")
Back to Journals » International Journal of Nanomedicine » Volume 18
Defeating Melanoma Through a Nano-Enabled Revision of Hypoxic and Immunosuppressive Tumor Microenvironment
Authors Yang W , Pan X , Zhang P, Yang X, Guan H, Dou H , Lu Q
Received 17 April 2023
Accepted for publication 19 June 2023
Published 6 July 2023 Volume 2023:18 Pages 3711—3725
DOI https://doi.org/10.2147/IJN.S414882
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Wenzhe Yang,1,2,* Xue Pan,2,* Peng Zhang,2 Xue Yang,1,2 Huashi Guan,1,2 Huan Dou,3 Qian Lu1,2
1Key Laboratory of Marine Drugs of Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao, Shandong Province, People’s Republic of China; 2Marine Traditional Chinese Medicine R&D Laboratory, Marine Biomedical Research Institute of Qingdao, Qingdao, Shandong Province, People’s Republic of China; 3Jiangsu Key Laboratory of Molecular Medicine, Medical School, Nanjing University, Nanjing, Jiangsu Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qian Lu, Key Laboratory of Marine Drugs of Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, 5 Yushan Road, Qingdao, Shandong Province, People’s Republic of China, Tel +86 15850672529, Fax +86 532 8506779, Email [email protected] Huan Dou, Jiangsu Key Laboratory of Molecular Medicine, Medical School, Nanjing University, 22 Hankou Road, Nanjing, Jiangsu Province, 210093, People’s Republic of China, Tel +86 13913832488, Fax +86 025 83686441, Email [email protected]
Rationale: Reversing the hypoxic and immunosuppressive tumor microenvironment (TME) is crucial for treating malignant melanoma. Seeking a robust platform for the effective reversion of hypoxic and immunosuppressive TME may be an excellent solution to revolutionizing the current landscape of malignant melanoma treatment. Here, we demonstrated a transdermal and intravenous dual-administration paradigm. A tailor-made Ato/cabo@PEG-TK-PLGA NPs were administrated transdermally to melanoma with the help of a gel spray containing a skin-penetrating material borneol. Nanoparticles encased Ato and cabo were released and thereby reversed the hypoxic and immunosuppressive tumor microenvironment (TME).
Methods: Ato/cabo@PEG-TK-PLGA NPs were synthesized through a self-assembly emulsion process, and the transdermal ability was assessed using Franz diffusion cell assembly. The inhibition effect on cell respiration was measured by OCR, ATP, and pO2 detection and in vivo photoacoustic (PA) imaging. The reversing of the immunosuppressive was detected through flow cytometry analysis of MDSCs and T cells. At last, the in vivo anti-tumor efficacy and histopathology, immunohistochemical analysis and safety detection were performed using tumor-bearing mice.
Results: The transdermally administrated Ato/cabo@PEG-TK-PLGA NPs successfully spread to the skin surface of melanoma and then entered deep inside the tumor with the help of a gel spray and a skin puncturing material borneol. Atovaquone (Ato, a mitochondrial-respiration inhibitor) and cabozantinib (cabo, a MDSCs eliminator) were concurrently released in response to the intratumorally overexpressed H2O2. The released Ato and cabo respectively reversed the hypoxic and immunosuppressive TME. The reversed hypoxic TME offered sufficient O2 for the intravenously administrated indocyanine green (ICG, an FDA-approved photosensitizer) to produce adequate amount of ROS. In contrast, the reversed immunosuppressive TME conferred amplified systemic immune responses.
Conclusion: Taken together, we developed a transdermal and intravenous dual-administration paradigm, which effectively reversed the hypoxic and immunosuppressive tumor microenvironment in the treatment of the malignant melanoma. We believe our study will open a new path for the effective elimination of the primary tumors and the real-time control of tumor metastasis.
Keywords: melanoma, tumor microenvironment, hypoxia, immunosuppression, transdermal administration
Introduction
Malignant melanoma, the most aggressive type of skin cancer, is highly resistant to traditional chemotherapy and radiotherapy.1–3 For this reason, various types of alternative treatments have been investigated. Mainly, photodynamic therapy (PDT), an emerging therapeutic modality capable of producing cytotoxic reactive oxygen species (ROS), has been extensively applied to treat melanoma due to the minimally invasive feature.4,5 Unlikely, the hypoxic tumor microenvironment (TME) could not afford sufficient oxygen (O2) for photosensitizers to generate effective amount of ROS, thus leading to the melanoma treatment failure.6 In addition to hypoxic TME, immunosuppression is also a typical hallmark of TME.7 The immunosuppressive TME, often brings high rates of recurrence and mortality to melanoma patients.8 In these regard, they are seeking a robust platform for the effective reversion of hypoxic and immunosuppressive TME may be an excellent solution to revolutionizing the current landscape of malignant melanoma treatment.
Currently, some strategies, including in situ O2 generations by the decomposition of tumor-overexpressed hydrogen peroxide (H2O2) by catalase (CAT) or CAT-like nanozymes, the delivery of O2 to tumor tissues by the usage of some O2-binding material and the normalization of tumor vasculature by the interference with some inhibitors or enzymes, have been reported to be able to alleviate hypoxic TME. However, the restricted amount of intratumoral H2O2, the high interstitial pressure, and the newly formed peri-tumoral vessels, greatly hinder tumor oxygenation. Most recently, a more effective strategy called mitochondrial-respiration inhibition has been verified to enable the effective inhibition of mitochondria-associated oxidative phosphorylation (OXPHOS).9 This strategy fundamentally suppresses intrinsic oxygen consumption by the rapid proliferation of tumor cells, thus showing tremendous potential to overcome hypoxia-driven therapeutic resistance.10
Myeloid-derived suppressor cells (MDSCs), defined as the CD11b+Gr-1+ phenotype in mice, have been reported to be the most crucial protector cell of the TME.11 They are the cornerstone of the immunosuppressive shield that protecting the tumor from the patient’s immune system and even immunotherapy. MDSCs induce and strengthen the immunosuppressive TME mainly from four aspects: 1) suppressing T cell activity and inducing regulatory T cells;12 2) driving the polarization of macrophages from anti-tumor M1-phenotype to the pro-tumor M2-phenotype;13 3) recruiting tumor-associated macrophages;14 4) converting the maturation of dendritic cells as well as macrophages.15
To this end, seeking a versatile platform that could simultaneously inhibit mitochondrial respiration and stop the immunosuppressive function of MDSCs will be a promising approach to reversing the hypoxic and immunosuppressive TME and promoting the malignant melanoma treatment.
The nanoparticles can work as a versatile platform which achieving precise drug delivery through active and passive targeting function.16,17 By enhancing permeation and retention, the nanoparticles can reach the diseased tissue (EPR effect).18 By specific binding with over-expressed receptors, the active targeted delivery of intracellular transport is achieved.19,20 Smart active delivery strategies are triggered responding to the typical of tumor microenvironment factors, such as pH, ROS, GSH and so on, which lead to more precise targeting.21
In this study, we demonstrated a transdermal and intravenous dual-administration paradigm for treating malignant melanoma (Scheme 1). The transdermal administration was accomplished by the mixture of a tailor-made nano-system (Ato/cabo@PEG-TK-PLGA NPs) and a cellular puncturing material borneol into a gel spray.22 Ato/cabo@PEG-TK-PLGA NPs were synthesized by the encasement of atovaquone (Ato, a mitochondrial-respiration inhibitor)23 and cabozantinib (cabo, a MDSCs eliminator)24 into a ROS-cleavable PEG-TK-PLGA. With the help of gel spray-featured sustained cargo-release function, the inner borneol (a natural transdermal enhancer extracted from Borneol Camphor)25 slowly spread to the skin surface of melanoma, changed skin microstructure, enlarged intercellular space, and enabled the transport of Ato/cabo@PEG-TK-PLGA NPs across skin.26 After meeting with the intratumorally overexpressed H2O2, Ato/cabo@PEG-TK-PLGA NPs were disintegrated and the encased Ato/cabo were gradually released, which dramatically reversed the hypoxic and immunosuppressive TME. Subsequently, indocyanine green (ICG, a FDA-approved photosensitizer) was intravenously administrated. The revision of hypoxic TME provided sufficient O2 for the intravenously administrated ICG to generate effective amount of ROS, thus facilitating the effective ablation of primary melanoma. Meanwhile, the revision of immunosuppressive TME, as verified by the decreased tumor-infiltrated MDSCs and the increased activation of cytotoxic T cells (CTLs), markedly prevented the tumor metastasis of melanoma. Taken together, we believe this work will provide a new option with higher safety for clinical melanoma treatment.
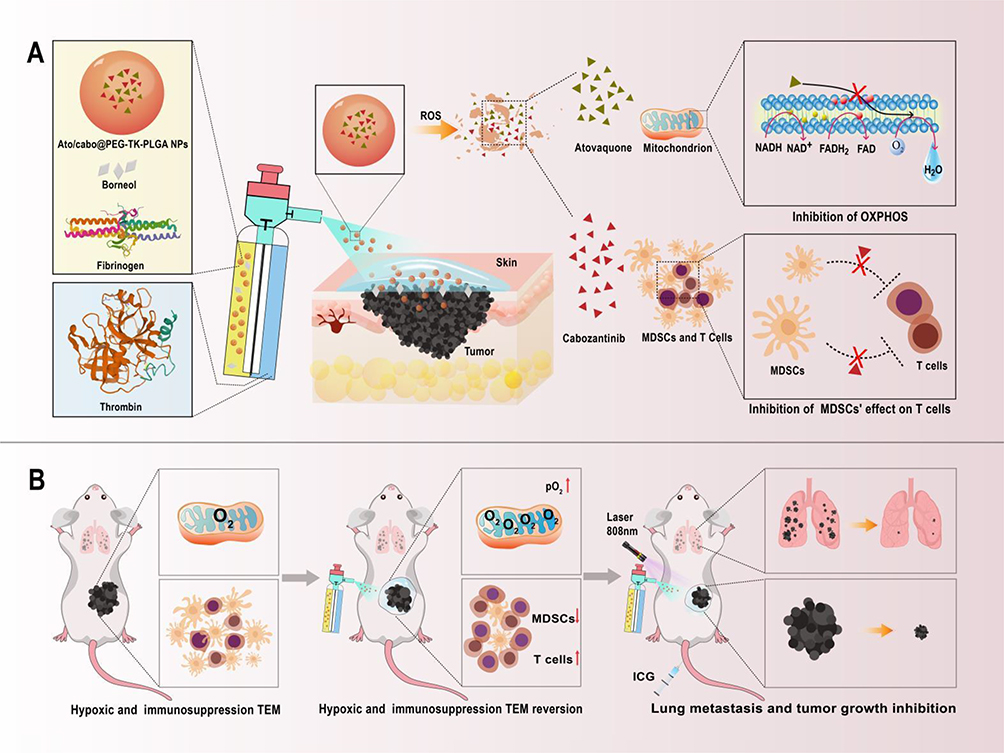 |
Scheme 1 A scheme illustrating the action mechanism of reversing the hypoxic and immunosuppressive TME with a transdermal and intravenous dual-administration paradigm for treating malignant melanoma. (A) Schematic showing the gel sprayed on the skin which containing Ato/cabo@PEG-TK-PLGA nanoparticles. ROS-responsible linkage triggered the release of Ato and cabo, which inhibit mitochondria-associated OXPHOS and MDSCs’ effect on T cells. (B) Schematic showing the PDT effect could be further enhanced by increasing tumor oxygen content and blocking immunosuppressive after transdermal absorption. What’s more, T cells were increased, which developed an effective immunogenic treatment against lung metastasis. |
Materials and Methods
Materials and Reagents
Atovaquone (Ato), and cabozantinib (cabo) were purchased from Aladdin Reagent (Shanghai, China), while mPEG (2000)-TK-PLGA (2000) were bought from Ruixi Biotech (Shanxi, China). A cellular puncturing material borneol was purchased from Lanzhou Wotelaisi Biology (Gansu, China). ICG was purchased from Dandong Yichuang Pharmaceutical co., LTD (Dandong, China). FcR blocking reagents, CD11b-PE Vio770 was purchased from Miltenyi Biotec (Köln, Germany). Gr-1-Alexa Fluor 532 was bought from Novus Biological (Colorado, USA). CD11c-BB700 was purchased from BD Pharmingen (New Jersey, USA). CD3-Alexa Fluor 532 was bought from eBioscience (San Diego, USA). CD80-Percp/Cy5.5, F4/80-PE/Dazzle 549, MHC II-APC/Cy7, CD4-PE/Cy5 and CD8-APC/Cy7 were purchased from BioLegend (San Diego, USA). Dulbecco’s modified Eagle’s medium (DMEM), Roswell Park Memorial Institute 1640 (RPMI-1640) growth medium, fetal bovine serum (FBS) and penicillin–streptomycin solution were purchased from Gibco BRL (Maryland, USA).
Synthesis of Ato/cabo@PEG-TK-PLGA NPs
Ato/cabo@PEG-TK-PLGA NPs were synthesized through a self-assembly emulsion process. In brief, mPEG (2000)-TK-PLGA (2000) (20 mg) was dissolved in deionized water (20 mL). Then, a dichloromethane suspension (2 mL) containing Ato (4 mg) and cabo (1 mg) was added to the mPEG (2000)-TK-PLGA suspension. Afterwards, the mixture was sonicated for 40–60 min by bath sonicator to completely volatilize dichloromethane. Eventually, the residue was removed by thrice centrifugation (15 min, 3000 g) and Ato/cabo@PEG-TK-PLGA NPs were collected.
Characterization
The morphologies of PEG-TK-PLGA NPs and Ato/cabo@PEG-TK-PLGA NPs were observed using the transmission electron microscope (TEM, JEOL-2100, Tokyo, Japan), while the size distribution of PEG-TK-PLGA NPs and Ato/cabo@PEG-TK-PLGA NPs were examined by using Zetasizer Nano ZS (Malvern, UK). The spectra of Ato, PE-labeled cabo, PEG-TK-PLGA, and Ato/cabo@PEG-TK-PLGA NPs were detected by ultraviolet visible spectrophotometer (UV3100, Shimadzu, Japan). The encapsulation efficiency (EE) and drug loading (DL) of Ato and PE-labeled cabo were determined by the reading number of supernatant and standard curve, which according to the concentration difference between initial solution and supernatant solution after loading drug. The in vitro drug release from Ato/cabo@PEG-TK-PLGA NPs was performed by dialysis method. 4 mL Ato/cabo@PEG-TK-PLGA NPs solution was placed in a dialysis bag (MWCO 2000 Da) and immersed in 50 mL PBS buffer with or without 0.2 mM H2O2. Then, the dialysis bag was incubated at 37 °C with continue stirring. 4 mL release medium was sampled and all release medium was replaced with an equal volume of PBS buffer. The cumulative release curves of Ato and cabo were drawn based on the standard curve (Ato: Y=0.1923X+0.018, R2=0.9991; PE-labeled cabo: Y=56323X-2373, R2=0.9993).
Cell Culture
Melanoma cell line B16F10 were obtained from KeyGEN BioTECH (Nanjing, China), and were cultured in RPMI-1640 (Gibco, USA) containing 10% fetal bovine serum (FBS; Gibco, USA) at 37°C with 5% CO2.
Cell Viability Assay
The cytotoxicity of PEG-TK-PLGA NPs and Ato/cabo@PEG-TK-PLGA NPs were assessed via MTT test. In brief, B16F10 cells were seeded in 96-well plates with concentration of 5000 cells/well. And then, the supernatant was replaced with culture medium containing samples with corresponding concentration (0.2, 0.4, 0.6, 0.8, 1.0 mg/mL) for 24 h. Add 20 μL MTT solution (5 mg/mL) to each well, and replaced the supernatant after 4 h. Afterwards, adding 150 μL DMSO to each well, and shake for 10 min at 37°C. A490 was detected with a multifunctional microplate reader and cell survival rate was calculated using the following formula. Cell viability (%) = OD (sample)/OD (control) × 100%, (OD (sample) was the absorbance treated with different concentration samples, OD (control) was the absorbance without sample treated).
The Investigation of the Transdermal Ability
Briefly, Ato/cabo@PEG-TK-PLGA NPs and borneol were mixed into a gel spray. Six weeks old female BALB/c mice were treated with 5% chloral hydrate and hair removal with the skin intact without damage. The mice were sacrificed the next day and the back skin was removed and fixed between the feeding and receiving pool. Adding 1.0 mL of Nile red (NiR, a hydrophobic fluorescent stain)-labeled sample solution to the feeding pool (NiR@PEG-TK-PLGA+Gel, NiR@PEG-TK-PLGA+Borneol, NiR@PEG-TK-PLGA NPs, NiR@PEG-TK-PLGA+Borneol+Gel). pH 7.4 PBS buffer was added to receiving pool, and the water temperature of the transdermal diffusion pool was maintained at 37°C with the speed of stirring was 250 rpm. 100 μL samples were taken from the receiving pool after 0.5, 1, 2, 4, 6, 10, 16, 24 and 30 h, and the fluorescence intensity of samples in the solution was measured (Ex 530 nm, Em 635 nm).
OCR, ATP, and pO2 Detection
To investigate the inhibition effect on cell respiration, Ato, Ato@PEG-TK-PLGA, Ato/cabo@PEG-TK-PLGA, Ato@PEG-TK-PLGA+0.2 mM H2O2, Ato/cabo@PEG-TK-PLGA+0.2 mM H2O2 treated B16F10 cells were conducted OCR and ATP assays by MitoXpress and ATP ELISA Kits, respectively. pO2 was measured by a JPB-607A dissolved oxygen meter, and the initial oxygen content in the medium was standardized as the base level of oxygen consumption.
In vivo Photoacoustic (PA) Imaging
B16F10 tumor-bearing mice were given saline, Borneol+Gel, Ato/cabo@PEG-TK-PLGA NPs, Ato/cabo@PEG-TK-PLGA+Borneol+Gel, respectively, to estimate the oxygen saturation of intratumoral blood vessels after different treatments. The applied instrument was a preclinical photoacoustic computed tomography scanner (Endra Nexus 128, USA) with detection wavelengths of 700 nm (Hb, deoxygenated hemoglobin) and 850 nm (HbO2, oxygenated hemoglobin).
Flow Cytometry Analysis of MDSCs and T Cells
Murine peripheral blood and spleen single-cell suspension were incubated with erythrocyte lysate for 2 min at room temperature, terminated with PBS. After centrifugation and counting, about 106 cells were pelleted in a tube and pre-incubated with FcR blocking reagent at room temperature for 15 min. Then the cells were stained with viability dye Zombie NIR for 30 min at 4°C in the dark. The population of MDSCs and functional markers were evaluated by staining cells with following anti-mouse antibodies: CD11b-PE Vio770, Gr-1-Alexa Fluor 532, CD80-Percp/Cy5.5, F4/80-PE/Dazzle 549, MHC II-APC/Cy7 and CD11c-BB700. T cells were estimated by staining with anti-mouse antibodies as CD3-Alexa Fluor 532, CD4-PE/Cy5 and CD8- APC/Cy7. Being kept on ice for 30 min in the dark, and cells were rinsed twice with PBS, and finally re-suspended in 200 μL buffer for subsequent detection by flow cytometry (CYTEK Aurora, USA). Data were analyzed using FlowJo software (Treestar, USA).
Bio-Distribution of ICG
A dose of 0.5 mg/kg of ICG was intravenously injected into B16F10-tumor-bearing mice. At 2, 4, 8, 12, 24 and 36 h, mice were anesthetized and imaged by Fluoromax-4 spectrofluorometer (Hori-baScientific, Edison, NJ, Ex 704 nm, Em 735 nm). The distribution of ICG over time in tumor-bearing mice was recorded, and the maximum difference between the tumor and surrounding normal tissues was determined as the optimal time point for PDT treatments.
In vivo Anti-Tumor Efficacy
The animal experiments were approved by the Institutional Animal Care and Use Committee of the Ocean University of China (No. OUC-SMP-2019-03-01) and were performed in accordance with the guidelines published by the National Institutes of Health. Specific pathogen-free (SPF)-grade Balb/c male mice (6–8 weeks old, 18–20 g) were obtained from Shanghai Slaccas Laboratory Animal Breeding Company (Shanghai, China) and maintained in an SPF-grade animal room with a temperature range of 20–22°C and a humidity of 40–60% containing 5 × 105 B16F10 cells was subcutaneously administrated into the right flank of the mice. 13 days later, B16F10 tumor-bearing mice were randomly divided into four groups, including Control group (saline), Transdermal group (Ato/cabo@PEG-TK-PLGA+Borneol+Gel only), Intravenous group (ICG injection only), Combinational group (treated with both ICG and Ato/cabo@PEG-TK-PLGA+Borneol+Gel) (n = 6 for each group). Mice were weighed, and the tumor sizes were measured using calipers every three days. Tumor volume was calculated according to the formula: volume (V) = Length (L) × Width (W)2/2. The therapeutic effects were estimated by the monitoring relative body weight and tumor size, tumor weight, representative tumor necropsy. On day 29, mice were sacrificed, tumors, blood and spleens were isolated, and specific indices were measured.
Treatment method for the lung metastasis model, B16F10 cells were injected into the tail vein before the initial treatment on day 13, and the treatment regimens were the same (n = 4 for each group). Mice were sacrificed on day 29 and the lung tissue was stripped to observe the tumor metastasis.
Histopathology and Immunohistochemical Analysis
After sacrificing the tumor-bearing mice on day 29, the tumor tissues were collected and stained with hematoxylin and eosin (H&E), TUNEL and Ki67 for histopathology and immunohistochemical analysis.
Long-Term in-vivo Safety Detection
After sacrificing the tumor-bearing mice on day 29, the vital organs (including heart, liver, spleen, lung and kidney) were collected for histopathology analysis. The blood was collected from eye orbit and detected several biochemical parameters including aminotransferase (AST), alanine transferase (ALT), creatinine (Cr), and blood urea nitrogen (UREA) to investigate long-term safety.
Statistics
All data were presented as mean ± standard error of mean (SEM), and each experiment included triplicate sets. The significant differences among groups with one independent variable were determined by one-way analysis of variance (ANOVA) with a Tukey’s multiple comparisons test for planned comparisons. P-value ≤ 0.05 was considered significant. GraphPad Prism 5 was used for data analysis (GraphPad Software Inc., CA, USA).
Results and Discussion
Characteristics of Ato/cabo@PEG-TK-PLGA NPs
TEM results indicate that PEG-TK-PLGA NPs and Ato/cabo@PEG-TK-PLGA NPs both have uniform and spherical morphology with diameter sizes of ~50 nm and ~75 nm (Figure 1A and B). DLS results (Figure 1C) reveal both PEG-TK-PLGA NPs and Ato/cabo@PEG-TK-PLGA NPs have greater hydrodynamic diameter sizes (~68 nm, ~91 nm, respectively) than that observed in TEM images. The UV-Vis-NIR absorption spectra of the cargos (Ato, PE-labeled cabo) and the vehicle (PEG-TK-PLGA) are monitored and displayed in Figure 1D. After the encasement of these cargos into the vehicle to form Ato/cabo@PEG-TK-PLGA NPs, the typical absorption peaks of these cargos and vehicles were all preserved, thus demonstrating the successful synthesis. The encapsulation efficiency of Ato and cabo was 80.12% and 82.36% (Table S1), respectively. Drug loading of Ato and cabo (Table S1) was 13.3% and 3.4%, respectively. Thioketal (TK) bond, as a ROS-responsible linkage, was introduced into the vehicle to give an on-demand payload release. From Figure 1E and F, we observed a H2O2-dependent release behavior of Ato and cabo, which gave a chance to mitigate the hypoxic and immunosuppressive TME. Additionally, PEG-TK-PLGA NPs and Ato/cabo@PEG-TK-PLGA NPs both displayed ignorable cytotoxicity to B16F10 cells (Figure S1), supporting the feasibility of Ato/cabo@PEG-TK-PLGA NPs for in vivo applications.
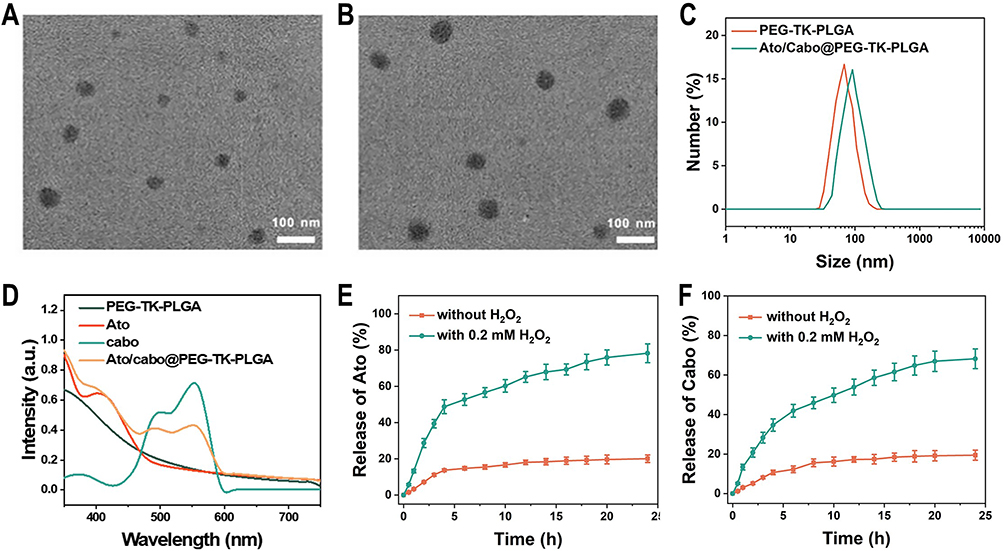 |
Figure 1 Characteristics of Ato/cabo@PEG-TK-PLGA NPs. TEM images of (A) PEG-TK-PLGA NPs and (B) Ato/cabo@PEG-TK-PLGA NPs. (C) DLS of PEG-TK-PLGA NPs and Ato/cabo@PEG-TK-PLGA NPs. (D) UV-VIS-NIR of PEG-TK-PLGA, Ato, cabo, and Ato/cabo@PEG-TK-PLGA NPs. Solvent: dichloromethane for Ato group and cabo; H2O2 for others. Release behaviors of (E) Ato and (F) cabo from Ato/cabo@PEG-TK-PLGA NPs treated with or without H2O2. |
The Investigation of the Transdermal Ability
For skin cancer, skin barrier function achieved by the stratum corneum limits the drug permeation.27 Micro-molecules can penetrate stratum corneum in small quantities, or directly enter the epidermis through sweat glands and pores, but for macro-molecules, penetration is still challenging.28 In recent years, transdermal administration has been reported to reduce unnecessary drug uptake and directly target tumor sites under the epidermis, which increases the effective drug concentration and amplifies the therapeutic outcome.29 Borneol has been widely used as a skin puncturing material in traditional Chinese medicine, which mechanism involved disrupting stratum corneum bilayer, increasing drug diffusion coefficient, and inducing the formation of transient pores.30 To take full advantage of this super benefit, Ato/cabo@PEG-TK-PLGA NPs, in this article, were also administrated via transdermal manner. In order to evaluate whether Ato/cabo@PEG-TK-PLGA NPs can be successfully delivered into skin by transdermal administration, NiR@PEG-TK-PLGA were synthesized by replacing Ato/cabo with Nile red (NiR, a hydrophobic fluorescent stain with excitation wavelength of 530 nm and emission wavelength of 635 nm). According to Figure 2A and B, the skin treated with NiR@PEG-TK-PLGA without the engagement of borneol showed invisible fluorescence while the treatment with NiR@PEG-TK-PLGA+Borneol displayed stronger red fluorescence, indicating the extraordinarily wonderful skin puncturing ability. Notably, NiR@PEG-TK-PLGA+Borneol+Gel treatment further amplifies the fluorescence signal, revealing the gel in NiR@PEG-TK-PLGA+Borneol+Gel treatment group can prolong the effective contact time between nanoparticles and skin due to gel state. In addition, the receiving cell solution was collected for fluorescence intensity measurement. By 30 h, the transdermal ability of NiR@PEG-TK-PLGA+Borneol+Gel treatment group was 1.2, 3.0 and 4.1 times of that of NiR@PEG-TK-PLGA+Borneol, NiR@PEG-TK-PLGA+Gel and NiR@PEG-TK-PLGA NPs solution treatment group, respectively (Figure 2C). All these results suggest that the borneol and gel can facilitate the effect transdermal delivery of PEG-TK-PLGA NPs, which may change the TME.
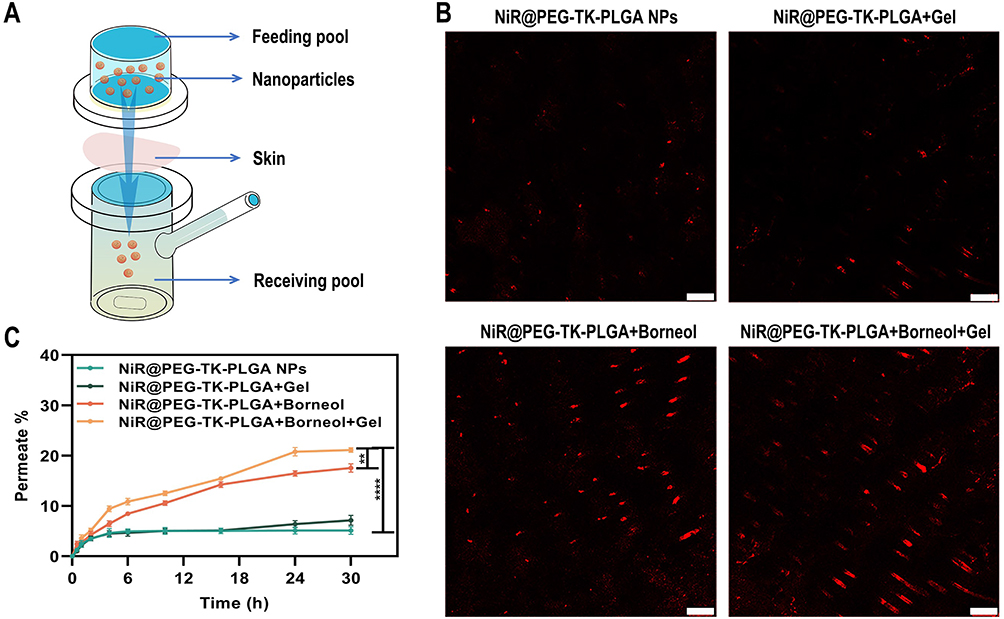 |
Figure 2 Transdermal administration of NPs. (A) Transdermal ability of nanoparticles test by Franz diffusion cell. (B) Laser confocal microscope fluorescence image and (C) Nile red transdermal curve of permeate fluorescence labeled nanoparticles. Scale bars: 100 μm. (**P < 0.01, ****P < 0.0001, n = 3). |
The Revision of Hypoxic Tumor Microenvironment
Ato has been verified to inhibit the mitochondria-associated OXPHOS of tumor cells and increase the cell-internal oxygen consumption, therefore relieving hypoxia tumor microenvironment.31,32 MitoXpress Kit was used to detect the tumor cells’ oxygen consumption rate (OCR) after different treatments, showing the effect on inhibition of cellular respiration.33 From Figure 3A, we found a concentration-dependent OCR reduction in Ato group and about 80% of OCR reduction was noticed when the concentration of Ato was 1 mM, proving Ato could robustly inhibit the cell respiration. Once the presence of H2O2, Ato-contained groups (Ato@PEG-TK-PLGA NPs, Ato/cabo@PEG-TK-PLGA NPs) also displayed similar concentration-dependent behavior in OCR reduction and cell respiration inhibition ability. Adenosine triphosphate (ATP) is synthesized during normal mitochondrial respiration. To determine whether cellular respiration inhibition occurred in mitochondria, we measured the amount of ATP in different groups. We found Ato@PEG-TK-PLGA NPs and Ato/cabo@PEG-TK-PLGA NPs markedly dwindled the ATP content inside B16F10 cells as long as the engagement of H2O2, attesting the cell respiration inhibition function of Ato occurred in mitochondria (Figure 3B). Due to the function of Ato, the mitochondrial respiratory chain in tumor cells was inhibited, resulting in the accumulation of oxygen. Therefore, the partial pressure of oxygen (pO2) in different treatment groups were measured. Compared with that in H2O2-free condition, pO2 in Ato/cabo@PEG-TK-PLGA group raise above fivefold with the assistance of H2O2 (Figure 3C), proving tremendous amount of oxygen was saved. Next, we leveraged non-invasive imaging or ex-vivo HIF-1ɑ staining to monitor the in vivo hypoxia condition of TME post different treatments. From the PA imaging as shown in Figure 3D, maximum oxyhemoglobin content (reflecting oxygen availability) was witnessed in mice received the transdermal administration of Ato/cabo@PEG-TK-PLGA. In contrast, the Borneol+Gel group (without the engagement of Ato/cabo@PEG-TK-PLGA) or the Ato/cabo@PEG-TK-PLGA group (without the addition of borneol and gel) displayed no obvious increase in oxyhemoglobin content with relative to control group, suggesting the significance of the successful transdermal administration of Ato/cabo@PEG-TK-PLGA to the reversion of hypoxic TME. Notably, the successful transdermal administration of Ato/cabo@PEG-TK-PLGA with the help of borneol and gel also displayed the lowest HIF-1ɑ level, reflecting the reduced hypoxia degree (Figure 3E).
 |
Figure 3 The revision of hypoxic TME. (A-C) Cell respiratory depression was evaluated via the relative OCR (A); the relative ATP content (B); pO2 (C) of B16F10 cells with indicated treatments. (D) Photoacoustic images were showing the ratio of oxygenated hemoglobin to deoxygenated hemoglobin in tumors after indicated treatments. (E) HIF-staining images showing the hypoxia degree in tumors after indicated treatments. Scale bars: 50 μm. (****P < 0.0001 compared with control group, n = 6) (####P < 0.0001, compared with other groups, n = 6). |
The Revision of Immunosuppressive Tumor Microenvironment
MDSC is considered instrumental for tumor-associated immune suppression.34 It has been reported that elevated MDSC is correlated with poorer outcomes in melanoma patients, making it difficult to cure.35–37 As such, we loaded MDSC inhibitor cabo into our nano-system. When analyzing the frequencies of MDSC in the peripheral blood and spleens, Ato/cabo@PEG-TK-PLGA+Borneol+Gel treated mice had a significant decrease in MDSC frequencies in the circulation and spleen (average inhibitory rate: 70.47%, 33.96%), while the treatment without the engagement of Ato/cabo@PEG-TK-PLGA (Borneol+Gel group) or without the addition of borneol and gel (Ato/cabo@PEG-TK-PLGA group) only contributed little to the down-regulation in MDSC proportion compared with control group (Figure 4A,B and Figure S2). F4/80, CD80, CD11c, and MHCII as the differentiation markers of MDSCs were also tested.38 The testing results in blood (Figure 4C-F) and spleen (Figure 4G-J) collectively demonstrated Ato/cabo@PEG-TK-PLGA+Borneol+Gel treatment promoted the maturation and differentiation of MDSC.
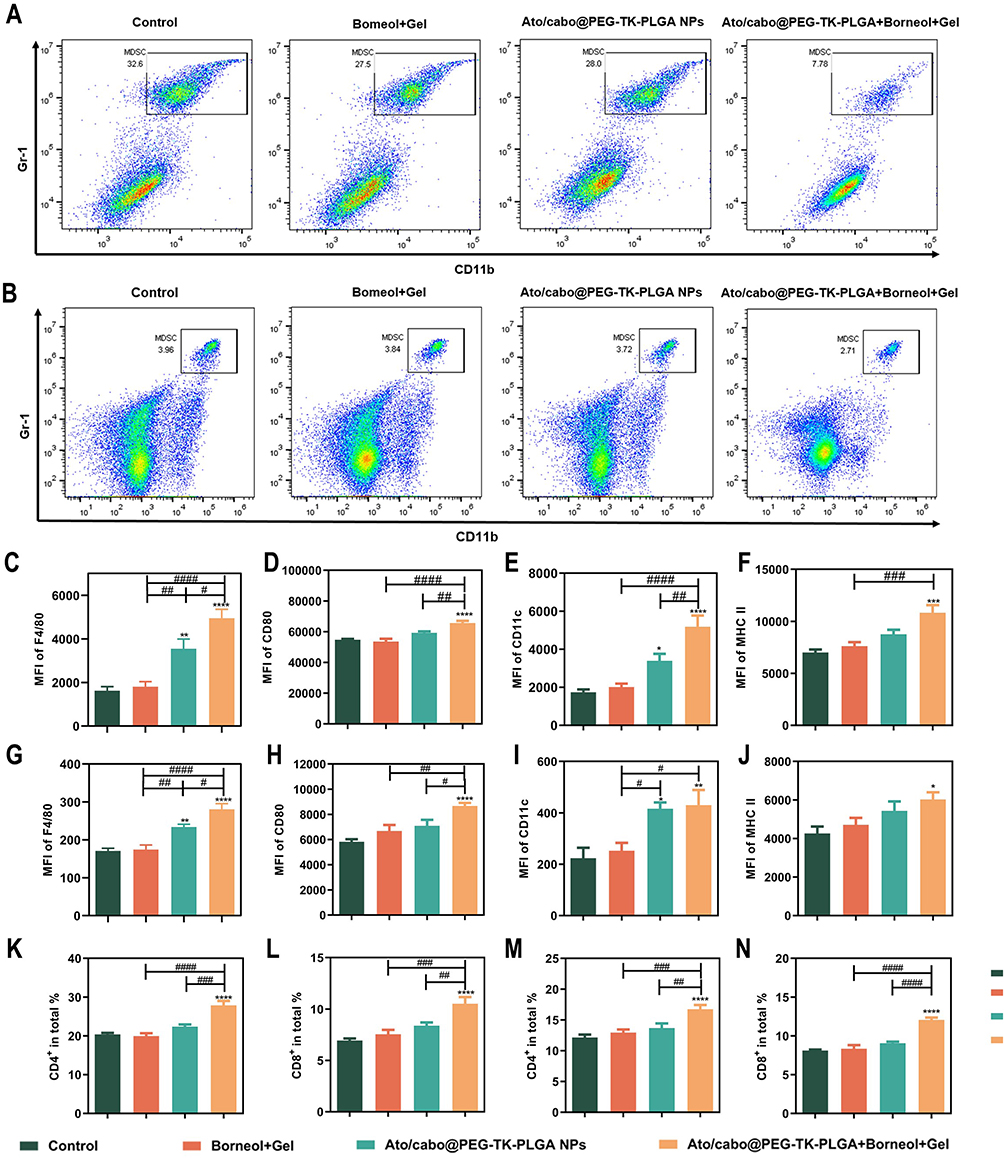 |
Figure 4 The revision of MDSC-mediated T cell suppression in tumor-bearing mice. Amount of MDSCs with different treatment in blood (A) and spleen (B). Differentiation markers of MDSCs in blood (C-F) and spleen (G-J). The two subsets of T cell in blood (K and L) and spleen (M and N) during treatment. (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared with control group, n = 6) (#P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001, compared with other groups, n = 6). |
MDSCs have been identified as one of the major suppressors of T cell proliferation and inducers of T cell tolerance.39–41 Thus, the proportion of total CD3+T cells, and two subsets (CD3+CD4+ and CD3+CD8+) were detected. From Figures 4K-N and Figure S3, we found Ato/cabo@PEG-TK-PLGA+Borneol+Gel treatment strikingly increased the proportion of CD3+, CD4+ and CD8+ T cells, suggesting Ato/cabo@PEG-TK-PLGA+Borneol+Gel treatment to regulate MDSC inhibitory function with enhanced T cell responses.
In vivo Anti-Tumor Efficacy
Before the study of in vivo anti-tumor effect, the bio-distribution of ICG was detected. As indicated in Figure S4, we found that ICG first distributed throughout the body, and the concentration of tumor tissue was relatively high at 8h of ICG injection and normal tissue was basically gradually metabolized, which was selected as PDT treatment time. Next, we evaluated the therapeutic effect of transdermal and intravenous treatments as the process displayed in Figure 5A. The Control group (saline), the tumor grew uncontrollably with the tumor volume reaching ~18.64-fold at the end, while the Transdermal group and Intravenous group both slowed down the uncontrollable tumor growth with the tumor volume reaching ~14.39-fold and ~7.74-fold at the end (Figure 5B and C). Interestingly, the combination of transdermal and intravenous treatments (Combinational) exerted dramatic therapeutic effect with the tumor volume reaching ~3.37-fold at the end (Figure 5B and C). The same trend was also seen in tumor weight of each group, without notable changes in body weight (Figure 5D and E). The representative photographs of tumors from the end of treatments (Figure S5). In addition, H&E staining, TUNEL staining, and Ki-67 immunohistochemical tests were performed to better understand the therapeutic outcome (Figure S6 and Figure 5F, G). According to the combinational treatment resulted into the maximum amount of necrotic cells, the minimum proliferation, and the largest area of tumor apoptosis, thus confirming the combination of transdermal and intravenous treatments is a robust treatment paradigm for melanoma.
 |
Figure 5 In vivo tumor inhibition of different treatment against melanoma. (A) Schematic illustration of treatment plan (Tumor model: B16F10 right flank inoculation only; Lung metastatic model: B16F10 inoculated on right flank and also tail vein injection). (B) Photographs of tumors obtained after treatment. (C) Variation of tumor during different treatment. (D) Relative average body weight of mice after treatment. (E) Tumor weight of different treated group during treatment. Representative TUNEL (F) and Ki67 (G) immunohistochemical test of tumor slices after different treatment. Scale bars: 50 μm. (*P < 0.05, **P < 0.01, ****P < 0.0001 compared with control group, n = 6) (#P < 0.05, ###P < 0.001 compared with other groups, n = 6). |
Considering urgency to develop an effective immunogenic treatment against lung metastasis,42 a mouse model of metastasis was established by the intravenous injection of B16F10 cells and the metastatic potential of primary tumor tissues were focused on to assess the anti-metastasis therapeutic effect. As shown in Figure 6A and B, the combinational group exhibited more efficiency in reducing pulmonary metastatic nodules and almost blocked melanoma lung metastasis, revealing the combinational paradigm possessing potent antitumor effects (metastasis). The same trend was also seen in histology H&E and PCNA staining (Figure 6C and D).
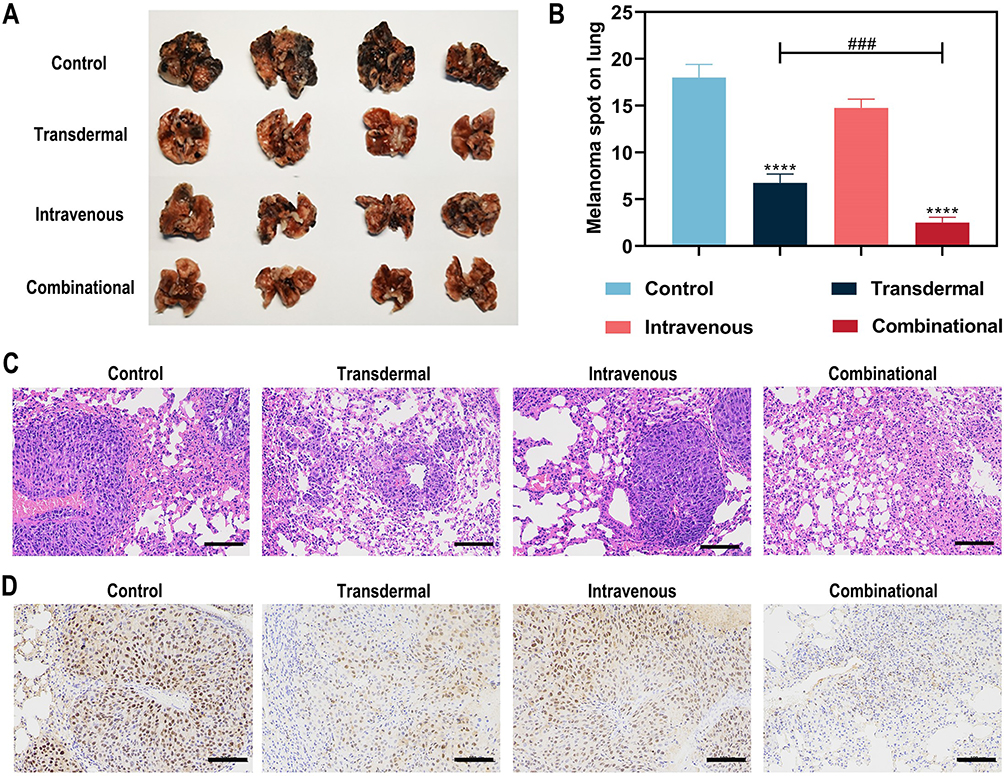 |
Figure 6 The efficiency of immunogenic treatment against lung metastasis. (A) Photographs of lungs obtained after treatment. (B)The number of melanoma spots on lungs during different treatments. Representative histology H&E (C) and PCNA staining (D) images of lung slices after different treatment. Scale bars: 100 μm. (****P < 0.0001 compared with control group, n = 4) (###P < 0.001 compared with other groups, n = 4). |
Long-Term in-vivo Safety Detection
Pathological assays and hematology assays were applied to assess the safety of this treatment paradigm. From the H&E staining in Figure 7A, all primary tissues (eg heart, lungs, livers, spleen and kidney) maintained the well-preserved structures except for a few infiltrating inflammatory immune cells. Several biochemical parameters including aminotransferase (AST), alanine transferase (ALT), creatinine (Cr), and blood urea nitrogen (UREA) were also detected. Undetectable changes were observed in these biochemical parameters after treatment, suggesting our paradigm do not adversely affect the body function (Figure 7B). Pathological and hematology analysis confirmed the biocompatibility of our treatment paradigm.
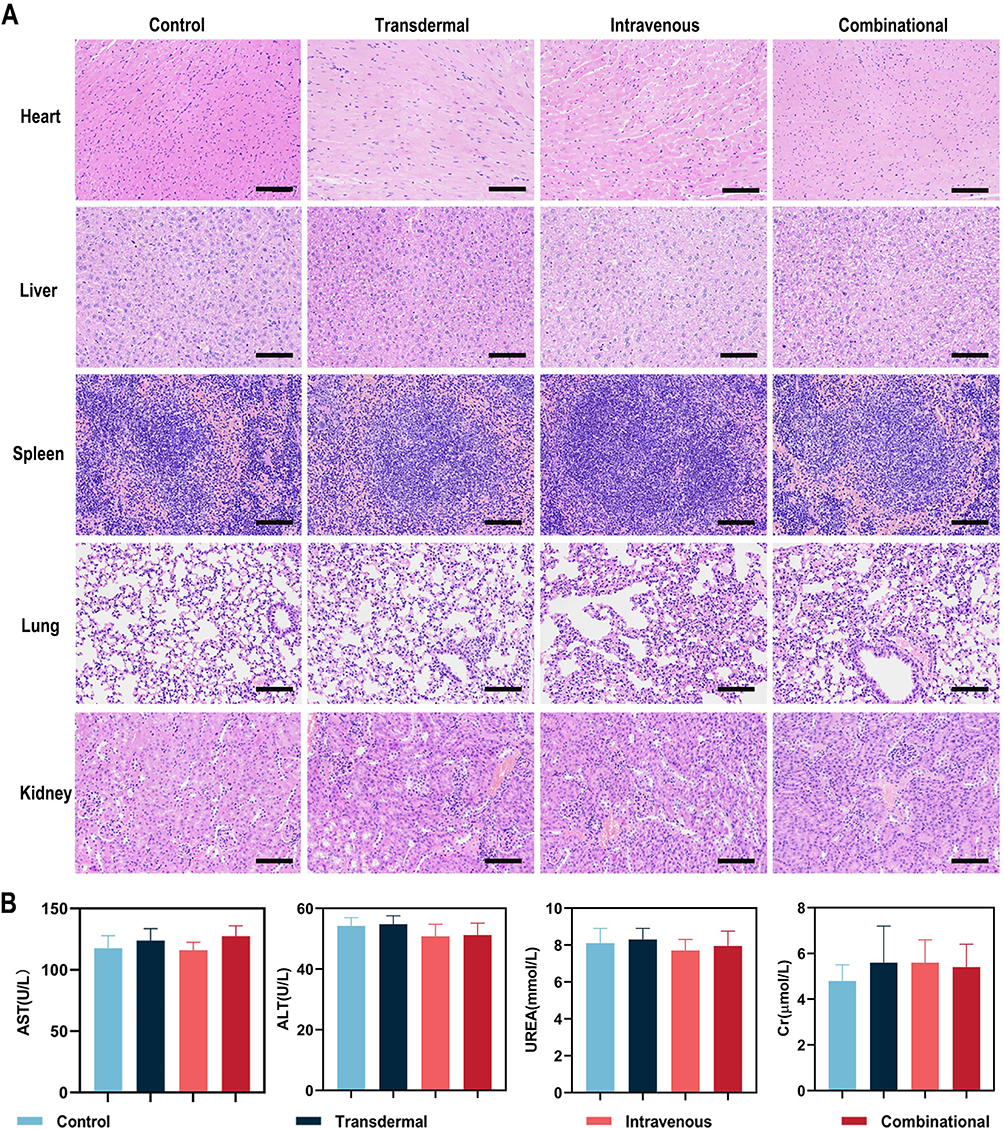 |
Figure 7 Long-term in-vivo safety (A) H&E staining of major organs after different treatments (Scale bars: 100 μm). (B) The variation of liver and kidney function showed safety after indicated treatment. |
Conclusion
In current study, we developed a transdermal and intravenous dual-administration paradigm to treat malignant melanoma. A tailor-made Ato/cabo@PEG-TK-PLGA NPs were administrated transdermally to melanoma with the help of a gel spray containing a natural penetration enhancer borneol. In response to the intratumorally overexpressed H2O2, encased Ato and cabo were released and thereby reversed the hypoxic and immunosuppressive TME. The reversed hypoxic TME provided sufficient O2 for the intravenously administrated ICG to generate effective amount of ROS. The reversed immunosuppressive TME conferred amplified systemic immune responses and prevented the tumor metastasis of melanoma. Long-term safety assay showed the excellent biocompatibility and rare systemic toxicity of this paradigm. In conclusion, the use of tumor microenvironment to accurately trigger drug release, which effectively reverse hypoxic TEM and enhance body immune function, thereby the tumor efficacy of PDT was increased. We believe this work will open a new avenue for the effective elimination of the primary malignant melanoma and the real-time control of tumor metastasis.
Acknowledgments
This work was supported by the grants from National Natural Science Foundation of China (81901843 and 32070883), Innovation & Development Joint Fund of Natural Science Foundation(ZR2022LSW009) and Key R&D Program (Major Innovation Project, 2021CXGC010510) of Shandong Province, Special Fund for Local Science and Technology Development Guided by the Central Committee (YDZX2021018) and Fundamental Research Funds for the Central Universities.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Guo W, Wang H, Li C. Signal pathways of melanoma and targeted therapy. Signal Transduction Targeted Therapy. 2021;6(1):424. doi:10.1038/s41392-021-00827-6
2. Lin W, Fisher D. Signaling and Immune Regulation in Melanoma Development and Responses to Therapy. Annu Rev Pathol. 2017;12:75–102. doi:10.1146/annurev-pathol-052016-100208
3. Zhang M, Qin X, Xu W, Wang Y, Luan Y. Engineering of a Dual-Modal Phototherapeutic Nanoplatform for Single NIR Laser-Triggered Tumor Therapy. J Colloid Interface Sci. 2021:46. doi:10.1016/j.jcis.2021.03.050
4. Hou Y, Yang X, Liu R, et al. Pathological Mechanism of Photodynamic Therapy and Photothermal Therapy Based on Nanoparticles. Int J Nanomedicine. 2020;15:6827–6838. doi:10.2147/ijn.s269321
5. Zhou S, Shang Q, Wang N, Li Q, Luan Y. Rational design of a minimalist nanoplatform to maximize immunotherapeutic efficacy: four birds with one stone. J Controlled Release. 2020;328. doi:10.1016/j.jconrel.2020.09.035
6. Wang W, Chen C, Ying Y, et al. Smart PdH@MnO Yolk-Shell Nanostructures for Spatiotemporally Synchronous Targeted Hydrogen Delivery and Oxygen-Elevated Phototherapy of Melanoma. ACS nano. 2022. doi:10.1021/acsnano.1c10450
7. Groth C, Hu X, Weber R, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120(1):16–25. doi:10.1038/s41416-018-0333-1
8. Umansky V, Sevko A. Melanoma-induced immunosuppression and its neutralization. Semin Cancer Biol. 2012;22(4):319–326. doi:10.1016/j.semcancer.2012.02.003
9. Xia D, Xu P, Luo X, et al. Overcoming Hypoxia by Multistage Nanoparticle Delivery System to Inhibit Mitochondrial Respiration for Photodynamic Therapy. Adv Funct Mater. 2019;29(13):1807294. doi:10.1002/adfm.201807294
10. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res. 2018;24(11):2482–2490. doi:10.1158/1078-0432.ccr-17-3070
11. Pawelec G, Verschoor CP, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: not only in tumor immunity. Front Immunol. 2019;101099. doi:10.3389/fimmu.2019.01099
12. Pu S, Qin B, He H, et al. Identification of early myeloid progenitors as immunosuppressive cells. Sci Rep. 2016;6:23115. doi:10.1038/srep23115
13. Sica A, Massarotti M. Myeloid suppressor cells in cancer and autoimmunity. J Autoimmun. 2017;85:117–125. doi:10.1016/j.jaut.2017.07.010
14. Rahma OE, Hodi FS. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin Cancer Res. 2019;25(18):5449–5457. doi:10.1158/1078-0432.ccr-18-1543
15. Salminen A. Activation of immunosuppressive network in the aging process. Ageing Res Rev. 2020;57:100998. doi:10.1016/j.arr.2019.100998
16. Akhter MH, Rizwanullah M, Ahmad J, Ahsan MJ, Mujtaba MA, Amin S. Nanocarriers in advanced drug targeting: setting novel paradigm in cancer therapeutics. Artif Cells, Nanomed Biotechnol. 2017;1:634. doi:10.1080/21691401.2017.1366333
17. Ahmad J, Ahmad M, Akhter H. Surface-engineered cancer nanomedicine: rational design and recent progress. Curr Pharm Des. 2020;26(11):1181–1190. doi:10.2174/1381612826666200214110645
18. Akhter MH, Beg S, Tarique M, Malik A, Hosawi S. Receptor-based targeting of engineered nanocarrier against solid tumors: recent progress and challenges ahead. Biochimica et Biophysica Acta. 2021;1865(2):129777. doi:10.1016/j.bbagen.2020.129777
19. Habban Akhter M, Sateesh Madhav N, Ahmad J. Epidermal growth factor receptor based active targeting: a paradigm shift towards advance tumor therapy. Artif Cells, Nanomed Biotechnol. 2018;46:1188–1198. doi:10.1080/21691401.2018.1481863
20. Akhter M, Amin S. An investigative approach to treatment modalities for squamous cell carcinoma of skin. Curr Drug Deliv. 2017;14(5):597–612. doi:10.2174/1567201801666160906104254
21. Taghipour Y, Zarebkohan A, Salehi R, et al. An update on dual targeting strategy for cancer treatment. J Control Release. 2022;349:67–96. doi:10.1016/j.jconrel.2022.06.044
22. Chen Q, Wang C, Zhang X. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat Nanotechnol. 2018;14:89. doi:10.1038/s41565-018-0319-4
23. Ashton TM, Fokas E, Kunz-Schughart LA, et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat Commun. 2016;7:12308. doi:10.1038/ncomms12308
24. Aggen D, Ager C, Obradovic A, et al. Blocking IL1 beta promotes tumor regression and remodeling of the myeloid compartment in a renal cell carcinoma model: multidimensional analyses. Clin Cancer Res. 2021;27(2):608–621. doi:10.1158/1078-0432.ccr-20-1610
25. Liu S, Long Y, Yu S, et al. Borneol in cardio-cerebrovascular diseases: pharmacological actions, mechanisms, and therapeutics. Pharmacol Res. 2021;169:105627. doi:10.1016/j.phrs.2021.105627
26. Zhang Y, Zhang N, Song H, et al. Design, characterization and comparison of transdermal delivery of colchicine via borneol-chemically-modified and borneol-physically-modified ethosome. Drug Deliv. 2019;26(1):70–77. doi:10.1080/10717544.2018.1559258
27. Bouwstra J, Honeywell-Nguyen P, Gooris G, Ponec M. Structure of the skin barrier and its modulation by vesicular formulations. Prog Lipid Res. 2003;42(1):1–36. doi:10.1016/s0163-7827(02)00028-0
28. Simon J, Jouanmiqueou B, Rols M, Flahaut E, Golzio M. Transdermal delivery of macromolecules using two-in-one nanocomposite device for skin electroporation. Pharmaceutics. 2021;13(11):645. doi:10.3390/pharmaceutics13111805
29. Shahzad Y, Louw R, Gerber M, du Plessis J. Breaching the skin barrier through temperature modulations. J Controlled Release. 2015;202:1–13. doi:10.1016/j.jconrel.2015.01.019
30. Wang R, Wu Z, Yang S, et al. A Molecular Interpretation on the Different Penetration Enhancement Effect of Borneol and Menthol towards 5-Fluorouracil. Int J Mol Sci. 2017;18(12):46. doi:10.3390/ijms18122747
31. Skwarski M, McGowan DR, Belcher E, et al. Mitochondrial inhibitor atovaquone increases tumor oxygenation and inhibits hypoxic gene expression in patients with non-small cell lung cancer. Clin Cancer Res. 2021;27(9):2459–2469. doi:10.1158/1078-0432.Ccr-20-4128
32. Chen D, Sun X, Zhang X, Cao J. Targeting mitochondria by anthelmintic drug atovaquone sensitizes renal cell carcinoma to chemotherapy and immunotherapy. J Biochem Mol Toxicol. 2018;32(9):e22195. doi:10.1002/jbt.22195
33. Papkovsky D, Zhdanov A. Cell energy budget platform for multiparametric assessment of cell and tissue metabolism. Methods Mol Biol. 2021;2276:305–324. doi:10.1007/978-1-0716-1266-8_23
34. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–119. doi:10.1038/s41590-017-0022-x
35. Jordan KR, Amaria RN, Ramirez O, et al. Myeloid-derived suppressor cells are associated with disease progression and decreased overall survival in advanced-stage melanoma patients. Cancer Immunol Immunother. 2013;62(11):1711–1722. doi:10.1007/s00262-013-1475-x
36. Butterfield LH, Zhao F, Lee S, et al. Immune Correlates of GM-CSF and Melanoma Peptide Vaccination in a Randomized Trial for the Adjuvant Therapy of Resected High-Risk Melanoma (E4697). Clin Cancer Res. 2017;23(17):5034–5043. doi:10.1158/1078-0432.ccr-16-3016
37. Sade-Feldman M, Kanterman J, Klieger Y, et al. Clinical Significance of Circulating CD33(+) CD11b(+)HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin Cancer Res. 2016;22(23):5661–5672. doi:10.1158/1078-0432.Ccr-15-3104
38. Albeituni SH, Ding C, Liu M, et al. Yeast-Derived Particulate beta-Glucan Treatment Subverts the Suppression of Myeloid-Derived Suppressor Cells (MDSC) by Inducing Polymorphonuclear MDSC Apoptosis and Monocytic MDSC Differentiation to APC in Cancer. J Immunol. 2016;196(5):2167–2180. doi:10.4049/jimmunol.1501853
39. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8(+) T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989–999. doi:10.4049/jimmunol.172.2.989
40. Gabrilovich D, Velders M, Sotomayor E, Kast W. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166(9):5398–5406. doi:10.4049/jimmunol.166.9.5398
41. Cauley LS, Miller EE, Yen M, Swain SL. Superantigen-induced CD4 T cell tolerance mediated by myeloid cells and IFN-gamma. J Immunol. 2000;165(11):6056–6066. doi:10.4049/jimmunol.165.11.6056
42. Han X, Shen S, Fan Q, et al. Red blood cell-derived nanoerythrosome for antigen delivery with enhanced cancer immunotherapy. Sci Adv. 2019;5(10):eaaw6870. doi:10.1126/sciadv.aaw6870
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.