Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
Decreased Sputum Type-2 Gene Expression in COPD Current Smokers
Authors Southworth T, Higham A, Beech A ![]() , Li J, Wolosianka S, Singh D
, Li J, Wolosianka S, Singh D
Received 12 June 2025
Accepted for publication 12 September 2025
Published 10 October 2025 Volume 2025:20 Pages 3377—3386
DOI https://doi.org/10.2147/COPD.S546528
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Min Zhang
Thomas Southworth,1,2 Andrew Higham,1 Augusta Beech,1,2 Jian Li,1 Sophie Wolosianka,2 Dave Singh1,2
1Division of Immunology, Immunity to Infection and Respiratory Medicine, School of Biological Sciences, Faculty of Biology, Medicine and Health, University of Manchester and Manchester University NHS Foundation Trust, Manchester, UK; 2Medicines Evaluation Unit, Manchester, UK
Correspondence: Thomas Southworth, Medicines Evaluation Unit, The Langley Building, Southmoor Road, Manchester, M23 9QZ, United Kingdom, Tel +44 161 946 4066, Email [email protected]
Background: Higher blood eosinophil counts in COPD patients are associated with a greater response to inhaled corticosteroid (ICS) treatment, with type 2 (T2) inflammation being the target for ICS in COPD. Current smokers have reduced responses to ICS treatment. We have investigated whether current smoking modulates the levels of T2 mediators in the airways, thereby influencing ICS responsiveness.
Methods: Induced sputum samples were collected from 73 COPD patients, including 41 ex-smokers. Twenty-six patients donated a second sputum sample, approximately 6 months after the initial sample. Sputum cell gene expressions of IL13, CLCA1, CCL26 and CST1 were assessed by quantitative RT-PCR. Differential cell counts were performed.
Results: Expression levels of all four genes significantly correlated with sputum eosinophil percentages. IL13 and CCL26 gene expression levels were significantly lower in COPD current versus ex-smokers (IL13 p< 0.0001; CCL26 p=0.005); there were no differences for CLCA1 or CST1. In repeat samples, IL13, CCL26 and CST1 expression showed good or very good consistency, while CLCA1 levels were more variable.
Conclusion: Sputum gene expression of IL13 and CCL26 is affected by the smoking status of COPD patients and have stable expression over time. These findings implicate IL-13 and CCL26 as key components of T2 inflammation in COPD but also suggest that current smoking skews the immune response away from a T2 profile.
Keywords: COPD, sputum, type 2 inflammation, smoking
Introduction
Inhaled corticosteroids (ICS) are the mainstay of anti-inflammatory treatment in chronic obstructive pulmonary disease (COPD), prescribed as part of combination treatment with one or two long-acting bronchodilators in order to prevent exacerbations.1 In COPD patients with a history of exacerbations, higher blood eosinophil counts (BEC) are associated with greater ICS effects on exacerbation prevention.2–4 BEC are used as a clinical biomarker to help identify COPD patients who are potentially more responsive to ICS treatment.5
BEC are associated with pulmonary eosinophil counts, although this relationship is not strong.6–8 Higher BEC are also associated with increased expression of type-2 (T2) genes in the bronchial epithelium and sputum cells, with IL13, CLCA1, CCL26 and CST1 levels reported as higher in different cohorts,9,10 although the association between BEC and sputum IL13 expression is not always consistent.11 Interleukin (IL)-13 is produced by T-helper 2 cells, type 2 innate lymphoid cells and mast cells and plays a role in eosinophilic inflammation, mucin production and airway remodeling.12 Calcium-activated chloride channel regulator 1 (CLCA1),13 CCL26, also known as eotaxin-3,14 and Cystatin SN (CST1)15 are all produced by airway epithelial cells in response to type 2 inflammatory stimulants, including IL-13. CLCA1 is associated with MUC5AC mucin production,13 CCL26 is an eosinophil chemokine,16 and CST1 is a cysteine protease inhibitor involved in maintaining airway epithelial integrity17 and has recently been shown to be associated with ICS response in COPD patients.18 Clinical trials have reported that the monoclonal antibody dupilimab, which targets the shared IL-13 and IL-4 receptor, reduced exacerbation rates in COPD patients with a history of exacerbations and BEC≥ 300 cells / µL,19 highlighting the ability of BEC to identify COPD patients with T2 inflammation.
The majority of airway sampling studies investigating ICS intervention in COPD have reported no modulation of airway eosinophil counts.20–23 In contrast, bronchial biopsy and airway epithelial brushing transcriptome analysis has shown increased T2 gene expression in COPD patients with higher airway eosinophil counts which was reduced by ICS and was associated with a greater clinical response.24,25 Mast cell numbers were also reduced by ICS treatment. Overall, it appears that ICS do not primarily exert their effects in COPD by reducing eosinophil numbers in the lungs, but by targeting other T2 inflammation mechanisms.
Evidence from randomized clinical trials shows that current smokers have reduced responses to ICS treatment,3,26,27 although this is not a consistent finding across all studies.28,29 Current smoking increases oxidative stress, which has multiple impacts on cell physiology and signalling pathways.30 COPD patients, who are current smokers, have lower levels of sputum supernatant innate immune mediators compared to ex-smokers, including IL-1β, IL-8, MCP-1, MIP-1α, MIP-1β and TNF-α, and lower numbers of sputum neutrophils.31,32 It is unclear whether current smoking also modulates the levels of T2 mediators in the airways, thereby influencing ICS responsiveness. Our previous COPD gene expression work using sputum and bronchoscopy samples had a limited sample size for this analysis of the effects of current smoking.9
Using a larger sample size than our previous study, we have investigated the effects of current smoking on airway IL13, CLCA1, CCL26 and CST1 gene expression in COPD. We also studied the stability of sputum T2 gene expression over 6 months.
Materials and Methods
Subjects
Sputum samples were collected from 73 COPD patients for expression analysis of IL13, CLCA1, CCL26 and CST1; gene expression was previously reported in 33 of these patients,9 while cell counts from 44 patients were part of a previous publication of the effects of current smoking on sputum neutrophil counts.8,32 COPD was diagnosed according to GOLD criteria,5 including a post bronchodilator forced expiratory volume in 1 second/forced vital capacity (FEV1 / FVC) ratio <0.7. All subjects had a smoking history of >10 pack years and were over 40 years of age. The smoking cessation period for ex-smokers was at least 1 year. Twenty-six COPD patients donated a second sputum sample, approximately 6 months after the initial sample. COPD patients were excluded if they had received oral corticosteroids or antibiotics within 6 weeks of sputum donation, or if they had a history of any other chronic respiratory disease. All patients provided written informed consent using protocols that complied with the Declaration of Helsinki and were approved by the local ethics committees (North West – Greater Manchester East [Ref: 05/Q1402/41], North West – Greater Manchester South [Ref: 10/H1003/108] and North West – Preston [Ref: 16/NW/0836]).
Sputum Induction and Processing
Induced sputum samples were collected and processed as previously reported.33 Sputum was processed for differential cell counts and gene expression.33 Briefly, selected sputum plugs were processed preferentially by homogenisation with phosphate-buffered saline (PBS). The sputum cell isolation following a two-step method using Dulbecco’s PBS, then a dithiothreitol step allowing for preparation of cytospins for differential cell counts and cell lysis in RLT buffer (Qiagen, Crawley, UK).
Quantification of Type-2 Inflammation
Following removal of the supernatants, the sputum cell pellet was re-suspended in RLT buffer plus β-mercaptoethanol. Total RNA was purified from cell lysates using RNeasy kits (Qiagen, Crawley, UK) according to manufacturer’s instructions. DNA contamination was prevented by on-column addition of DNase (Qiagen, Crawley, UK) according to manufacturer’s instructions. RNA purity was assessed by spectrophotometry, with samples being excluded if the 260/280nm ratio was outside the 1.7–2.0 range. RNA integrity was not assessed. Reverse transcription was performed on 50 ng of RNA using the Verso cDNA kit (Thermo Fisher Scientific). cDNA was reacted with ABsolute blue qPCR mix (Thermo Fisher Scientific) in 25 µL reactions containing premade ABI Taqman gene expression assays (Catalogue no: 4331182) for IL13 (Hs00174379_m), CLCA1 (Hs00976287_m1), CCL26 (Hs00171146_m1), CST1 (Hs00606961_m1), or the endogenous control glyceraldehyde-3phosphate dehydrogenase (GAPDH) (Catalogue no: 4352934E) (Life Technologies, Parsley, UK). No template control showed there was no amplification. Thermal cycling was carried out on an Agilent MX3005P (Agilent Technologies, West Lothian, UK). Relative expression levels were determined using the 2−ΔCt (cycle threshold of gene of interest minus cycle threshold of GAPDH). Samples with a cycle threshold value of >40 were classed as having undetectable levels of gene expression. To allow for statistical analysis and presentation of data on a logarithmic scale, samples with undetectable levels were given the arbitrary value of 1×10−8; to ensure statistical analysis was not affected, 2−ΔCt values for samples with detectable levels were increased by 1×10−8.
Statistical Analysis
Distribution of data assessed by D’Agostino & Pearson normality test. The proportion of subjects with detectable levels of each gene were compared by Fisher’s exact test, with associations between the genes being assessed by Spearman correlation. The effects of smoking and inhaled corticosteroid treatment on gene expression in COPD patients were assessed by Mann–Whitney test. Comparisons between repeat samples were by Spearman’s rank tests and interclass correlation coefficients (ICCs), following log-transformation to achieve parametric distribution of the data. Sputum data were normalised via a Log(x + 1) transformation to account for zero values. All analysis was performed using Graphpad Prism version 9.2.0 (San Diego, California), except for ICCs, which were assessed using SPSS version 25.0 (IBM, Armonk, New York) and based on an absolute agreement, two-way mixed effects model.
Results
The clinical characteristics of the patients are summarised in Table 1; the mean age was 65.9 years with 32 current smokers (43.8%). The mean FEV1 was 59.3% predicted. COPD ex-smokers had a higher median sputum neutrophil percentage (84.4% vs 67.3%, p=0.02), higher median neutrophil numbers per gram of sputum (7.31 vs 3.44 neutrophils ×106/g, p=0.002), higher total cell numbers per gram of sputum (10.29 vs 5.79 cells ×106/g, p=0.0011) and lower median macrophage percentage (12.6% vs 27.5%, p=0.0009) compared to current smokers. Sputum eosinophil and columnar epithelial cell percentages and numbers per gram of sputum were similar between current and ex-smokers.
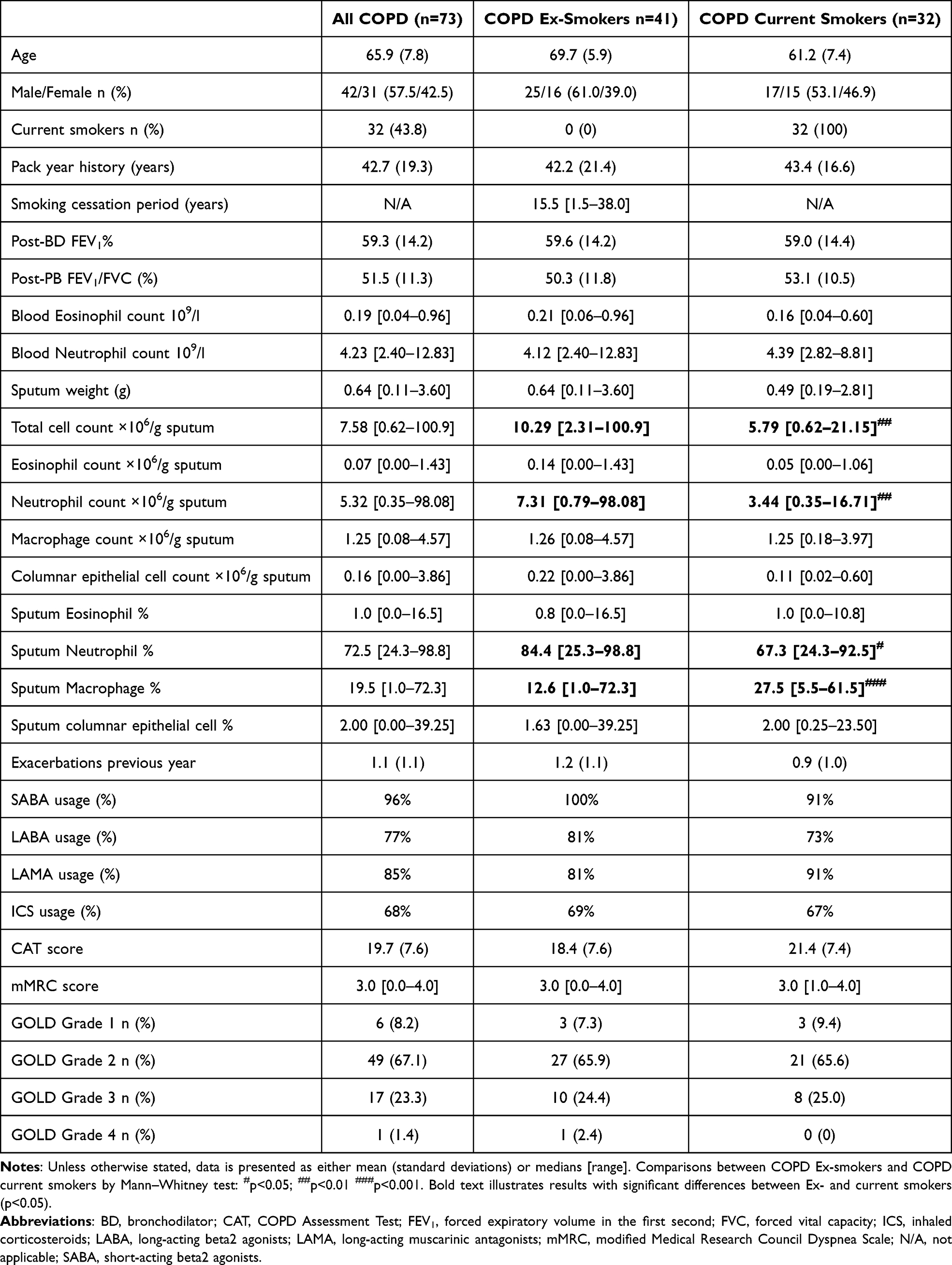 |
Table 1 Subject Clinical Characteristics |
The proportion of subjects with detectable gene expression levels varied across the 4 genes, with IL13 and CCL26 being detected in 89.1% (65/73) of patients, while CLCA1 and CST1 were detected in 35.6% (26/73) and 63.0% (46/73), respectively. The proportion of subjects with detectable levels of IL13 and CCL26 were significantly higher compared to CLCA1 and CST1 (p<0.001 for both comparisons). There were correlations between levels of IL13 and CCL26 (rho: 0.666, p<0.0001), CST1 and CLCA1 (rho: 0.403, p=0.0004), and CST1 and CCL26 (rho: 0.287, p=0.0137) (Supplementary Figure 1 and Supplementary Table 1). Expression levels of all four genes significantly correlated with sputum eosinophil percentages (Figure 1). CST1 (Rho: 0.356, p=0.002) and CLCA1 (Rho: 0.300, p=0.011) showed significant correlation with sputum columnar epithelial percentages, while no correlations were seen for IL13 (Rho: 0.085; p=0.479) or CCL26 (Rho: 0.056; p=0.642). ICS use did not affect gene expression levels (Supplementary Figure 2).
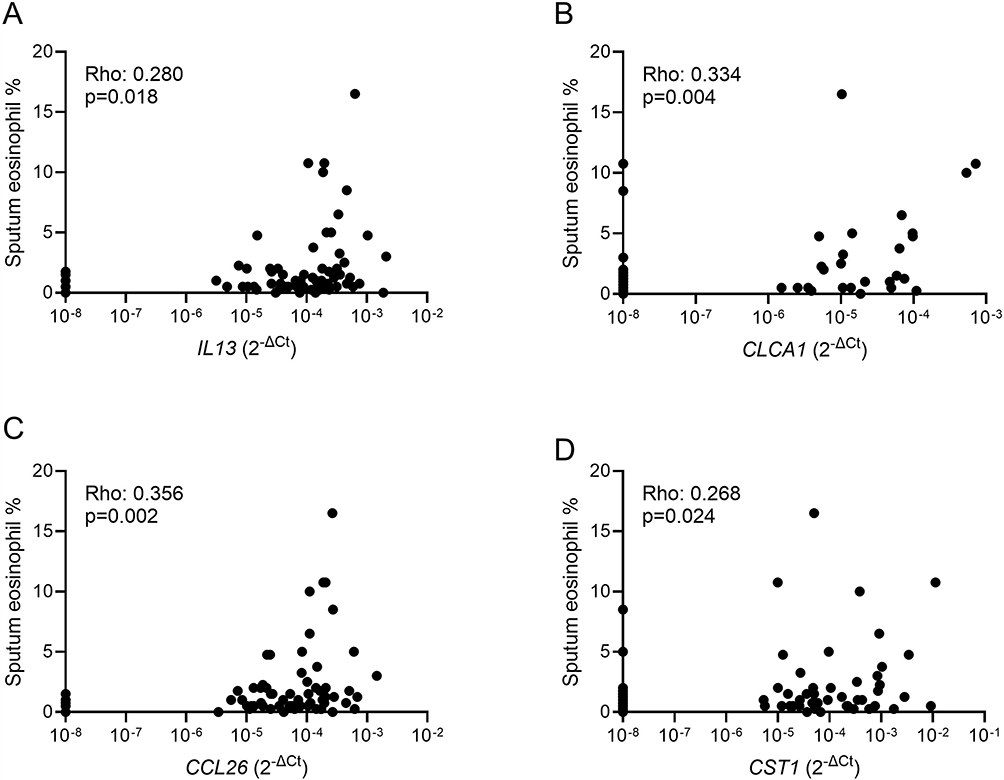 |
Figure 1 Correlations of sputum eosinophil percentages with T2 gene expression in the whole cohort. Associations between eosinophil percentages and IL13 (A), CLCA1 (B), CCL26 (C) and CST1 (D) were assessed by Spearman’s rank test with Rho and p-values are presented for each analysis. |
IL13 and CCL26 gene expression levels were significantly lower in COPD current versus ex-smokers (IL13 p<0.0001; CCL26 p=0.005; Figure 2); there were no differences for CLCA1 or CST1. Statistically significant correlations between sputum eosinophil percentages and the expression of all four genes were observed in ex-smokers, but not current smokers (Table 2 and Supplementary Figure 3).
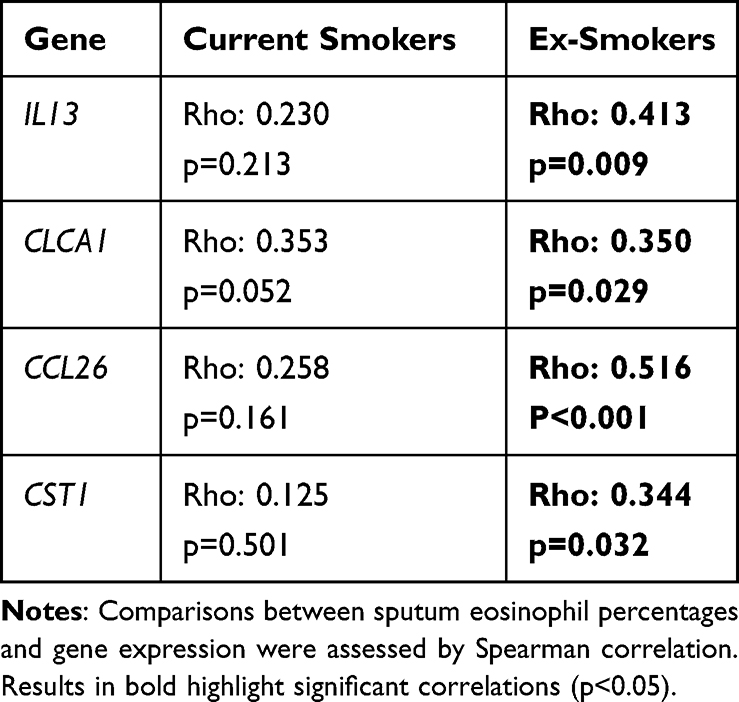 |
Table 2 Correlations Between Sputum Eosinophil Percentages and T2 Gene Expression in Current and Ex-Smokers |
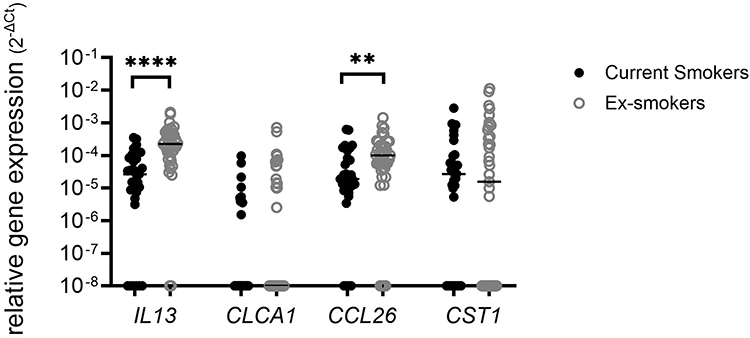 |
Figure 2 Sputum T2 gene expression in current and ex-smoking COPD patients. Gene expression was quantified by RT-PCR. Comparisons between current and ex-smokers were by Mann–Whitney test: **p<0.01; ****p<0.0001. |
As sputum columnar epithelial cell numbers were associated with higher levels of CLCA1 and CST1 expressions, the smoking effect on CLCA1 and CST1 were assessed in a subset of samples with the highest proportion of columnar epithelial cells (≥2% median columnar epithelial level; n=36). In this subset, CLCA1 (median 2−ΔCT: current- 1×10−8; ex- 5.2×10−6; p=0.201) and CST1 (median 2−ΔCT: current- 3.2×10−5; ex- 1.3×10−4; p=0.277) levels were similar in current (n=18) and ex-smokers (n=18).
Gene expression for repeated sampling at 6 months (n=26) is shown in Figure 3. There were significant (p<0.05) associations between repeated samples for IL13, CCL26 and CST1 but not CLCA1. The ICC for CCL26 and CST1 demonstrated good consistency (ICC > 0.5) while for IL13 there was very good consistency (ICC > 0.8) (Figure 3). Significant associations between repeat sputum cell counts were observed in the same samples (Supplementary Figure 4).
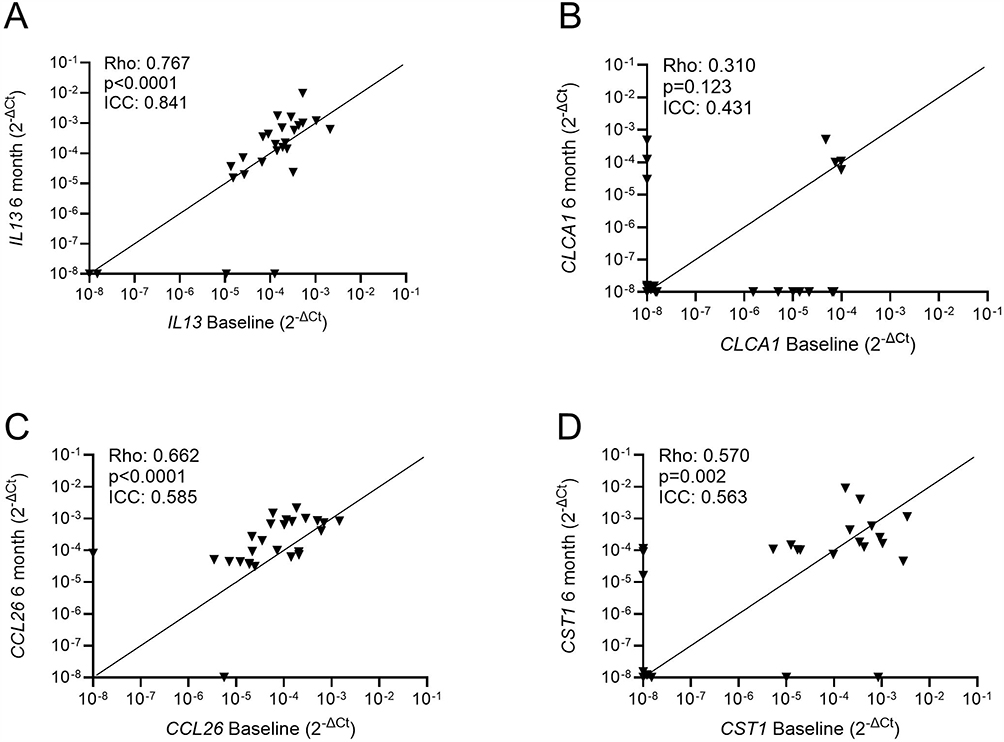 |
Figure 3 Repeat analysis of sputum T2 gene expression in COPD patients. Sputum samples were collected from 26 COPD patients at baseline and at 6 months. Gene expressions for IL13 (A), CLCA1 (B), CCL26 (C) and CST1 (D) were quantified by RT-PCR. Associations between baseline and 6-month measurements were assessed by Spearman’s rank test and Interclass coefficient analysis. Rho, p-values and ICC values are presented for each analysis. The number of patients with undetectable levels at both baseline and 6 months were n=2 for IL13, n=12 for CLCA1, n=0 for CCL26 and n=4 for CST1. |
Discussion
Sputum gene expression of IL13 and CCL26 was lower in COPD current versus ex-smokers. Associations between T2 gene expression and sputum eosinophil counts were present in ex-smokers, but mostly absent in current smokers. IL13 and CCL26 expression were present in more individuals compared to CLCA1 or CST1, indicating that IL13 and CCL26 are more commonly expressed T2 genes in COPD sputum. IL13, CCL26 and CST1 expression showed good to very good consistency when repeated at 6 months.
Higher blood and sputum eosinophil counts are associated with a greater effect of ICS in COPD patients.2–4 This ICS effect seems to be mediated through modulation of T2 inflammation.24,25 Analyses of COPD clinical trials have shown that current smoking reduces the benefit of ICS.34,35 The findings reported here suggest this phenomenon could be due to a reduction in the levels of T2 inflammation in the airways by current smoking. However, smoking does not completely blunt the T2 inflammatory response in COPD as dupilumab, a monoclonal antibody targeting both IL-4 and IL-13 signalling, was effective at reducing exacerbation rates in both current and ex-smokers.19,36
The repeat sampling at 6 months showed at least good consistency (by ICC analysis) for all 4 genes studied, with the strongest correlations (Rho > 0.6) for IL13 and CCL26, which were also the two most highly expressed genes. The consistency in T2 gene expression in the sputum cells was matched with good stability of neutrophil, macrophage and eosinophil cell counts in the same samples. Overall, these results show that IL13 and CCL26 show relatively high expression levels that are consistent over time and sensitive to the effects of current smoking.
Unlike IL13 and CCL26 expression, sputum eosinophil counts did not differ between current and ex-smoking COPD patients, which matches previous findings in sputum studies.37,38 This lack of effect of current smoking on sputum eosinophil counts, in contrast to a reduction of sputum T2 gene expression, explains the loss of correlation between these parameters in current smokers. We have recently reported that the numbers of small airway intra-epithelial eosinophils are lower in COPD current compared to ex-smokers.39 This may be explained by current smoking providing signals that promote the movement of eosinophils across the epithelium into the airway lumen; in support, it has been shown that broncho-alveolar lavage eosinophil counts are increased in COPD current versus ex-smokers.37 The site of sampling is important, as the peripheral lung small airways may give different results to sputum samples from the more proximal airways.
Airway mast cells are a major source of IL-13.40 We have recently shown an association between the expression of the four T2 genes investigated here and mast cell gene signature in sputum cells from COPD patients with high eosinophil counts.41 IgE-independent activation of mast-cells occurs through various stimuli, including eosinophil derived major basic protein and eosinophil cationic protein, and typically occurs through activation of G-protein coupled receptors.42,43 The G-protein subunit Gi3α, which has been linked to IgE-independent activation mechanisms in mast cells,43 is down regulated by cigarette smoke.44 This is one possible mechanism by which T2 gene expression is modulated by cigarette smoke. Alternatively, IL-13 is also secreted by type 2 innate leukocyte cells after IL-33 stimulation.45 Bronchial epithelial expression of IL-33, at both gene and protein levels, is lower in COPD current smokers.46
Fractional exhaled nitric oxide levels are higher in COPD patients compared to healthy subjects, but levels are reduced in COPD patients who continue to smoke.47 IL-13 drives the upregulation of inducible nitric oxide synthase, and the cigarette smoke induced down regulation of IL-13 reported here is likely to be a mechanism relevant to the downregulation of FeNO levels in COPD current smokers.
CCL26 expression can be directly induced by IL-13,48 accounting for the correlation that we observed between these cytokines. The current smoking effect on CCL26 may simply be a downstream response to downregulation of IL13 expression. CCL26 is a potent chemokine for eosinophils.16 However, as sputum eosinophil numbers did not differ between current and ex-smokers, CCL26 may not be the dominant driver of eosinophilia in COPD.
Unlike IL13 and CCL26, CST1 and CLCA1 expression levels did not differ in current and ex-smokers. In a prior asthma study, increased CLCA1 expression was only observed in sputum samples with higher columnar epithelial cell composition.49 We also observed that levels of CLCA1, and CST1, expression correlated with sputum columnar epithelial cell percentage. However, a smoking effect on CST1 and CLCA1 expression was still not observed in an epithelial-high enriched sample subset. In airway epithelial cells, IL-13 stimulates expression of CLCA1, with this induction being cigarette smoke sensitive.50 This mechanism may not be relevant in sputum cells. While IL13 and CCL26 were more commonly expressed compared to CST1 and CLCA1, all 4 genes showed associations with sputum eosinophil counts, implicating all these genes in T2 inflammation in COPD although the extent may vary between patients. Furthermore, our analysis was based on sputum samples, and different results could be obtained from other airway samples. For example, CLCA1 is expressed in bronchial brushings and is typically associated with mucin production from airway epithelial cells.9,51
CST1 stimulates IL-5 production in nasal polyp tissue,52 providing mechanistic support to explain the correlation between CST1 expression and eosinophil numbers in COPD patients. Recombinant CST1 induces expression of eosinophil cationic protein (ECP) and eosinophil peroxidase (EPX) in human blood eosinophils, as well as increasing cell surface expression of CD69,52 suggesting a role for CST1 in eosinophil activation and degranulation. While CST1 expression was stable, detectable levels of CST1 and CLCA1 were lower compared to IL13 and CCL26. CLCA1 had the lowest expression of the 4 genes assessed with inconsistency in detection. Assay sensitivity at low gene expression levels can contribute to the lower consistency during repeat sampling.
A limitation of this study is that analysis of T2 gene expression was restricted to sputum only. Other cell types in the lung, such as airway epithelial cells, also express T2 genes, such as IL13.25 The effect of smoking status on T2 gene expression in these cell types requires further investigation. Expression of IL13 was affected by smoking status, so it would be of interest to assess smoking effects on the other helper-2 T-cell cytokines IL4 and IL5. This was not possible in this investigation as it was an extension of a previous study which did not assess IL4 and IL5 and there were insufficient RNA samples remaining to enable additional analysis. Only a single reference gene, GAPDH, was used to assess T2 gene expression. The use of multiple and alternative references genes53 may have revealed other differences in gene expression.
Conclusions
We have shown that sputum gene expression of IL13 and CCL26 are affected by the smoking status of COPD patients but are readily detected and have stable expression over time. These findings further implicate IL-13 as a key component of T2 inflammation in COPD and suggest that the level of T2 inflammation is modified by current smoking.34,35
Abbreviations
BEC, Blood eosinophil counts; CAT, COPD Assessment Test; CCL26, C-C Motif Chemokine Ligand 26; CD69, Cluster of differentiation 69; cDNA, Copy deoxyribonucleic acid; CLCA1, Calcium-activated chloride channel regulator 1; COPD, Chronic Obstructive Pulmonary Disease; CST1, Cystatin SN; DNA, Deoxyribonucleic acid; DNase, Deoxyribonuclease; ECP, Eosinophil cationic protein; EPX, Eosinophil peroxidase; FeNO, Fractional Exhaled Nitric Oxide; FEV1, Forced expiratory volume in the first second; FVC, Forced vital capacity; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; GOLD, Global Initiative for Chronic Obstructive Lung Disease; ICC, Intraclass correlation coefficient; ICS, Inhaled corticosteroids; IgE, Immunoglobulin E; IL-13, Interleukin 13; IL-1β, Interleukin 1 beta; IL-33, Interleukin 33; IL-5, Interleukin 5; IL-8, Interleukin 8; LABA, Long-acting beta2 agonists; LAMA, Long-acting muscarinic antagonists; MCP-1, Monocyte chemoattractant protein-1; MIP-1β, Macrophage inflammatory protein 1 beta; mMRC, modified Medical Research Council Dyspnea Scale; NHS, National Health Service; NIHR, National institute for Healthy research; PBS, Phosphate buffered saline; qPCR, Quantifiable polymerase chain reaction; RNA, Ribonucleic acid; RT-PCR, Reverse transcription-polymerase chain reaction; SABA, Short-acting beta2 agonists; T2, Type-2 inflammation; TNFα, Tumor necrosis factor alpha.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
This research was supported by the Medicines Evaluation Unit, the North West Lung Centre Charity, Manchester, UK and Astra Zeneca. GB and DS are supported by the National Institute for Health and Care Research (NIHR) Manchester Biomedical Research Centre (BRC) (NIHR203308). This report is independent research, and the views expressed in this publication are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Disclosure
DS reports grants from AstraZeneca during the conduct of the study, personal consulting fees from Advocate, Aerogen, Almirall, Apogee, Arrowhead, AstraZeneca, Bial, Boehringer Ingelheim, Chiesi, Cipla, Connect Pharma, Covis, CSL Behring, DevPro Biopharma, Elpen, Empirico, EpiEndo, Genentech, Generate Biomedicines, GlaxoSmithKline, Glenmark, Gossamerbio, Kamada, Kinaset Therapeutics, Kymera, Menarini, MicroA, Novarti, O M Pharma, Orion, Pieris Pharmaceuticals, Pulmatrix, Revolo, Roivant Sciences, Sanofi, Synairgen, Tetherex, Teva, Theravance Biopharm, Upstream and Verona outside of the submitted work. TS reports grants from AstraZeneca during the conduct of the study. AB was supported by AstraZeneca during the conduct of the study. SW reports grants from AstraZeneca outside this work. The authors declare no other competing interests for this work.
References
1. Singh D. Pharmacological treatment of stable chronic obstructive pulmonary disease. Respirology. 2021;26(7):643–651. doi:10.1111/resp.14046
2. Singh D, Bafadhel M, Brightling CE, et al. Blood eosinophil counts in clinical trials for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2020;202(5):660–671. doi:10.1164/rccm.201912-2384PP
3. Bafadhel M, Peterson S, De Blas MA, et al. Predictors of exacerbation risk and response to budesonide in patients with chronic obstructive pulmonary disease: a post-hoc analysis of three randomised trials. Lancet Respir Med. 2018;6(2):117–126. doi:10.1016/S2213-2600(18)30006-7
4. Singh D, Agusti A, Martinez FJ, et al. Blood eosinophils and chronic obstructive pulmonary disease: a global initiative for chronic obstructive lung disease science committee 2022 review. Am J Respir Crit Care Med. 2022;206(1):17–24. doi:10.1164/rccm.202201-0209PP
5. Agusti A, Celli BR, Criner GJ, et al. Global initiative for chronic obstructive lung disease 2023 report: GOLD executive summary. Eur Respir J. 2023;61(4):2300239. doi:10.1183/13993003.00239-2023
6. Hastie AT, Martinez FJ, Curtis JL, et al. Association of sputum and blood eosinophil concentrations with clinical measures of COPD severity: an analysis of the SPIROMICS cohort. Lancet Respir Med. 2017;5(12):956–967. doi:10.1016/S2213-2600(17)30432-0
7. Singh D, Kolsum U, Brightling CE, et al. Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J. 2014;44(6):1697–1700. doi:10.1183/09031936.00162414
8. Beech A, Jackson N, Singh D. Identification of COPD inflammatory endotypes using repeated sputum eosinophil counts. Biomedicines. 2022;10(10):2611. doi:10.3390/biomedicines10102611
9. Higham A, Beech A, Wolosianka S, et al. Type 2 inflammation in eosinophilic chronic obstructive pulmonary disease. Allergy. 2021;76(6):1861–1864. doi:10.1111/all.14661
10. George L, Taylor AR, Esteve-Codina A, et al. Blood eosinophil count and airway epithelial transcriptome relationships in COPD versus asthma. Allergy. 2020;75(2):370–380. doi:10.1111/all.14016
11. Fricker M, McDonald VM, Winter NA, et al. Molecular markers of type 2 airway inflammation are similar between eosinophilic severe asthma and eosinophilic chronic obstructive pulmonary disease. Allergy. 2021;76(7):2079–2089. doi:10.1111/all.14741
12. Marone G, Granata F, Pucino V, et al. The intriguing role of interleukin 13 in the pathophysiology of asthma. Front Pharmacol. 2019;10:1387. doi:10.3389/fphar.2019.01387
13. Alevy YG, Patel AC, Romero AG, et al. IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J Clin Invest. 2012;122(12):4555–4568. doi:10.1172/JCI64896
14. Provost V, Langlois A, Chouinard F, et al. Leukotriene D4 and interleukin-13 cooperate to increase the release of eotaxin-3 by airway epithelial cells. PLoS One. 2012;7(8):e43544. doi:10.1371/journal.pone.0043544
15. Imoto Y, Tokunaga T, Matsumoto Y, et al. Cystatin SN upregulation in patients with seasonal allergic rhinitis. PLoS One. 2013;8(8):e67057. doi:10.1371/journal.pone.0067057
16. Provost V, Larose MC, Langlois A, Rola-Pleszczynski M, Flamand N, Laviolette M. CCL26/eotaxin-3 is more effective to induce the migration of eosinophils of asthmatics than CCL11/eotaxin-1 and CCL24/eotaxin-2. J Leukoc Biol. 2013;94(2):213–222. doi:10.1189/jlb.0212074
17. Yao L, Yuan X, Fu H, et al. Epithelium-derived cystatin SN inhibits house dust mite protease activity in allergic asthma. Allergy. 2023;78(6):1507–1523. doi:10.1111/all.15739
18. Fang J, Wolters JC, Rafie K, et al. Extracellular vesicles from bronchoalveolar lavage fluid provide insights into the inhaled corticosteroids treatment response in COPD. Respir Res. 2025;26(1):254. doi:10.1186/s12931-025-03330-6
19. Bhatt SP, Rabe KF, Hanania NA, et al. Dupilumab for COPD with type 2 inflammation indicated by eosinophil counts. N Engl J Med. 2023;389(3):205–214. doi:10.1056/NEJMoa2303951
20. Brightling CE, McKenna S, Hargadon B, et al. Sputum eosinophilia and the short term response to inhaled mometasone in chronic obstructive pulmonary disease. Thorax. 2005;60(3):193–198. doi:10.1136/thx.2004.032516
21. Reid DW, Wen Y, Johns DP, Williams TJ, Ward C, Walters EH. Bronchodilator reversibility, airway eosinophilia and anti-inflammatory effects of inhaled fluticasone in COPD are not related. Respirology. 2008;13(6):799–809. doi:10.1111/j.1440-1843.2008.01380.x
22. Yildiz F, Kaur AC, Ilgazli A, et al. Inhaled corticosteroids may reduce neutrophilic inflammation in patients with stable chronic obstructive pulmonary disease. Respiration. 2000;67(1):71–76. doi:10.1159/000029466
23. Lea S, Higham A, Beech A, Singh D. How inhaled corticosteroids target inflammation in COPD. Eur Respir Rev. 2023;32(170):230084. doi:10.1183/16000617.0084-2023
24. Christenson SA, Steiling K, van den Berge M, et al. Asthma-COPD overlap. Clinical relevance of genomic signatures of type 2 inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(7):758–766. doi:10.1164/rccm.201408-1458OC
25. Leung C, Park HY, Li X, et al. Transcriptomic profiling of the airway epithelium in COPD links airway eosinophilia to type 2 inflammation and corticosteroid response. Eur Respir J. 2025;65:2401875. doi:10.1183/13993003.01875-2024
26. Pascoe S, Barnes N, Brusselle G, et al. Blood eosinophils and treatment response with triple and dual combination therapy in chronic obstructive pulmonary disease: analysis of the IMPACT trial. Lancet Respir Med. 2019;7(9):745–756. doi:10.1016/S2213-2600(19)30190-0
27. Bardsley S, Criner GJ, Halpin DMG, et al. Single-inhaler triple therapy fluticasone furoate/umeclidinium/vilanterol versus dual therapy in current and former smokers with COPD: IMPACT trial post hoc analysis. Respir Med. 2022;205:107040. doi:10.1016/j.rmed.2022.107040
28. Bafadhel M, Han MK, Singh D, Jenkins M, Dorinsky P, Patel M. A post-hoc analysis of the effect of smoking status on COPD exacerbation reductions with budesonide/glycopyrrolate/formoterol fumarate in patients with COPD in the ETHOS study [abstract]. Am J Respir Crit Care Med. 2022;205:A1451.
29. Papi A, Vestbo J, Fabbri L, et al. Extrafine inhaled triple therapy versus dual bronchodilator therapy in chronic obstructive pulmonary disease (TRIBUTE): a double-blind, parallel group, randomised controlled trial. Lancet. 2018;391(10125):1076–1084. doi:10.1016/S0140-6736(18)30206-X
30. Cha SR, Jang J, Park SM, Ryu SM, Cho SJ, Yang SR. Cigarette smoke-induced respiratory response: insights into cellular processes and biomarkers. Antioxidants. 2023;12(6). doi:10.3390/antiox12061210
31. Mulvanny A, Pattwell C, Beech A, Southworth T, Singh D. Validation of sputum biomarker immunoassays and cytokine expression profiles in COPD. Biomedicines. 2022;10(8):1949. doi:10.3390/biomedicines10081949
32. Higham A, Beech A, Jackson N, Lea S, Singh D. Sputum cell counts in COPD patients who use electronic cigarettes. Eur Respir J. 2022;59(5):2103016. doi:10.1183/13993003.03016-2021
33. Beech A, Lea S, Li J, Jackson N, Mulvanny A, Singh D. Airway bacteria quantification using polymerase chain reaction combined with neutrophil and eosinophil counts identifies distinct COPD endotypes. Biomedicines. 2021;9(10):1337. doi:10.3390/biomedicines9101337
34. Bhatt SP, Anderson JA, Brook RD, et al. Cigarette smoking and response to inhaled corticosteroids in COPD. Eur Respir J. 2018;51(1):1701393. doi:10.1183/13993003.01393-2017
35. Sonnex K, Alleemudder H, Knaggs R. Impact of smoking status on the efficacy of inhaled corticosteroids in chronic obstructive pulmonary disease: a systematic review. BMJ Open. 2020;10(4):e037509. doi:10.1136/bmjopen-2020-037509
36. Bhatt SP, Rabe KF, Hanania NA, et al. Dupilumab for COPD with blood eosinophil evidence of type 2 inflammation. N Engl J Med. 2024;390(24):2274–2283. doi:10.1056/NEJMoa2401304
37. Martinez CH, Li SX, Hirzel AJ, et al. Alveolar eosinophilia in current smokers with chronic obstructive pulmonary disease in the SPIROMICS cohort. J Allergy Clin Immunol. 2018;141(1):429–432. doi:10.1016/j.jaci.2017.07.039
38. Wen Y, Reid DW, Zhang D, Ward C, Wood-Baker R, Walters EH. Assessment of airway inflammation using sputum, BAL, and endobronchial biopsies in current and ex-smokers with established COPD. Int J Chron Obstruct Pulmon Dis. 2010;5:327–334. doi:10.2147/COPD.S11343
39. Beech A, Booth S, Higham A, Singh D. Current smoking reduces small airway eosinophil counts in COPD. ERJ Open Res. 2023. (in press).
40. Saha SK, Berry MA, Parker D, et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;121(3):685–691. doi:10.1016/j.jaci.2008.01.005
41. Higham A, Dungwa J, Pham TH, McCrae C, Singh D. Increased mast cell activation in eosinophilic chronic obstructive pulmonary disease. Clin Transl Immunol. 2022;11(9):e1417. doi:10.1002/cti2.1417
42. Ogasawara H, Furuno M, Edamura K, Noguchi M. Peptides of major basic protein and eosinophil cationic protein activate human mast cells. Biochem Biophys Rep. 2020;21:100719. doi:10.1016/j.bbrep.2019.100719
43. Munitz A, Piliponsky AM, Levi-Schaffer F. IgE-independent activation of human mast cells indicates their role in the late phase reaction of allergic inflammation. Cell Tissue Bank. 2003;4(1):25–28. doi:10.1023/A:1026307812980
44. Orouk I, Bellehsen L, Levi-Shaffer F. Does exposure to cigarette smoke compromise mast cell function?: implications for chronic lung inflammation and host defense against pathogens. Internet J Asthma Allergy Immunol. 2006;6(1).
45. Kato A. Group 2 innate lymphoid cells in airway diseases. Chest. 2019;156(1):141–149. doi:10.1016/j.chest.2019.04.101
46. Faiz A, Mahbub RM, Boedijono FS, et al. IL-33 expression is lower in current smokers at both transcriptomic and protein level. Am J Respir Crit Care Med. 2023;208:1075–1087. doi:10.1164/rccm.202210-1881OC
47. Higham A, Beech A, Dean J, Singh D. Exhaled nitric oxide, eosinophils and current smoking in COPD patients. ERJ Open Res. 2023;9:00686–2023. (in press). doi:10.1183/23120541.00686-2023
48. Banwell ME, Tolley NS, Williams TJ, Mitchell TJ. Regulation of human eotaxin-3/CCL26 expression: modulation by cytokines and glucocorticoids. Cytokine. 2002;17(6):317–323. doi:10.1006/cyto.2002.1021
49. Qin L, Gibson PG, Simpson JL, et al. Dysregulation of sputum columnar epithelial cells and products in distinct asthma phenotypes. Clin Exp Allergy. 2019;49(11):1418–1428. doi:10.1111/cea.13452
50. Mertens TCJ, van der Does AM, Kistemaker LE, Ninaber DK, Taube C, Hiemstra PS. Cigarette smoke differentially affects IL-13-induced gene expression in human airway epithelial cells. Physiol Rep. 2017;5(13):e13347. doi:10.14814/phy2.13347
51. Iwashita H, Fujimoto K, Morita S, Nakanishi A, Kubo K. Increased human Ca(2)(+)-activated Cl(-) channel 1 expression and mucus overproduction in airway epithelia of smokers and chronic obstructive pulmonary disease patients. Respir Res. 2012;13(1):55. doi:10.1186/1465-9921-13-55
52. Yan B, Lou H, Wang Y, et al. Epithelium-derived cystatin SN enhances eosinophil activation and infiltration through IL-5 in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2019;144(2):455–469. doi:10.1016/j.jaci.2019.03.026
53. Moermans C, Deliege E, Pirottin D, et al. Suitable reference genes determination for real-time PCR using induced sputum samples. Eur Respir J. 2019;54(6):1800644. doi:10.1183/13993003.00644-2018
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.