Back to Journals » Open Access Journal of Clinical Trials » Volume 7
Deep brain stimulation targeting the fornix for mild Alzheimer dementia: design of the ADvance randomized controlled trial
Authors Holroyd K, Fosdick L, Smith G, Leoutsakos J, Munro C, Oh E, Drake K, Rosenberg P, Anderson W, Salloway S, Pendergrass JC, Burke A, Wolk D, Tang-Wai D, Ponce F, Asaad W, Sabbagh M, Okun M, Baltuch G, Foote K, Targum S, Lozano A, Lyketsos C
Received 25 January 2015
Accepted for publication 10 April 2015
Published 10 July 2015 Volume 2015:7 Pages 63—76
DOI https://doi.org/10.2147/OAJCT.S81542
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Greg Martin
Kathryn B Holroyd,1 Lisa Fosdick,2 Gwenn S Smith,1 Jeannie-Marie Leoutsakos,1 Cynthia A Munro,1 Esther S Oh,1 Kristen E Drake,2 Paul B Rosenberg,1 William S Anderson,1 Stephen Salloway,3–5 J Cara Pendergrass,6 Anna D Burke,7 David A Wolk,8 David F Tang-Wai,9–11 Francisco A Ponce,12 Wael F Asaad,13,14 Marwan N Sabbagh,15 Michael S Okun,16 Gordon Baltuch,17 Kelly D Foote,18 Steven D Targum,2,6 Andres M Lozano,10,11 Constantine G Lyketsos1
1Johns Hopkins University Memory and Alzheimer's Treatment Center, Baltimore, MD, 2Functional Neuromodulation Ltd, Minneapolis, MN, 3Department of Neurology, Butler Hospital, 4Department of Neurology, Rhode Island Hospital, 5Department of Neurology, Warren Alpert Medical School of Brown University, Providence, RI, 6Clintara LLC, Boston, MA, 7Banner Alzheimer's Institute, Phoenix, AZ, 8Penn Memory Center, Department of Neurology, University of Pennsylvania, Philadelphia, PA, USA; 9Department of Neurology, 10Department of Neurosurgery, University of Toronto, 11Division of Neurology, University Health Network Memory Clinic, Toronto, ON, Canada; 12Division of Neurological Surgery, Barrow Neurological Institute, St Joseph's Hospital and Medical Center, Phoenix, AZ, 13Department of Neurosurgery, Rhode Island Hospital, 14Department of Neurosurgery, Warren Alpert Medical School of Brown University, Providence, RI, 15Banner Sun Health Research Institute, Sun City, AZ, 16Center for Movement Disorders and Neurorestoration, Department of Neurology, University of Florida – Gainsville, Gainsville, FL, 17Center for Functional and Restorative Neurosurgery, University of Pennsylvania, Philadelphia, PA, 18Department of Neurosurgery, Center for Movement Disorders and Neurorestoration, University of Florida, Gainsville, FL, USA
Background: There are currently few available treatments and no cure for Alzheimer disease (AD), a growing public health burden. Animal models and an open-label human trial have indicated that deep brain stimulation (DBS) of memory circuits may improve symptoms and possibly slow disease progression. The ADvance trial was designed to examine DBS of the fornix as a treatment for mild AD.
Methods: ADvance is a randomized, double-blind, placebo-controlled, delayed-start, multicenter clinical trial conducted at six sites in the US and one site in Canada. Eighty-five subjects initially consented to be screened for the trial. Of these, 42 subjects who met inclusion and exclusion criteria were implanted with DBS leads anterior to the columns of the fornix bilaterally. They were randomized 1:1 to DBS “off” or DBS “on” groups for the initial 12 months of follow-up. After 1 year, all subjects will have their devices turned “on” for the remainder of the study. Postimplantation, subjects will return for 13 follow-up visits over 48 months for cognitive and psychiatric assessments, brain imaging (up to 12 months), and safety monitoring. The primary outcome measures include Alzheimer's Disease Assessment Scale – cognitive component (ADAS-cog-13), Clinical Dementia Rating sum of boxes (CDR-SB), and cerebral glucose metabolism measured with positron emission tomography. This report details the study methods, baseline subject characteristics of screened and implanted participants, and screen-to-baseline test–retest reliability of the cognitive outcomes.
Results: Implanted subjects had a mean age of 68.2 years, were mostly male (55%), and had baseline mean ADAS-cog-13 and CDR-SB scores of 28.9 (SD, 5.2) and 3.9 (SD, 1.6), respectively. There were no significant differences between screened and implanted or nonimplanted subjects on most demographic or clinical assessments. Implanted subjects had significantly lower (better) ADAS-cog-11 (17.5 vs 21.1) scores, but did not differ on CDR-SB. Scores on the major outcome measures for the trial were consistent at screening and baseline.
Conclusion: ADvance was successful in enrolling a substantial group of patients for this novel application of DBS, and the study design is strengthened by rigorous subject selection from seven sites, a double-blind placebo-controlled design, and extensive open-label follow-up.
Keywords: deep brain stimulation, Alzheimer disease, fornix, methods, clinical trials
Introduction
Alzheimer disease (AD) is the most common neurodegenerative dementia, affecting an estimated 5.4 million people1 and costing as much as $214 billion annually in the US alone.2 New cases of AD continue to increase at an alarming rate worldwide, and its economic and health burden are projected to expand substantially in the next 50 years.3 Currently, there is no cure for AD. US Food and Drug Administration (FDA)-approved medications, including three cholinesterase inhibitors and one noncompetitive N-methyl-d-aspartic acid receptor antagonist, temporarily ameliorate some symptoms in a subset of patients.1 Phase II and III trials of putative disease-modifying therapies targeting the amyloid protein aggregations observed in AD have largely been unsuccessful in preventing disease progression.4–7 Additionally, the long-term safety of these treatments remains to be determined. Clearly, there is a great need for more specific and efficacious therapies for AD.
Converging evidence from neuropathological and in vivo imaging studies has demonstrated the vulnerability of the hippocampus and heteromodal association cortices in AD, as well as decreased functional connectivity in cortico-cortical and cortico-hippocampal circuits.8,9 Approaching AD as a dysfunction in cortico-cortical and cortico-hippocampal circuits suggests that modulating neuronal activity within these networks may be a viable treatment option for early AD. Deep brain stimulation (DBS) is a neurosurgical technique that has shown success modulating pain,10 motor dysfunction,11,12 and mood13,14 circuits in conditions for which pharmacologic treatments are not effective. The importance of developing circuitry-based therapeutic approaches to modulate cortical and hippocampal networks affected in AD was the impetus for a Phase I study of DBS of the fornix in early AD patients.15
The fornix is a white matter bundle that connects the hippocampus with other components of the limbic system,16,17 forming circuits that have been implicated in episodic memory.18,19 Recent diffusion tensor imaging (DTI) studies have shown white matter reductions in the fornix of individuals with AD.20 In fact, fornix atrophy may precede both hippocampal degeneration and clinical symptoms of AD and predict conversion from mild cognitive impairment (MCI) to AD.21,22 This growing body of evidence, combined with rodent studies showing that DBS of the Papez circuit improves memory and promotes neurogenesis,23,24 indicates that DBS targeting of the fornix region (DBS-f) may be an effective treatment for AD symptoms. In the open-label Phase I trial of six AD subjects treated with DBS-f for 12 months, positron emission tomography (PET) scans showed an increase in cortical glucose metabolism and increased cortical functional connectivity over 1 year15 in contrast to the decreases in cortical metabolism and functional connectivity typically observed in AD.25–27
Building on the aforementioned collective observations, ADvance was designed as a double-blind placebo-controlled clinical trial to preliminarily assess the safety and efficacy of DBS-f for the treatment of very mild AD, as well as the utility of prespecified clinical and neuroimaging baseline measures as predictors of response. While other targets along the Papez circuit could, at least in theory, be suitable or perhaps more optimal, we chose a target within the hypothalamus which contains both the descending portion of the fornix and the mammillary body. This was done based on our empirical observation of acute memory effects in a patient with hypothalamic stimulation for obesity28 and the preliminary observations in the six AD patients in the pilot study of DBS for AD.15 Other potential targets would require validation of both safety and efficacy, something that was beyond the scope of our trial. Here, we describe the design and methods of the ADvance clinical trial and discuss the reliability of the major clinical outcome measures used in the study.
Methods
Overview
ADvance (ClinicalTrials.gov identifier: NCT01608061) is a feasibility clinical trial funded by the National Institute on Aging (R01AG042165) and Functional Neuromodulation Ltd (FNM). Six sites in the United States and one in Canada participated in enrollment (see “Acknowledgments” section). Sites were chosen based on previous experience with clinical research related to AD, neuroimaging capabilities, ethical approvals, and experience with DBS research.
ADvance is a 12-month double-blind, placebo-controlled, masked clinical trial, as shown in Figure 1. Consenting individuals participated in a screening visit followed by a baseline visit to assess eligibility and collect baseline cognitive and other clinical assessments, historical documents, laboratory values, and neuroimaging. DBS device implantation took place within 60 days of consent. Two weeks following implantation, participants were randomized into two groups, DBS “on” or DBS “off”, for a 12-month blinded period followed by a 12-month open-label extension during which all subjects will be programmed to DBS “on”. Additional long-term clinical follow-up for up to 48 months will be provided.
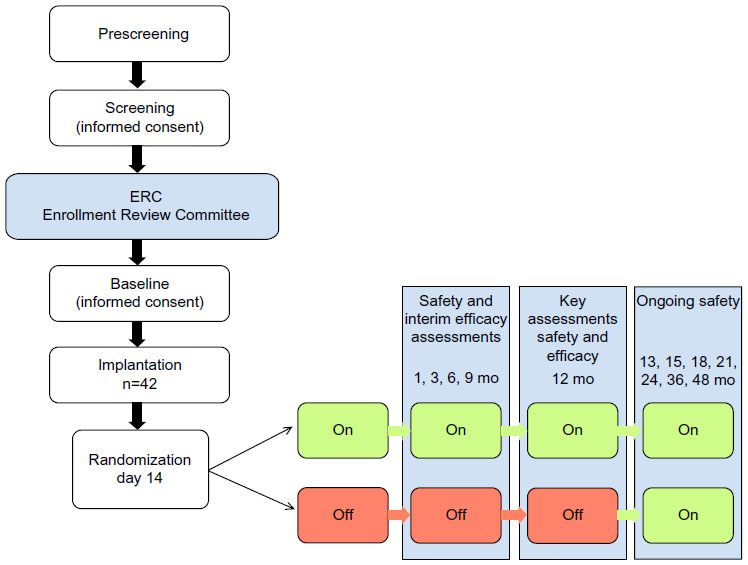 | Figure 1 Flow diagram depicting an overview of the ADvance trial schedule. Major visits are shown along with the number of participants at implantation (n=42). Green indicates DBS “on”, red indicates DBS “off ”. |
Participants: inclusion and exclusion criteria
Inclusion
- Informed consent signed by the subject, caregiver, and a surrogate
- Nonchildbearing/postmenopausal women, and men between 45 and 85 years of age
- Met criteria for probable AD according to the National Institute of Aging/Alzheimer’s Association criteria29
- Clinical Dementia Rating scale (CDR global) rating of 0.5 or 1 at screening30
- Alzheimer’s Disease Assessment Scale – cognitive component (ADAS-cog-11) score of 12–24, inclusive, with a score ≥4 on ADAS-cog item 1 (immediate recall) at both screening and baseline visits30,31
- General medical health rating ≥3 (good or excellent)32
- Living at home with an available caregiver or informant to report on daily activities and function throughout the study
- Fluent in English
- Good surgical candidate for placement of a deep brain stimulator, as determined by the neurosurgical team
- On a stable dose of a acetylcholinesterase inhibitor (AChEI) donepezil, galantamine, or rivastigmine for at least 60 days prior to signing informed consent, without intention to modify this medication dose throughout the entirety of the study.
Exclusions
- Neuropsychiatric Inventory (NPI)33 total score ≥10 or score ≥4 in any domain except apathy at screening
- Modified Hachinski ischemic score >4 at screening34
- Young Mania Rating Scale (YMRS) ≥11 at screening35
- Attempted suicide in the 2 years prior to signing consent
- Risk for suicide as determined by an answer of “yes” to “suicidal ideation” or “yes” to any items in the suicidal behavior section with reference to the 3-month period prior to screening on the Columbia Suicide Severity Rating Scale (C-SSRS)36
- Current major psychiatric disorder
- Score >10 on the Cornell Scale for Depression and Dementia (CSDD) at screening37
- History of head trauma in the 2 years prior to signing consent
- History of brain tumor, subdural hematoma, or other clinically significant space-occupying lesions on CT (computed tomography) or MRI (magnetic resonance imaging)
- Mental retardation
- Current alcohol or substance use disorder as defined by Diagnostic and Statistical Manual of Mental Disorders, fourth edition – text revision (DSM-IV-TR)
- Exclusions for PET and MRI, including claustrophobia, metal implanted in the body (MRI), and insulin-dependent diabetes (PET)
- Radiation exposure in the year prior to consent that added to exposure in the study would exceed 5 rem over 12 months
- Any abnormal laboratory results, cardiovascular or neurovascular disorders, or currently prescribed non-AD medications that would preclude participation in the study
- Unstable doses of any medication prescribed for the treatment of memory loss or AD
- Unwilling or unable to comply with the protocol
- Life expectancy of <1 year
- Actively enrolled in another concurrent clinical trial.
Recruitment and consent
Participants were recruited from memory and geriatric psychiatry clinics, advertising, and community outreach activities at each of the seven sites. Prospective participants were first assessed for their ability to provide consent through clinical interviews. Clinicians experienced in dementia research and DBS surgery evaluated each participant’s ability to comprehend the consent form as well as understand the personal consequences of what would and could happen during the study. Assessments and consent procedures took place in the presence of a caregiver who cosigned the consent form as a witness. Voluntary written informed consent by each subject and his or her caregiver was required at the beginning of both the screening and baseline visits and prior to surgical implant procedure (if all entry requirements were met).
Eligibility screening
Enrollment was defined as the time a subject signed the screening informed consent to participate and was followed by an initial screening visit at which medical and neuropsychiatric information was gathered (Table 1). In order to move forward with implantation, a site-independent Enrollment Review Committee (ERC) reviewed the data collected to determine if subjects met inclusion and exclusion criteria. For each subject, the ERC reviewed historical documentation of early AD as well as scores on cognitive tests conducted during the screening visit to confirm the diagnosis of probable AD. They also verified the absence of concomitant medical or psychiatric conditions or medications, and any surgical risks that might affect DBS surgery. Additionally, US trial sites audio-recorded selected psychometric assessments, and site-independent ERC raters dually scored a randomly selected sample of ADAS-cog and CDR interviews to confirm scoring accuracy and rater consistency.
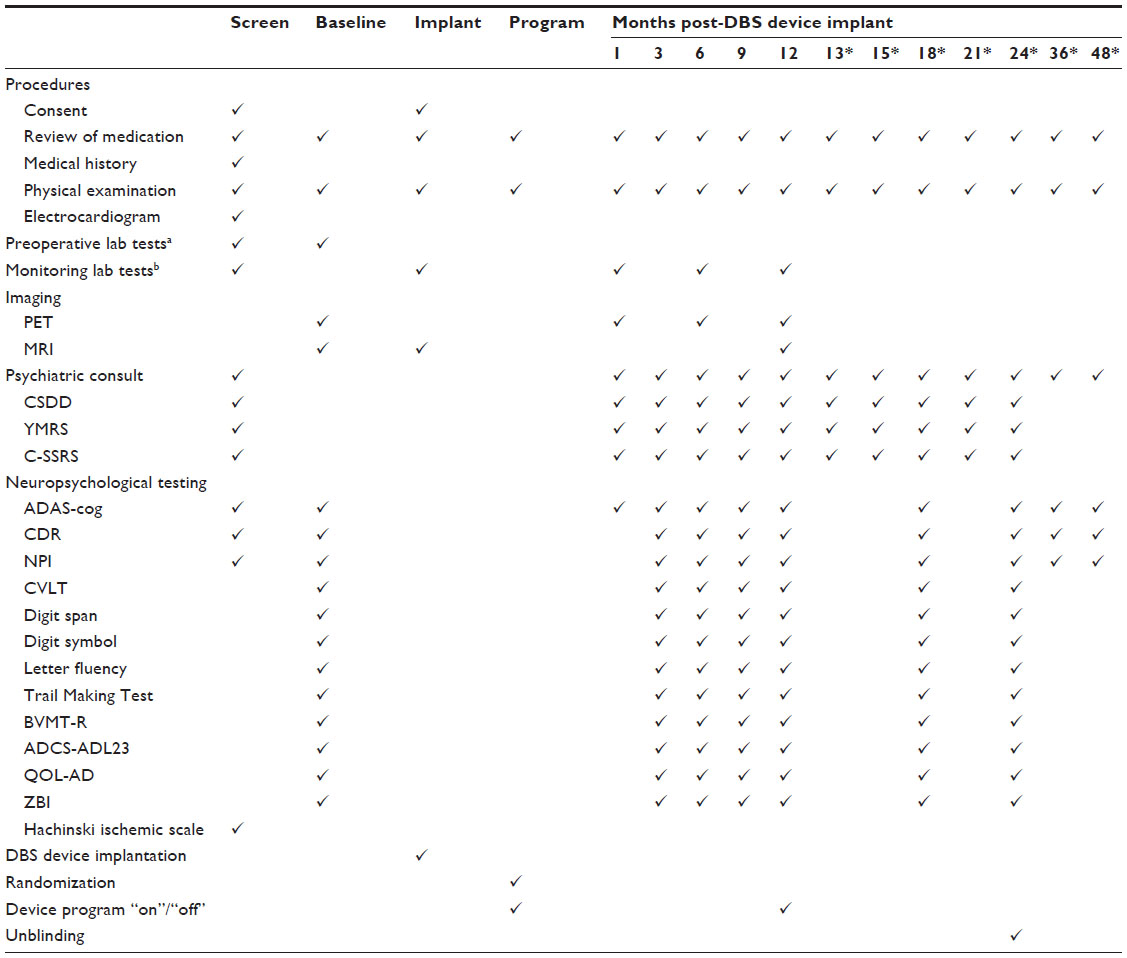 | Table 1 ADvance visit schedule |
Baseline visit and DBS device implantation
Baseline visit and surgery
A baseline visit was scheduled ≤59 days postconsent at which baseline medical, laboratory, neuropsychological, and imaging data (PET and MRI) were obtained (Table 1).
Implantation surgery took place within 60 days following screening consent if all study requirements were met. High-resolution, stereotactic MRI scans of the brain were used to directly target the bilateral postcommissural fornices. More specifically, the DBS electrode was implanted 2 mm anterior and parallel to the vertical portion of the fornix within the hypothalamus (Figure 2). The most ventral contact of the DBS lead (Medtronic model 3387) was typically placed posteromedial to the optic tract at a depth such that the dorsal most contact was intraparenchymal, approximately at the level of the midcommisural plane. Laterally, the target corresponded to the midpoint of the medial/lateral extent of the fornix in the coronal plane to maximize the proximity of the four DBS contacts to the descending column of the fornix. A burr hole was placed approximately 2.5 cm lateral to the midline at or just anterior to the coronal suture with adjustments made so that the lead trajectory would avoid sulci and deflection from the wall of the frontal horn of the lateral ventricle. Intraoperative stimulation was performed at the discretion of the surgeon to evaluate contact position. After surgery and prior to hospital discharge, an MRI was conducted to confirm the position of the leads. If necessary, the implanting surgeon repositioned the leads during the same hospitalization, followed by a second MRI. A third-party-blinded determination of lead position was obtained by sending the postimplant MRI scan to a neurosurgeon not involved with surgical implantation of devices for the study, who subsequently performed stereotactic analysis of the MRI on a DBS planning station.
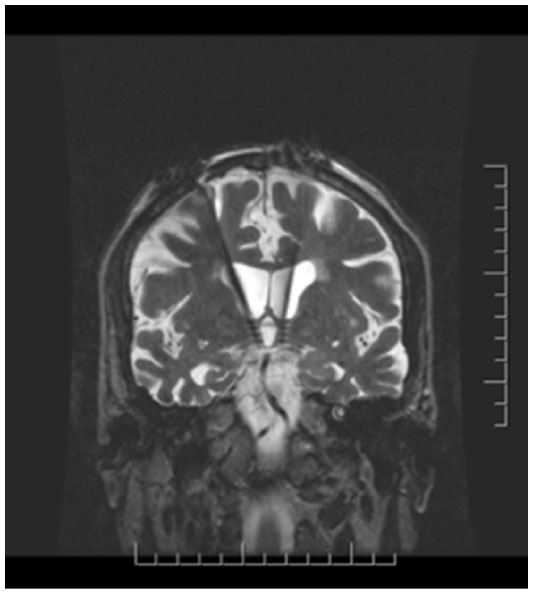 | Figure 2 Coronal MRI demonstrating placement of the DBS electrodes anterior and parallel to the vertical portion of the fornix within the hypothalamus bilaterally. |
Surgical devices and programming
The DBS system used in the study includes the Model 37601 Activa® PC stimulator, Model 3387 Lead, and Model 37085 extension (Medtronic, Inc., Minneapolis, MS, USA). All implantable devices, external control devices, and accessories (Model 8840 N’Vision® programmer with 8870 Activa applications software and Model 37022 External neurostimulator) are approved by the FDA for DBS treatment of Parkinson’s disease and essential tremor. The Activa PC neurostimulator (Medtronic, Inc.) is a dual-channel device capable of delivering bilateral stimulation. It contains a nonrechargeable battery and microelectronic circuitry that delivers controlled electrical pulses to specifically targeted brain areas. The device was implanted subcutaneously just inferior to the clavicle, connected to an extension run subcutaneously along the head, neck, and shoulder and connected to the implanted leads. Quadripolar DBS leads (3387) are made of four thin, insulated coiled wires bundled with polyurethane insulation. Each lead has four 1.5 mm electrodes at the tip spaced 1.5 mm apart. Stimulation can be delivered using one electrode or a combination of electrodes. The N’Vision programmer (8840) is an external component that noninvasively reviews and adjusts the neurostimulator’s output parameters.
Randomization, blinding, and DBS-f dosing
The overall study design was a delayed-start trial, in which all subjects received DBS stimulator implantation and were randomly allocated to begin DBS-f soon after implantation or 1 year later. Patients were randomized 2 weeks after implantation in a 1:1 allocation to DBS-f “on” or DBS-f “off”. Random blocks were used to generate randomization for each site, and the randomization assignment was provided via phone call to the unblinded technician responsible for programming the implanted device. Study subjects, the implanting surgeon, study coordinators, the principal investigator, and follow-up clinicians responsible for administering questionnaires and outcome assessments remain blinded to treatment assignment until all subjects complete the 12-month visit or until each subject’s 24-month visit, whichever occurs first. Instances of emergency unblinding are disclosed to site monitors and the national primary investigators (CGL and AML). These procedures are only initiated in cases of compromised subject welfare and are, whenever possible, reviewed by the national and site primary investigators and reported to the commercial sponsor (FNM).
At a follow-up visit 11–17 days following surgery, the DBS device was programmed according to the randomization assignment by an unblinded clinician (Table 1). Starting with the most ventral contact, monopolar stimulation was delivered at a frequency of 130 Hz with a 90-microsecond pulse width. The initial amplitude was set to 1 V and was increased incrementally by 1 V every 30–60 seconds to a maximum until the subject reported experiential phenomena, including memory-related phenomena or autonomic symptoms (eg, increased heart rate). Each contact was individually tested, and the contact on each side that produced an experiential or autonomic-related event at the lowest voltage was chosen as the therapeutic contact. If no experiential phenomena were produced with stimulation, the contact that had the highest threshold for autonomic adverse effects was chosen. This was usually contact 2 or 3, the two most superior of the four contacts on the DBS electrodes. For each subject randomized to the DBS-f “on” group, the chosen contact on each side was set to a voltage of 50% of that eliciting a stimulation-related event or of 3.5 V, whichever was lower. If a stimulation-related event was reported with bilateral stimulation, the voltage was turned down in 0.2 V increments on each lead until the event was no longer experienced. For each subject randomized to the DBS-f “off” group, all leads were set at 0 V. The programming protocol was completed in full regardless of DBS-f “on” or DBS-f “off” randomization status to prevent unblinding of participants, with the only difference being the final voltage of the DBS-f electrodes. The unblinded clinician at each site is responsible for ensuring that the parameters remain consistent throughout the 24-month follow-up unless there are stimulation-associated side effects or safety concerns. Following the end of the 24-month follow-up visit, programming will be left to the discretion of the site physician.
Follow-up during the double-blind period
Follow-up visits are scheduled for 1, 3, 6, 9, and 12 months after implantation. These visits include a physical examination, psychiatric consultation, neuropsychological testing, blood tests (6, 12 months), PET scans (1, 6, 12 months), and brain MRI (12 months). All subjects, including patients randomized to DBS-f “off”, have the implanted device programmed “on” after the 12-month visit for a subsequent open-label treatment phase. Open-label follow-up visits are scheduled for 13, 15, 18, 21, 24, 36, and 48 months after implantation. These visits consist of safety monitoring, clinical updates, physical examination, psychiatric consultation, and/or neuropsychological testing (18, 24 months). There will be additional phone follow-ups at months 27, 30, 33, 39, 42, and 45 to assess any adverse events (AEs) or changes in cognition or personality. Subjects will exit the study at the conclusion of their 48-month visit (Table 1).
PET and MRI
MRI and PET protocols were implemented based on the AD Neuroimaging Initiative (ADNI) protocols that have been used extensively to measure longitudinal changes in gray matter volumes (MRI) and cerebral glucose metabolism across different MRI and PET scanners and study sites.38,39 PET scans with the radiotracer [18F]-2-deoxy-2-fluoro-d-glucose ([18F]-FDG) to measure regional cerebral glucose metabolism are acquired preoperatively and at 1, 6, and 12 months. PET scans are performed on a PET/CT scanner at each site using a uniform protocol and postprocessing methods designed to obtain comparable measurements of cerebral glucose metabolism across scanners/sites.40 During radiotracer uptake, subjects are maintained in a quiet, dimly lit room, with eyes open and ears unoccluded. Thirty minutes after a 5 mCi ±10% radiotracer injection, patients are positioned in the scanner, and a 20-minute emission scan is obtained, followed by a transmission scan. The second, 10-minute frame of the emission scan (40 minutes after [18F]-FDG administration) is used for quantitative analysis. The MRI protocol was designed to detect focal pathology (eg, tumors or strokes) and for MRI-PET registration to define regions of interest. The MRI scans were performed prior to the first PET scan, 1–2 days postoperatively, and are repeated at 12 months. MRI scans are acquired on 1.5 T scanners at each site. The sequences were implemented from the ADNI protocols that were developed for each MRI scanner to obtain comparable quantitative measurements, including gray matter volumes.38,39 The MRI sequences include 3-plane localizer, volumetric sequence (magnetization-prepared rapid gradient-echo or spoiled gradient recalled echo), T2 sequences for electrode localization (postoperative scan only), fast spin-echo inversion recovery (FSE-IR), DTI (when available), and resting-state functional MRI (when available).
Safety monitoring
An independent Clinical Events Committee (CEC) consisting of a multidisciplinary team of physicians from geriatrics, neurology, and neurosurgery who are not investigators in the study was created. The CEC conducts reviews of all AEs reported for study subjects. Each AE is adjudicated for its relatedness to the study, surgical procedure, implantable pulse generator, leads (electrodes), and programming. AEs are categorized as general medical, psychiatric, surgical, or programming in nature. In addition, adjudications are made regarding whether an event is a serious AE or an unanticipated adverse device effect. Psychiatric AEs are assessed at each follow-up visit using measures such as the C-SSRS,36 CSDD,37 and YMRS.35
A separate independent Data Safety and Monitoring Board (DSMB) of one practicing neurosurgeon, two neurologists and/or psychiatrists, and one biostatistician not connected to the sponsor or participating investigators was established. The DSMB reviews CEC-adjudicated AEs, occurrences of serious AEs and unanticipated adverse device effects, as well as safety and efficacy trends and makes recommendations regarding the continuation, suspension, or termination of the study. Following each review by the DSMB, a summary of results is sent to the FDA and filed with all overseeing institutional review boards.
Outcome measures and analytic plan
Safety outcome assessment
Doctors and clinical staff at local sites are in regular contact with participants and study partners to monitor for AEs, as described earlier. As soon as an AE is detected, the local team obtains as much clinical information as possible and rapidly reports to the study principal investigator, the medical monitor, and the coordinating center. The coordinating center, working as a team, then reviews, requests additional information, notifies other sites, and notifies DSMB, institutional review boards, and FDA as spelled out in study procedures. AEs will be presented as a measure of the safety of DBS-f surgery and treatment for mild AD.
Clinical outcome assessment
One primary goal of this study is to examine the acute and long-term safety of DBS-f for mild AD. Acute safety is assessed by the rate of serious device- or procedure-related AEs from the date of implant through the date of randomization as well as serious procedure-related events through 30 days postimplant. Long-term safety is assessed by the rate of serious therapy (programming) related AEs from the date of randomization through the date of the 12-month visit.
The second goal of this study is to preliminarily examine the efficacy of DBS-f for mild AD. The two primary outcomes are change from baseline in the ADAS-cog and CDR sum of boxes (CDR-SB) scores. The ADAS-cog is one of the more commonly used measures to assess cognitive symptoms associated with AD in clinical trials. The ADAS-cog is able to differentiate individuals with nonimpaired cognition from those with impaired cognition and has demonstrated reliability in assessing the extent of cognitive impairment in individuals.41 The standard ADAS-cog consists of 11 subscales designed to assess memory, language, and praxis, and scoring is based on the number of errors made on each item, with a higher score indicating greater impairment. Previous clinical trials have indicated that a four-point change on the ADAS-cog total score is suggestive of a clinically important difference.42,43 However, systematic analysis of double-blind placebo-controlled trials of cholinesterase inhibitors demonstrated an average −2.7 improvement at 6 months and 1 year.44 The CDR was developed for the evaluation of staging severity of dementia.45 The CDR characterizes cognitive and functional performance by assessing the subject in six domains including memory, orientation, and problem solving. The CDR has consistently demonstrated good reliability46,47 and has been validated against neuropathological findings.48,49 A global CDR score is computed via an algorithm based on the input of the ratings of the six domains and is useful for characterizing and tracking a subject’s level of impairment and stage of dementia severity,45 with values between 0 (normal) and 3 (severe dementia). The CDR-SB score is obtained by summing each of the six domain ratings. CDR-SB scores range from 0 to 18, with higher scores reflecting more severe impairment.46
Secondary outcomes include change from baseline to 12 months in scores on other cognitive tests: California Verbal Learning Test, second edition (CVLT-II50); Verbal Fluency;51 Brief Visuospatial Memory Test – revised version;52 and Trail Making Test.53 Other measures include Quality of Life – AD, which is a rating of the patient’s quality of life both from the patient and the caregiver,54 AD Cooperative Study – Activities of Daily Living Inventory, Zarit Burden Interview to assess caregiver distress,55 and NPI to assess the presence of psychiatric symptoms and behaviors.33
Neuroimaging outcome assessment
The primary neuroimaging outcome measure is a regional change in glucose metabolism from baseline to 12 months, measured by PET. Previous research using PET measures of cerebral glucose metabolism has identified a specific pattern in AD of hypometabolism in the parietal and temporal heteromodal association cortices.56 This pattern has been found in over 85% of pathologically confirmed AD cases56 and has been correlated with dementia severity.56 FDG-PET is sensitive to AD clinical progression and to effects of pharmacotherapy, including DBS.15,26,57–59
A secondary imaging outcome is bilateral hippocampal volume measured using volumetric methods, decreases in which have been correlated with the progression of AD.21,22 An additional imaging tool of interest is fornix integrity measured with DTI for which the fornix is manually drawn (with high reliability) as a region of interest. However, we do not plan to use DTI obtained after implantation analytically due to concerns about interference by the implanted electrodes.
Clinical measure analytic plan
All analyses will be conducted according to the prespecified statistical analysis plan for the study. Descriptive statistics compare treatment group on baseline demographics. Categorical variables are analyzed using frequency, incidence, and event rate. For continuous variables, analyses include mean, median, standard deviation, and range.
For the acute safety end point, rate and 95% confidence interval will be presented. For long-term safety end points, rate and 95% confidence interval will be compared according to randomization group. Other data summaries will include a detailed summary and rate estimation of all serious AEs, as well as Kaplan–Meier estimates of the cumulative rates over time.
For the two clinical efficacy outcomes of particular interest (ADAS-cog score and CDR score), all analyses will follow intent-to-treat (ITT) or modified-ITT principles. The mean change from baseline (preimplant) to 12 months postimplant will be calculated in each group. Differences between randomized groups in mean change on each of these primary outcomes will be calculated, along with corresponding two-sided 95% confidence intervals. In addition, within group improvements will be assessed relative to a null change of zero. Additional analyses will include assessments of change over time in mixed model regression estimations with repeated measurements. Site will be used as a covariate in this model.
In addition to ADAS-cog-13 total score, this method will also be used to examine derived Word Recall Total score, Word Recognition scores, and the subset of five ADAS-cog items shown to be most sensitive to memantine. The subsetting methodology described by Ihl et al60 will be applied to this data set to assess the relative strength of treatment effect across other subtests of the ADAS-cog. Further analyses will be conducted to determine the impact of baseline ADAS-cog scores on outcome (ie, to assess whether the treatment effect diminishes in the more advanced population), including mixed model regression analyses with baseline ADAS-cog score as a predictor, and fit with an interaction term. Within-group outcomes, by baseline ADAS-cog score also will be summarized. CDR analyses will include global score, SB, and memory domain score. In addition we will analyze the CDR-SB score relative to the overall severity classification from the ADAS-cog to determine the agreement of categorization of mild symptom severity at baseline with how the scores change together over time.
Neuroimaging measure analytic plan
The end point is the mean, per-subject, percent glucose metabolism change in prespecified regions of interest, including the subdivisions of the temporal and parietal association cortex and the hippocampus. The primary analysis will be conducted on an ITT basis. The percent improvement for each subject will be determined by subtracting the baseline value for glucose metabolism in each region of interest (ROI-BL) from the 12-month glucose metabolism (ROI-12) and dividing the result by ROI-BL ([ROI-12 − ROI-BL]/ROI-BL). This quantity will be averaged across all subjects, and mean improvement will be compared across randomization groups in a two-sample t-test evaluated at the 0.05 significance level. Supportive analyses will only evaluate those subjects with complete data and/or those who are compliant with the protocol.
Exploratory analyses will be conducted to evaluate the relationship between changes in regional glucose metabolism and changes in clinical outcomes (eg, ADAS-cog, CVLT, etc). Exploratory analyses will be performed to identify baseline structural and functional neuroimaging predictors of DBS clinical response. These analyses may include, but not be limited to, regional glucose metabolism, hippocampal volumes, and regional white matter functional integrity (DTI). These analyses will fit multivariate linear regression models, with the candidate predictor and randomization assignment, and their interaction as independent variables, and clinical outcome (eg, ADAS-cog-11, CDR-SB, CVLT) as the dependent variable. Covariates with apparent effect on outcome will then be fit in a multivariate regression model with stepwise selection procedure using significance level of 0.10 as a cutoff.
Power estimation
The power to detect a treatment effect depends on the size of the effect, sample size, and the configuration of the within-person variance–covariance matrix. For the ADAS-cog, we conducted a power simulation using ADNI data. Assuming a mixed effects model with random intercept and slope, no missing data, variance of intercept =(4.68)2, variance of slope =(0.37)2, correlation between random intercept and random slope of 0 (model with unstructured covariance failed to converge), variance of residuals =(5.87)2, time vector = c (0,3,6,9,12), there is 84% power to detect a difference in change scores of 7.53 points with 42 subjects, which translates to a difference in slopes of 0.63. We will have 80% power to detect a difference in slope of 0.588 points/month or 7.06 points/year. Using the same method, the power to detect a difference in a change of score of 4.0 points (a typical effect size) was calculated to be 36%, which translates to a difference in slopes of 0.33.
Statistical power for the neuroimaging hypothesis listed previously was estimated using PASS 2008 software under a two-sided, two-sample t-test and the assumption of 40 randomized subjects in a 1:1 allocation. Assuming a significance level (α) of 5%, mean improvement (DBS) of 10%, standard deviation (DBS and control) of 12%, and mean improvement (control) of 0.0%, a total of 40 randomized subjects provides at least 80% power to statistically evaluate the end point of glucose metabolism as specified earlier.
Results
Characterization of the baseline study population
Following recruitment, 85 potential subjects across the seven study sites signed the initial consent and participated in a screening visit. Forty-eight (56.5%) met all inclusion and exclusion criteria at screening and were approved by ERC reviewers to proceed in the trial. Forty-two of these 48 patients (49.4% of consenting potential subjects) met all criteria following the baseline visit, signed a second consent form, and underwent DBS-f device implantation. The six patients who were cleared at screening but did not proceed to implant did so for two reasons: two declined to proceed and did not sign consent for baseline testing, and four failed to meet inclusion/exclusion criteria when ADAS-cog was repeated at baseline, with scores out of range.
We compared (Table 2) consenting subjects who were screened and successfully implanted (N=42) to those who were screened but not implanted with a DBS device (N=43). We saw no difference between these groups in demographic information (age, sex, and elapsed time since initial AD diagnosis) gathered at screening. We observed significantly lower (ie, better) ADAS-cog-11 (17.5±3.6 vs 21.1±9.8, P=0.03) and CDR global scores (67% vs 38%, with a score of 0.5, P=0.05) in subjects who were implanted. This is explained by the inclusion and exclusion criteria designed to limit participation to subjects with very mild AD. Approximately 40% of patients who failed screening did so because symptom severity was too high. No significant difference was found in CDR-SB score, Hachinski ischemic scale score, or psychiatric assessment measures, including the NPI, C-SSRS, CSDD, or the YMRS in implanted compared to not implanted patients (Table 2).
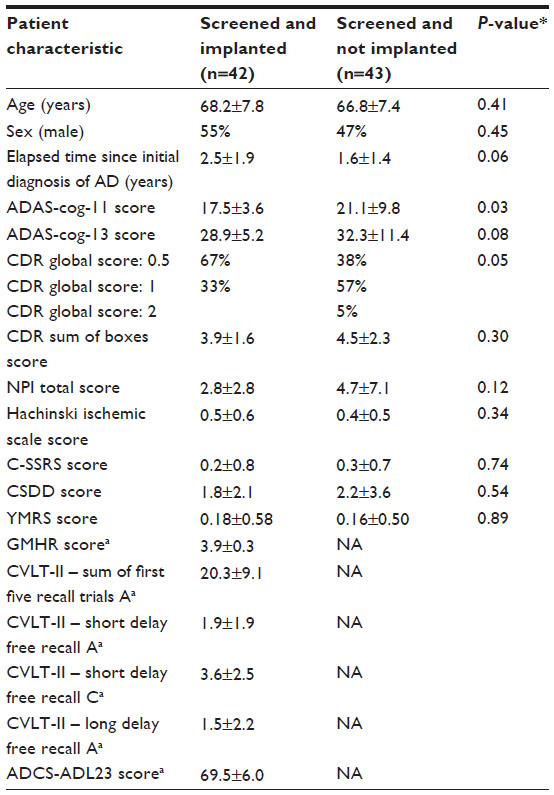 | Table 2 Demographics and screening measures by implantation status |
Table 2 summarizes the scores of implanted subjects on additional cognitive and psychiatric tests from the baseline visit. In addition to inclusion criteria measures such as the CDR, scores on the CVLT-II, a verbal memory measure, were similar to published scores of individuals with MCI who later progressed to AD.61 Note that these additional scores include all 48 subjects who were assessed at baseline, not only the 42 who were implanted.
Reliability of primary outcome measures
We examined test–retest reliability of the main outcome measures by comparing score stability between the screening and baseline visits for the 42 implanted subjects. There was a moderate correlation between screening and baseline visit scores for ADAS-cog-11 and a high correlation for ADAS-cog-13, CDR global score, CDR-SB, and NPI (Table 3). The stability in scores seen within subjects across initial visits suggests that our primary clinical outcome measures are reliable for the population in this study and can be used to measure disease progression over time in this trial.
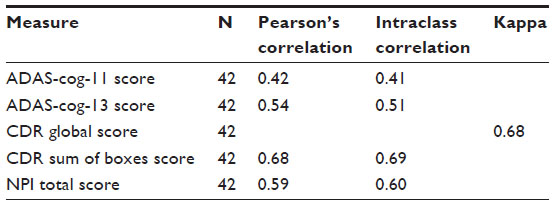 | Table 3 Correlation* between screening and baseline value |
Discussion
ADvance is the first multicenter, randomized, double-blind controlled clinical trial evaluating the efficacy of DBS-f for mild AD. This paper outlines the methods used in the ADvance trial and characterizes the study population. Subjects demonstrate cognitive test scores indicative of mild AD, and implanted participants differ from consenting nonimplanted participants only on average ADAS-cog-11 and CDR global scores. Furthermore, we confirm the reliability of the primary outcome measures used in the ADvance study population by demonstrating consistency across two visits.
ADvance joins three smaller DBS studies conducted in AD patients published thus far62 and builds on data from approximately 25 animal studies.23,24,28,63 ADvance was designed based on an open-label Phase I study of DBS-f conducted in six patients with mild AD, as previously discussed. Interestingly, patients with less severe prestimulation cognitive dysfunction and less severe metabolic deficits were more likely to benefit from DBS-f in this trial.15 This study provided the basis for the inclusion criteria used in ADvance, which focus on selection of participants with very mild AD. Implanted participants were younger, more likely to be male, and had lower baseline ADAS-cog-11 scores than the mean scores in recently reported Phase III trials of amyloid-lowering agents in AD.64–66 Mean ADAS-cog-11 was 17.5 for implanted ADvance participants, and ranged from 19 to 23 in recent Phase III trials. Thus, our participants were younger and less impaired than those in these Phase III trials, but the differences were relatively small.
Fontaine et al67 recruited 110 patients with Alzheimer dementia or MCI to be screened for a study assessing DBS for cognitive decline, and one subject met criteria, consented, and continued on to implantation. In the current study, 42 of 85 screened subjects consented and underwent implantation. One possible explanation for this difference may be that the Fontaine et al67 study required subjects to have received a DSM-IV AD diagnosis within 2 years and have a mini–mental state examination score between 20 and 24, which was not required in the ADvance study. It also is possible that the 1-year open-label follow-up phase used in ADvance may have contributed to improved participation.
Another study in 2014 examined open-label DBS targeting the nucleus basalis of Meynert in six patients meeting criteria for mild-to-moderate AD. Although ADAS-cog scores worsened by an average of 3 points after 1 year of stimulation, the authors noted that this was less than the average 6- to 7-point worsening seen in prior reports of patients with AD. Further, global increases in metabolism in amygdalohippocampal and temporal regions were seen in three out of the four patients who underwent PET scans of cerebral glucose metabolism.68
Taken in the context of prior research, ADvance is a novel, important step in studying DBS-f as a treatment for mild AD. The subject pool is much larger than prior studies and involves 1 year of double-blind, controlled cognitive testing and neuroimaging. The 1-year duration of the controlled comparison increases the likelihood of identifying significant differences between the stimulated (treatment arm) and nonstimulated (control arm) cohorts in this slowly progressive disorder. The 1-year blinded, controlled phase of the study is followed by up to 3 years of continued open-label follow-up with all patients being stimulated, which affords systematic pre- and postassessment of outcomes in about half the participants, and 2 years of additional follow-up. While the primary objective of the study is to evaluate safety, the larger subject population and controlled design affords preliminary testing of the efficacy of DBS-f in delaying cognitive decline and improving cortical glucose metabolism.
The ADvance methodology outlined here is strengthened by recruitment from seven sites with experienced investigators and rigorous subject selection by an independent ERC. The primary clinical outcome measures are standardized and validated and have been widely used so that the results of the current study can be evaluated in comparison to studies that tested other symptomatic treatments such as cholinesterase inhibitors.69 The neuroimaging outcome measures, especially cerebral glucose metabolism, have also been well studied in AD56,57,70,71 and enable us to examine changes in brain function throughout the trial. Limitations of ADvance include limited sample size. Although larger than prior DBS studies, there are only 21 patients in each of the masked treatment groups. The goal is that the results of ADvance will inform a larger-scale clinical trial focused primarily on efficacy, rather than on safety, of DBS-f as a treatment for mild AD.
Research elucidating the neurobiological mechanism of DBS treatment for neurodegenerative diseases such as AD remains limited. While we hypothesize that this treatment works by stimulation of fornix–hippocampal–cortical circuits, and potentially even promotes neurogenesis in the hippocampus, this remains unproven. The memory circuits we are examining may be undergoing degeneration at varying rates in individual study participants, limiting the effectiveness of DBS-f to slow cognitive decline and our ability to examine the efficacy of DBS-f within our study population.
In summary, the ADvance trial was successful in enrolling appropriate patients for a novel application of DBS, and we believe several unique design aspects outlined here could be considered in future clinical trials of DBS targeting AD and other cognitive disorders.
Acknowledgments
This research is supported by a grant from the National Institute on Aging, R01AG042165, and a vendor grant received from Functional Neuromodulation Ltd, the sponsor of the ADvance study.
The ADvance study team includes Todd Langevin, Lisa Fosdick, Kristen Drake, Donald E Reymers, Robyn Moxon, Dan O’Connell, Vince Owens, Cara Pendergrass, Susan Klees, Steven D Targum, and the seven participating clinical trial sites.
Chair’s office at Johns Hopkins University and University of Toronto: Constantine G Lyketsos, MD, MHS, coprincipal investigator; Elizabeth Plank Althouse, professor and chair of psychiatry at Johns Hopkins Bayview; Andres M Lozano, MD, PhD, FRCSC, FACS, co-principal investigator, professor, and Chair of Neurosurgery, Tasker Chair of Functional Neurosurgery; Gwenn Smith, PhD, imaging core director, professor of psychiatry and behavioral sciences, Johns Hopkins University; Cynthia Munro, PhD, neuropsychologist, associate professor of psychiatry and behavioral sciences, Johns Hopkins University; Esther Oh, MD, medical monitor, assistant professor of geriatric medicine, Johns Hopkins University; Jeannie Sheppard Leoutsakos, PhD, data core leader, assistant professor of psychiatry and behavioral sciences, Johns Hopkins University.
Clinical trial sites
Johns Hopkins University School of Medicine, Baltimore, MD: Paul Rosenberg, MD, associate professor, associate director, Memory and Alzheimer’s Treatment Center; William S Anderson, MD, associate professor of neurosurgery.
University of Toronto/Toronto Western Hospital: Andres M Lozano, MD, PhD, FRCSC, FACS, professor of neurosurgery, Tasker Chair of Functional Neurosurgery; David F Tang-Wai, MDCM FRCPC, associate professor of neurology.
Banner Alzheimer’s Institute, Phoenix: Anna Burke, MD, geriatric psychiatrist, dementia specialist; Francisco A Ponce, MD, associate professor of neurosurgery, director of Barrow Center for Neuromodulation.
Banner Sun Health Research Institute, Sun City: Marwan Sabbagh, MD, director, Banner Sun Health Research Institute; Francisco A Ponce, MD, associate professor of neurosurgery, director of Barrow Center for Neuromodulation.
Brown University, Rhode Island Hospital, Butler Hospital: Stephen Salloway, MD/MS, professor of neurology, director of neurology and memory and aging program; Rees Cosgrove, MD/PhD, chair of neurosurgery; Wael Asaad, MD/PhD, assistant professor of neurosurgery.
University of Florida – Gainesville: Michael S Okun, MD, professor of neurology, codirector of the Center for Movement Disorders and Neurorestoration; Kelly D Foote, MD, professor of neurosurgery, codirector of the Center for Movement Disorders and Neurorestoration.
University of Pennsylvania: David A Wolk, MD, associate professor of neurology, assistant director of Penn Memory Center; Gordon Baltuch, MD/PhD, professor of neurosurgery, director of the Center for Functional and Neurorestorative Neurosurgery.
Disclosure
Steven D Targum: Has received consultation fees, retainers, or vendor grants from the following: Acadia Pharmaceuticals, Acumen, Alcobra, Alkermes Inc., AstraZeneca, BioMarin, BrainCells Inc., CeNeRx Pharmaceuticals, Clintara, LLC, Civitas, Eli Lilly and Company, EnVivo (Forum) Pharmaceuticals, Functional Neuromodulation Inc., Johnson & Johnson PRD, Intracellular Therapies Inc., Methylation Sciences Inc., Mitsubishi Tanabe, Neurophage, Nupathe, Pfizer Inc., Prana Biotechnology Ltd, ReViva Pharmaceuticals, Roche Labs, Sophiris, Sunovion, Targacept, Theravance, Transcept.
Kristen Drake: Has no conflicts of interests other than being an employee of Functional Neuromodulation Inc.
J Cara Pendergrass: Has no conflicts of interests other than being an employee of Clintara LLC.
Gwenn S Smith: Has received grant support as follows: NIH RO1 MH086881, AG038893, AG041633.
Paul B Rosenberg: Has received consultation fees from Pfizer, Janssen, Global Exchange for Alzheimer’s Research, Abbvie, Lundbeck; research grant support from Lilly, Merck, Pfizer, Elan, Janssen, Functional Neuromodulation Inc., American Federation for Aging Research, and the National Institute on Aging.
Michael S Okun: Has served as a consultant for the National Parkinson Foundation and has received research grants from NIH, NPF, the Michael J Fox Foundation, the Parkinson Alliance, Smallwood Foundation, the Bachmann-Strauss Foundation, the Tourette Syndrome Association, and the UF Foundation. Dr Okun has previously received honoraria, but has received no support from industry in more than 60 months. Dr Okun has received royalties for publications with Demos, Manson, Amazon, Smashwords, Books$Patients, and Cambridge (movement disorders books). Dr Okun is an associate editor for the New England Journal of Medicine Journal Watch Neurology. Dr Okun has participated in CME and educational activities on movement disorders (in the last 36 months) sponsored by PeerView, Prime, Quantia, Henry Stewart, and by Vanderbilt University. The institution and not Dr Okun receives grants from Medtronic, Abbvie, and ANS/St Jude, and the PI has no financial interest in these grants. Dr Okun has participated as a site PI and/or co-I for several NIH-, foundation-, and industry-sponsored trials over the years but has not received honoraria.
Marwan Sabbagh: Has received grants, honoraria, travel support from the following: Amarantus, Avanir, Avid, Biogen-Idec, BSHRI: DART, Eli Lilly and Company, Functional Neuromodulation Inc., Genentech, Merck, Neuronix, Navidea, Piramal, Roche, Takeda; has received royalties from Tenspeed/Random House.
Andres M Lozano: Is cofounder of Functional Neuromodulation and has received consultation fees from the following: Aleva, Boston Scientific, Brainstorm, Codman, Elekta, Eli Lilly and Company, Insightec, Michael J Fox Foundation, Medtronic, Neuronova, Neurophage, Neuropace, St Jude, Schering.
Constantine G Lyketsos: Has received grant support from the following: AstraZeneca, Associated Jewish Federation of Baltimore, Bristol-Myers, Forest, GlaxoSmithKline, Eisai, Elan, Functional Neuromodulation, Lilly, National Football League, NIMH, NIA, Novartis, Ortho-McNeil, Pfizer Inc., Weinberg Foundation; has received consulting fees, honoraria, or travel support from the following: Abbvie, Adlyfe, AstraZeneca, Avanir, Bristol-Myers, Eisai, Elan, Eli Lilly and Company, Forest, Genentech, GlaxoSmithKline, Health Monitor, Janssen, Lundbeck, Merz, NFL Players Association, NFL Benefits Office, Novartis, Orion, Pfizer Inc., Supernus, Takeda, Wyeth, Zinfandel.
The authors report no other conflicts of interest in this work.
References
Lanctôt KL, Rajaram RD, Herrmann N. Therapy for Alzheimer’s disease: how effective are current treatments? Ther Adv Neurol Disord. 2009;2(3):163–180. | |
Latest Facts and Figures Report. 2014. Accessed October 1, 2014. | |
Wimo A, Prince M. World Alzheimer Report 2010: The Global Economic Impact of Dementia. London, United Kingdom: Alzheimer’s Disease International (ADI); 2010. | |
Aisen PS, Saumier D, Briand R, et al. A phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology. 2006;67(10):1757–1763. | |
Doody RS, Gavrilova SI, Sano M, et al. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372(9634):207–215. | |
Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–2070. | |
Green RC, Schneider LS, Amato DA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302(23):2557–2564. | |
Andrews-Hanna JR, Snyder AZ, Vincent JL, et al. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56(5):924–935. | |
Buckner RL, Sepulcre J, Talukdar T, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J Neurosci. 2009;29(6):1860–1873. | |
Davis KD, Kiss ZH, Luo L, Tasker RR, Lozano AM, Dostrovsky JO. Phantom sensations generated by thalamic microstimulation. Nature. 1998;391(6665):385–387. | |
Deep-Brain Stimulation for Parkinson’s Disease Study Group. Deep-brain stimulation of the subthalamic nucleus or the pars interna of the globus pallidus in Parkinson’s disease. N Engl J Med. 2001;345(13):956–963. | |
Weaver FM, Follett K, Stern M, et al. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63–73. | |
Lozano AM, Mayberg HS, Giacobbe P, Hamani C, Craddock RC, Kennedy SH. Subcallosal cingulate gyrus deep brain stimulation for treatment-resistant depression. Biol Psychiatry. 2008;64(6):461–467. | |
Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651–660. | |
Laxton AW, Tang-Wai DF, McAndrews MP, et al. A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann Neurol. 2010;68(4):521–534. | |
Hopper MW, Vogel FS. The limbic system in Alzheimer’s disease. A neuropathologic investigation. Am J Pathol. 1976;85(1):1–20. | |
Yasmin H, Aoki S, Abe O, et al. Tract-specific analysis of white matter pathways in healthy subjects: a pilot study using diffusion tensor MRI. Neuroradiology. 2009;51(12):831–840. | |
Rudebeck SR, Scholz J, Millington R, Rohenkohl G, Johansen-Berg H, Lee AC. Fornix microstructure correlates with recollection but not familiarity memory. J Neurosci. 2009;29(47):14987–14992. | |
Gaffan EA, Gaffan D, Hodges JR. Amnesia following damage to the left fornix and to other sites. A comparative study. Brain. 1991; 114(Pt 3):1297–1313. | |
Oishi K, Mielke MM, Albert M, Lyketsos CG, Mori S. The fornix sign: a potential sign for Alzheimer’s disease based on diffusion tensor imaging. J Neuroimaging. 2012;22(4):365–374. | |
Fletcher E, Raman M, Huebner P, et al. Loss of fornix white matter volume as a predictor of cognitive impairment in cognitively normal elderly individuals. JAMA Neurol. 2013;70(11):1389–1395. | |
Mielke MM, Kozauer NA, Chan KC, et al. Regionally-specific diffusion tensor imaging in mild cognitive impairment and Alzheimer’s disease. Neuroimage. 2009;46(1):47–55. | |
Toda H, Hamani C, Fawcett AP, Hutchison WD, Lozano AM. The regulation of adult rodent hippocampal neurogenesis by deep brain stimulation. J Neurosurg. 2008;108(1):132–138. | |
Hamani C, Stone SS, Garten A, Lozano AM, Winocur G. Memory rescue and enhanced neurogenesis following electrical stimulation of the anterior thalamus in rats treated with corticosterone. Exp Neurol. 2011;232(1):100–104. | |
Smith GS, Laxton AW, Tang-Wai DF, et al. Increased cerebral metabolism after 1 year of deep brain stimulation in Alzheimer disease. Arch Neurol. 2012;69(9):1141–1148. | |
Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer’s disease treatment studies. Am J Psychiatry. 2002;159(5):738–745. | |
Damoiseaux JS, Prater KE, Miller BL, Greicius MD. Functional connectivity tracks clinical deterioration in Alzheimer’s disease. Neurobiol Aging. 2012;33(4):828. e819–e830. | |
Hamani C, McAndrews MP, Cohn M, et al. Memory enhancement induced by hypothalamic/fornix deep brain stimulation. Ann Neurol. 2008;63(1):119–123. | |
Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13. | |
Berg L. Clinical Dementia Rating (CDR). Psychopharmacol Bull. 1988;24(4):637–639. | |
Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141(11):1356–1364. | |
Lyketsos CG, Galik E, Steele C, et al. The General Medical Health Rating: a bedside global rating of medical comorbidity in patients with dementia. J Am Geriatr Soc. 1999;47(4):487–491. | |
Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308–2314. | |
Rosen WG, Terry RD, Fuld PA, Katzman R, Peck A. Pathological verification of ischemic score in differentiation of dementias. Ann Neurol. 1980;7(5):486–488. | |
Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429–435. | |
Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. | |
Alexopoulos GS, Abrams RC, Young RC, Shamoian CA. Cornell scale for depression in dementia. Biol Psychiatry. 1988;23(3):271–284. | |
Jack CR, Bernstein MA, Fox NC, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27(4):685–691. | |
Jack CR, Bernstein MA, Borowski BJ, et al. Update on the magnetic resonance imaging core of the Alzheimer’s disease neuroimaging initiative. Alzheimers Dement. 2010;6(3):212–220. | |
Jagust WJ, Bandy D, Chen K, et al. The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6(3):221–229. | |
Benge JF, Balsis S, Geraci L, Massman PJ, Doody RS. How well do the ADAS-cog and its subscales measure cognitive dysfunction in Alzheimer’s disease? Dement Geriatr Cogn Disord. 2009;28(1):63–69. | |
Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289(21):2819–2826. | |
Farlow M, Potkin S, Koumaras B, Veach J, Mirski D. Analysis of outcome in retrieved dropout patients in a rivastigmine vs placebo, 26-week, Alzheimer disease trial. Arch Neurol. 2003;60(6):843–848. | |
Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006(1):CD005593. | |
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–2414. | |
O’Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer’s research consortium study. Arch Neurol. 2008;65(8):1091–1095. | |
Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. 1997;9(Suppl 1):173–176; discussion 177–178. | |
Morris JC, Storandt M, McKeel DW, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology. 1996;46(3):707–719. | |
Berg L, McKeel DW, Miller JP, Baty J, Morris JC. Neuropathological indexes of Alzheimer’s disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50(4):349–358. | |
Delis DC, Kramer JH, Kaplan E, Ober BA, [Inventors]. California Verbal Learning Test – Second Edition. 2000. | |
Borkowski JG, Benton AL, Spreen O. Word fluency and brain damage. Neuropsychologia. 1967;5:135–140. | |
Benedict R. Brief Visuospatial Learning Test-Revised, Professional Manual. Lutz, FL: Psychological Assessment Resources; 1997. | |
Reitan RM. Validity of the Trail Making test as an indicator of organic brain damage. Perceptual Motor Skills. 1958;8:271–276. | |
Logsdon RG, Gibbons LE, McCurry SM, Teri L. Assessing quality of life in older adults with cognitive impairment. Psychosom Med. 2002;64(3):510–519. | |
Robert P, Ferris S, Gauthier S, Ihl R, Winblad B, Tennigkeit F. Review of Alzheimer’s disease scales: is there a need for a new multi-domain scale for therapy evaluation in medical practice? Alzheimers Res Ther. 2010;2(4):24. | |
Silverman DH, Small GW, Chang CY, et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA. 2001;286(17):2120–2127. | |
Smith GS, de Leon MJ, George AE, et al. Topography of cross-sectional and longitudinal glucose metabolic deficits in Alzheimer’s disease. Pathophysiologic implications. Arch Neurol. 1992;49(11):1142–1150. | |
Kadir A, Andreasen N, Almkvist O, et al. Effect of phenserine treatment on brain functional activity and amyloid in Alzheimer’s disease. Ann Neurol. 2008;63(5):621–631. | |
Stefanova E, Wall A, Almkvist O, et al. Longitudinal PET evaluation of cerebral glucose metabolism in rivastigmine treated patients with mild Alzheimer’s disease. J Neural Transm. 2006;113(2):205–218. | |
Ihl R, Ferris S, Robert P, Winblad B, Gauthier S, Tennigkeit F. Detecting treatment effects with combinations of the ADAS-cog items in patients with mild and moderate Alzheimer’s disease. Int J Geriatr Psychiatry. 2012;27(1):15–21. | |
Pozueta A, Rodríguez-Rodríguez E, Vazquez-Higuera JL, et al. Detection of early Alzheimer’s disease in MCI patients by the combination of MMSE and an episodic memory test. BMC Neurol. 2011;11:78. | |
Hardenacke K, Shubina E, Bührle CP, et al. Deep brain stimulation as a tool for improving cognitive functioning in Alzheimer’s dementia: a systematic review. Front Psychiatry. 2013;4:159. | |
Hescham S, Lim LW, Jahanshahi A, et al. Deep brain stimulation of the forniceal area enhances memory functions in experimental dementia: the role of stimulation parameters. Brain Stimul. 2013;6(1):72–77. | |
Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341–350. | |
Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–321. | |
Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014; 370(4):322–333. | |
Fontaine D, Deudon A, Lemaire JJ, et al. Symptomatic treatment of memory decline in Alzheimer’s disease by deep brain stimulation: a feasibility study. J Alzheimers Dis. 2013;34(1):315–323. | |
Kuhn J, Hardenacke K, Lenartz D, et al. Deep brain stimulation of the nucleus basalis of Meynert in Alzheimer’s dementia. Mol Psychiatry. 2015;20(3):353–360. | |
Gillette-Guyonnet S, Andrieu S, Nourhashemi F, et al. Long-term progression of Alzheimer’s disease in patients under antidementia drugs. Alzheimers Dement. 2011;7(6):579–592. | |
Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol. 1997;42(1):85–94. | |
Mosconi L, Tsui WH, De Santi S, et al. Reduced hippocampal metabolism in MCI and AD: automated FDG-PET image analysis. Neurology. 2005;64(11):1860–1867. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.