")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Delivery of baicalein and paclitaxel using self-assembled nanoparticles: synergistic antitumor effect in vitro and in vivo
Authors Wang W, Xi M, Duan X, Wang Y, Kong F
Received 4 January 2015
Accepted for publication 18 February 2015
Published 22 May 2015 Volume 2015:10(1) Pages 3737—3750
DOI https://doi.org/10.2147/IJN.S80297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Lei Yang
Wei Wang,1,* Mei Xi,2,* Xuezhong Duan,1 Yong Wang,3 Fansheng Kong4
1Department of Chinese Medicine Integrated Traditional Chinese Medicine and Western Medicine, General Hospital of Ji’nan Command, People’s Liberation Army, 2Emergency Department, The Fourth People’s Hospital of Ji’nan, Medical School, Tai Shan Medical College, 3Department of Rehabilitation and Physiotherapy, General Hospital of Ji’nan Command, People’s Liberation Army, 4Department of Hematology, General Hospital of Ji’nan Command, People’s Liberation Army, Ji’nan, People’s Republic of China
*These authors contributed equally to this work
Purpose: Combination anticancer therapy is promising to generate synergistic anticancer effects to maximize the treatment effect and overcome multidrug resistance. The aim of the study reported here was to develop multifunctional, dual-ligand, modified, self-assembled nanoparticles (NPs) for the combination delivery of baicalein (BCL) and paclitaxel (PTX) prodrugs.
Methods: Prodrug of PTX and prodrug of BCL, containing dual-targeted ligands of folate (FA) and hyaluronic acid (HA), were synthesized. Multifunctional self-assembled NPs for combination delivery of PTX prodrug and BCL prodrug (PTX-BCL) were prepared and the synergistic antitumor effect was evaluated in vitro and in vivo. The in vitro transfection efficiency of the novel modified vectors was evaluated in human lung cancer A549 cells and drug-resistant lung cancer A549/PTX cells. The in vivo antitumor efficiency and systemic toxicity of different formulations were further investigated in mice bearing A549/PTX drug-resistant human lung cancer xenografts.
Results: The size of the PTX-BCL NPs was approximately 90 nm, with a positive zeta potential of +3.3. The PTX-BCL NPs displayed remarkably better antitumor activity over a wide range of drug concentrations, and showed an obvious synergism effect with CI50 values of 0.707 and 0.513, indicating that double-ligand modification and the co-delivery of PTX and BCL prodrugs with self-assembled NPs had remarkable superiority over other formulations.
Conclusion: The prepared PTX-BCL NP drug-delivery system was proven efficient by its targeting of drug-resistant human lung cancer cells and delivering of BCL and PTX prodrugs. Enhanced synergistic anticancer effects were achieved by PTX-BCL NPs, and multidrug resistance of PTX was overcome by this promising targeted nanomedicine.
Keywords: combination chemotherapy, prodrug-based nano-drug delivery system, multidrug resistance, self-assembled nanoparticles
Introduction
Combination chemotherapy for anticancer treatment is a promising strategy to generate synergistic anticancer effects, reduce individual drug-related toxicity, suppress multidrug resistance (MDR) through different mechanisms of action, and reduce the dose of each agent required.1,2 It refers to the simultaneous administration of two or more active agents to modulate different signaling pathways in tumor cells, to induce cell cycle arrest in a different phase of the cell cycle, to maximize the treatment effect, and to overcome MDR.3 Recently, there have been several reviews concerning nano-sized carriers designed for combination drug delivery in cancer chemotherapy.4–7 Most of them focused only on conventional nanocarriers, such as micelles,8,9 liposomes,10 and polymeric nanoparticles (NPs).11 Prodrug-based nano-drug delivery systems (P-N-DDSs) in the field of combination chemotherapy are still an advanced and pioneering field. The strategy – the combination of two polymer-drug conjugates, each carrying a single therapeutic agent – has been explored preclinically.12
P-N-DDSs for combination chemotherapy can be divided into four types: (1) amphiphilic polymer conjugates of two different antitumor drugs utilized to construct a P-N-DDS;7 (2) one free antitumor drug carried by a P-N-DDS formed by the amphiphilic polymer conjugate of another antitumor drug;13,14 (3) self-assembling conjugates composed of two different antitumor drugs bound together through suitable spacers;15 and (4) self-assembling conjugates composed of two different antitumor drugs, each of which is conjugated with different polymers separately to achieve dual targets. Among these, the fourth kind of P-N-DDS is a pioneering field and as far as we are aware, few researchers are concerned with it. The reasons for pursuing research into this fourth kind of P-N-DDS include the following: polymer prodrug is not only amphiphilic but also has a targeting effect and dual-target NPs can enhance a synergistic effect and carry a greater amount of drug to the tumor site compared with a single target.
Various targeting molecules have been applied to target paclitaxel (PTX) to tumor tone, such as folate (FA),16 arginylglycylaspartic acid,17 biotin,18 and hyaluronic acid (HA).19 As far as we are aware, few publication refer to targeting the delivery of baicalein (BCL). Therefore, we designed multifunctional NPs for the combination delivery of FA-mediated PTX and HA-mediated BCL. HA has been extensively used as a targeting moiety in HA self-assembled NPs for the cancer-targeting delivery of chemotherapy drugs.20–24 HA is a polymer of disaccharides, composed of D-glucuronic acid and D-N-acetylglucosamine, linked via alternating β-1,4 and β-1,3 glycosidic bonds. HA can bind to cluster of differentiation (CD) 44 and HAMM (CD168) receptors, which are overexpressed in various kinds of cancer cells, such as lung, breast, ovarian, colon.19,25–27 FA has become a popular molecule for targeting attached drugs to cancer cells due to its high binding affinity, low immunogenicity, ease of modification, etc. FA receptors are also overexpressed in many human cancer cells, including cancer cells of the lung, breast, ovary, brain, kidney, etc. To date, many chemical and biological therapeutic agents have been successfully conjugated to FA, most of which have demonstrated significantly enhanced delivery to folate receptor-positive tumor cells both in vitro and in vivo.28,29 Based on the synergistic effect of HA and FA, a dual-receptor targeting drug-delivery system, a novel strategy for targeted drug delivery, was designed.30
Baicalein (5,6,7-trihydroxyflavone; BCL), a bioactive flavonoid derived from the root of the traditional Chinese medicinal herb Scutellaria baicalensis Georgi, is reported to have anti-inflammatory, antioxidant, and antitumor effects on cancers of lung, breast, stomach, etc.31–34 The broad antitumor activity of BCL is due to the induction of cell cycle arrest, apoptosis in the G0/S phase, and inhibition of signal pathways.35 Apoptosis-associated proteins can be regulated by BCL, which can reduce the level of B-cell lymphoma,36,37 interleukin-6 (IL-6) and messenger RNA expression,38 cyclin-dependent kinase-1 (CDK-1),39 nuclear factor kappa B,38 12-lipoxygenase,35 hypoxia-inducible factor-1 alpha (HIF-1α),40 and promote levels of p53.41 Shortcomings of BCL, which lead to poor clinical effect in vivo compared with its powerful efficacy in vitro, have been found with further research. The shortcomings are extensive first-pass metabolism, low bioavailability, short half-life (t1/2, 10 minutes), poor water solubility, and that it is oxidized easily.42–45 Therefore, nanostructured lipid carriers have been developed to solve the issues mentioned.31
PTX, derived from the dark of the Pacific yew tree Taxus brevifolia, represents a new class of microtubule antiproliferative agents. It has been extensively used for the treatment of various types of cancers in clinic, such as lung, breast, ovarian, esophageal. The antitumor mechanism of PTX is the blocking mitosis, which leads to G2/M cell cycle arrest and apoptosis.46 Therefore, combination therapy with PTX and BCL will have a synergistically enhanced antitumor effect due to the combining of the anticancer mechanisms of each drug.47 Overexpression of B-cell lymphoma,48 IL-6,49 and CDK-150 has been shown to inhibit PTX-induced apoptosis and result in MDR. Therefore, the mentioned antitumor mechanisms of BCL can inhibit the drug resistance of PTX.
In the study reported here, novel prodrugs of FA-valine (V)-PTX and HA-lysine (L)-BCL were synthesized. Then, multifunctional and self-assembled NPs for the combination delivery of FA-V-PTX and HA-L-BCL were prepared by a dialysis method. The in vitro and in vivo antitumor efficacy was assessed in human lung cancer A549 cells and drug-resistant lung cancer A549/PTX cells. This system was expected to achieve the stable and targeted drug delivery of the BCL and PTX prodrugs separately, to enhance synergistic anticancer effects and suppress the MDR of PTX.
Materials and methods
Materials
BCL was purchased from Xi’an Ciyuan Biochemical Technology Co, Ltd (Xi’an, People’s Republic of China). PTX was provided by Hisun Pharmaceutical Co Ltd (Zhejiang, People’s Republic of China). FA was purchased from Sigma-Aldrich Co (St Louis, MO, USA). N-Butoxycarbonyl-L-lysine (N-Boc-L-lysine) was purchased from TCI (Shanghai, People’s Republic of China). N-alpha-Fmoc-L-valine (Fmoc-Val) was purchased from Shanghai Fortune Biotech Co, Ltd (Shanghai, People’s Republic of China). HA was provided by Shandong Freda Biochem Co, Ltd (Ji’nan, People’s Republic of China). Poly(D,L-lactic-co-glycolic acid) (PLGA, 50:50, Mw 25,000) was obtained from the Shandong Institute of Medical Instruments (Ji’nan, People’s Republic of China).
Cell culture
Human lung cancer A549 cells (A549 cells) and drug-resistant lung cancer A549/PTX cells (A549/PTX cells) were obtained from the American Type Culture Collection (Manassas, VA, USA). All other chemicals were of analytical grade or higher.
A549 cells and A549/PTX cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% heat-inactivated fetal bovine serum and 100 U/mL penicillin and 100 μg/mL streptomycin under 5% CO2 at 37°C for 24 hours. To maintain the drug resistance, A549/PTX cells were cultured in the presence of 4 μM cisplatin for 1 week in a drug-free medium before the experiment.
Animals
Kunming mice (4–6 weeks old, 18–22 g weight) were purchased from the Medical Animal Test Center of Shandong Province (Jinan, People’s Republic of China). All animal experiments complied with the Animal Management Rules of the Ministry of Health of the People’s Republic of China.
Synthesis of FA-V-PTX
FA-V-PTX was synthesized by three-step reaction. The synthesis scheme is depicted in Figure 1.
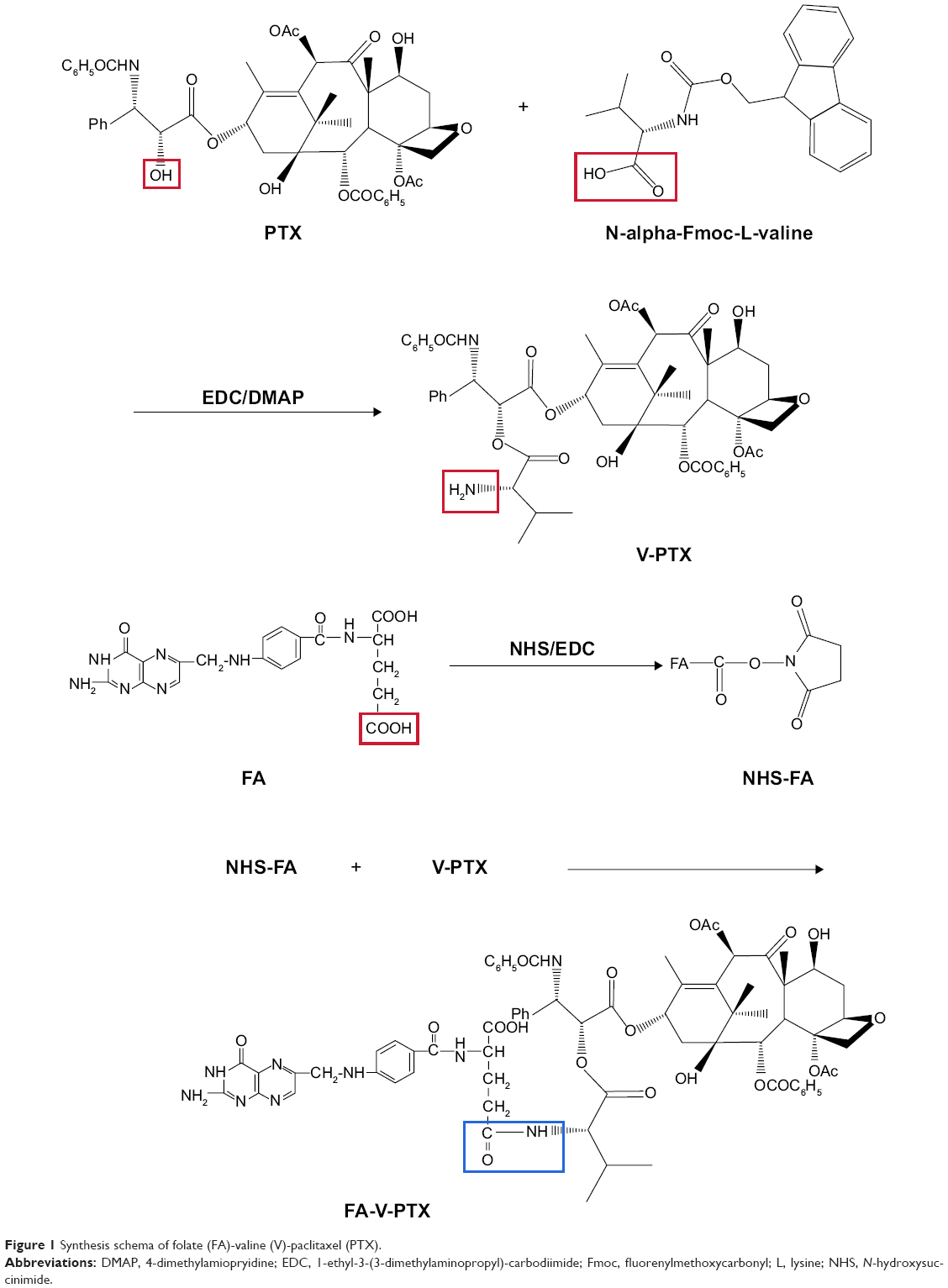 | Figure 1 Synthesis schema of folate (FA)-valine (V)-paclitaxel (PTX). |
First, V-PTX was synthesized. PTX (1 equivalent) and an excess amount of Fmoc-Val were dissolved in dichloromethane with the addition of 4-dimethylamiopryidine (DMAP), and the reaction was allowed at 0°C for 1 hour under stirring. 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) was added to react for 1 hour at 0°C. Then, the mixture was reacted for another 8 hours at room temperature (RT). HCl solution (0.05 N) was added to wash and separate the organic phase. Dichloromethane was used to extract the aqueous phase. The extraction solution was mixed with the organic phase. Then, the concentrated solution was obtained via absorption by anhydrous magnesium sulfate. Tetrahydropyridine and dichloromethane were added to the concentrated solution. Finally, V-PTX was obtained by removal of the solvent and precipitation in n-hexane.
Second, N-hydroxysuccinimide (NHS)-FA was synthesized. FA (1 g) was dissolved in dimethyl sulfoxide (DMSO; 15 mL). Then NHS and EDC were added to react at RT for 40 hours in the dark. The NHS-FA was obtained by filter and dried under vacuum.
Finally, FA-V-PTX was obtained as follows: V-PTX and NHS-FA were dissolved in DMSO separately, and mixed to synthesize for 48 hours at RT. Then, the mixture was purified by dialysis with a 10,000 MV cutoff membrane for 48 hours to remove excess NHS-FA and other impurities. The final product, FA-V-PTX, was obtained by lyophilization.
Synthesis of HA-L-BCL
HA-L-BCL was synthesized by three-step reaction. The synthesis scheme is depicted in Figure 2.
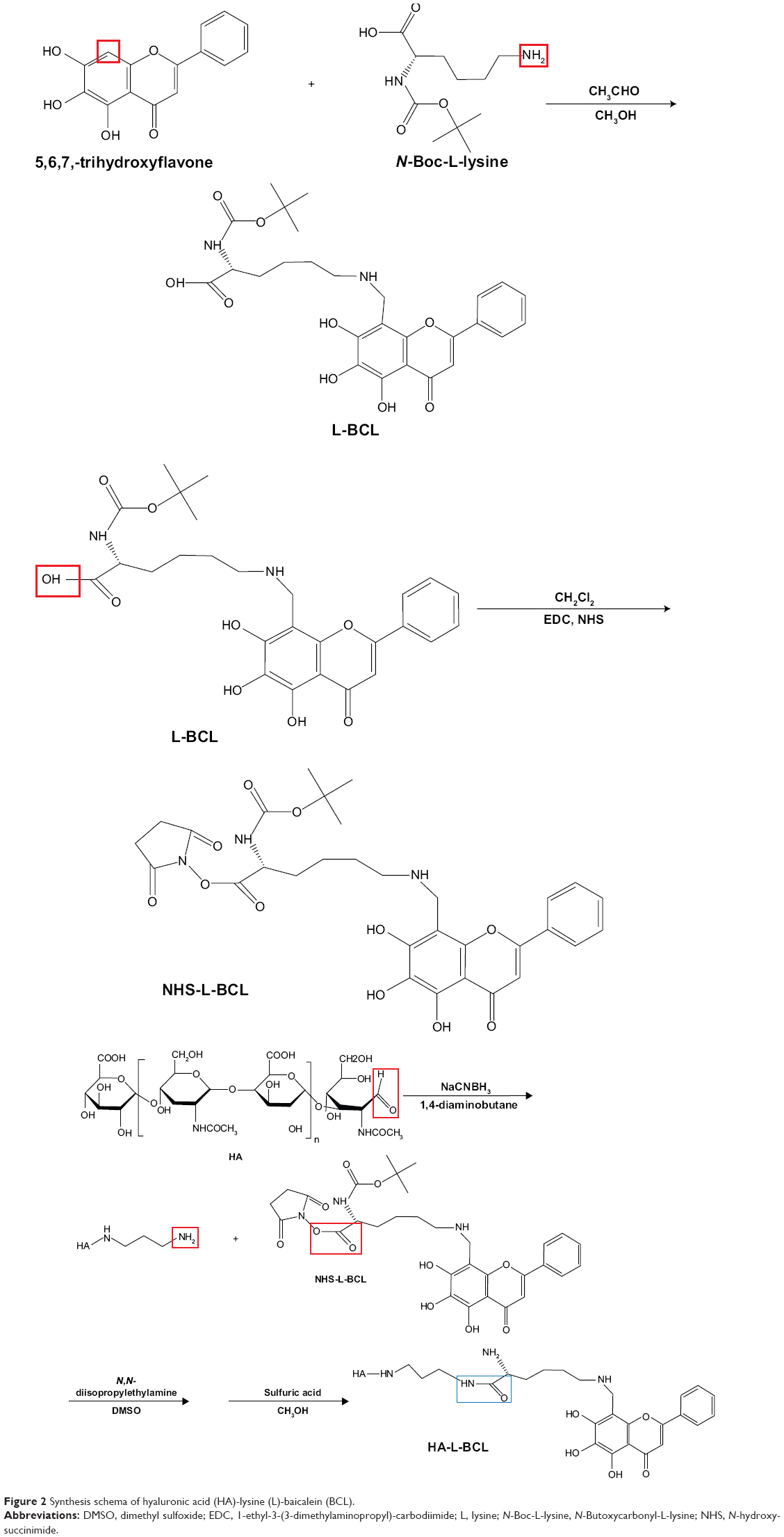 | Figure 2 Synthesis schema of hyaluronic acid (HA)-lysine (L)-baicalein (BCL). |
First, L-BCL was synthesized by the Mannich reaction. Briefly, 0.1 g BCL was dissolved in 10 mL CH3OH and 0.03 mL CH3CHO aqueous solution (36%, w/w), and heated to 60°C until the reaction solution was clear. Then 0.2 g N-Boc-L-lysine was dissolved in chloroform and added slowly and stirred for 2 hours. Finally, L-BCL was obtained via rotary evaporation and purified by reversed-phase high-performance liquid chromatography (HPLC).
Second, NHS-L-BCL was obtained as follows: L-BCL (0.3 g) was dissolved in dimethylformamide (10 mL) then 48 mg NHS and 80 mg EDC were added and the mixture was allowed to react at RT for 24 hours. Finally, NHS-L-BCL was obtained via rotary evaporation and purified by reversed-phase HPLC.
Finally, HA-L-BCL was synthesized. HA was reacted with 1,4-diaminobutane (C4H12N2) in the presence of 0.1 g sodium cyanoborohydride (NaCNBH3) in 0.5 mL acetate buffer at pH 5.0. Then NHS-L-BCL was dissolved in DMSO and 10 μL N, N-diisopropylethylamine (C8H19N) was added. Trifluoroacetic acid and methanol (1:1, v/v) were used to detach the Boc group of N-Boc-L-lysine. Then, the mixture was purified by dialysis (Spectra/Por® 4 dialysis tubing; MWCO 12,000–14,000) against water for 48 hours to remove the excess HA and other micromolecule impurities. The final product, HA-L-BCL, was obtained by lyophilization.
Proton nuclear magnetic resonance (1H NMR) spectra and infrared (IR) spectra of FA-V-PTX and HA-L-BCL were analyzed on a nuclear magnetic resonance (NMR) spectrometer (300 Hz, Bruker Advance AVII-500 NMR spectrometer; Bruker Corporation, Billerica, MA, USA) and Fourier transform IR spectrometer (TANGO FT-NIR Spectrometer; Bruker Corporation), respectively.
Preparation of FA-V-PTX and HA-L-BCL NPs (PTX-BCL NPs)
PTX-BCL NPs were prepared by the nanoprecipitation method.51 The amphiphilic nature of FA-V-PTX and HA-L-BCL makes it possible for them to self-assemble into NPs with PTX and BCL as the inner core, FA and HA as the hydrophilic shell (Figure 3). Briefly, PLGA (50 mg)-acetone (3 mL) solution was prepared. FA-V-PTX and HA-L-BCL (1/5 and 1/2, wPTX/wBCL) were dissolved in acetone–water solution (50:50, w/w). Then, PLGA, FA-V-PTX, and HA-L-BCL solution were simultaneously and separately added dropwise into the 0.05% poloxamer 188 containing aqueous solution. The mixture was stirred at 600 rpm at RT until complete evaporation of the organic solvent occurred. The redundant stabilizers were removed by centrifugation at 1,000 × g and 4°C for 30 minutes. The pellet was vortexed and resuspended in Milli-Q® water, washed several times, filtered through a 0.45 μm membrane, and adjusted to pH 7.0±0.1 with sodium hydroxide. The obtained PTX-BCL NPs were stored at 2°C–8°C.
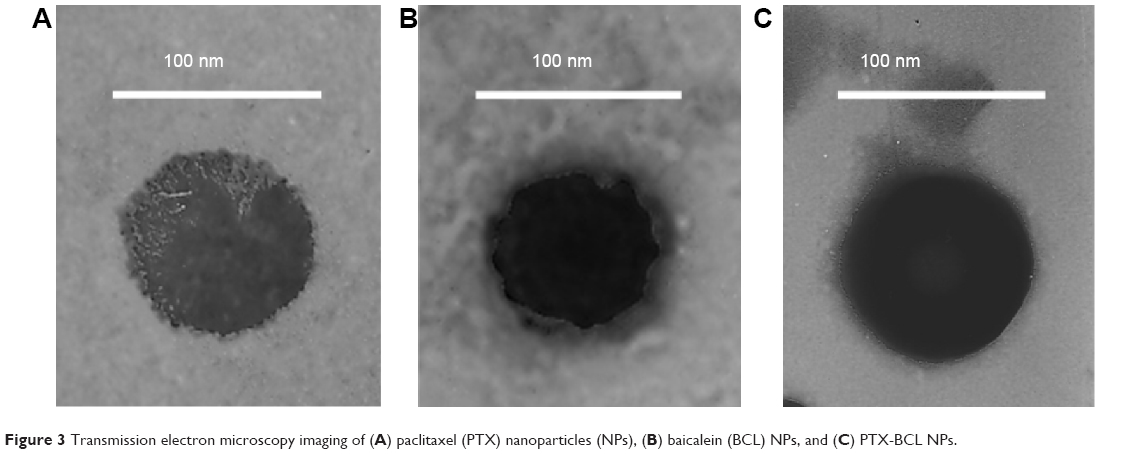 | Figure 3 Transmission electron microscopy imaging of (A) paclitaxel (PTX) nanoparticles (NPs), (B) baicalein (BCL) NPs, and (C) PTX-BCL NPs. |
Single-drug NPs were prepared using the same method without the presence of another prodrug. For instance, FA-V-PTX NPs (PTX NPs) were formulated without HA-L-BCL. HA-L-BCL NPs (BCL NPs) were prepared with no FA-V-PTX.
Characterization of NPs
The surface morphologies of the NPs were examined by transmission electronic microscopy. The particle size, polydispersity index (PDI), and zeta potential of the NPs were analyzed using photon correlation spectroscopy with a Zetasizer 3000 (Malvern Instruments, Malvern, England). The average particle size is expressed as volume mean diameter and the reported value is represented as mean ± standard deviation (n=3).
The entrapment efficiency (EE) of FA-V-PTX and HA-L-BCL in the NP formulations was detected by HPLC (LC-20A; Shimadzu Corporation, Kyoto, Japan). Chromatographic separations were carried out using an Inertsil® ODS-3V (SHIMADZU Group, Kyoto, Japan; 250.0×4.6 mm) octadecylsilane column. For PTX, the mobile phase consisted of a mixture of acetonitrile and water (55:45, v/v). The flow rate was kept at 1 mL/min and the system was maintained at 35°C. Detection was carried out at λ=227 nm, at which FA does not absorb. The injection volume was 20 μL. For BCL, the mobile phase consisted of a mixture of acetonitrile and 0.01 M 1-pentanesulfonic acid sodium salt in water (30:70, v/v). The flow rate was kept at 1 mL/min and the system was maintained at 35°C. Detection was carried out at λ=275 nm, at which HA does not absorb. The injection volume was 20 μL. The EE was calculated according to Equation 1:
|
|
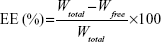
in which Wtotal is the weight of FA-V-PTX or HA-L-BCL added when preparing the NPs and Wfree is the weight of the FA-V-PTX or HA-L-BCL measured in the supernatant.
Cytotoxicity
The cytotoxicity of the PTX solution, BCL solution, PTX + BCL mixed solution (PTX-BCL solution), PTX NPs, BCL NPs, and PTX-BCL NPs was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. PTX solution consisted of a mixture of polyoxyethylated castor oil and ethanol (1:1, v/v). BCL solution consisted of 10% N, N-dimethylacetamide, 20% polyethylene glycol (PEG) 400, 20% tetraglycol, and 50% distilled water.31 In brief, A549 cells and A549/PTX cells were seeded in separate 96-well plates at 1×104 cells/well. After overnight attachment, cells were incubated with 0.9% saline (the control group), PTX solution, BCL solution, PTX-BCL solution (PTX/BCL =1/10, 1/5, 1/2, 1/1, 2/1, 5/1, 10/1 w/w), PTX NPs, BCL NPs, and PTX-BCL NPs at various concentrations for 48 hours at 37°C in a 5% CO2 atmosphere. Culture medium was used as the blank group. Then, MTT solution (5 mg/mL) was added to each well and cells were incubated for 4 hours. DMSO (200 μL) was added to each well to dissolve the MTT formazan crystals. The optical density (OD) of the formazan product was measured using a microplate reader (Model 680, Bio-Rad Laboratories Inc, Hercules, CA, USA) at 570 nm. The relative cell viability (CV) was calculated according to Equation 2:
|
|
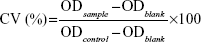
The half maximal inhibitory concentration (IC50) values of the different formulations were calculated using SPSS software (v 18; IBM Corp, Armonk, NY, USA).
To further study the synergy in the PTX-BCL solution and PTX-BCL NPs, combination index (CI) was determined.52 CI analysis provides qualitative information on the nature of drug interaction. CI was calculated according to Equation 3:
|
|
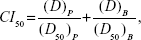
in which “(D50)P” and “(D50)B” represent the IC50 value of PTX alone and BCL alone, respectively. “(D)P” and “(D)B” represent the concentration of PTX and BCL, respectively, in the combination system at the IC50 value. A CI value <1 represents synergism, =1 represents addition, and >1 represents antagonism.
In vivo antitumor efficiency
We evaluated the efficacy of the combination therapy of PTX-BCL solution and PTX-BCL NPs using models of A549/PTX cells developed by injection of these cells into the right armpits of Kunming mice. When the tumor volume reached 90–100 mm3, transplanted mice were randomly divided into seven groups (ten per group). The seven groups were treated as follows: (1) 0.9% saline as control; (2) PTX solution (10 mg/kg); (3) BCL solution (50 mg/kg); (4) PTX-BCL solution (10 mg PTX/kg, 50 mg BCL/kg); (5) PTX NPs (10 mg/kg); (6) BCL NPs (50 mg/kg); and (7) PTX-BCL NPs (10 mg PTX/kg, 50 mg BCL/kg). The mice of each group were given the treatments by tail vein injection once every 4 days. After 24 days, all the mice were sacrificed by cervical dislocation and the tumor tissue samples were taken out.53
The volumes of the solid tumors were measured with a digital caliper every 4 days, and were calculated according to Equation 4:54
|
|
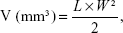
in which L and W represent the largest diameter and the smallest diameter, respectively.
The antitumor efficacy of each formulation was evaluated by tumor inhibition rate (TIR), which was calculated using the following formula (Equation 5):55
|
|
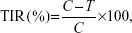
in which C and T represent the tumor weight of the control and treated groups, respectively.
The body weights of the mice and tumor sizes were also measured every 4 days.
Statistical analysis
All studies were repeated three times and all measurements were carried out in triplicate. Results are reported as mean ± standard deviation. Statistical significance was tested by two-tailed Student’s t-test or one-way analysis of variance. Differences between experimental groups were considered significant when the P-value was less than 0.05.
Results and discussion
Structure confirmation of FA-V-PTX and HA-L-BCL
The structures of FA-V-PTX and HA-L-BCL were confirmed by IR and 1H NMR spectroscopy.
- FA-V-PTX: IR ν/cm−1: 3,439 (–OH, –NH–); 3,079 (–CH2–, –CH–); 1,623 (–NH–CO–, –CO–O–); 1,471 (–C6H5); 1,366 (–CO–O–); 708 (–C6H5). 1H NMR (DMSO-d6, 300 mHz), δ (ppm): 1.05–1.81 (s, –CH3), 2.10 (s, –NH2), 4.01 (t, –CO–O–), 4.15 (t, –CO–O–), 4.31 (s, –NH–), 7.46–8.10 (m, –C6H5, Benzene ring of PTX), 12.18 (s, –OH). The presence of –NH–CO– and –CO–O– peaks confirmed the formation of the FA-V-PTX.
- HA-L-BCL: IR ν/cm−1: 3,424 (–OH, –NH–); 3,021 (–CH2–, –CH–); 1,635 (–NH–CO–, –NH–); 1,489 (–C6H5); 1,096 (–CH2–O–CH2–); 720 (–C6H5). 1H NMR (DMSO-d6, 300 mHz), δ (ppm): 0.82 (t, –CH3), 1.13–1.95 (s, –CH3), 2.20 (s, –NH2), 3.03–5.21 (m, –CH2–O–CH2–, glucosidic bond of HA), 4.23 (s, –NH–CH2–), 7.46–8.10 (m, –C6H5, Benzene ring of BCL), 12.49 (s, –OH). The presence of –NH–CO– and –NH–CH2– peaks confirmed the formation of the HA-L-BCL.
Valine and lysine were chosen as the connection between drugs and ligands to obtain the prodrugs. The amino acid linkages are relatively weak bonds; under certain conditions, the chemical bonds could break and release the drugs easily.56 PEG as linker for the production of prodrugs has been widely investigated.57,58 However, in vivo experiments have shown that a PEG drug system has lower efficacy than the drug alone.59 In contrast, an amino acid used as a spacer could adjust the drug-release behavior and achieve better therapeutic efficiency.60
Characterization of PTX-BCL NPs
FA-V-PTX and HA-L-BCL are amphiphilic compounds. PTX and BCL are the lipophilic heads, while FA and HA are the hydrophilic tails. The nature of FA-V-PTX and HA-L-BCL makes it possible for them to self-assemble into NPs, with PTX and BCL as the hydrophobic inner core and FA and HA as the hydrophilic shell. The transmission electron micrograph pictures of the PTX NPs, BCL NPs, and PTX-BCL NPs are shown in Figure 4. The PTX NPs, BCL NPs, and PTX-BCL NPs appear as white sphere-shaped particles with dark coats, but each type of NP has a slightly different appearance. The particle size, PDI, zeta potential, and EE of the PTX NPs, BCL NPs, and PTX-BCL NPs were analyzed and are summarized in Table 1.
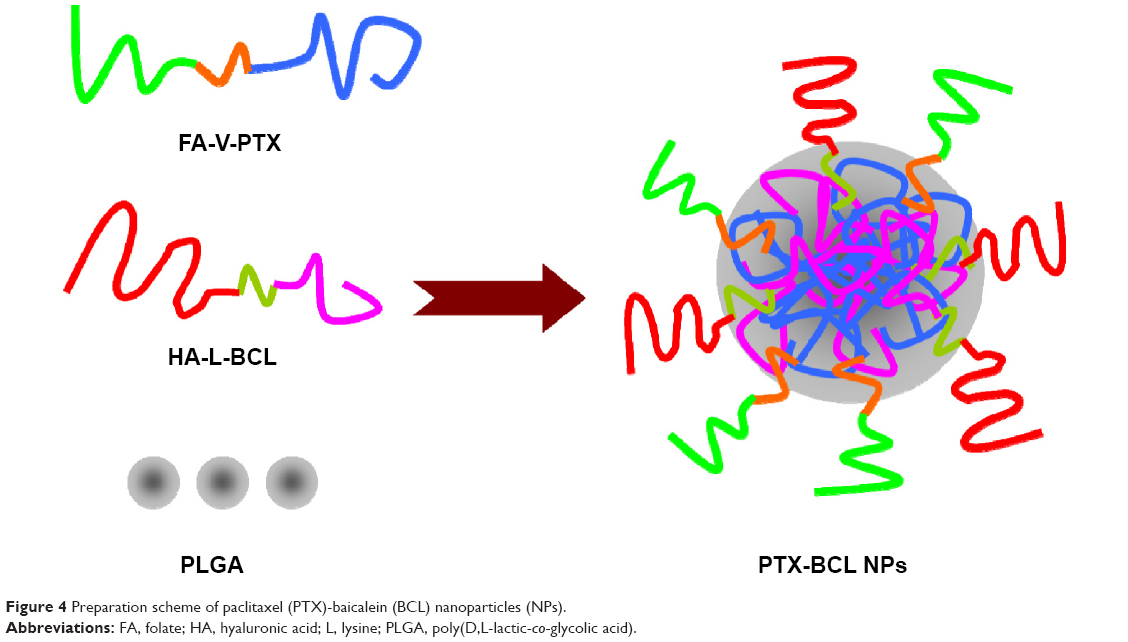 | Figure 4 Preparation scheme of paclitaxel (PTX)-baicalein (BCL) nanoparticles (NPs). |
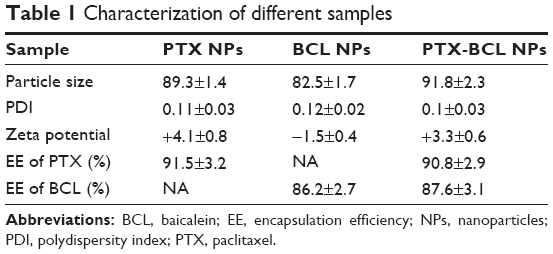 | Table 1 Characterization of different samples |
The size of the PTX-BCL NPs was approximately 90 nm, with a narrow size distribution (PDI =0.1). The size of a single prodrug-containing NP was between 80 and 90 nm. Particle size is a key effect that can influence the in vivo distribution of carriers.61 The great advantages of NPs include decreased uptake by the liver, prolonged blood circulation time, and improved bioavailability.62,63 PDI exhibits the size distribution of NPs. The PDI of the PTX-BCL NPs, PTX NPs, and BCL NPs was approximately 0.1, showing the uniformity of the NP systems.64,65 The zeta potential of the PTX-BCL NPs was +3.3. The positive charge of NPs could exploit the negative charge present at the cell surface, increase the residence time of the NPs, and promote cell penetration, thus the internalization process would be facilitated.66,67
The EE of PTX and BCL in the PTX-BCL NPs was 91% and 88%, respectively. EE is an important parameter in the determination of drug therapeutic effect.61 The high EE achieved could offer advantages in in vitro cytotoxicity studies and also have good effects on in vivo antitumor efficacy.
Cytotoxicity assays
In vitro cytotoxicity of the NPs was evaluated by MTT assay in A549 and A549/PTX cells. Figure 5 shows the viability of A549 cells treated with the different formulations. Significantly, the toxicity of of PTX-BCL solution, PTX NPs, BCL NPs, and PTX-BCL NPs on lung tumor cells was observed at the concentration of 50–200 μM, conforming to dose-dependent cell proliferation inhibition behavior. The cytotoxicity of the PTX-BCL NPs was significantly higher than that of the PTX-BCL solution, PTX NPs, and BCL NPs (P<0.05). The PTX NPs and BCL NPs showed significantly higher cytotoxicity than the PTX-BCL solution at the concentrations of 100 and 200 μM (P<0.05), while no obvious higher cytotoxicity was observed at the lower concentrations (P>0.05). For A549/PTX cells, PTX solution showed no significant effect on the cells (Figure 6). The PTX NPs showed lower cytotoxicity than the BCL NPs at the concentration of 50–200 μM (P<0.05). A significant obstacle to successful chemotherapy with PTX is MDR in tumor cells.68 In our study, the presence of BCL was expected to overcome MDR. PTX-BCL solution and PTX-BCL NPs exhibited better antitumor activity over a wide range of drug concentrations compared to their PTX counterparts.
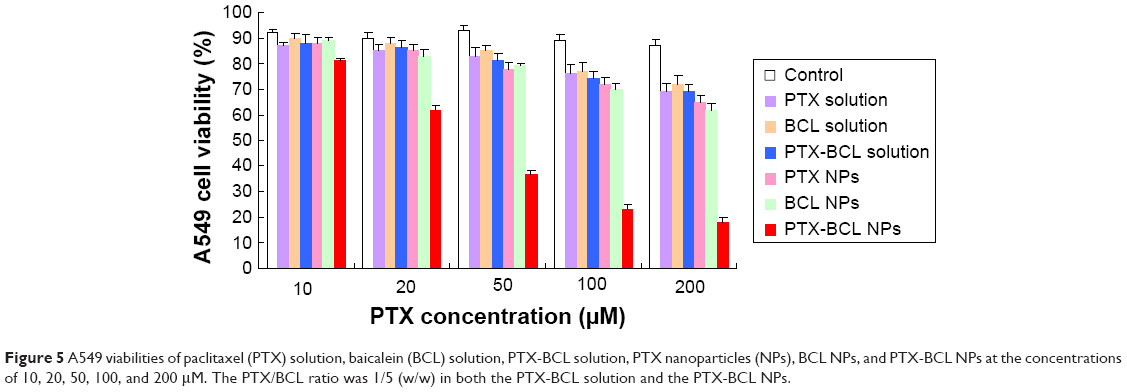 | Figure 5 A549 viabilities of paclitaxel (PTX) solution, baicalein (BCL) solution, PTX-BCL solution, PTX nanoparticles (NPs), BCL NPs, and PTX-BCL NPs at the concentrations of 10, 20, 50, 100, and 200 μM. The PTX/BCL ratio was 1/5 (w/w) in both the PTX-BCL solution and the PTX-BCL NPs. |
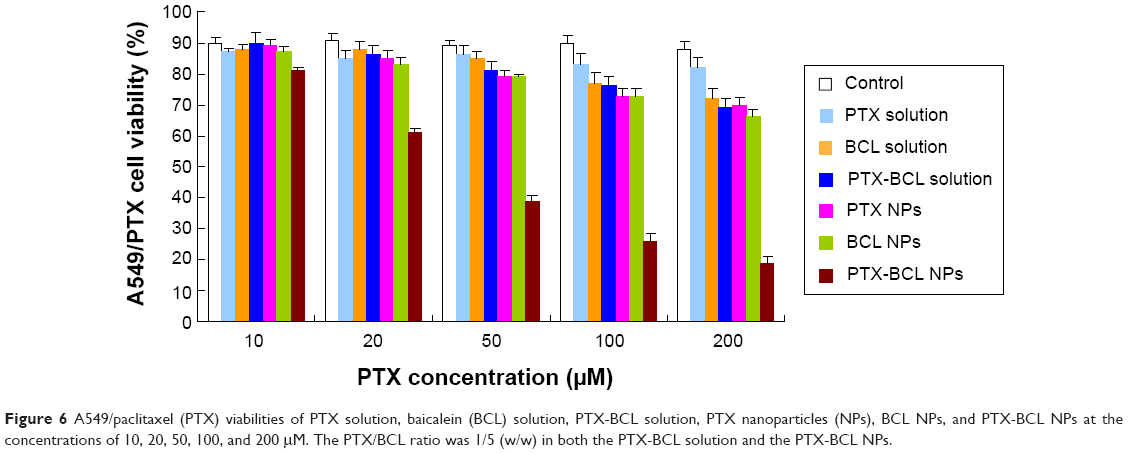 | Figure 6 A549/paclitaxel (PTX) viabilities of PTX solution, baicalein (BCL) solution, PTX-BCL solution, PTX nanoparticles (NPs), BCL NPs, and PTX-BCL NPs at the concentrations of 10, 20, 50, 100, and 200 μM. The PTX/BCL ratio was 1/5 (w/w) in both the PTX-BCL solution and the PTX-BCL NPs. |
To determine the optimal ratio of PTX to BCL in the PTX-BCL NPs for in vitro and in vivo treatment, free PTX and free BCL combinations at various weight ratios (PTX/BCL =1/10, 1/5, 1/2, 1/1, 2/1, 5/1, 10/1 w/w) were applied in A549 cells and A549/PTX cells. The IC50 values of the free drugs and the CI50 values were calculated and are shown in Tables 2 and 3. The IC50 values of PTX and BCL in the mixed PTX-BCL solution are lower than those of the PTX and BCL solutions. CI analysis provides qualitative information on the nature of drug interaction and also provides a quantitative measure of the extent of drug interaction.69 A CI value lower than, higher than, or equal to 1 indicates synergism, antagonism, or addition, respectively. According to the results of the free PTX and free BCL combination solution, the combination therapy was more synergistic when the PTX/BCL ratio was 1/5 and 1/2 (CI <1) in both A549 and A549/PTX cells. These results suggest that PTX/BCL at 1/5 or 1/2 may be the optimal prescription in the combination therapy.
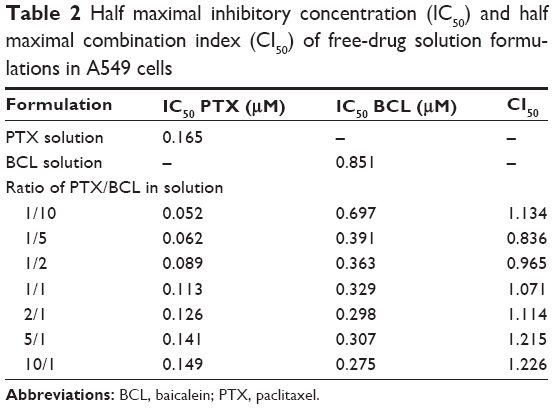 | Table 2 Half maximal inhibitory concentration (IC50) and half maximal combination index (CI50) of free-drug solution formulations in A549 cells |
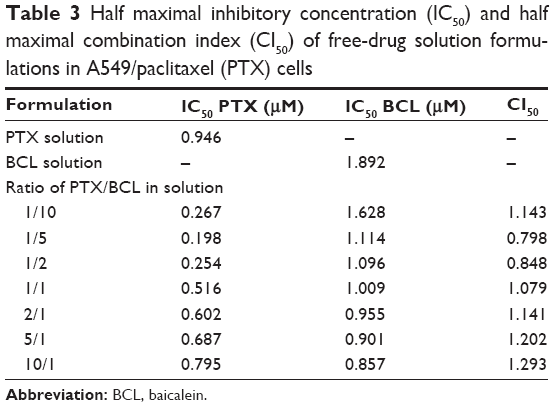 | Table 3 Half maximal inhibitory concentration (IC50) and half maximal combination index (CI50) of free-drug solution formulations in A549/paclitaxel (PTX) cells |
With the help of the combination ratio determined by the drug solutions, PTX-BCL NPs with the ratios of 1/5 and 1/2 were then prepared. The IC50 and CI50 values of the single drug solutions and combination PTX-BCL NPs were calculated and are shown in Tables 4 and 5. The CI50 value of the PTX-BCL NPs (PTX/BCL =1/2) in A549 cells was 1.05, showing no synergism in A549 cells. The CI50 value of the NPs with the PTX/BCL ratio of 1/5 and 1/2 in A549/PTX cells was 0.513 and 0.889, respectively. The PTX-BCL NPs with a PTX/BCL ratio of 1/5 had the better CI value, indicating that PTX-BCL NPs with a PTX/BCL ratio of 1/5 would provide the optimal synergistic combination of PTX and BCL. This ratio was then used for the following in vivo experiments.
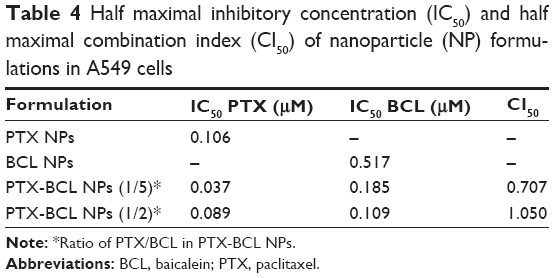 | Table 4 Half maximal inhibitory concentration (IC50) and half maximal combination index (CI50) of nanoparticle (NP) formulations in A549 cells |
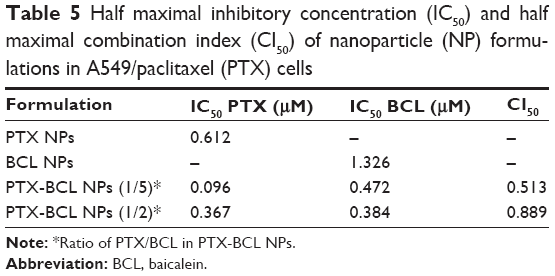 | Table 5 Half maximal inhibitory concentration (IC50) and half maximal combination index (CI50) of nanoparticle (NP) formulations in A549/paclitaxel (PTX) cells |
In vivo antitumor efficiency
Before we discuss the in vivo antitumor efficiency of the formulations, we would like to analyze how the prodrugs are released from the complex and act on cancer cells. HA on the surface of the complex can bind to CD44 and CD168 receptors, which are overexpressed in lung cancer cells. FA can also bind to the folate receptor, which is also overexpressed in cancer cells of the lung. After the ligands bound to the receptors and let the complexes into the cancer cells, FA-V-PTX and HA-L-BCL were released from the complex, and then the amino acid links were detached and released the drugs PTX and BCL.
Based on the in vitro cytotoxicity results, the in vivo antitumor efficiency and systemic toxicity of the different formulations were further investigated in mice bearing A549/PTX drug-resistant human lung cancer xenografts. As shown in Figure 7, there were no statistically significant differences between the PTX solution and 0.9% saline control (P>0.05) groups, or between the PTX NP and BCL NP groups (P>0.05). Tumor growth was significantly inhibited by the NP formulations compared with the single-drug solutions (P<0.05).
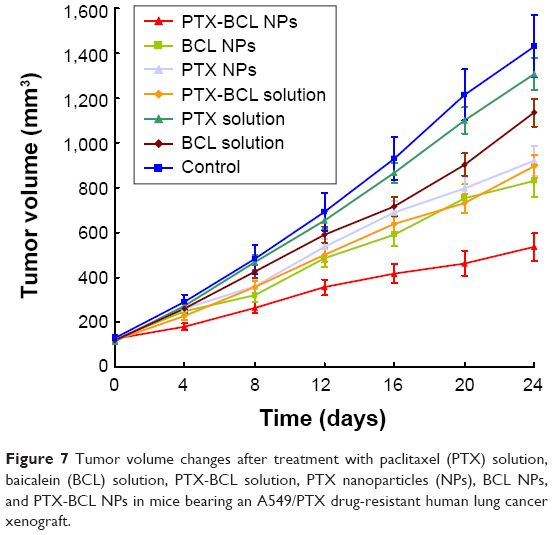 | Figure 7 Tumor volume changes after treatment with paclitaxel (PTX) solution, baicalein (BCL) solution, PTX-BCL solution, PTX nanoparticles (NPs), BCL NPs, and PTX-BCL NPs in mice bearing an A549/PTX drug-resistant human lung cancer xenograft. |
We summarize the tumor inhibition behavior of the free-drug solutions and the NP formulations as follows: first, the combination of PTX and BCL was more effective than the use of a single drug; second, the drug-loaded NPs showed better antitumor effect than their free drug counterparts. The most obvious tumor regressions were observed in the PTX-BCL NP group, in which the tumor growth was prominently inhibited, demonstrating the best antitumor activity of the dual-ligand-modified, co-delivered multifunctional self-assembled NPs.
Systemic toxicity is a key factor during cancer therapy. Targeted nanocarriers were designed to conquer this problem. Body weight loss is an indicator of systemic toxicity. In this experiment, the body weights of the mice in the 0.9% saline, PTX NP, and BCL NP groups showed slight increase, which might be due to the tumor growth and low toxicity of the NP formulations (Figure 8). Noticeable body weight loss was observed in the PTX solution and PTX-BCL solution groups, denoting the severe toxicity of PTX. Comparatively, the PTX-BCL NPs did not lead to any significant body weight loss, demonstrating the reduced systemic toxicity.
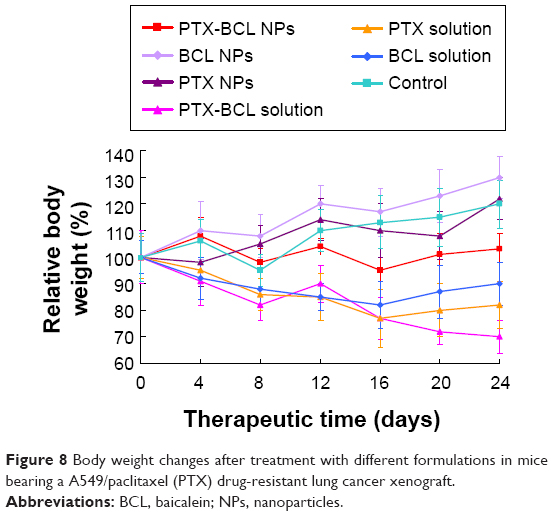 | Figure 8 Body weight changes after treatment with different formulations in mice bearing a A549/paclitaxel (PTX) drug-resistant lung cancer xenograft. |
The high anticancer efficiency and low systemic toxicity of the dual-drug-loaded, targeted nanosystem developed in this study may lead to a promising strategy in lung cancer combination therapy.
Conclusion
In this study, prodrugs of PTX and BCL, containing dual-targeted ligands of FA and HA, were synthesized. Multifunctional self-assembled NPs for the combination delivery of PTX prodrug and BCL prodrug were prepared and the synergistic antitumor effect was evaluated in vitro and in vivo. This system was proven to be efficient in delivering BCL prodrug and PTX prodrug, and in targeting drug-resistant human lung cancer cells in mice. Enhanced synergistic anticancer effects were achieved with PTX-BLC NPs and the MDR of PTX was overcome by this active targeting nanomedicine. Further studies will be required to investigate the optimal doses of both anticancer drugs to achieve the maximal anticancer efficiency but minimum systemic toxicity, and the application of this system to treat different cancers.
Disclosure
The authors report no conflicts of interest in this work.
References
Wang H, Zhao Y, Wu Y, et al. Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG-PLGA copolymer nanoparticles. Biomaterials. 2011;32(32):8281–8290. | ||
Parhi P, Mohanty C, Sahoo SK. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discov Today. 2012;17(17–18):1044–1052. | ||
Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61(13):1203–1213. | ||
Eldar-Boock A, Polyak D, Scomparin A, Satchi-Fainaro R. Nano-sized polymers and liposomes designed to deliver combination therapy for cancer. Curr Opin Biotechnol. 2013;24(4):682–689. | ||
Lammers T, Subr V, Ulbrich K, Hennink WE, Storm G, Kiessling F. Polymeric nanomedicines for image-guided drug delivery and tumor-targeted combination therapy. Nano Today. 2010;5(3):197–212. | ||
Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61(13):1203–1213. | ||
Luo C, Sun J, Sun B, He Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol Sci. 2014;35(11): 556–566. | ||
Gill KK, Kaddoumi A, Nazzal S. Mixed micelles of PEG(2000)-DSPE and vitamin-E TPGS for concurrent delivery of paclitaxel and parthenolide: enhanced chemosenstization and antitumor efficacy against non-small cell lung cancer (NSCLC) cell lines. Eur J Pharm Sci. 2012;46(1–2):64–71. | ||
Zhu C, Jung S, Luo S, et al. Co-delivery of siRNA and paclitaxel into cancer cells by biodegradable cationic micelles based on PDMAEMA-PCL-PDMAEMA triblock copolymers. Biomaterials. 2010;31(8): 2408–2416. | ||
Dai W, Jin W, Zhang J, et al. Spatiotemporally controlled co-delivery of anti-vasculature agent and cytotoxic drug by octreotide-modified stealth liposomes. Pharm Res. 2012;29(10):2902–2911. | ||
Duan J, Mansour HM, Zhang Y, et al. Reversion of multidrug resistance by co-encapsulation of doxorubicin and curcumin in chitosan/poly(butyl cyanoacrylate) nanoparticles. Int J Pharm. 2012;426(1–2):193–201. | ||
Shiah JG, Sun Y, Kopecková P, Peterson CM, Straight RC, Kopecek J. Combination chemotherapy and photodynamic therapy of targetable N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin/mesochlorin e(6)-OV-TL 16 antibody immunoconjugates. J Control Release. 2001;74(1–3):249–253. | ||
Kolishetti N, Dhar S, Valencia PM, et al. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107(42):17939–17944. | ||
Duan X, Xiao J, Yin Q, et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7(7):5858–5869. | ||
Xiao H, Li W, Qi R, et al. Co-delivery of daunomycin and oxaliplatin by biodegradable polymers for safer and more efficacious combination therapy. J Control Release. 2012;163(3):304–314. | ||
Zhao P, Wang H, Yu M, et al. Paclitaxel-loaded, folic-acid-targeted and TAT-peptide-conjugated polymeric liposomes: in vitro and in vivo evaluation. Pharm Res. 2010;27(9):1914–1926. | ||
Zhan C, Gu B, Xie C, Li J, Liu Y, Lu W. Cyclic RGD conjugated poly(ethylene glycol)-co-poly(lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J Control Release. 2010;143(1):136–142. | ||
Heo DN, Yang DH, Moon HJ, et al. Gold nanoparticles surface-functionalized with paclitaxel drug and biotin receptor as theranostic agents for cancer therapy. Biomaterials. 2012;33(3):856–866. | ||
Yang XY, Li YX, Li M, Zhang L, Feng LX, Zhang N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013;334(2):338–345. | ||
Cho HJ, Yoon HY, Koo H, et al. Self-assembled nanoparticles based on hyaluronic acid-ceramide (HA-CE) and Pluronic® for tumor-targeted delivery of docetaxel. Biomaterials. 2011;32(29):7181–7190. | ||
Han HS, Lee J, Kim HR, et al. Robust PEGylated hyaluronic acid nanoparticles as the carrier of doxorubicin: mineralization and its effect on tumor targetability in vivo. J Control Release. 2013;168(2): 105–114. | ||
Mizrahy S, Raz SR, Hasgaard M, et al. Tumor targeting profiling of hyaluronan-coated lipid based-nanoparticles. J Control Release. 2011; 156(2): 231–238. | ||
El-Dakdouki MH, Zhu DC, El-Boubbou K, et al. Development of multifunctional hyaluronan-coated nanoparticles for imaging and drug delivery to cancer cells. Biomacromolecules. 2012;13(4):1144–1151. | ||
Mizrahy S, Goldsmith M, Leviatan-Ben-Arye S, et al. Tumor targeting profiling of hyaluronan-coated lipid based-nanoparticles. Nanoscale. 2014;6(7):3742–3752. | ||
Ganesh S, Iyer AK, Morrissey DV, Amiji MM. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials. 2013;34(13):3489–502. | ||
Choi KY, Min KH, Yoon HY, et al. PEGylation of hyaluronic acid nanoparticles improves tumor targetability in vivo. Biomaterials. 2011; 32(7):1880–1889. | ||
Yoon HY, Kim HR, Saravanakumar G, et al. Bioreducible hyaluronic acid conjugates as siRNA carrier for tumor targeting. J Control Release. 2013;172(3):653–661. | ||
Lu Y, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 2002;54(5):675–693. | ||
Jain K, Gupta U, Jain NK. Dendronized nanoconjugates of lysine and folate for treatment of cancer. Eur J Pharm Biopharm. 2014;87(3): 500–509. | ||
Liu Y, Sun J, Cao W, et al. Dual targeting folate-conjugated hyaluronic acid polymeric micelles for paclitaxel delivery. Int J Pharm. 2011; 421(1):160–169. | ||
Tsai MJ, Wu PC, Huang YB, et al. Baicalein loaded in tocol nanostructured lipid carriers (tocol NLCs) for enhanced stability and brain targeting. Int J Pharm. 2012;423(2):461–470. | ||
Lee HZ, Leung HW, Lai MY, Wu CH. Baicalein induced cell cycle arrest and apoptosis in human lung squamous carcinoma CH27 cells. Anticancer Res. 2005;25(2A):959–964. | ||
Lee DH, Kim C, Zhang L, Lee YJ. Role of p53, PUMA, and Bax in wogonin-induced apoptosis in human cancer cells. Biochem Pharmacol. 2008;75(10):2020–2033. | ||
Tong WG, Ding XZ, Adrian TE. The mechanisms of lipoxygenase inhibitor-induced apoptosis in human breast cancer cells. Biochem Biophys Res Commun. 2002;296(4):942–948. | ||
Leung HW, Yang WH, Lai MY, Lin CJ, Lee HZ. Inhibition of 12-lipoxygenase during baicalein-induced human lung nonsmall carcinoma H460 cell apoptosis. Food Chem Toxicol. 2007;45(3):403–411. | ||
Huang ST, Wang CY, Yang RC, Chu CJ, Wu HT, Pang JH. Wogonin, an active compound in Scutellaria baicalensis, induces apoptosis and reduces telomerase activity in the HL-60 leukemia cells. Phytomedicine. 2010;17(1):47–54. | ||
Takahashi H, Chen MC, Harris DM, et al. Anti-apoptotic BCL-2 family protein MCL-1 plays a critical role in baicalein-induced apoptosis in human pancreatic cancer cells. Gastroenterology. 2011;140(5) Suppl 1:S-684. | ||
Nakamura N, Hayasaka S, Zhang XY, et al. Effects of baicalin, baicalein, and wogonin on interleukin-6 and interleukin-8 expression, and nuclear factor-kappab binding activities induced by interleukin-1beta in human retinal pigment epithelial cell line. Exp Eye Res. 2003; 77(2):195–202. | ||
Zhang Y, Song L, Cai L, Wei R, Hu H, Jin W. Effects of baicalein on apoptosis, cell cycle arrest, migration and invasion of osteosarcoma cells. Food Chem Toxicol. 2013;53:325–333. | ||
Song X, Yao J, Wang F, et al. Wogonin inhibits tumor angiogenesis via degradation of HIF-1α protein. Toxicol Appl Pharmacol. 2013;271(2): 144–155. | ||
Lee DH, Kim C, Zhang L, Lee YJ. Role of p53, PUMA, and Bax in wogonin-induced apoptosis in human cancer cells. Biochem Pharmacol. 2008;75(10):2020–2033. | ||
Zhang L, Lin G, Chang Q, Zuo Z. Role of intestinal first-pass metabolism of baicalein in its absorption process. Pharm Res. 2005;22(7): 1050–1058. | ||
Fong YK, Li CR, Wo SK, et al. In vitro and in situ evaluation of herb-drug interactions during intestinal metabolism and absorption of baicalein. J Ethnopharmacol. 2012;141(2):742–753. | ||
Seo MJ, Choi HS, Jeon HJ, Woo MS, Lee BY. Baicalein inhibits lipid accumulation by regulating early adipogenesis and m-TOR signaling. Food Chem Toxicol. 2014;67:57–64. | ||
Tsai TH, Liu SC, Tsai PL, Ho LK, Shum AY, Chen CF. The effects of the cyclosporin A, a P-glycoprotein inhibitor, on the pharmacokinetics of baicalein in the rat: a microdialysis study. Br J Pharmacol. 2002; 137(8):1314–1320. | ||
McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta. 2008;1785(2):96–132. | ||
Akmal Y, Senthil M, Yan J, et al. Combination of a natural compound (baicalein) and paclitaxel results in synergistic apoptosis in mouse breast cancer cells. J Surg Res. 2011;165(2):218–219. | ||
Kanakkanthara A, Miller JH. MicroRNAs: novel mediators of resistance to microtubule-targeting agents. Cancer Treat Rev. 2013; 39(2): 161–170. | ||
Duan Z, Lamendola DE, Penson RT, Kronish KM, Seiden MV. Overexpression of IL-6 but not IL-8 increases paclitaxel resistance of U-2OS human osteosarcoma cells. Cytokine. 2002;17(5):234–242. | ||
Flint MS, Kim G, Hood BL, Bateman NW, Stewart NA, Conrads TP. Stress hormones mediate drug resistance to paclitaxel in human breast cancer cells through a CDK-1-dependent pathway. Psychoneuroendocrinology. 2009;34(10):1533–1541. | ||
Jiang Z, Sun C, Yin Z, et al. Comparison of two kinds of nanomedicine for targeted gene therapy: premodified or postmodified gene delivery systems. Int J Nanomedicine. 2012;7:2019–2031. | ||
Lv S, Tang Z, Li M, et al. Co-delivery of doxorubicin and paclitaxel by PEG-polypeptide nanovehicle for the treatment of non-small cell lung cancer. Biomaterials. 2014;35(23):6118–6129. | ||
Xiao H, Song H, Yang Q, et al. A prodrug strategy to deliver cisplatin(IV) and paclitaxel in nanomicelles to improve efficacy and tolerance. Biomaterials. 2012;33(27):6507–6519. | ||
Tabatt K, Sameti M, Olbrich C, Müller RH, Lehr CM. Effect of cationic lipid and matrix lipid composition on solid lipid nanoparticle-mediated gene transfer. Eur J Pharm Biopharm. 2004;57(2):155–162. | ||
Shi F, Yang G, Ren J, Guo T, Du Y, Feng N. Formulation design, preparation, and in vitro and in vivo characterizations of β-Elemene-loaded nanostructured lipid carriers. Int J Nanomedicine. 2013;8: 2533–2541. | ||
Sugahara S, Kajiki M, Kuriyama H, Kobayashi TR. Paclitaxel delivery systems: the use of amino acid linkers in the conjugation of paclitaxel with carboxymethyldextran to create prodrugs. Biol Pharm Bull. 2002;25(5):632–641. | ||
Zhang L, Liu M, Jamil S, Han R, Xu G, Ni Y. PEGylation and pharmacological characterization of a potential anti-tumor drug, an engineered arginine deiminase originated from Pseudomonas plecoglossicida. Cancer Lett. 2014;357(1):346–354. | ||
Ruan S, Yuan M, Zhang L, et al. Tumor microenvironment sensitive doxorubicin delivery and release to glioma using angiopep-2 decorated gold nanoparticles. Biomaterials. 2014;37:425–435. | ||
Greenwald RB, Pendri A, BoIikal D. Highly water soluble taxol derivatives: 7-polyethylene glycol carbamates and carbonates. J Org Chem. 1995;60(2):331–336. | ||
Feng X, Yuan YJ, Wu JC. Synthesis and evaluation of water-soluble paclitaxel prodrugs. Bioorg Med Chem Lett. 2002;12(22):3301–3303. | ||
Rahman HS, Rasedee A, How CW, et al. Zerumbone-loaded nanostructured lipid carriers: preparation, characterization, and antileukemic effect. Int J Nanomedicine. 2013;8:2769–2781. | ||
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. | ||
Fang YP, Lin YK, Su YH, Fang JY. Tryptanthrin-loaded nanoparticles for delivery into cultured human breast cancer cells, MCF7: the effects of solid lipid/liquid lipid ratios in the inner core. Chem Pharm Bull (Tokyo). 2011;59(2):266–271. | ||
Cai H, Wen X, Wen L, et al. Enhanced local bioavailability of single or compound drugs delivery to the inner ear through application of PLGA nanoparticles via round window administration. Int J Nanomedicine. 2014;9:5591–5601. | ||
Yang H, Cai W, Xu L, et al. Nanobubble-Affibody: Novel ultrasound contrast agents for targeted molecular ultrasound imaging of tumor. Biomaterials. 2014;37C:279–288. | ||
Harush-Frenkel O, Rozentur E, Benita S, Altschuler Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules. 2008;9(2):435–443. | ||
Wang CZ, Fu YC, Jian SC, et al. Synthesis and characterization of cationic polymeric nanoparticles as simvastatin carriers for enhancing the osteogenesis of bone marrow mesenchymal stem cells. J Colloid Interface Sci. 2014;432:190–199. | ||
Wang Y, Yu L, Han L, Sha X, Fang X. Difunctional Pluronic copolymer micelles for paclitaxel delivery: synergistic effect of folate-mediated targeting and Pluronic-mediated overcoming multidrug resistance in tumor cell lines. Int J Pharm. 2007;337(1–2):63–73. | ||
Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res. 2004;10(23):7994–8004. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.