Back to Journals » Journal of Pain Research » Volume 12
Deep sequencing analysis to identify novel and rare variants in pain-related genes in patients with acute postoperative pain and high morphine use
Authors Loke MF ![]() , Wei H, Yeo J
, Wei H, Yeo J ![]() , Sng BL
, Sng BL ![]() , Sia AT, Tan EC
, Sia AT, Tan EC ![]()
Received 29 April 2019
Accepted for publication 1 August 2019
Published 19 September 2019 Volume 2019:12 Pages 2755—2770
DOI https://doi.org/10.2147/JPR.S213869
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Katherine Hanlon
Mun-Fai Loke,1 Heming Wei,1 Junjie Yeo,2 Ban-Leong Sng,3 Alex T Sia,3 Ene-Choo Tan1
1Research Laboratory, KK Women’s & Children’s Hospital, Singapore, Singapore; 2Duke-NUS Medical School, Singapore, Singapore; 3Department of Women’s Anaesthesia, KK Women’s & Children’s Hospital, Singapore, Singapore
Correspondence: Ene-Choo Tan
KK Research Centre, KK Women’s and Children’s Hospital, Singapore 229899, Singapore
Tel +65 6 394 3792
Fax +65 6 394 1618
Email [email protected]
Purpose: Most of the genetic variants that are reported to be associated with common pain phenotypes and analgesic use are common polymorphisms. The objective of our study was to identify new variants and investigate less common genetic variants that are usually not included in either small single-gene studies or high-throughput genotyping arrays.
Patients and methods: From a cohort of 1075 patients who underwent a scheduled total abdominal hysterectomy, 92 who had higher self-rated pain scores and used more morphine were selected for the re-sequencing of 105 genes.
Results: We identified over 2400 variants in 104 genes. Most were intronic with frequencies >5%. There were 181 novel variants, of which 30 were located in exons: 17 nonsynonymous, 10 synonymous, 2 non-coding RNA, and 1 stop-gain. For known variants that are rare (population frequency <1%), the frequencies of 54 exonic variants and eight intronic variants for the sequenced samples were higher than the weighted frequencies in the Genome Aggregation Database for East and South Asians (P-values ranging from 0.000 to 0.046). Overall, patients who had novel and/or rare variants used more morphine than those who only had common variants.
Conclusion: Our study uncovered novel variants in patients who reported higher pain and used more morphine. Compared with the general population, rare variants were more common in this group.
Keywords: postoperative pain, genetic variants, next-generation sequencing, morphine
Introduction
Acute pain is the body’s mechanism to signal tissue injury and danger. Although pain helps to protect against further tissue damage by altering host behavior, prolonged and persistent pain has little biological value. On the contrary, it has an adverse impact on a person’s psychosocial well-being. The anticipation of pain can also influence the patient’s willingness to undergo potentially beneficial medical treatments that may be perceived as painful. Any pain that persists after surgery or injury carries adverse health and socioeconomic impacts, reduces the quality of life, increases health care cost and decreases work attendance.1
Pain perception is highly subjective with wide inter-individual variability in its sensitivity and tolerance. Known biological factors that impact this perception include age, race, gender, physiology, and social and psychological status.2–5 Pain is also a heritable phenotype, with multiple lines of evidence from Mendelian pain disorders, twin studies and increased risk for chronic pain conditions in individuals with family history. For instance, mutations in SCN9A and related genes have been identified in autosomal recessive congenital indifference to pain (MIM #243000) and autosomal dominant Marsili syndrome (MIM#147430).6–9 For less extreme and more complex pain phenotypes, the genetic contribution to sensitivity variation for different types of pain varies from 22% to 60%.10–12 For chronic pain conditions, twin studies suggest heritability of 39–58% for neuropathic pain,13–15 46% for chronic pelvic pain16 and as high as 70% for low back pain.17
Candidate gene studies have uncovered the contribution of variants of genes in the pain pathways across different types of pain in multiple populations. With the advent of genome-wide association studies (GWAS), the number of variants and chromosomal loci associated with pain has been further expanded.18–20 Published results from various pain studies are captured in several online databases such as the "Pain Genes Database of pain-related transgenic knockout studies" (PainGenes db)21 and the "Human Pain Genetics Database".22 Due to the study design and limitations in statistical power, most of the identified variants have been common genetic polymorphisms. These common variants tend to have only small to moderate impact on the difference in quantitative measures of pain. Furthermore, rare and low-frequency variants have been suggested to account for the remaining heritability.23–26
To uncover novel and rare variants that might be enriched in individuals who experienced more intense pain, we re-sequenced 105 genes in 92 patients who self-reported higher postoperative pain or used more morphine. They were selected from a cohort of patients who underwent total abdominal hysterectomy in our hospital. Our results showed that these patients had higher frequencies of rare variants in pain-related genes compared with those from population databases.
Patients and methods
Our study was approved by the SingHealth Central Institutional Review Board and conducted in accordance with the Declaration of Helsinki. Written informed consent for genetic study was obtained from all patients prior to surgical procedure.
Subjects characteristics, pain assessment and sample collection
The study protocol for this prospectively recruited cohort of 1075 women who underwent planned total hysterectomy at the KK Women’s and Children’s Hospital has been described previously.27 Briefly, pain sensitivity and tolerance were determined preoperatively using the blood pressure cuff of a sphygmomanometer. The cuff was placed around the patient’s upper arm and inflated until she indicated pain. The mercury reading (in mm) at that point was taken as the pain threshold. Pain tolerance was recorded as the mercury reading at which the patient requested for the deflation of the cuff. Immediately after surgery completion, the patient was fitted with a patient-controlled analgesia pump (PCA) that was set to deliver an intravenous bolus of 1 mg morphine on demand, with lockout interval of 5 mins, no basal infusion and a maximum hourly dose of 10 mg morphine. At 4-hourly intervals, patients were asked to rate their pain according to the VAS (0=no pain, 10=worst pain imaginable), as well as pruritus and nausea on a scale of 0–3 (0=none, 1=mild, 2=moderate, 3=severe).
For sequencing analysis, we selected from 1047 patients who had complete morphine data and pain scores for the 24-hr postoperative period. Tukey fence analysis was applied to select patients with outlier acute pain profiles. Since our interest was on higher pain scores, we only selected the upper fence. Fourteen outliers were selected based on acute pain scores at 4 hrs and average pain scores. To increase the sample size, we selected additional 50 patients whose acute pain scores at 4 hrs were greater than the third quartile. Despite not reporting pain scores greater than the third quartile, additional 4 patients were included based on higher outlier morphine consumption. Lastly, we also included 41 patients with 4-hr pain scores in the third quartile and had 8-hr pain scores that were less than the first quartile. We assumed that these patients had higher acute pain but also rapid resolution. Our final list had 109 patients arranged according to the date of surgery. Of these, the first 92 on the list with adequate good quality DNA were used for preparing sequencing libraries. The demographic and clinical characteristics of the 92 patients who were sequenced and those who were not are shown in Table 1. From the medical record, 35 of the 91 patients had one or more chronic conditions, of which the most common was hypertension (19 patients) followed by diabetes (7 patients). Only one patient had a pain condition (migraine). None of the patients were on opioid medication.
 |
Table 1 Characteristics of samples selected and not selected for sequencing |
DNA sequencing
Genomic DNA was extracted in batches from frozen whole blood samples in EDTA tubes using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany). DNA was checked for quantity and purity using the Quawell Q5000 UV-Vis Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
The 105 target genes (Table 2) were selected based on published literature and the maximum target size for the chosen sequencing platform. SureSelect and HaloPlex advanced wizards (Agilent Technologies, Santa Clara, CA, USA) were used to design the capture probes for target regions. Genomic coordinates for specified targets were obtained from RefSeq, Ensembl, CCDS, Gencode, VEGA, SNP, and CytoBand genome annotation databases, using the H. sapiens hg19 (GRCh37) as the reference sequence. All coding exons with minimum extensions of 10 bases from both 3ʹ and 5ʹ ends of each exon were included. The design covered 99.47% of the target region using 12,776 amplicons. The total size of the amplicons was 637,374 kilobases (kb), with total analyzable target of 234,538 kb.
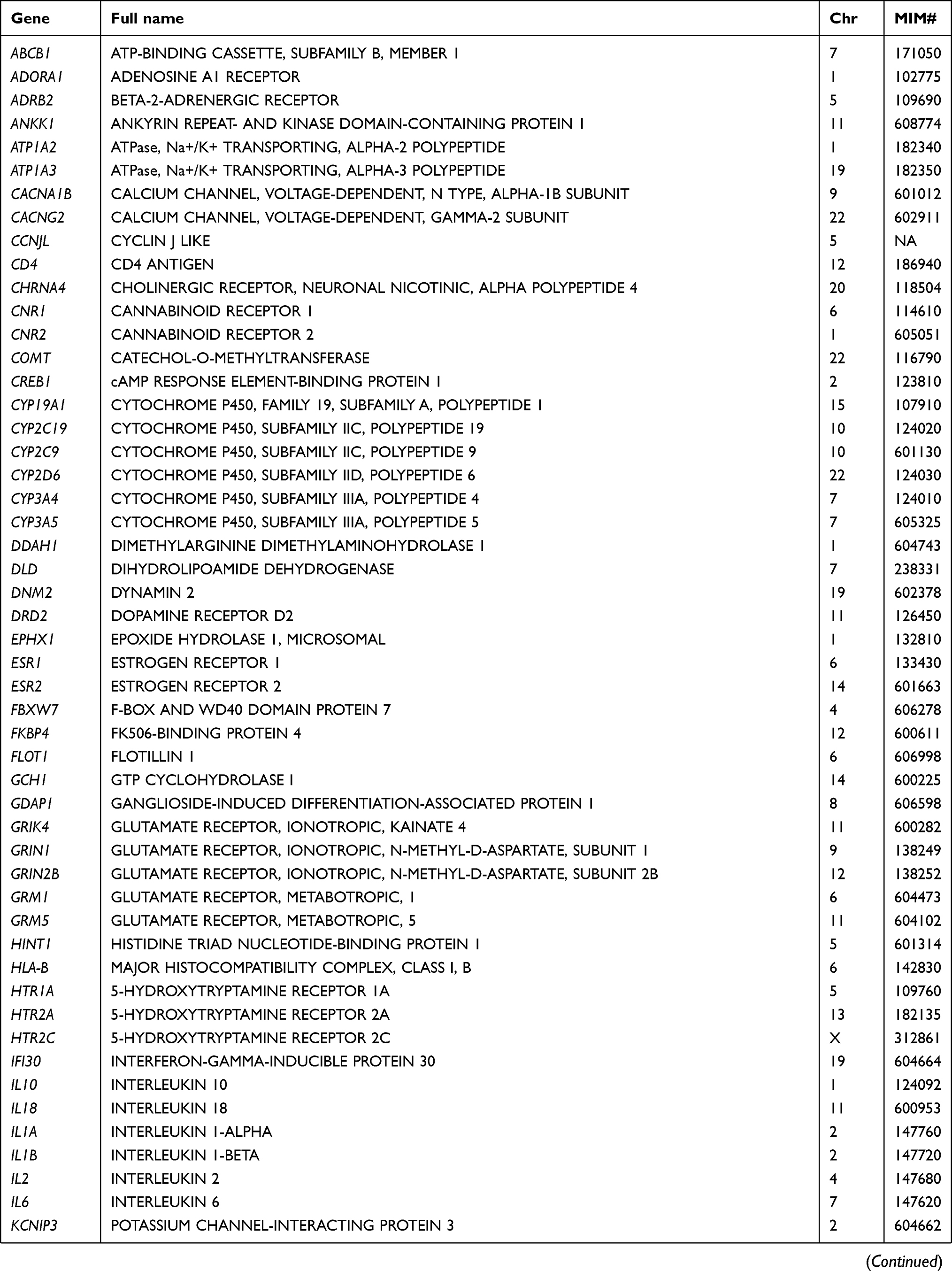 | 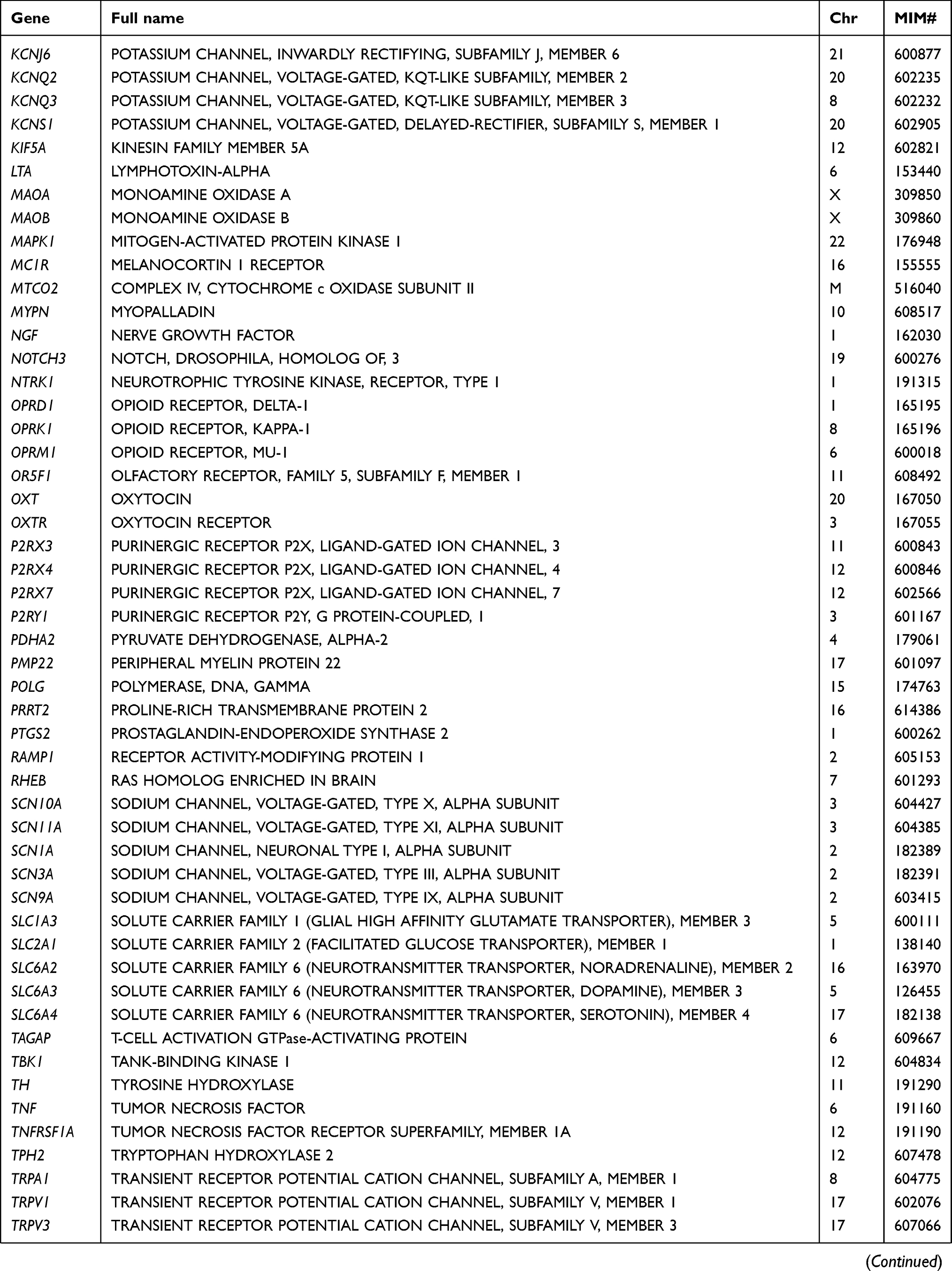 | 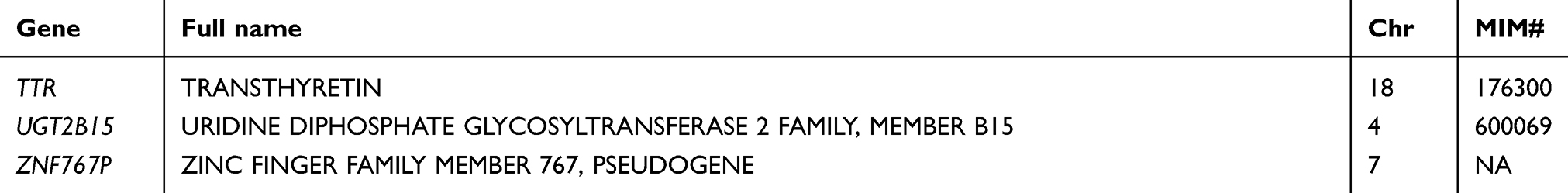 |
Table 2 List of pain-related genes sequenced in this study |
The HaloPlex Target Enrichment System (version F1) was used to index the samples and amplify the target regions according to the manufacturer’s instruction (Agilent Technologies). Libraries produced from the 92 samples were sequenced using 250 bp paired-end sequencing (600-cycle) on one MiSeq Reagent Kit (v3) on a MiSeq System (Illumina, San Diego, CA, USA).
Data processing and analysis
Bases were called using the on-instrument MiSeq Reporter software (version 2.6). Alignment processing and variant calling were performed with reference to human genome GRCh37 (hg19). The variant call format file generated was annotated and prioritized using wANNOVAR.28 Variants were considered novel if they were not previously reported in Genome Aggregation Database (gnomAD), Exome Sequencing Project, Human Genetic Variation Database, ClinVar, 1000 Genomes, or Human Gene Mutation Database databases, and not documented in scientific literature.
Consequences of sequence changes were assessed using Alamut Visual software version 2.10 (Interactive Biosoftware, Rouen, France) that included in silico prediction algorithms for likely effect on amino acid substitutions (SIFT v6.2.0, and PolyPhen-2 v2.2.2r398). Nonsynonymous variants with SIFT scores of <0.05 were classified as “deleterious”.29 For PolyPhen‐2, scores of >0.85 were classified as “probably damaging”, and scores of 0.15–0.85 were considered as “possibly damaging”.30
Two programs (MaxEnt and NNSPLICE) were used to evaluate the potential effect on splicing. Variants were considered positive if one or both programs had variation in the splice site score greater than the cutoff value of 10% from that of the reference allele.31
For rare variants (population frequencies of <1%), only exonic variants that are not synonymous, and intronic variants with predicted splice effects were compared with corresponding frequencies in gnomAD r2.0.2. Since our sequenced samples comprised 69 Chinese (75.0%), 18 Malays (19.6%) and 5 Indians (5.4%), analysis on statistically significant difference was performed with weighted gnomAD frequencies calculated from both the East Asian and the South Asian populations with respective weightings of 94.6% and 5.4%.
Interaction network and enrichment analyses
For genes with identified rare and/or novel variants of functional consequence, their involvement in biological pathways was queried using STRING database (version 10.5) (https://string-db.org/)32 that contains known and predicted protein interactions. We used Kyoto Encyclopedia of Genes and Genomes (KEGG) to assess network representation and for biological interpretation of the network nodes. Pathways with P-values <0.001 after false discovery rate adjustment were considered statistically enriched.
Statistical analyses
One-way ANOVA test was used to compare quantitative variables between groups, with Tukey post hoc test for comparison of more than two groups. Chi-square or Fisher’s exact test was used to compare frequencies for categorical variables. Post hoc Bonferroni test for multiple comparisons was performed for the comparison of the 62 rare variants for P-value correction. All statistical analyses were performed using IBM SPSS Statistics 19, with P-values ≤0.05 considered as statistically significant. For association analysis with rare variants, the P-value cutoff would be 0.00083 after applying Bonferroni correction for multiple testing.
Results
Quality of next-generation sequencing
Of the 92 samples, one failed to produce sequence data output. For the remaining 91 samples, 97.63% of the reads aligned to the reference genome (GRCh37/hg19) and 95.11% of the reads mapped to the targeted regions, with mean region coverage depth of 157.1× (Table S1). The mean coverage of targeted bases was 88.15% and 67.22% at 20× and 50×, respectively (Table S2).
At the gene level, all 105 targeted genes had mean coverage of at least 30× even for the gene with the lowest coverage. Eighty-one genes had a mean of >100×. Except for TBK1 which had the lowest mean coverage of ~49×, the remaining 104 genes had mean coverage of at least 62×. The mitochondrial gene MTCO2 had the highest mean coverage (>13,000×), followed by CNR2 (349×) (Table S3). Despite the high mean target gene coverage, amplification failed in at least one sample for 6 of 1014 target regions. Five genes (ADRB2, CHRNA4, HLA-B, TNFRSF1A, and TRPV3) had at least one region that was not amplified and therefore not sequenced. There were also 18 target regions from 13 genes (ADRB2, ATP1A3, CYP2C19, GRM5, RAMP1, SCN1A, SCN3A, SCN9A, SLC1A3, TBK1, TNFRSF1A, TRPA1, UGT2B15) with read depth of <20×.
Summary of genetic variants identified
In total, 2466 variants were identified from 104 genes. Only the mitochondrial gene MTCO2 had no variant. Most of the variants were common (population frequency >5%) and low-frequency polymorphisms (frequency 1–5%), the remaining comprised 608 rare (frequency <1%) and 181 novel variants (defined as those with no Reference SNP numbers and not documented in databases or published literature). In terms of location, the largest number of 1477 were found in introns, followed by 771 in protein-coding exons, 123 in 3ʹ untranslated regions (or trailer sequences), 45 in 5ʹ untranslated regions (or leader sequences), 35 in upstream regions of genes, 12 in the downstream regions, and the remaining 3 in intergenic regions. Overall, there was an average of 27.1 variants per patient.
For single-nucleotide substitutions located in the exons, 386 were synonymous while 350 were missense variants. There were also 5 stop-gain variants and 1 stop-loss variant. For changes involving multiple nucleotides, there were 2 non-frameshift insertions, 1 frameshift insertion, and 4 non-frameshift deletions. In addition, there were 21 exonic non-coding RNA variants. The position of a putative OPRK1 variant (chr8:54141824:C>T) within the gene could not be determined.
Analysis of novel and rare variants
There were 181 novel variants in 70 genes, most of which were in the introns. Of the 30 variants found in exons, 17 were missense, 10 synonymous, 1 stop-gain, and 2 were non-coding RNA. The list of 30 exonic variants and two intronic variants with their predicted consequences are listed in Table 3, along with the number of reads for novel/alternate alleles and their corresponding reference alleles. The 32 novel variants were from 28 patients. The numbers of reads for the 2 alleles were mostly balanced. Hence, we did not perform Sanger validation.
 |
Table 3 List of novel exonic and intronic variants (with splicing effect) identified in this population |
Rare variants were found in 102 genes. All but one (IFI30) of the 70 genes with novel variants also had rare variants. Three genes (CYP19A1, IL2, MTCO2) had no such variants, while another 5 (ADORA1, HINT1, HTR2A, OXT, TTR) had no variant in either the exonic or intronic regions. Five genes (ADRB2, HINT1, HLA-B, IL1B, and PRRT2) had only one such variant. The 2 genes with the highest number of rare variants were CACNA1B with 47 and POLG with 29. Both NTRK1 and SCN10A had 21 while DNM2, KIF5A, and NOTCH3 had 20 variants. The remaining genes had 2–19 rare variants.
All 91 patients had at least 3 novel or rare variants (inclusive of intronic variants), or an average of 7.5 each. The highest number was 26 (one patient), followed by 25 (one patient) and 24 (one patient). There were two patients with 23 and another two with 19 variants. Three patients had 18 and the remaining 81 (89.0%) had between 3 and 17 variants each.
Rare variants that were enriched in the study population
Among the identified rare exonic variants (frequencies <1%) that are not synonymous, 54 had frequencies that were statistically significantly (P-value ≤0.05) higher than the corresponding frequencies for East/South Asians in the Genome Aggregation Database (gnomAD). Two of the 54 were in-frame: a 3-nucleotide insertion and a 3-nucleotide deletion. Of the 52 missense variants, 21 were predicted by both Polyphen-2 and SIFT to have a significant consequence on the encoded proteins, while another 13 were predicted to have a damaging effect by one of the two programs (Table 4). Two of the exonic variants (NOTCH3 c.3141C>G and POLG c.2069C>T) were also putative splice variants. For intronic variants that were rare, there were 8 with higher frequencies than those in gnomAD, and all were predicted to affect splicing. After Bonferroni correction for multiple testing, statistically significant difference remained for one exonic (POLG c.125_127dupGGC:p.(Arg42dup); corrected P-value of 0.017) and one intronic variant (CYP3A5 c.433-1G>C; corrected P-value of 0.017).
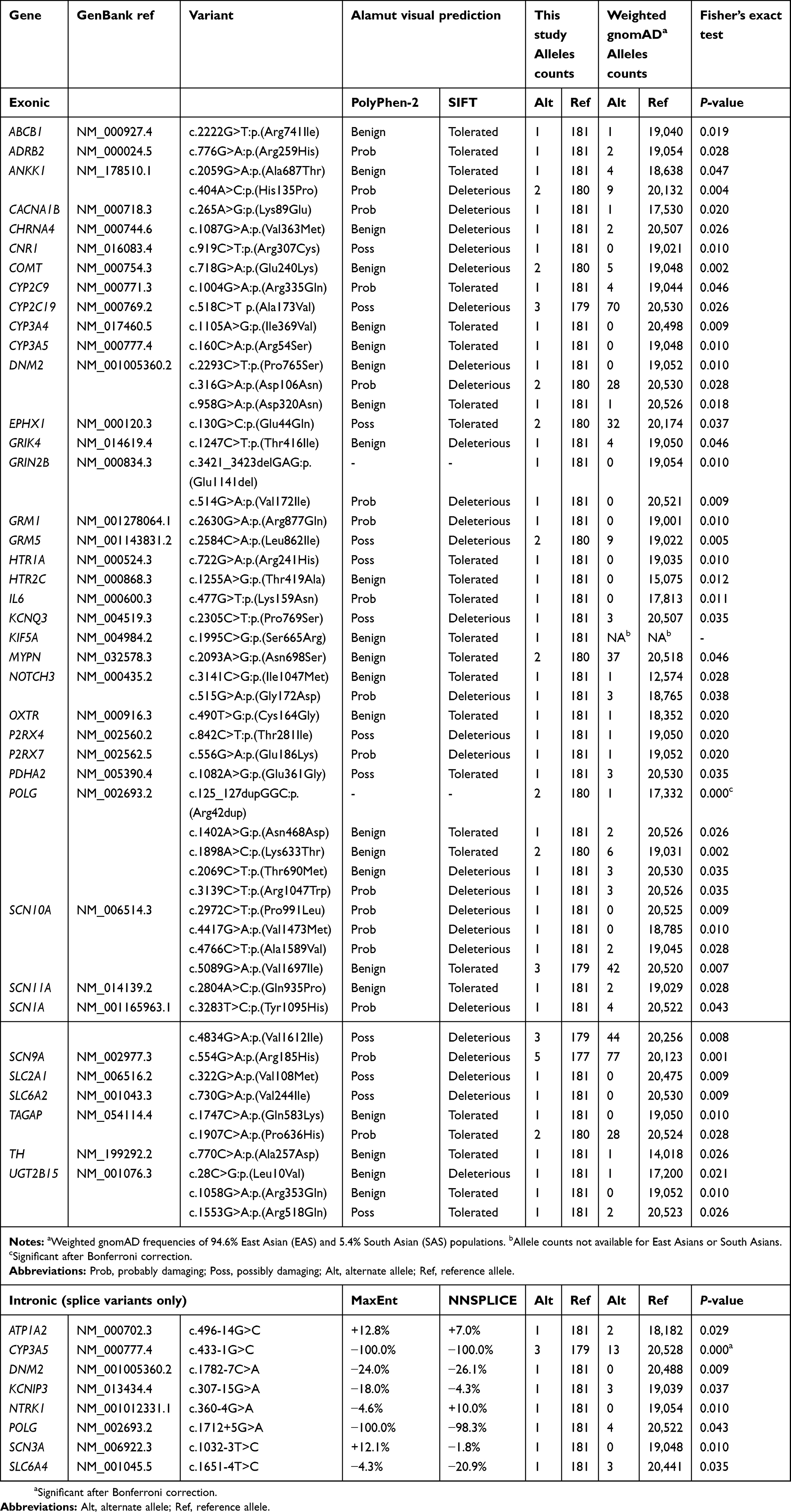 |
Table 4 List of rare non-synonymous or intronic variants with frequencies significantly higher than expected data in gnomAD |
The 62 rare variants in Table 4 (comprising 54 exonic that are non-synonymous and eight intronic-splice variants) were identified from 46 patients; 18 of whom also had novel variants of functional significance (non-synonymous or splicing variants). The highest number per patient was 6 (one patient) while another patient had 5. There were 2 patients with 4 such variants and 6 patients with 3. The remaining 36 patients had either 1 or 2 rare variants while 10 patients had only novel variants. Thirty-five patients did not have any novel or rare variants of functional significance that had higher frequencies than the general population.
Pathway analysis
Twenty-one of the genes that had either novel or rare variants with higher frequencies in this high-pain population were found to be involved in 7 non-redundant pathways in the STRING database (P-values of <0.001 after correcting for false discovery rate). The significantly enriched pathways include neuroactive ligand-receptor interaction, dopaminergic synapse and cocaine addiction, metabolism of xenobiotics by cytochrome P450 and morphine addiction, serotonergic synapse, and bile secretion, all known to be pain related (Table 5).
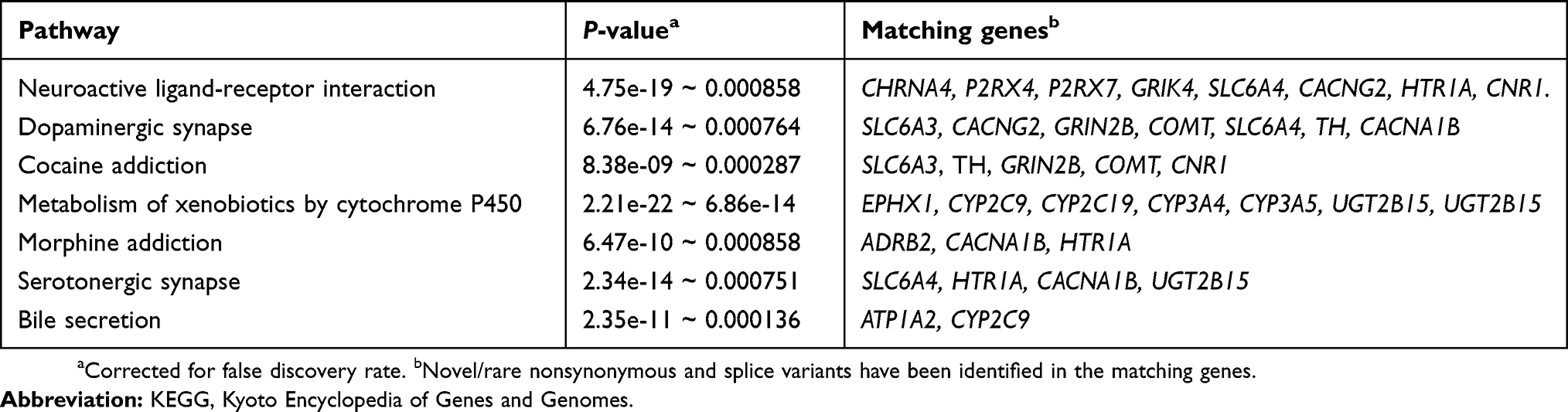 |
Table 5 KEGG pathways identified for genes with novel or rare nonsynonymous or splice variants identified in the study population |
Association of morphine usage with the presence of novel and rare variants
The patients were further grouped based on whether they carried the novel (listed in Table 3) and/or rare exonic variants (listed in Table 4). Their morphine usage was further compared with those who only had common variants. Although there was statistically significant difference only for the 20-hr PCA morphine, the trend was similar across all time-points (Table 6). The group with novel variants used more morphine compared with the group carrying rare variants. This in turn resulted in higher mean morphine dosage than the group of 35 patients with only common variants. There was no statistically significant difference in terms of age, BMI and self-reported pain scores between the groups.
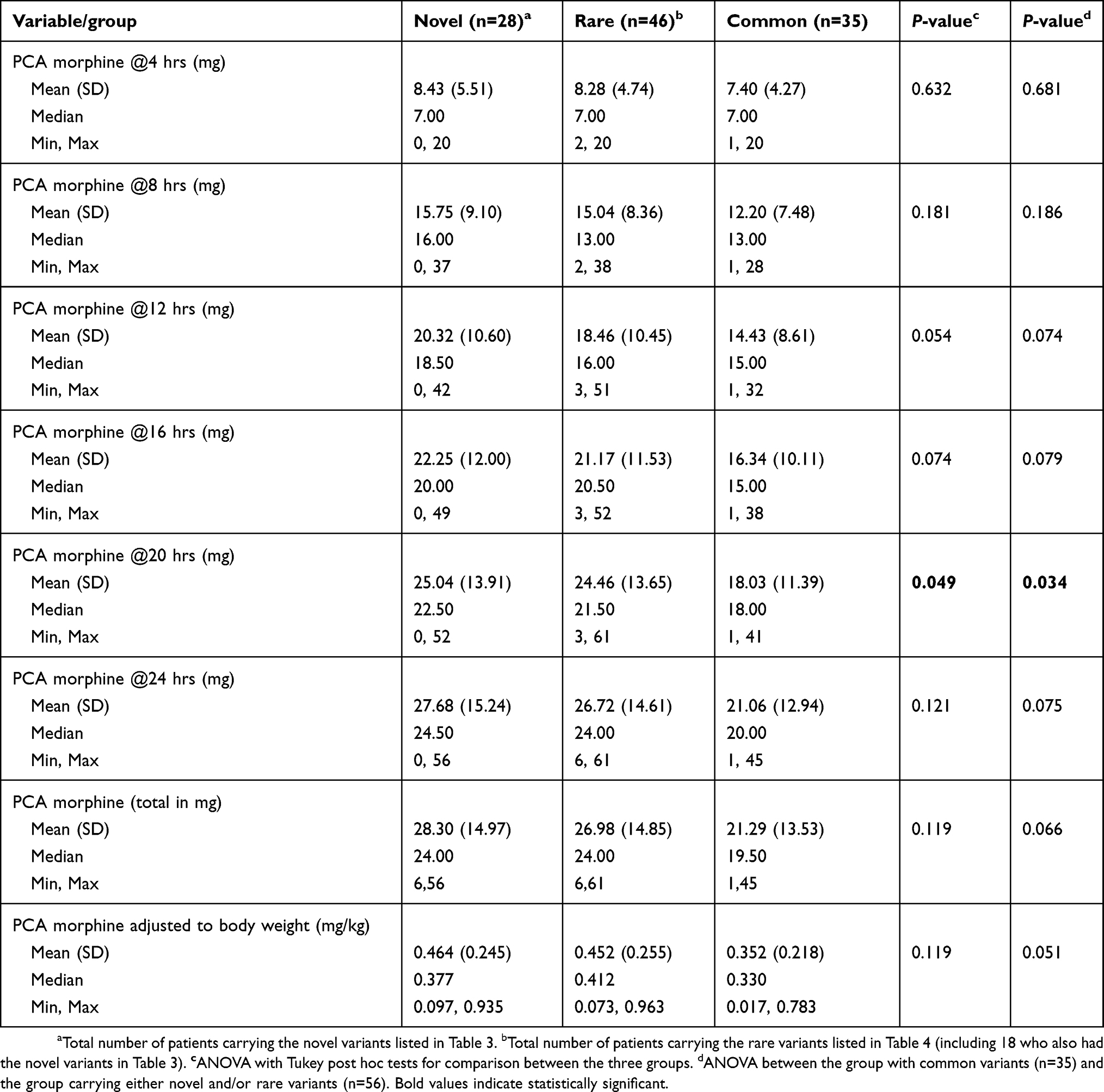 |
Table 6 Comparison of morphine usage for patients carrying variants of different frequencies |
Discussion
The advent of high-throughput genotyping technologies has led to the identification of genetic variants associated with many complex diseases and traits. In particular, GWAS had uncovered many common variants associated with various phenotypes. However, it is not designed to detect association involving variants of very low frequencies. Since NGS has become more cost-efficient, it is now feasible to genotype by resequencing, thereby uncovering the rare variants that may be important. By resequencing 105 known genes related to pain in our cohort of high postoperative pain patients, we were able to detect variants that were either absent or reported at very low frequencies in the general population.
The most interesting novel variant was the stop-gain in SCN3A. Pathogenic mutations in this gene have been linked to focal epilepsy. However, there was no record of this condition in the patient. On the other hand, there were 10 synonymous variants that were novel. Although synonymous variants are generally well tolerated and most have no functional consequence, novel variants are important in the context of rate of replication error, position within the gene and base changes tolerated. In addition, not all synonymous variants are insignificant as they might change the secondary structure of the mRNAs and influence their stability. In cases where the synonymous substitution affects RNA–RNA interaction, it may alter translation efficiency which is important in the biological regulation of gene expression and transcriptome complexity. Some synonymous substitutions are also known to lead to aberrant splicing.33–35
Among the genes with novel and rare variants that had higher frequencies in this sample set, 21 were mapped to the pain-related KEGG pathways. These genes likely play key roles in processes related to pain response and signaling, as well as drug metabolism. Recent animal studies have confirmed that neuroactive ligand-receptor influenced disease-related pain and its severity, duration, and relief.36,37 It is not surprising that variants in the cytochrome P450 pathway are more common in this high-pain population as opioids are metabolized through two major enzyme systems: CYP450 and, to a lesser extent, UDP-glucuronosyltransferases. Although more than 50 CYP450 enzymes are known, CYP1A2, CYP2C9, CYP2D6, CYP3A4, and CYP3A5 account for the metabolism of up to 90% of drugs.38,39 Increased cytochrome P450 enzyme activities may result in faster metabolism and shorter half-life, which may potentially decrease a drug’s pharmacologic effect.
Five of the genes in Table 5 are involved in the cocaine addiction pathway. Based on experimental studies and some indirect clinical evidence, dopamine has been suggested to have anti-nociceptive effect.40–42 Cocaine increases the level of dopamine and cocaine addiction is related to pain.43 In addition, serotonergic synapse44 and bile secretion45 pathways are also linked to pain. Variants in any of these genes might have affected the functions of the encoded proteins, resulting in the enhanced and prolonged postoperative pain experienced by our study subjects.
Genes with novel or rare variants that had higher frequencies than the general population include POLG, SCN10A, and DNM2. POLG (DNA polymerase gamma) encodes a polymerase responsible for the replication of human mitochondrial DNA. Mutations in the gene have been linked to mitochondrial diseases, such as myocerebrohepatopathy spectrum disorders, Alpers-Huttenlocher syndrome, myoclonic epilepsy myopathy sensory ataxia, ataxia neuropathy spectrum, and progressive external ophthalmoplegia.46,47 POLG mutations are associated with peripheral neuropathy and a potentially painful, axonal/mixed, mainly sensory polyneuropathy48 and muscle pain.49 This gene had the highest number of identified variants (six rare and two novel), including one missense variant found in 10 patients. Another gene SCN10A had four rare variants which were more prevalent in the study population. It encodes a component of the Nav1.8 sodium channel and is associated with peripheral neuropathy.50 The other gene which had multiple variants with higher frequencies is DNM2. This gene codes for Dynamin-2, one of the subfamilies of GTP-binding proteins. DNM2 has been associated with pain flare in patients who received palliative radiation therapy for painful bone metastases.51
Although our study uncovered novel and rare variants from patients who reported higher pain and used more morphine, it has several limitations. First, sequencing was only performed in <10% of a patient cohort, on those with the highest pain burden (self-reported pain scores and high morphine use). Second, the frequency comparison was done with data from population databases. In addition, functional effects were based on in silico predictions, and no in vivo or in vitro studies were carried out for validation. Lastly, although the number of reads for reference and alternate alleles were similar, the variants were also not Sanger validated. Therefore, further studies are warranted to address these limitations.
In summary, our results showed that some rare variants were more common in patients who reported more pain and used more PCA morphine. We also identified several novel variants that were predicted to either result in amino acid substitutions or affect splicing. Carriers of such variants tend to use more morphine over the first 24 hrs of the postoperative period. Whether the novel variants affect the sensitivity and tolerance to pain remain to be investigated. The cost of genomic technologies has become more affordable, and the analysis of sequencing data is also amenable to automated pipelines. Thus, it is possible to incorporate genotyping or sequencing for a set of gene variants that account for a significant portion of the inter-individual variation. The genetic information could be combined with other predictive factors in patient risk stratification. This will enable early intervention and timely modulation of nociception that has been shown to reduce the incidence of persistent pain and improve patient recovery.
Acknowledgments
This work was supported by grants NMRC/CG/006/2013 and NMRC/CG/M003/2017 from the Singapore Ministry of Health’s National Medical Research Council. We thank Dr Eddy Saputra Leman (Duke-NUS Medical School) for his editorial support. Mun-Fai Loke and Heming Wei are co-first authors for this study.
Disclosure
All authors report no conflicts of interest in this work.
References
1. Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain. 2012;13:715–724. doi:10.1016/j.jpain.2012.03.009
2. Kim HJ, Yang GS, Greenspan JD, et al. Racial and ethnic differences in experimental pain sensitivity: systematic review and meta-analysis. Pain. 2017;158:194–211. doi:10.1097/j.pain.0000000000000731
3. Peters ML. Emotional and Cognitive Influences on Pain Experience. Mod Trends Pharmacopsychiatry. 2015;30:138–152. doi:10.1159/000435938
4. Rahim-Williams B, Riley JL
5. Tan EC, Lim Y, Teo YY, Goh R, Law HY, Sia AT. Ethnic differences in pain perception and patient-controlled analgesia usage for postoperative pain. J Pain. 2008;9:849–855. doi:10.1016/j.jpain.2008.04.004
6. Goldberg YP, Pimstone SN, Namdari R, et al. Human Mendelian pain disorders: a key to discovery and validation of novel analgesics. Clin Genet. 2012;82:367–373. doi:10.1111/j.1399-0004.2012.01942.x
7. Nahorski MS, Chen YC, Woods CG. New Mendelian Disorders of Painlessness. Trends Neurosci. 2015;38:712–724. doi:10.1016/j.tins.2015.08.010
8. Habib AM, Matsuyama A, Okorokov AL, et al. A novel human pain insensitivity disorder caused by a point mutation in ZFHX2. Brain. 2018;141:365–376. doi:10.1093/brain/awx326
9. Waxman SG, Merkies ISJ, Gerrits MM, et al. Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol. 2014;13:1152–1160. doi:10.1016/S1474-4422(14)70150-4
10. Angst MS, Phillips NG, Drover DR, et al. Pain sensitivity and opioid analgesia: a pharmacogenomic twin study. Pain. 2012;153:1397–1409. doi:10.1016/j.pain.2012.02.022
11. Nielsen CS, Stubhaug A, Price DD, Vassend O, Czajkowski N, Harris JR. Individual differences in pain sensitivity: genetic and environmental contributions. Pain. 2008;136:21–29. doi:10.1016/j.pain.2007.06.008
12. Norbury TA, MacGregor AJ, Urwin J, Spector TD, McMahon SB. Heritability of responses to painful stimuli in women: a classical twin study. Brain. 2007;130:3041–3049. doi:10.1093/brain/awm233
13. Veluchamy A, Hebert HL, Meng W, Palmer CNA, Smith BH. Systematic review and meta-analysis of genetic risk factors for neuropathic pain. Pain. 2018;159:825–848. doi:10.1097/j.pain.0000000000001164
14. Momi SK, Fabiane SM, Lachance G, Livshits G, Williams FM. Neuropathic pain as part of chronic widespread pain: environmental and genetic influences. Pain. 2015;156:2100–2106. doi:10.1097/j.pain.0000000000000277
15. Zorina-Lichtenwalter K, Parisien M, Diatchenko L. Genetic studies of human neuropathic pain conditions: a review. Pain. 2018;159:583–594. doi:10.1097/j.pain.0000000000001099
16. Vehof J, Zavos HMS, Lachance G, Hammond CJ, Williams FMK. Shared genetic factors underlie chronic pain syndromes. Pain. 2014;155:1562–1568. doi:10.1016/j.pain.2014.05.002
17. Malkin I, Williams FM, LaChance G, Spector T, MacGregor AJ, Livshits G. Low back and common widespread pain share common genetic determinants. Ann Hum Genet. 2014;78:357–366. doi:10.1111/ahg.12074
18. Cook-Sather SD, Li J, Goebel TK, Sussman EM, Rehman MA, Hakonarson H. TAOK3, a novel genome-wide association study locus associated with morphine requirement and postoperative pain in a retrospective pediatric day surgery population. Pain. 2014;155:1773–1783. doi:10.1016/j.pain.2014.05.032
19. Kim H, Ramsay E, Lee H, Wahl S, Dionne RA. Genome-wide association study of acute post-surgical pain in humans. Pharmacogenomics. 2009;10:171–179. doi:10.2217/14622416.10.2.171
20. Takahashi K, Nishizawa D, Kasai S, et al. Genome-wide association study identifies polymorphisms associated with the analgesic effect of fentanyl in the preoperative cold pressor-induced pain test. J Pharmacol Sci. 2018;136:107–113. doi:10.1016/j.jphs.2018.02.002
21. Lacroix-Fralish ML, Ledoux JB, Mogil JS. The pain genes database: an interactive web browser of pain-related transgenic knockout studies. Pain. 2007;131(3):e1–4. doi:10.1016/j.pain.2007.04.041
22. Meloto CB, Benavides R, Lichtenwalter RN, et al. Human pain genetics database: a resource dedicated to human pain genetics research. Pain. 2018;159:749–763. doi:10.1097/j.pain.0000000000001135
23. Diatchenko L, Nackley AG, Tchivileva IE, Shabalina SA, Maixner W. Genetic architecture of human pain perception. Trends Genet. 2007;23:605–613. doi:10.1016/j.tig.2007.09.004
24. De La Vega F, Bustamante CD, Leal SM. Genome-wide association mapping and rare alleles: from population genomics to personalized medicine - session introduction. Pac Symp Biocomput. 2011;74–75.
25. Bomba L, Walter K, Soranzo N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017;18:77. doi:10.1186/s13059-017-1212-4
26. Wagner MJ. Rare-variant genome-wide association studies: a new frontier in genetic analysis of complex traits. Pharmacogenomics. 2013;14:413–424. doi:10.2217/pgs.13.36
27. Sia AT, Lim Y, Lim EC, et al. Influence of mu-opioid receptor variant on morphine use and self-rated pain following abdominal hysterectomy. J Pain. 2013;14:1045–1052. doi:10.1016/j.jpain.2013.03.008
28. Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–1566. doi:10.1038/nprot.2015.105
29. Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi:10.1101/gr.212802
30. Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat. 2011;32:661–668. doi:10.1002/humu.21490
31. Tang R, Prosser DO, Love DR. Evaluation of bioinformatic programmes for the analysis of variants within splice site consensus regions. Adv Bioinformatics. 2016;2016:5614058. doi:10.1155/2016/5614058
32. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi:10.1093/nar/gku1003
33. Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet. 2014;30:308–321. doi:10.1016/j.tig.2014.04.006
34. Shabalina SA, Spiridonov NA, Kashina A. Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 2013;41:2073–2094. doi:10.1093/nar/gks1205
35. Sharma Y, Miladi M, Dukare S, et al. A pan-cancer analysis of synonymous mutations. Nat Commun. 2019;10:2569. doi:10.1038/s41467-019-10489-2
36. Su S, Shao J, Zhao Q, et al. MiR-30b attenuates neuropathic pain by regulating voltage-gated sodium channel Nav1.3 in rats. Front Mol Neurosci. 2017;10:126. doi:10.3389/fnmol.2017.00126
37. Deng X, Wang D, Wang S, Wang H, Zhou H. Identification of key genes and pathways involved in response to pain in goat and sheep by transcriptome sequencing. Biol Res. 2018;51:25. doi:10.1186/s40659-018-0176-5
38. Slaughter RL, Edwards DJ. Recent advances: the cytochrome P450 enzymes. Ann Pharmacother. 1995;29:619–624. doi:10.1177/106002809502900612
39. Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352:2211–2221. doi:10.1056/NEJMra032424
40. Lu Y, Doroshenko M, Lauzadis J, et al. Presynaptic inhibition of primary nociceptive signals to dorsal horn lamina I neurons by dopamine. J Neurosci. 2018;38:8809–8821. doi:10.1523/JNEUROSCI.0323-18.2018
41. Chakraborty S, Rebecchi M, Kaczocha M, Puopolo M. Dopamine modulation of transient receptor potential vanilloid type 1 (TRPV1) receptor in dorsal root ganglia neurons. J Physiol. 2016;594:1627–1642. doi:10.1113/JP271198
42. Treister R, Pud D, Ebstein RP, et al. Associations between polymorphisms in dopamine neurotransmitter pathway genes and pain response in healthy humans. Pain. 2009;147:187–193. doi:10.1016/j.pain.2009.09.001
43. Ahmadian-Moghadam H, Sadat-Shirazi MS, Zarrindast MR. Cocaine- and amphetamine-regulated transcript (CART): a multifaceted neuropeptide. Peptides. 2018;110:56–77. doi:10.1016/j.peptides.2018.10.008
44. Bannister K, Dickenson AH. The plasticity of descending controls in pain: translational probing. J Physiol. 2017;595:4159–4166. doi:10.1113/JP274165
45. Alemi F, Kwon E, Poole DP, et al. The TGR5 receptor mediates bile acid-induced itch and analgesia. J Clin Invest. 2013;123:1513–1530. doi:10.1172/JCI64551
46. Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. 2019;15:40–52. .
47. Stumpf JD, Saneto RP, Copeland WC. Clinical and molecular features of POLG-related mitochondrial disease. Cold Spring Harb Perspect Biol. 2013;5:a011395. doi:10.1101/cshperspect.a011395
48. Mancuso M, Orsucci D, Angelini C, et al. “Mitochondrial neuropathies”: a survey from the large cohort of the Italian Network. Neuromuscul Disord. 2016;26:272–276. doi:10.1016/j.nmd.2016.02.008
49. Horvath R, Hudson G, Ferrari G, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain. 2006;129:1674–1684. doi:10.1093/brain/awl088
50. Dib-Hajj SD, Geha P, Waxman SG. Sodium channels in pain disorders: pathophysiology and prospects for treatment. Pain. 2017;158(Suppl 1):S97–S107. doi:10.1097/j.pain.0000000000000854
51. Furfari A, Wan BA, Ding K, et al. Genetic biomarkers associated with pain flare and dexamethasone response following palliative radiotherapy in patients with painful bone metastases. Ann Palliat Med. 2017;6:S240–S247. doi:10.21037/apm.2017.09.04
Supplementary materials
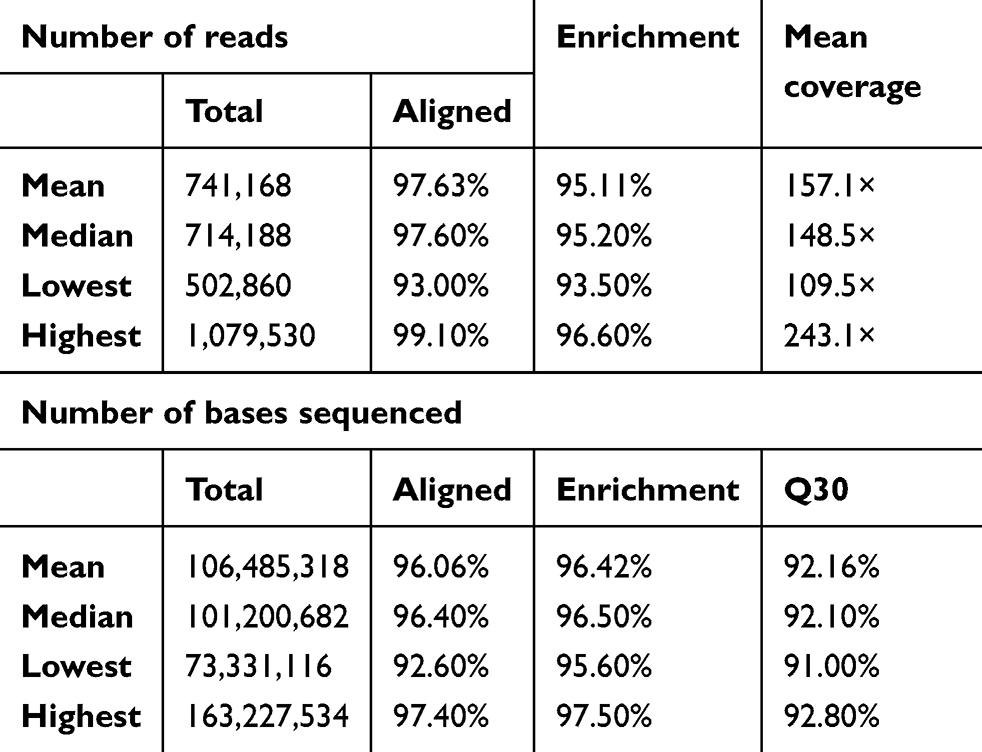 |
Table S1 Summary of sequencing quality and output for the 91 samples sequenced |
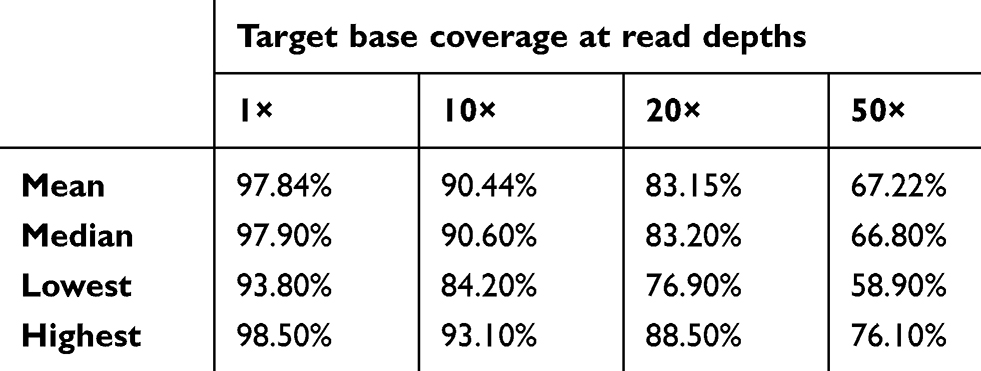 |
Table S2 Percentage of bases sequenced at the different read depths |
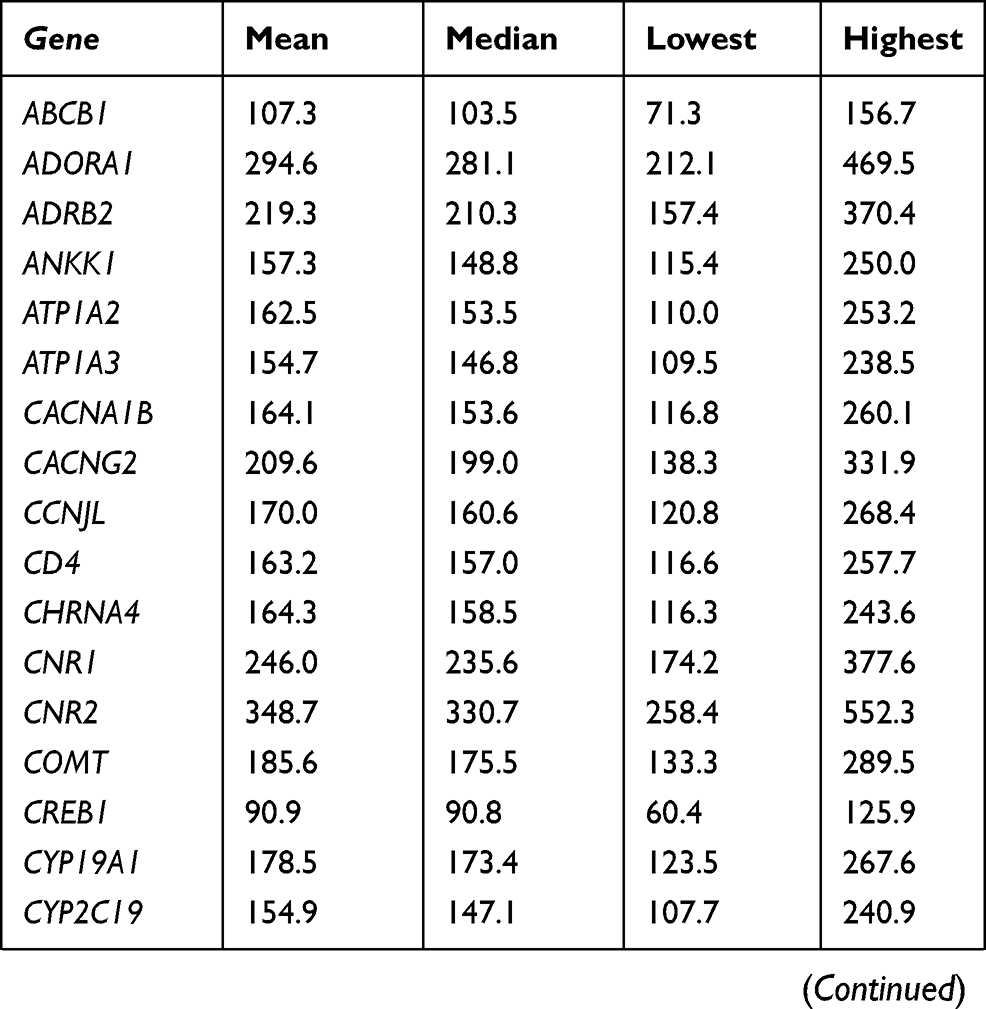 | 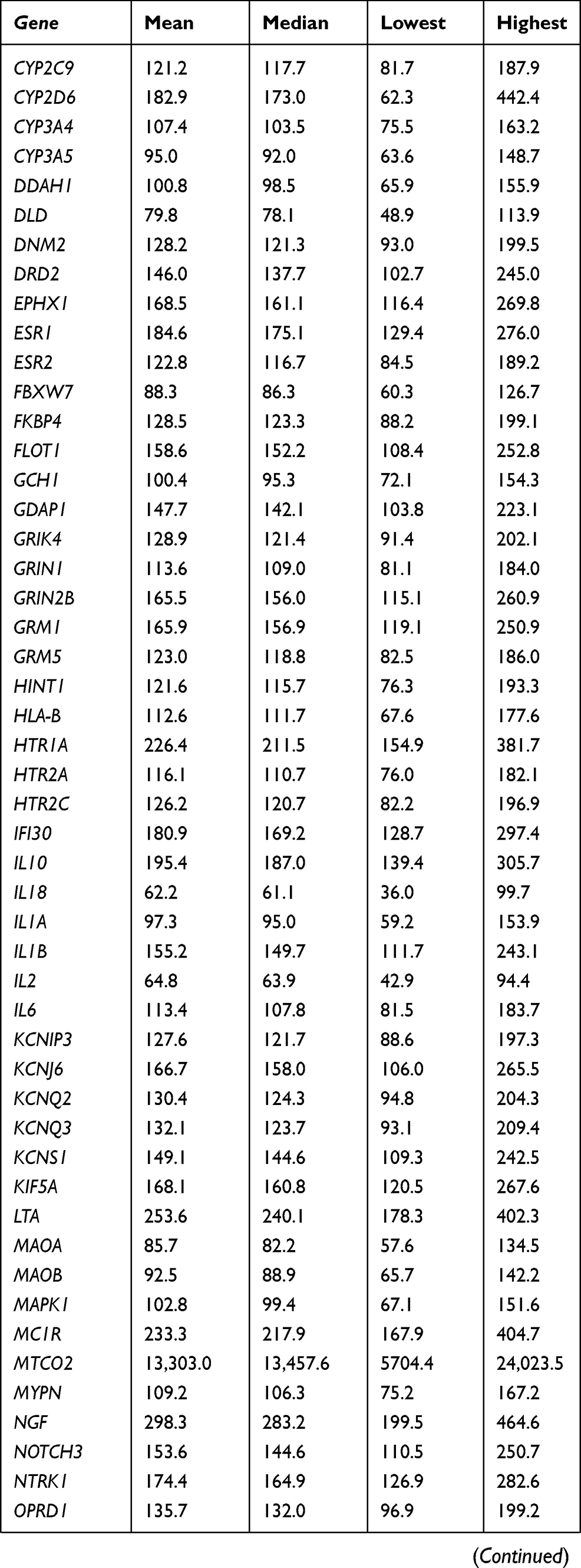 | 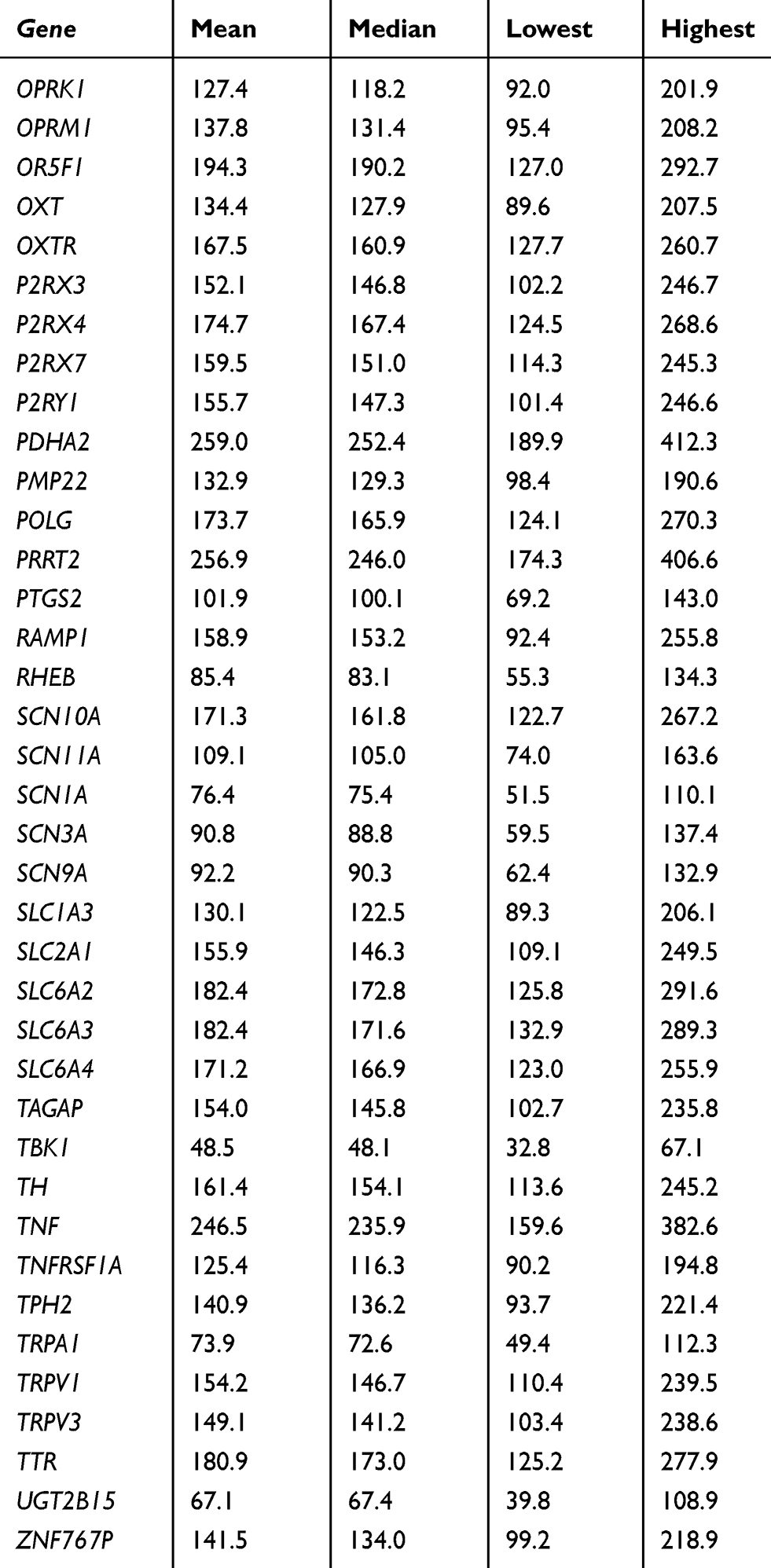 |
Table S3 Coverage details for each gene |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.