD8+ T cell production strongly influences tumor antigen recognition and age-dependent glioma mortality. J Immunol. 2003;171(9):4927–4933. | |
Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in Patients with malignant glioma. Cancer Res. 2004;64(14):4973–4979. | |
De Vleeschouwer S, Van Calenbergh F, Demaerel P, et al. Transient local response and persistent tumor control in a child with recurrent malignant glioma: treatment with combination therapy including dendritic cell therapy. Case report. J Neurosurg. 2004;100(5 Suppl Pediatrics):492–497. | |
Rutkowski S, De Vleeschouwer S, Kaempgen E, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer. 2004;91(9):1656–1662. | |
Yamanaka R, Homma J, Yajima N. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Dev Immunol. 2005;11(11):4160–4167. | |
Wheeler CJ, Black KL, Liu G, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68(14):5955–5964. | |
De Vleeschouwer S, Fieuws S, Rutkowski S, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14(10):3098–3104. | |
Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N Engl J Med. 2008;359(5):539–541. | |
Ardon H, Van Gool S, Lopes IS, et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: a pilot study. J Neurooncol. 2010;99(2):261–272. | |
Ardon H, De Vleeschouwer S, Van Calenbergh F, et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr Blood Cancer. 2010;54(4):519–525. | |
Fadul CE, Fisher JL, Hampton TH, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011;34(4):382–389. | |
Elens I, De Vleeschouwer S, Pauwels F, Van Gool S. Resection and Immunotherapy for Recurrent Grade III Glioma. ISRN Immunol. 2012;2012:1–9. | |
Ardon H, Van Gool SW, Verschuere T, et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG-2006 phase I/II trial. Cancer Immunol Immunother. 2012;61(11):2033–2044. | |
De Vleeschouwer S, Ardon H, Van Calenbergh F, et al. Stratification according to HGG-IMMUNO RPA model predicts outcome in a large group of patients with relapsed malignant glioma treated by adjuvant postoperative dendritic cell vaccination. Cancer Immunol Immunother. 2012;61(11):2105–2112. | |
Pellegatta S, Eoli M, Frigerio S, et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology. 2013;2(3):e23401. | |
Lasky JL, Panosyan EH, Plant A, et al. Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas. Anticancer Res. 2013;33(5):2047–2056. | |
Liau LM, Black KL, Martin NA, et al. Treatment of a glioblastoma patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Case report. Neurosurg Focus. 2000;9(6):e8. | |
Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61(3):842–847. | |
Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial t-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–5525. | |
Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2010;8(10):2773–2779. | |
Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330–336. | |
Iwami K, Shimato S, Ohno M, et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor α2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy. 2012;14(6):733–742. | |
Akiyama Y, Oshita C, Kume A, et al. α-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: a phase I clinical trial. BMC Cancer. 2012;12(1):623. | |
Phuphanich S, Wheeler CJ, Rudnick JD, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62(1):125–135. | |
Caruso DA, Orme LM, Neale AM, et al. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro Oncol. 2004;6(3):236–246. | |
Vik-Mo EO, Nyakas M, Mikkelsen BV, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62(9):1499–1509. | |
Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. J Clin Neurosci. 2008;15(2):114–121. | |
Chang C-N, Huang Y-C, Yang D-M, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011;18(8):1048–1054. | |
Jie X, Hua L, Jiang W, Feng F, Feng G, Hua Z. Clinical application of a dendritic cell vaccine raised against heat-shocked glioblastoma. Cell Biochem Biophys. 2012;62(1):91–99. | |
Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50(7):337–344. | |
Kikuchi T, Akasaki Y, Abe T, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. Immunotherapy. 2004;27(6):452–459. | |
Okada H, Lieberman FS, Walter KA, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J Transl Med. 2007;5:67. | |
Behin A, Hoang-Xuan K, Carpentier AF, Delattre J-Y. Primary brain tumours in adults. Lancet. 2012;379(9830):1984–1996. | |
Lee Y, Scheck AC, Cloughesy TF, et al. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med Genomics. 2008;1:52. | |
Fagnoni FF, Vescovini R, Passeri G, et al. Shortage of circulating naive CD8(+) T cells provides new insights on immunodeficiency in aging. Blood. 2000;95(9):2860–2868. | |
Mirimanoff R-O, Gorlia T, Mason W, et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 phase III randomized trial. J Clin Oncol. 2006;24(16):2563–2569. | |
Girndt M, Sester U, Harald K, Hünger F, Köhler H. Glucocorticoids inhibit activation-dependent expression of costimulatory molecule B7-1 in human monocytes. Transplantation. 1998;66(3):370–375. | |
Brenner AV, Linet MS, Fine HA, et al. History of allergies and autoimmune diseases and risk of brain tumors in adults. Int J Cancer. 2002;99(2):252–259. | |
Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. 1999;100(1–2):216–232. | |
Rodrigues JC, Gonzalez GC, Zhang L, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010;12(4):351–365. | |
Raychaudhuri B, Rayman P, Ireland J, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13(6):591–599. | |
Siddiqui M, Ristow K, Markovic SN, et al. Absolute lymphocyte count predicts overall survival in follicular lymphomas. Br J Haematol. 2006;134(6):596–601. | |
Ege H, Gertz MA, Markovic SN, et al. Prediction of survival using absolute lymphocyte count for newly diagnosed patients with multiple myeloma: a retrospective study. Br J Haematol. 2008;141(6):792–798. | |
Lissoni P, Brivio F, Fumagalli L, et al. Efficacy of cancer chemotherapy in relation to the pretreatment number of lymphocytes in patients with metastatic solid tumors. Int J Biol Markers. 2004;19(2):135–140. | |
Rochet N, Kottschade L, Grotz T, Porrata L, Markovic S. The prognostic role of the preoperative absolute lymphocyte count and absolute monocyte count in patients with resected advanced melanoma. Am J Clin Oncol. Epub June 14, 2013. | |
Iwase R, Shiba H, Haruki K, et al. Post-operative lymphocyte count may predict the outcome of radical resection for galbladder carcinoma. Anticancer Res. 2013;33(8):3439–3444. | |
Schueneman AJ, Sugar EA, Uram J, et al. Low total lymphocyte count is associated with poor survival in patients with resected pancreatic adenocarcinoma receiving a GM-CSF secreting pancreatic tumor vaccine. Ann Surg Oncol. 2013;20 Suppl 3:S725–S730. | |
Grossman SA, Ye X, Lesser G, et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res. 2011;17(16):5473–5480. | |
Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13(3):324–333. | |
Dummer W, Niethammer AG, Baccala R, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110(2):157–159. | |
Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen H-J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7(5):392–401. | |
Stummer W, Reulen H-J, Meinel T, et al. Extent of resection and survival in glioblastoma multiforme: identification of and adjustment for bias. Neurosurgery. 2008;62(3):564–576. | |
Sanai N, Polley M-Y, McDermott MW, Parsa AT, Berger MS. An extent of resection threshold for newly diagnosed glioblastomas. J Neurosurg. 2011;115(1):3–8. | |
Huse JT, Phillips HS, Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011;59(8):1190–1199. | |
Barajas R Jr, Phillips JJ, Parvataneni R, et al. Regional variation in histopathologic features of tumor specimens from treatment-naive glioblastoma correlates with anatomic and physiologic MR Imaging. Neuro Oncol. 2012;14(7):942–954. | |
Hodges TR, Choi BD, Bigner DD, Yan H, Sampson JH. Isocitrate dehydrogenase 1: what it means to the neurosurgeon: a review. J Neurosurg. 2013;118(6):1176–1180. | |
Yan H, Williams Parsons D, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. | |
Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19(4):764–772. | |
Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. | |
Rutledge WC, Kong J, Gao J, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res. 2013;19(18):4951–4960. | |
Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. | |
Freije WA, Castro-Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–6510. | |
Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. | |
Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. | |
Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Dev Immunol. 2011;17(6):1603–1615. | |
Naeini KM, Pope WB, Cloughesy TF, et al. Identifying the mesenchymal molecular subtype of glioblastoma using quantitative volumetric analysis of anatomic magnetic resonance images. Neuro Oncol. 2013;15(5):626–634. | |
Dunn GP, Dunn IF, Curry WT. Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human glioma. Cancer Immun. 2007;7:12. | |
Palma L, Di Lorenzo N, Guidetti B. Lymphocytic infiltrates in primary glioblastomas and recidivous gliomas. J Neuroimmunol. 1978;49(6):854–861. | |
Rossi M, Jones N, Candy E, et al. The mononuclear cell infiltrate compared with survival in high-grade astrocytomas. Acta Neuropathol. 1989;78(2):189–193. | |
Safdari H, Hochberg FH, Richardson EP. Prognostic value of round cell (lymphocyte) infiltration in malignant gliomas. Surg Neurol. 1985;23(3):221–226. | |
Yang I, Tihan T, Han SJ, et al. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci. 2010;17(11):1381–1385. | |
Lohr J, Ratliff T, Huppertz A, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin cancer Res. 2011;17(13):4296–4308. | |
Sonabend AM, Rolle CE, Lesniak MS. The role of regulatory T cells in malignant glioma. Anticancer Res. 2008;28(2B):1143–1150. | |
Ardon H, Verbinnen B, Maes W, Beez T, Van Gool S, De Vleeschouwer S. Technical advancement in regulatory T cell isolation and characterization using CD127 expression in patients with malignant glioma treated with autologous dendritic cell vaccination. J Immunol Methods. 2010;352(1–2):169–173. | |
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res. 2008;14(16):5166–5172. | |
Fecci PE, Mitchell Da, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66(6):3294–3302. | |
El Andaloussi A, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol. 2006;8(3):234–243. | |
Jacobs JFM, Idema AJ, Bol KF, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol. 2010;225(1–2):195–199. | |
Maes W, Rosas GG, Verbinnen B, et al. DC vaccination with anti-CD25 treatment leads to long-term immunity against experimental glioma. Neuro Oncol. 2009;11(5):529–542. | |
Grauer OM, Sutmuller RPM, van Maren W, et al. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int J Cancer. 2008;122(8):1794–1802. | |
Wainwright DA, Balyasnikova I V, Chang AL, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–6121. | |
Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting Tregs in malignant brain cancer: overcoming IDO. Front Immunol. 2013;4:116. | |
Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59(8):1169–1180. | |
Yang I, Han SJ, Kaur G, Crane C, Parsa AT. The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci. 2010;17(1):6–10. | |
Da Fonseca ACC, Badie B. Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol. 2013;2013:264124. | |
Kushchayev SV, Kushchayeva YS, Wiener PC, Badie B, Preul MC. Monocyte-derived cells of the brain and malignant gliomas: the double face of Janus. World Neurosurg. Epub November 23, 2012. | |
Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. | |
Fong B, Jin R, Wang X, et al. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS One. 2012;7(4):e32614. | |
Vrabec M, Van Cauter S, Himmelreich U, et al. MR perfusion and diffusion imaging in the follow-up of recurrent glioblastoma treated with dendritic cell immunotherapy: a pilot study. Neuroradiology. 2011;53(10):721–731. | |
Ferguson PM, Slocombe A, Tilley RD, Hermans IF. Using magnetic resonance imaging to evaluate dendritic cell-based vaccination. PLoS One. 2013;8(5):e65318. | |
Engell-Noerregaard L, Hendel HW, Johannesen HH, Alslev L, Svane IM. FDG PET scans as evaluation of clinical response to dendritic cell vaccination in patients with malignant melanoma. Cancer Immunol Immunother. 2013;62(1):17–25. | |
Byrne HM. Dissecting cancer through mathematics: from the cell to the animal model. Nat Rev Cancer. 2010;10(3):221–230. | |
Baldock AL, Rockne RC, Boone AD, et al. From patient-specific mathematical neuro-oncology to precision medicine. Front Oncol. 2013;3:62. | |
Georgiadi EC, Dionysiou DD, Graf N, Stamatakos GS. Towards in silico oncology: adapting a four dimensional nephroblastoma treatment model to a clinical trial case based on multi-method sensitivity analysis. Comput Biol Med. 2012;42(11):1064–1078. | |
Stamatakos GS, Kolokotroni E, Dionysiou D, et al. In silico oncology: exploiting clinical studies to clinically adapt and validate multiscale oncosimulators. Conf Proc IEEE Eng Med Biol Soc. 2013;2013:5545–5549. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.