")
Back to Journals » International Journal of Nanomedicine » Volume 18
Delivery of Plasmid DNA by Ionizable Lipid Nanoparticles to Induce CAR Expression in T Cells
Authors Prazeres PHDM , Ferreira H , Costa PAC, da Silva W, Alves MT, Padilla M, Thatte A, Santos AK , Lobo AO , Sabino A , Del Puerto HL, Mitchell MJ, Guimaraes PPG
Received 27 June 2023
Accepted for publication 13 October 2023
Published 18 October 2023 Volume 2023:18 Pages 5891—5904
DOI https://doi.org/10.2147/IJN.S424723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Farooq A. Shiekh
Pedro Henrique Dias Moura Prazeres,1,2 Heloísa Ferreira,2 Pedro Augusto Carvalho Costa,2 Walison da Silva,2 Marco Túllio Alves,2 Marshall Padilla,3 Ajay Thatte,3 Anderson Kenedy Santos,4,5 Anderson Oliveira Lobo,6 Adriano Sabino,7 Helen Lima Del Puerto,1 Michael J Mitchell,3 Pedro Pires Goulart Guimaraes1,2
1Department of Pathology, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil; 2Department of Physiology and Biophysics, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil; 3Department of Bioengineering, University of Pennsylvania, Philadelphia, PA, USA; 4Department of Pediatrics/Gastroenterology and Hepatology, Yale School of Medicine, New Haven, CT, USA; 5Department of Genetics, Yale School of Medicine, New Haven, CT, USA; 6Department of Materials Engineering, Federal University of Piauí, Teresina, PI, Brazil; 7Department of Clinical and Toxicological Analysis, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil
Correspondence: Pedro Pires Goulart Guimaraes, Department of Physiology and Biophysics, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil, Email [email protected]
Introduction: Chimeric antigen receptor (CAR) cell therapy represents a hallmark in cancer immunotherapy, with significant clinical results in the treatment of hematological tumors. However, current approved methods to engineer T cells to express CAR use viral vectors, which are integrative and have been associated with severe adverse effects due to constitutive expression of CAR. In this context, non-viral vectors such as ionizable lipid nanoparticles (LNPs) arise as an alternative to engineer CAR T cells with transient expression of CAR.
Methods: Here, we formulated a mini-library of LNPs to deliver pDNA to T cells by varying the molar ratios of excipient lipids in each formulation. LNPs were characterized and screened in vitro using a T cell line (Jurkat). The optimized formulation was used ex vivo to engineer T cells derived from human peripheral blood mononuclear cells (PBMCs) for the expression of an anti-CD19 CAR (CAR-CD19BBz). The effectiveness of these CAR T cells was assessed in vitro against Raji (CD19+) cells.
Results: LNPs formulated with different molar ratios of excipient lipids efficiently delivered pDNA to Jurkat cells with low cytotoxicity compared to conventional transfection methods, such as electroporation and lipofectamine. We show that CAR-CD19BBz expression in T cells was transient after transfection with LNPs. Jurkat cells transfected with our top-performing LNPs underwent activation when exposed to CD19+ target cells. Using our top-performing LNP-9-CAR, we were able to engineer human primary T cells to express CAR-CD19BBz, which elicited significant specific killing of CD19+ target cells in vitro.
Conclusion: Collectively, our results show that LNP-mediated delivery of pDNA is a suitable method to engineer human T cells to express CAR, which holds promise for improving the production methods and broader application of this therapy in the future.
Keywords: lipid nanoparticles, pDNA delivery, CAR T cells, cell engineering, cancer immunotherapy
Introduction
Immunotherapy, using T cells expressing the chimeric antigen receptor (CAR) targeting CD19, has been approved since 2017 by the FDA for the treatment of B-cell acute lymphoblastic leukemia (B-ALL),1,2 and later for large B-cell lymphoma (BCL)3 and multiple myeloma (MM).4,5 Due to the high remission rates obtained by this treatment, CAR T cells are a useful and efficient approach for the treatment of hematologic cancers.6,7 Despite its specificity, they are still inaccessible to many patients due to the high cost of available treatments, such as Axicabtagene ciloleucel (Yescarta, Gilead)8 and Tisagenlecleucel-T (Kymriah, Novartis).9 The associated costs of this therapy are especially troublesome in developing countries, such as Brazil, where the costs of treatment, hospital procedures, and diagnostics are financed by the public healthcare system for a total of R$ 3.5 billion.10
Approved CAR T cell products use viral vectors to engineer T cells, and their production requires expensive quality controls, trained personnel, and a production facility that is difficult to scale up. When combined, these factors restrict the broad application of CAR T cell therapy.8,11 Another bottleneck is the time required for the activation, engineering, and expansion of CAR T cells, which can take up to 22 days for Axicabtagene ciloleucel.8 This extensive ex vivo culturing procedure influences the phenotype of CAR T cells, which in turn may affect their ability to engraft and be effective upon reinfusion to patients.12 Furthermore, viral transduction systems are associated with immunogenicity against CAR T cells and B cell aplasia due to constitutive expression of CAR in effector cells.13,14 Despite these limitations associated with T cell engineering, studies using CAR T cells are ongoing, with durable responses against refractory multiple myeloma15 and several solid tumors.16 Taken together, this indicates that CAR T cells will be largely applied in the future, highlighting the need for novel production methods that are easy to scale-up and manufacture in accordance with the safety and effectiveness profile required by this therapy.
Non-viral delivery methods have been studied for the production of CAR T cells. Reports using transposon-transposase systems delivered by electroporation have shown promising results in pre-clinical mouse models of leukemia.17,18 Although this strategy aims at maintaining T cells in culture for shorter periods, it still uses an integrative system, which is related to adverse effects observed due to the long-term expression of CAR.19–21 Chemical agents such as polyethylenimine (PEI) and poly(2-dimethylamino) ethyl methacrylate (PDMAEMA) can be used as non-viral delivery methods to T cells, despite the difficulties in DNA uptake by primary T cells. Additionally, this method severely impacts cell viability, as also observed after electroporation.22
As an alternative to viral vectors or electroporation methods to engineer CAR T cells, nanomaterials such as ionizable lipid nanoparticles (LNPs) have been explored as they form systems that protect nucleic acids from nucleases and facilitate intracellular delivery.23,24 In contrast with viral vectors, LNPs can be rapidly optimized, do not require complex production plants, accommodate different cargos, and are able to scale up.25,26 Compared to other non-viral methods, such as polymeric nanosystems, LNPs are more clinically applicable, with approved products that efficiently deliver mRNA and interference RNA to different cell types in vivo.27–29 Using LNPs also overcomes common barriers of viral vectors, since there are fewer limitations to the amount of genetic material that can be delivered by this system.30 Previous reports from our group show the ability of LNPs formulated with C12-200 to transfect primary cardiomyocytes with DNA31 as well as their optimization potential for targeted in vivo delivery of both DNA and mRNA.32 The delivery of mRNA by LNPs to T cells has recently been described. These observations confirm that LNPs can be optimized for the production of CAR T cells. More importantly, CAR T cells produced by this approach transiently express CAR, which is an important feature as a non-integrative system.33,34 Therefore, LNPs can be used as a platform to manufacture cellular immunotherapeutics, although their potential to deliver plasmid DNA (pDNA) to engineer T cells needs further investigation.
In this context, we sought to evaluate the delivery of pDNA to T cells using LNPs. Thus, we synthesized and screened a mini-library of pDNA-loaded LNPs (Figure 1A and B) and evaluated their transfection efficiency in Jurkat cells, an immortalized human T cell line (Figure 1C). By varying the lipid ratios in each formulation, we identified a lead LNP that induced high expression of CAR in this cell line, with low toxicity when compared to standard transfection methods. Further investigation revealed the activation of transfected Jurkat cells after co-plating with Raji cells (CD19+ Burkitt’s lymphoma cell line). Additionally, specific killing was assessed after co-plating CAR T cells with CD19+ cells at different ratios, demonstrating the effectiveness of the transfected cells (Figure 1D).
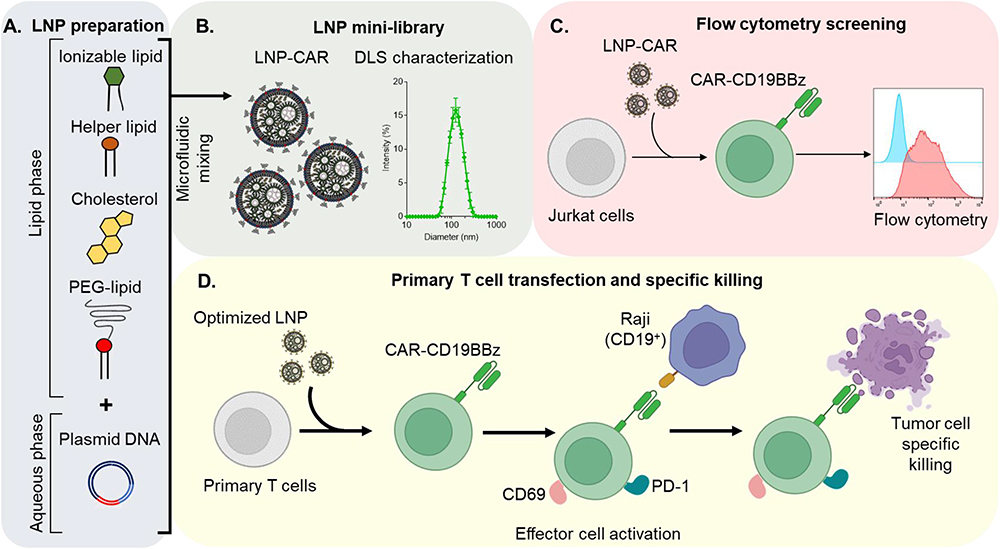 |
Figure 1 Schematic workflow for the optimization of DNA-loaded LNPs for the expression of CAR-CD19BBz. (A) Schematic representation of the components used for LNP synthesis by microfluidic. (B) Representation of the mini-library of LNPs (left) and DLS measurement of the optimized LNP-9-CAR. (C) Schematic representation of LNP screening using Jurkat cells. (D) Representation of the transfection of T cells using the optimized LNP for the expression of CAR-CD19BBz and co-culture experiments with Raji cells to assess effector cell activation and tumor cell specific killing. |
Materials and Methods
Synthesis and Characterization of Lipid Nanoparticles
To formulate ionizable lipid nanoparticles (LNPs), an ethanolic phase containing ionizable lipids and excipients was mixed with an aqueous phase of pDNA in a customized microfluidic device made in-house as previously described.35 Stock solutions of the ionizable lipid C12-200, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, Avanti Polar Lipids), cholesterol (Avanti Polar Lipids), and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (ammonium salt) (C14-PEG 2000, Avanti Polar Lipids) (Figure S1) were prepared in ethanol and mixed at predetermined molar ratios as shown in Table 1 and Table 2. The aqueous phase containing the plasmid DNA of interest was prepared in 10 mM citrate buffer, pH 3.0 (Teknova). Both inlets were connected to programmable syringe pumps (Harvard Apparatus), and the solutions were injected into the inlet of the microfluidic devices at a 2.5:1 ratio. LNPs were collected and then dialyzed against PBS in a 20,000 MWCO dialysis cassette (Thermo Scientific) for 2h. Afterwards, the LNPs were passed through a 0.22 µm filter (Merck Millipore). LNPs were diluted in a PBS containing 1% Triton before quantification of dsDNA using NanoDrop (Thermo Scientific). Qubit dsDNA HS Assay Kit (Thermo Scientific) was used to determine the encapsulation efficiency (EE) of the LNP-CAR library as described previously.36
 |
Table 1 Composition and Characterization of pDNA-ZsGreen Encapsulated in LNPs with Different DOPE and Cholesterol Ratios |
 |
Table 2 Composition and Characterization of pDNA-CAR-CD19BBz Encapsulated LNPs with Different DOPE and Cholesterol Ratios |
LNP hydrodynamic diameter, polydispersity index (PDI), and zeta potential were measured using a Zetasizer Nano ZS machine (Malvern Instrument). The structural analysis of the optimized LNP was performed via cryogenic-transmission electron microscopy (Cryo-TEM). Vitrified samples were examined using Tecnai G2-20 - FEI SuperTwin 200 kV at the Microscopy Center of UFMG. The apparent pKa of the optimized LNP was assessed by the 6-(p-Toluidino)-2-naphthalenesulfonic acid sodium salt (TNS) binding assay as previously described.31
Cell Culture
Jurkat cells (ATCC, TIB-152) were purchased from the Rio de Janeiro Cell Bank (BCRJ), and Raji cells (ATCC, CCL-86) were acquired by collaborators from Fiocruz-Rio and INCA. All cell lines were cultured in RPMI-164 supplemented with 2 mM L-glutamine, 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, 10 mM HEPES, and 1 mM sodium pyruvate.
Plasmid DNA Delivery to Jurkat Cells in vitro
Jurkat cells were plated in triplicate in 96-well plates at a density of 6 × 104 cells/well in 100 µL and treated with established doses of pDNA encapsulated in LNPs diluted in complete culture medium. As positive controls of transfection, Jurkat cells were treated with the same dose of pDNA using lipofectamine and electroporation. For lipofectamine transfection, pDNA was mixed with Lipofectamine 2000 (Invitrogen) reagent in OptiMEM and incubated for 20 minutes at room temperature, as per manufacturer’s instructions, before adding the mixture to the cells. For electroporation, 2 × 106 Jurkat cells were transferred to 2 mm cuvettes with 100 µL of OptiMEM with the determined dose of pDNA. Cells were then electroporated using an Amaxa Nucleofector II Device (Lonza). Cells transfected with LNPs, Lipofectamine 2000, or electroporation were incubated at 37 °C for 72 hours. Cells were then collected, centrifuged (400 xg, 5 minutes, 4 °C), and resuspended in PBS. The expression of CAR-CD19BBz in transfected cells was assessed by the enhanced green fluorescent protein (eGFP) present in the plasmid construct, as previously described.37,38 Briefly, this pDNA presents a P2A ribosomal skipping sequence separating the CAR-CD19BBz and eGFP sequences, allowing the detection of CAR-expressing cells without the need of additional markers.37 To exclude dead cells from the analysis, propidium iodide (PI) was added to the cell suspension at a concentration of 0.5 µg/mL immediately before acquisition in an FACS Calibur. The expression of ZsGreen or CAR-CD19BBz was carried out gating “live cells” determined as PI−.
Activation of Jurkat Cells Expressing CAR-CD19BBz
Raji cells, a CD19+ Burkitt’s lymphoma cell line, were co-plated with Jurkat cells transfected with different LNPs at a 1:1 effector-to-target ratio. After 24 hours, cells were collected and labeled with an antibody mix to determine effector cell activation and target cell death. Activation of CAR+ Jurkat cells was determined by CD69 and PD-1 mean fluorescence intensity (MFI). Similarly, Raji cells were plated at a density of 3 × 104 cells/well and Jurkat cells transfected with either LNP-9-CAR or LNP-9-GN1 were plated at a serial dilution at varying effector-to-target ratios.
Ex vivo pDNA Delivery to Primary Human T Cells
Blood from healthy donors was collected after informed consent, in accordance with the Research Ethics Committee of the Federal University of Minas Gerais (COEP-UFMG) under the approval number 02177612.0.0000.5149. Peripheral blood mononuclear cells (PBMC) were collected by centrifugation using a density gradient (Ficoll, GE). Primary T cells (CD3+) were purified from fresh PBMC using negative selection beads (Thermo Scientific), then stimulated overnight with human T cell activator beads CD3/CD28 (Thermo Scientific) at a 1:3 bead-to-cell ratio. Stimulated T cells were expanded in vitro for 6 days by adding 100 U/mL of recombinant human IL-2 (rhIL-2). Following activation and in vitro expansion, T cells were transfected using the optimized LNP. Cells were periodically counted by Trypan Blue viability staining so the cell density did not exceed 1 × 106 cells/mL.
Functional Assays Using Engineered Human CAR T Cells
CAR T cells were co-plated with Raji cells at varying effector-to-target ratios. Non-transfected T cells were co-plated at the same ratios as the controls. After 24 hours, cells were collected and labeled with surface markers to identify CD19+ cells and with the viability markers Annexin V and 7AAD (Thermo Scientific) as per manufacturer’s instructions. Specific killing was expressed by the frequency of CD19+ cells positively labeled for both Annexin V and 7AAD.
Statistics
Data are expressed as mean ± SD. Analyses were performed using Prism 7 (GraphPad Software Inc). ANOVA tests followed by Bonferroni post-tests were applied for comparison of more than 2 groups. Student's t-test was used for analysis between 2 groups. In all cases, p values <0.05 indicate statistical significance.
Results
In vitro Transfection of Jurkat Cells by DNA-Loaded LNPs
A mini-library of LNPs encapsulating a plasmid DNA (pDNA, Addgene no. 54702) encoding for ZsGreen (LNP-GN1) was synthesized using an ionizable lipid (C12-200), a PEG–lipid conjugate (C14-PEG2000), phospholipid (DOPE) and cholesterol at varying ratios (Table 1 and Figure S2A).31 LNPs were added to Jurkat cells and the frequency of ZsGreen-expressing cells, as well as cell viability, were assessed after 3 days by flow cytometry (Figure S2B). Our LNPs were characterized by size, polydispersity index (PDI), and zeta potential (Figure S2C and D). Although, all LNPs induced ZsGreen expression in Jurkat cells, LNP-5-GN1 performed better than the other formulations without affecting cell viability (Figure S2E and F). Electroporation and Lipofectamine 2000, were used as controls and induced higher cytotoxicity compared to the LNPs (Figure S2E and F). The lead formulation LNP-5-GN1 also induced higher ZsGreen expression, as measured by mean fluorescence intensity (MFI) in Jurkat cells (Figure S2G). Furthermore, it was found that increased DOPE molar ratio in the LNP formulation improved transfection efficiency in Jurkat cells (Figure S2H). Next, we sought to analyze how the lead formulation LNP-5 would perform delivering pDNA in Jurkat cells for the expression of CAR.
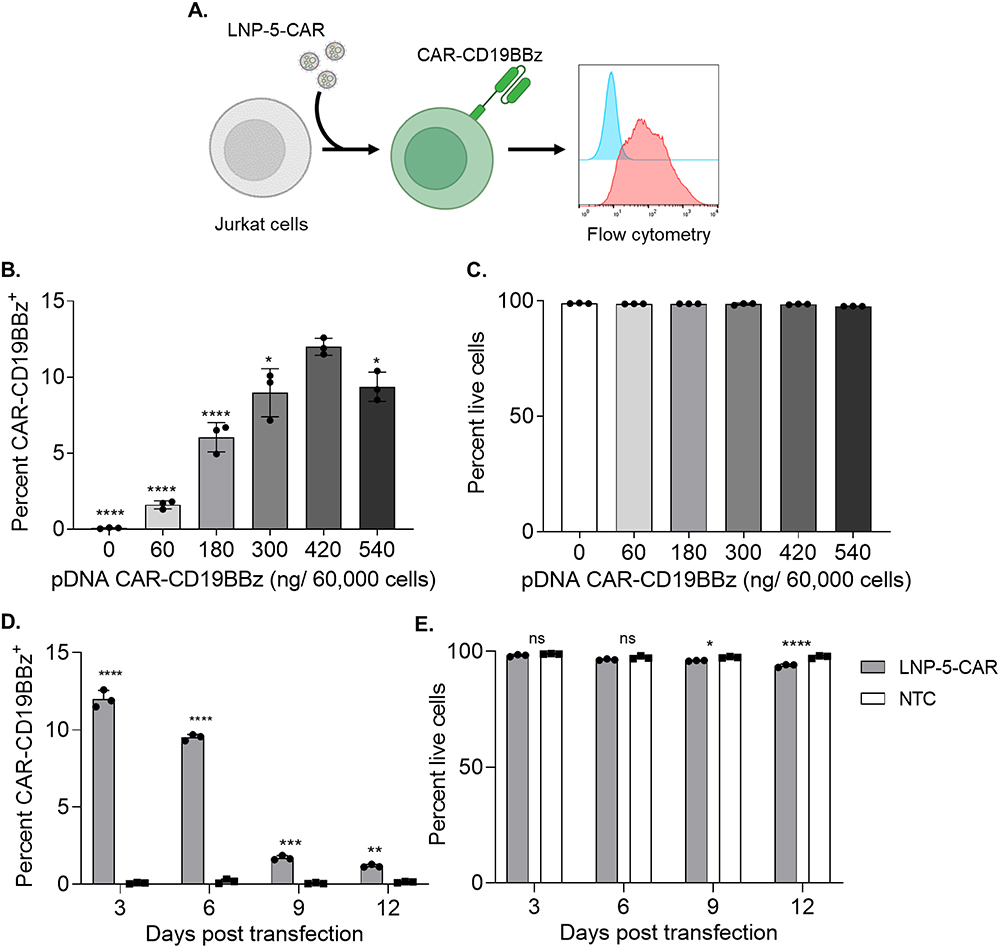 |
Figure 2 DNA loaded LNPs induce transient expression of CAR-CD19BBz in Jurkat cells. (A) Schematic of LNP-CAR mediated transfection of Jurkat cells. (B) CAR-CD19BBz expression in live (PI−) Jurkat cells 3 days after transfection with LNP-5-CAR with different amounts of pDNA identifying the best concentration to be used. * p<0.05; **** p<0.0001 versus 420 ng/ 60,000 cells evaluated by one-way ANOVA with n=3. (C) Cell viability of Jurkat cells assessed by the frequency of PI− cells after transfection with LNP-5-CAR. (D) CAR-CD19BBz expression over time in Jurkat cells treated with 420 ng/60,000 cells of LNP-5-CAR confirming the transient expression of CAR in these cells. ** p<0.01; ***p<0.001; ****p<0.0001 versus NTC on the same timepoint evaluated by one-way ANOVA with n=3. (E) Cell viability of Jurkat cells over time. * p<0.05; ****p<0.0001; Not significant (ns) p>0.05 versus NTC on the same timepoint evaluated by one-way ANOVA with n=3. |
Screening of LNPs for pDNA-CAR Delivery to Jurkat Cells
We encapsulated a pDNA encoding for a second-generation CAR targeting CD19 (CAR-CD19BBz, Addgene no. 135992) in our lead formulation LNP-5 (LNP-5-CAR). Jurkat cells were treated with different amounts of pDNA, and the expression of CAR-CD19BBz was assessed after 3 days by flow cytometry (Figure 2A). We determined that the optimal pDNA concentration for transfection of Jurkat cells was 420 ng of pDNA per 60,000 cells (Figure 2B). Cell viability was not affected after treatment with LNP-5-CAR at different concentrations (Figure 2C). We then evaluated the expression of CAR for up to 12 days using the optimal pDNA concentration, as a means to assess the CAR expression over time. We found higher CAR-CD19BBz expression 3 days after transfection with LNP-5-CAR, and from day 6 onwards the frequency of cells expressing CAR decreased significantly (Figure 2D). In addition, we observed a slight decrease in cell viability along the culture period, despite cell viability being maintained above 90% at all timepoints (Figure 2E).
To optimize our lead formulation, we formulated a new library of LNPs encapsulating pDNA-CAR-CD19BBz varying the molar ratios of DOPE and cholesterol, and screened these formulations using Jurkat cells (Table 2, Figure 3A and B). The hydrodynamic diameter of all LNPs ranged from 99.15 to 129.80 nm with PDI values ranging from 0.06 to 0.22 and slightly negative or neutral zeta potential (Figure 3C and D). As assessed by cryo-TEM, LNPs showed spherical morphology with an electron-dense core (Figure 3E). Our screening on Jurkat cells identified four top-performing LNPs, which induced higher frequency of cells expressing CAR-CD19BBz when compared to non-transfected control, electroporation and Lipofectamine 2000 (Figure 3F). When analyzing the effects of different amounts of excipient lipids in delivering pDNA to Jurkat cells, we found improved transfection with increased molar ratios of DOPE (35% to 42.5%), associated with reduced molar ratios of Cholesterol (30% to 20%) (Figure S3A and B). These formulations showed over 80% of encapsulation efficiency (Table 2). TNS assay showed a pKa of 7.45 for LNP-5-CAR, 7.54 for LNP-7-CAR, 7.42 for LNP-9-CAR and 7.47 for LNP-11-CAR (Figure S4A–D). Importantly, these top-performing LNPs induced enhanced CAR-CD19BBz expression compared to electroporation and Lipofectamine 2000, with significantly reduced cytotoxicity (Figure 3G). Expression of CAR-CD19BBz was similar after transfection with different LNPs and was significantly higher than non-transfected cells and cells transfected by electroporation and Lipofectamine 2000 (Figure 3H and I). After determining the transfection efficiency, we assessed the activation of Jurkat cells expressing CAR-CD19BBz after transfection with the four top-performing LNPs.
 |
Figure 3 Higher DOPE molar ratios facilitate efficient transfection with CAR-CD19BBz in Jurkat cells. (A) Schematic of the components used to prepare LNPs by microfluidic mixing. (B) Schematic of LNP-CAR screening using Jurkat cells. (C) Hydrodynamic diameter (Size) and polydispersity index (PDI) of different LNPs encapsulating CAR-CD19BBz pDNA. (D) Zeta potential of different LNPs. (E) Representative cryo-TEM of LNPs encapsulating pDNA. (F) CAR-CD19BBz expression in live (PI−) Jurkat cells 3 days after transfection with different LNPs identifying top-performing LNPs. **** p<0.0001 versus NTC evaluated by one-way ANOVA with n=3. (G) Cell viability of Jurkat cells assessed by the frequency of PI− cells after transfection with top-performing LNPs **** p<0.0001 versus NTC; evaluated by one-way ANOVA with n=3. (H) Representative histograms of CAR-CB19BBz expression after transfection. NTC = non-transfected control; EP = Electroporation; Lipo = Lipofectamine. Graphs represent mean ± SD. (I) CAR-CD19BBz expression measured by mean fluorescence intensity (MFI) after transfection with top-performing LNPs and controls. **p<0.01; ****p<0.0001; Not significant (ns) p>0.05 versus NTC evaluated by one-way ANOVA with n=3. “a” = p<0.001 compared to EP; “b” = p<0.001 compared to Lipo evaluated by one-way ANOVA with n=3. NTC = non-transfected control; Graphs represent mean ± SD. Abbreviations: EP, Electroporation; Lipo, Lipofectamine. |
Activation of Jurkat Cells Transfected with Top-Performing LNPs
According to the literature, Jurkat cells expressing CAR-CD19BBz recognize target cells expressing CD19, in a co-culture model and upregulate the expression of activation markers such as CD69 and PD-1. This is a fast and easy method that allows high-throughput evaluation of different CAR constructs.37 Thus, we evaluated whether Jurkat cells transfected with our top-performing LNPs underwent activation when exposed to CD19+ target cells (Figure 4A). After transfection with top-performing LNPs, Jurkat cells were co-plated with Raji cells at a 1:1 or 1:0 ratio, and the expression of activation markers was assessed by flow cytometry (Figure 4B and C). Jurkat cells transfected with LNP-9-CAR showed higher levels of CD69 and PD-1 than cells transfected with the control formulation LNP-9-GN1 (Figure 4D and E). These markers were not upregulated in CAR-expressing Jurkat cells in the absence of target cells, indicating a specific activation upon antigen binding of CAR-CD19BBz (Figure 4D and E).
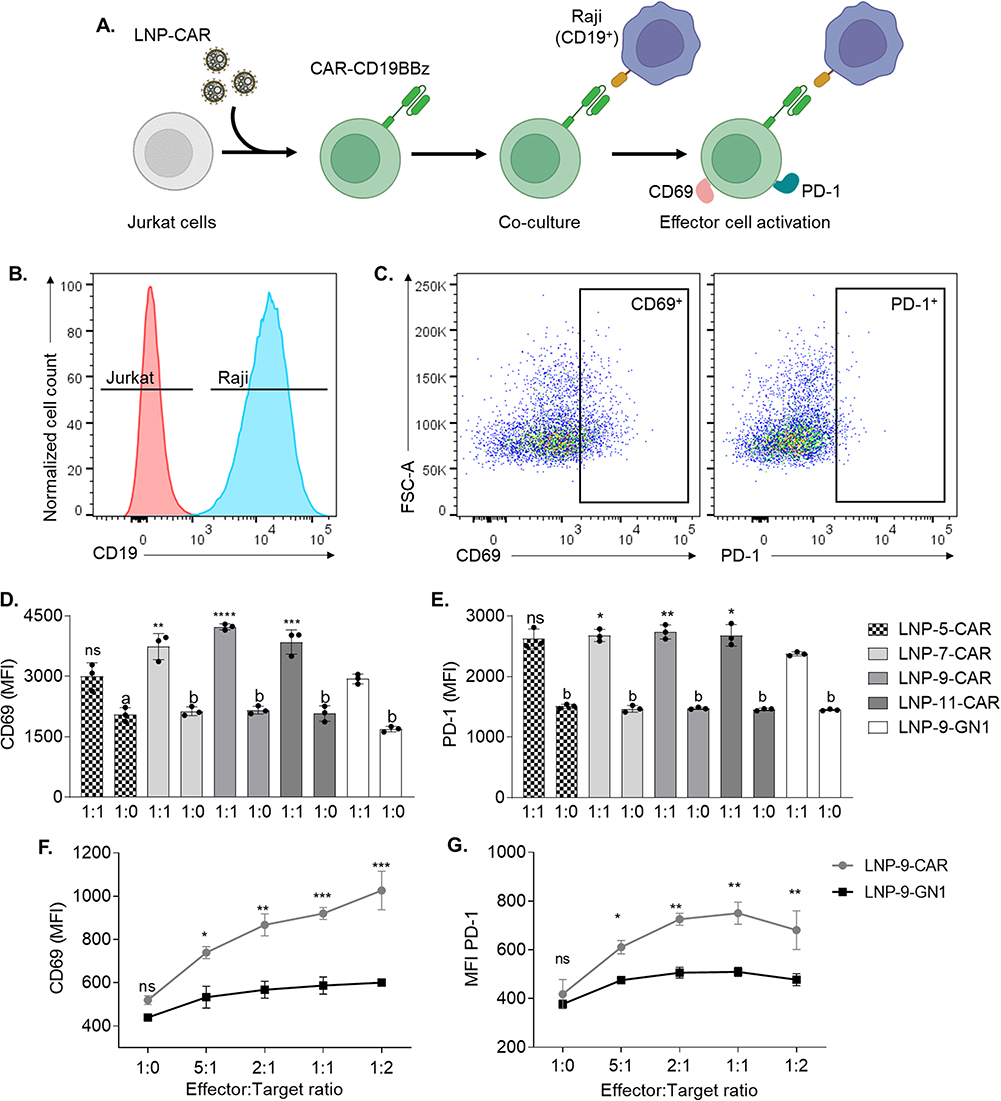 |
Figure 4 Top-performing LNPs generate CAR-Jurkat cells with high efficiency. Jurkat cells were transfected with top-performing LNPs using 420 ng/60,000 cells, collected after 3 days and co-cultured at 1:1 or 1:0 ratios with Raji cells. (A) Schematic of Jurkat cell transfection with LNP-CAR and effector cell activation after co-culture with Raji cells. (B) Representative histogram of Jurkat and Raji cells distinguished based on the expression of CD19. (C) Expression of activation markers PD-1 and CD69 in Jurkat cells expressing CAR-CD19BBz. (D and E) Activation of Jurkat cells expressing CAR-CD19BBz measured by mean fluorescence intensity (MFI) of CD69 (D) and PD-1 (E) after 24 hours co-cultured with Raji cells. * p<0.05; ** p<0.01; ***p<0.001; **** p<0.0001 Not significant (ns) p>0.05 versus LNP-9-GN1; “a” = p<0.001 compared to 1:1; “b” = p<0.0001 compared to 1:1; evaluated by one-way ANOVA with n=3. (F and G) Activation of Jurkat cells transfected with the optimized formulation LNP-9 24 hours after co-culture with Raji cells at different effector-to-target ratios. Activation was assessed by the expression of CD69 (F) and PD-1 (G). *p<0.05; **p<0.01; ***p<0.001; Not significant (ns) p>0.05 evaluated by two-way ANOVA with n=3. Graphs represent mean ± SD. |
We then analyzed whether the activation of Jurkat cells expressing CAR-CD19BBz was dependent on the ratio between target and effector cells. After transfecting Jurkat cells with the optimized LNP-9-CAR or the control LNP-9-GN1 (Figure S5A and B), we co-plated these cells with Raji cells at varying effector-to-target ratios. We demonstrated that Jurkat cells expressing CAR-CD19BBz upregulate the expression of both CD69 and PD-1 in a ratio-dependent manner (Figure 4F and G). Thus, our LNP system was able to generate CAR-expressing cells that properly respond to CD19+ target cells. Despite being a good model to analyze cell activation, Jurkat cells are not able to secrete all cytokines required to induce target cell death,37 which can be assessed in vitro by co-plating target cells with human primary T cells expressing CAR.
Ex vivo Engineering of Human Primary CAR T Cells
To assess the DNA delivery of our optimized formulation, human primary T cells were transfected with LNP-9-CAR, and the expression of CAR-CD19BBz was assessed by flow cytometry after 3 days (Figure 5A). Our LNP system was able to transfect T cells without affecting cell viability (Figure 5B–D). Additionally, both CD4+ and CD8+ T cells were transfected by the LNP, with a higher frequency of CD4+ T cells expressing CAR-CD19BBz (Figure 5E and F).
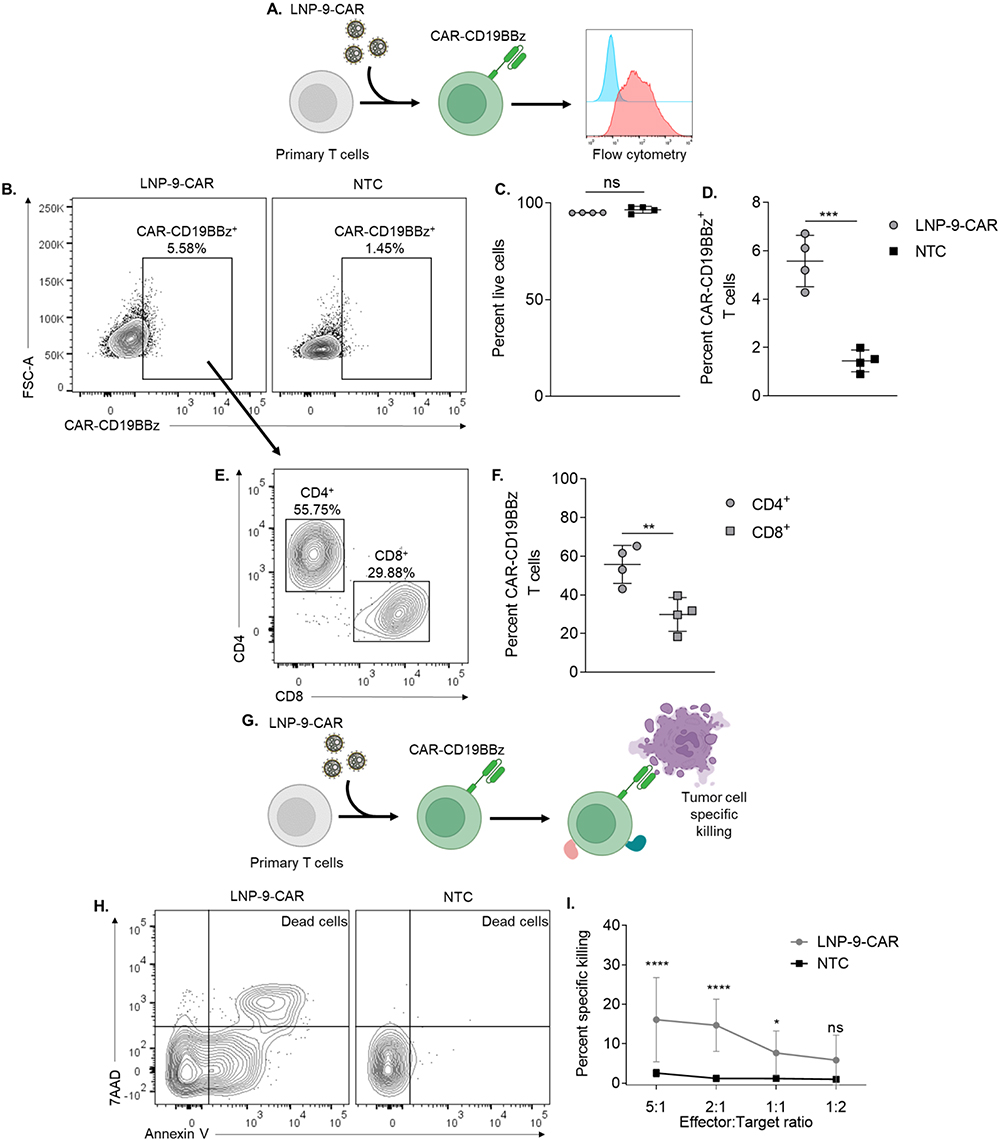 |
Figure 5 LNP-9-CAR promotes functional delivery of pDNA to primary human T cells. (A) Schematic of primary human T cell transfection with LNP-9-CAR (B) Representative plots of CAR-CD19BBz expression 3 days after transfection of primary human T cells with LNP-9-CAR. (C) Cell viability of CD3+ cells 3 days after transfection with LNP-9-CAR. Not significant (ns) p>0.05 evaluated by unpaired t-test compared to NTC. (D) Frequency of Live/CD3+/CAR-CD19BBz+ T cells analyzed 3 days after transfection with LNP-9-CAR. ***p<0.001 evaluated by unpaired t-test compared to NTC. (E) Representative plots of T cell subsets expressing CAR-CD19BBz 3 days after transfection with LNP-9-CAR identified by the expression of CD4 and CD8. (F) Frequency of Live/CD3+/CAR-CD19BBz+ CD4+ and CD8+ T cells analyzed 3 days after transfection with LNP-9-CAR. **p<0.01 evaluated by unpaired t-test compared to NTC. (G) Schematic of T cell transfection with LNP-9-CAR and tumor cell specific killing in co-culture with Raji cells. (H) Representative plots of Annexin V/7AAD labeled cells gated on the CD3−/CD19+ population. Dead cells are labeled as double positive for both markers. (I) Results of specific killing of CD19+ 24 hours after coplating with CAR T cells or non-transfected cells at different ratios. *p<0.05; ****p<0.0001; Not significant (ns) p>0.05 evaluated by two-way ANOVA with n=4 donors. NTC = non-transfected control. Graphs represent mean ± SD. |
Specific Killing of Target Cells by Engineered CAR T Cells
The effectiveness of T cells transfected by LNP-9-CAR was evaluated by co-plating CAR T cells or control non-transfected cells with CD19+ cells for 24 hours at varying effector-to-target ratios (Figure 5G). Specific killing of CD19+ cells was determined by labeling the cells with Annexin V and 7AAD and expressed as the frequency of double positive cells within the CD19+ population (Figure 5H). Our results demonstrate that CAR T cells most effectively killed CD19+ cells at the highest effector-to-target ratio (5:1 and 2:1). Specific killing of target cells was still observed even at a 1:1 ratio, indicating the efficacy of CAR T cells engineered using LNPs (Figure 5I).
Discussion
Significant advances have been made in CAR T cell therapy in terms of regulatory aspects, production methods, and application against different malignancies.34,39,40 Although durable remissions are induced by this therapy, long-term adverse effects such as B-cell aplasia,14 cytokine release syndrome,41,42 and neurotoxicity43 are commonly observed. One of the factors that contribute to the long-term adverse effects is the integration of the CAR construct to the genome of the T cells, which induces constitutive expression of CAR. Therefore, novel methodologies are needed to overcome the development of severe long-term adverse effects associated with CAR T cell therapy, as well as to improve the manufacturing process, reducing the time and cost of the final product.
LNPs have been investigated as potential delivery methods for both DNA and mRNA. Recently, the potential of LNPs was demonstrated by their wide application in COVID-19 vaccines,44 as well as by their ability to transfect primary cells,31 reassuring their potential in cell engineering. LNPs were also used to produce CAR T cells in vitro, with a screening showing the performance of different ionizable lipids in delivering mRNA to T cells and highlighting the importance of optimizing a formulation for T cell engineering.33,34
In this study, LNPs were formulated and characterized focusing on the enhanced pDNA delivery to T cells. In all formulations, the ionizable lipid-to-pDNA ratio was fixed in 10:1, and the molar ratios of two excipient lipids were varied. A primary screening showed that increasing the DOPE molar ratio favors the expression of ZsGreen in Jurkat cells, which are considered hard to transfect by non-viral methods.45 In this screening, we identified a lead formulation, which induced a significantly higher frequency of transfected cells when compared to other formulations as well as to standard transfection methods such as electroporation and Lipofectamine.
Using the lead formulation, LNP-5, encapsulating a pDNA for the expression of CAR-CD19BBz, we showed enhanced frequency of CAR-expressing cells 3 days after transfection. This system does not likely lead to incorporation of the CAR construct to the genome of the cell, since a significant reduction in the frequency of CAR-expressing cells was observed after 9 days of culture with no effects on cell viability. Similar results have been achieved after transfection of Jurkat cells with mRNA encoding for Luciferase, with maximum luciferase expression after 24 hours followed by a significant reduction after 48 hours.34 The differences in mRNA and pDNA expression rely not only on the delivery method but also on the intracellular processing of these molecules. Following pDNA delivery, transcription only occurs after nuclear translocation of the construct, whereas mRNA is readily transcribed by the cellular machinery once it is released into the cytoplasm.25 A second screening was performed to optimize our lead formulation, formulating new LNPs varying the molar ratios of excipient lipids. Four new formulations with high transfection efficiency were identified. These results highlighted that pDNA delivery can be improved by increasing the molar ratio of DOPE in the LNP. In fact, other studies using RNA loaded LNPs have reported that increasing DOPE percentages in the formulation favor the in vitro transfection of Jurkat cells,46 as well as in vivo targeting and transfection of immune cells.47,48 However, formulations with more than 50% of DOPE do not perform as well in T cells.
Despite Jurkat cells being a good model to study T cells, their cytokine repertoire is limited.49 However, Jurkat cells expressing CAR upregulate the expression of activation markers, especially CD69, when co-plated with target cells. In a screening setting, this serves as an indicator that the construct, or cell engineering method, is able to generate functional CAR-expressing cells.37 By co-plating transfected Jurkat cells with CD19+ cells, we demonstrated that the activation of CAR-expressing cells was significantly higher in cells transfected with our optimized formulation, LNP-9. Additionally, we showed that transfection with LNP-9 generated CAR-expressing Jurkat cells that were activated in an effector-to-target ratio-dependent manner.
In a short culture period (3 days), we were able to engineer human primary T cells after in vitro activation. Our analysis shows that both CD4+ and CD8+ T cells were successfully engineered to express CAR-CD19BBz, with a higher frequency of CD4+ cells. Some variability in transfection efficiency was observed across the four donors, which is expected even in the clinical setting.50 When co-plating these CAR T cells with CD19+ cells, we observed higher specific killing in effector-to-target ratios with more effector cells. Specific killing was observed up to the 1:1 ratio, corroborating previous reports which show that higher numbers of target cells reduce specific killing induced by CAR T cells.17,34,42 The presence of a CD4+ CAR-expressing T cell population has clinical importance, with reports showing a decade-long persistence of this subset51 and their participation in cytokine release syndrome and long-term responses52 after CAR T cell treatment. Though clinical trials and reports show that the CD4:CD8 ratio should be determined beforehand to induce better responses, each subset has their importance in the success of CAR T cell therapy for hematological malignancies.53,54
Although in vivo studies are needed to determine the engraftment and efficacy of CAR T cells generated by LNP mediated pDNA delivery, our results suggest that this LNP platform can be used as a fast and non-toxic approach for T cell engineering. Further optimization may be performed based on our LNP-9 formulation opening ways for in vivo T cell targeted delivery as well as an alternative method to manufacture CAR T cells with different constructs targeting solid tumors.
Conclusion
In this work, we developed optimized LNP formulations to efficiently deliver pDNA to human primary T cells. This allows the engineering of CAR T cells targeting CD19, which were capable of inducing specific killing of lymphoma cells in vitro. The transient expression of CAR-CD19BBz induced by our system could be an interesting approach for CAR T cell immunotherapy. Further studies should evaluate the effect of this transient expression of CAR on the onset of adverse effects associated with this therapy. Additionally, our results provide new venues for pDNA delivery to T cells using non-viral transfection methods and could be applied for the treatment of solid tumors and other diseases.
Abbreviations
CAR, Chimeric Antigen Receptor; B-ALL, B-cell Acute Lymphoblastic Leukemia; BCL, B-cell Lymphoma; MM, Multiple Myeloma; PEI, Polyethylenimine; PDMAEMA, Poly(2-dimethylamino) ethyl methacrylate; LNP, Lipid Nanoparticle; pDNA, Plasmid DNA; DOPE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, C14-PEG, 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (ammonium salt), PDI, Polydispersity Index; MFI, Mean Fluorescence Intensity; COEP-UFMG, Ethics Research Committee of the Federal University of Minas Gerais; PBMC, Peripheral Blood Mononuclear Cells; rhIL-2, Recombinant Human Interleukin 2.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded by National Council for Scientific and Technological Development-CNPq (442731/2020-5; 305932/2022-5), PRPq-UFMG, CAPES (88887.506690/2020-00), FAPEMIG (APQ-00826-21; RED-00202-22 Rede de Pesquisa em Imunobiológicos e Biofármacos para terapias avançadas e inovadoras), Brazilian Ministry of Health – MoH, Brazilian National Programme of Genomics and Precision Health – Genomes Brazil, Decit/SCTIE/MS, DGITIS/SCTIE/MS. P.P.G.G. is supported by CNPq (442731/2020-5; 305932/2022-5) and FAPEMIG (APQ-00826-21).
Disclosure
Mr Pedro Henrique Dias Moura Prazeres, Mrs Heloísa Ferreira and Prof. Dr Pedro Pires Goulart Guimaraes report a patent BR1020230120431 pending. The authors report no other conflicts of interest in this work.
References
1. Liu Y, Chen X, Han W, Zhang Y. Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drugs Today. 2017;53(11):597–608. doi:10.1358/dot.2017.53.11.2725754
2. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222
3. Roschewski M, Longo DL, Wilson WH. CAR T-cell therapy for large B-cell lymphoma — who, when, and how? N Engl J Med. 2021;386(7):692–696. doi:10.1056/NEJMe2118899
4. Teoh PJ, Chng WJ. CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J. 2021;11(4):84. doi:10.1038/s41408-021-00469-5
5. Rendo MJ, Joseph JJ, Phan LM, DeStefano CB. car t-cell therapy for patients with multiple myeloma: current evidence and challenges. Blood Lymphat Cancer. 2022;12:119–136. doi:10.2147/BLCTT.S327016
6. Mougiakakos D, Krönke G, Völkl S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med. 2021;385(6):567–569. doi:10.1056/NEJMc2107725
7. Reinhard K, Rengstl B, Oehm P, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367(6476):446–453. doi:10.1126/science.aay5967
8. Papathanasiou MM, Stamatis C, Lakelin M, Farid S, Titchener-Hooker N, Shah N. Autologous CAR T-cell therapies supply chain: challenges and opportunities? Cancer Gene Ther. 2020;27(10):799–809. doi:10.1038/s41417-019-0157-z
9. Yip A, Webster RM. The market for chimeric antigen receptor T cell therapies. Nat Rev Drug Discov. 2018;17(3):161–162. doi:10.1038/nrd.2017.266
10. INCA MdS. Estimativa 2020 - Incidência de Câncer no Brasil. 2020.
11. Tang J, Hubbard-Lucey VM, Pearce L, O’Donnell-Tormey J, Shalabi A. The global landscape of cancer cell therapy. Nat Rev Drug Discov. 2018;17(7):465–466. doi:10.1038/nrd.2018.74
12. Ghassemi S, Nunez-Cruz S, O’Connor RS, et al. Reducing ex vivo culture improves the antileukemic activity of Chimeric Antigen Receptor (CAR) T cells. Cancer Immunol Res. 2018;6(9):1100–1109. doi:10.1158/2326-6066.CIR-17-0405
13. Seow Y, Wood MJ. Biological gene delivery vehicles: beyond viral vectors. Mol Ther. 2009;17(5):767–777. doi:10.1038/mt.2009.41
14. Wagner DL, Fritsche E, Pulsipher MA, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. 2021;18:1–15.
15. J-Q M, Zhao W, Jing H, et al. Phase II, open-label study of ciltacabtagene autoleucel, an anti–B-cell maturation antigen chimeric antigen receptor–T-cell therapy, in Chinese patients with relapsed/refractory multiple myeloma (CARTIFAN-1). J Clin Oncol. 2023;41(6):1275–1284.
16. Patel U, Abernathy J, Savani BN, Oluwole O, Sengsayadeth S, Dholaria B. CAR T cell therapy in solid tumors: a review of current clinical trials. EJHaem. 2022;3:24–31. doi:10.1002/jha2.356
17. Chicaybam L, Abdo L, Viegas M, et al. Transposon-mediated generation of CAR-T cells shows efficient anti B-cell leukemia response after ex vivo expansion. Gene Ther. 2020;27(1–2):85–95. doi:10.1038/s41434-020-0121-4
18. de Macedo Abdo L, Barros LRC, Saldanha Viegas M, et al. Development of CAR-T cell therapy for B-ALL using a point-of-care approach. Oncoimmunology. 2020;9(1):1752592. doi:10.1080/2162402X.2020.1752592
19. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. doi:10.1038/mto.2016.11
20. Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Mirzaei HR, Hadjati J. Prolonged persistence of chimeric antigen receptor (CAR) T cell in adoptive cancer immunotherapy: challenges and ways forward. Front Immunol. 2020;11:702.
21. Schubert ML, Schmitt M, Wang L, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. 2021;32(1):34–48. doi:10.1016/j.annonc.2020.10.478
22. Riedl SAB, Kaiser P, Raup A, Synatschke CV, Jérôme V, Freitag R. Non-viral transfection of human T lymphocytes. Processes. 2018;6(10):188. doi:10.3390/pr6100188
23. El-Mayta R, Zhang Z, Hamilton AG, Mitchell MJ. Delivery technologies to engineer natural killer cells for cancer immunotherapy. Cancer Gene Ther. 2021;28(9):947–959. doi:10.1038/s41417-021-00336-2
24. Han X, Mitchell MJ, Nie G. Nanomaterials for therapeutic RNA delivery. Matter. 2020;3(6):1948–1975. doi:10.1016/j.matt.2020.09.020
25. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–555. doi:10.1038/nrg3763
26. Shepherd SJ, Warzecha CC, Yadavali S, et al. Scalable mRNA and siRNA lipid nanoparticle production using a parallelized microfluidic device. Nano Lett. 2021;21(13):5671–5680. doi:10.1021/acs.nanolett.1c01353
27. Garber K. Alnylam launches era of RNAi drugs. Nat Biotechnol. 2018;36(9):777–778. doi:10.1038/nbt0918-777
28. Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol. 2020;20(5):321–334. doi:10.1038/s41577-019-0269-6
29. Vogel AB, Kanevsky I, Che Y, et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature. 2021;592(7853):283–289. doi:10.1038/s41586-021-03275-y
30. Basarkar A, Singh J. Nanoparticulate systems for polynucleotide delivery. Int J Nanomedicine. 2007;2(3):353–360.
31. Scalzo S, Santos AK, Ferreira HA, et al. Ionizable lipid nanoparticle-mediated delivery of plasmid DNA in cardiomyocytes. Int J Nanomedicine. 2022;17:2865–2881. doi:10.2147/IJN.S366962
32. Guimaraes PPG, Zhang R, Spektor R, et al. Ionizable lipid nanoparticles encapsulating barcoded mRNA for accelerated in vivo delivery screening. J Control Release. 2019;316:404–417. doi:10.1016/j.jconrel.2019.10.028
33. Billingsley MM, Hamilton AG, Mai D, et al. Orthogonal design of experiments for optimization of lipid nanoparticles for mRNA engineering of CAR T cells. Nano Lett. 2022;22(1):533–542. doi:10.1021/acs.nanolett.1c02503
34. Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 2020;20(3):1578–1589. doi:10.1021/acs.nanolett.9b04246
35. Chen D, Love KT, Chen Y, et al. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J Am Chem Soc. 2012;134(16):6948–6951. doi:10.1021/ja301621z
36. Au - El-Mayta R, Au - Padilla MS, Au - Billingsley MM, Au - Han X, Au - Mitchell MJ. Testing the in vitro and in vivo efficiency of mRNA-lipid nanoparticles formulated by microfluidic mixing. JoVE. 2023;20(191):e64810.
37. Bloemberg D, Nguyen T, MacLean S, et al. A high-throughput method for characterizing novel chimeric antigen receptors in Jurkat cells. Mol Ther Methods Clin Dev. 2020;16:238–254. doi:10.1016/j.omtm.2020.01.012
38. McComb S, Nguyen T, Shepherd A, et al. Programmable attenuation of antigenic sensitivity for a nanobody-based EGFR chimeric antigen receptor through hinge domain truncation. Front Immunol. 2022;2022:13.
39. ANVISA. 2022. https://www.gov.br/anvisa/pt-br/assuntos/noticias-anvisa/2022/anvisa-aprova-produto-de-terapia-avancada-para-tratamento-de-cancer.
40. Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T cells in solid tumors: blueprints for building effective therapies. Front Immunol. 2018;9:1740. doi:10.3389/fimmu.2018.01740
41. Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol. 2018;11:1–12.
42. Liu Y, Fang Y, Chen X, et al. Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020;5(43):eaax7969. doi:10.1126/sciimmunol.aax7969
43. Zheng PP, Kros JM, Wang G. Elusive neurotoxicity in T cell-boosting anticancer therapies. Trends Immunol. 2019;40(4):274–278. doi:10.1016/j.it.2019.02.005
44. Guerrini G, Magrì D, Gioria S, Medaglini D, Calzolai L. Characterization of nanoparticles-based vaccines for COVID-19. Nat Nanotechnol. 2022;17(6):570–576. doi:10.1038/s41565-022-01129-w
45. Zhao N, Qi J, Zeng Z, et al. Transfecting the hard-to-transfect lymphoma/leukemia cells using a simple cationic polymer nanocomplex. J Control Release. 2012;159(1):104–110. doi:10.1016/j.jconrel.2012.01.007
46. LoPresti ST, Arral ML, Chaudhary N, Whitehead KA. The replacement of helper lipids with charged alternatives in lipid nanoparticles facilitates targeted mRNA delivery to the spleen and lungs. J Control Release. 2022;345:819–831. doi:10.1016/j.jconrel.2022.03.046
47. Dilliard SA, Cheng Q, Siegwart DJ. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. P Natl Acad Sci USA. 2021;118(52). doi:10.1073/pnas.2109256118
48. Oberli MA, Reichmuth AM, Dorkin JR, et al. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017;17(3):1326–1335. doi:10.1021/acs.nanolett.6b03329
49. Inoue T, Swain A, Nakanishi Y, Sugiyama D. Multicolor analysis of cell surface marker of human leukemia cell lines using flow cytometry. Anticancer Res. 2014;34(8):4539–4550.
50. Jiang J, Ahuja S. Addressing patient to patient variability for autologous CAR T therapies. J Pharm Sci. 2021;110(5):1871–1876. doi:10.1016/j.xphs.2020.12.015
51. Melenhorst JJ, Chen GM, Wang M, et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature. 2022;602(7897):503–509. doi:10.1038/s41586-021-04390-6
52. Bove C, Arcangeli S, Falcone L, et al. CD4 CAR-T cells targeting CD19 play a key role in exacerbating cytokine release syndrome, while maintaining long-term responses. J Immunother Cancer. 2023;11(1):e005878. doi:10.1136/jitc-2022-005878
53. Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+: CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. doi:10.1172/JCI85309
54. Shah NN, Highfill SL, Shalabi H, et al. CD4/CD8 T-cell selection affects Chimeric Antigen Receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol. 2020;38(17):1938–1950. doi:10.1200/JCO.19.03279
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.