")
Back to Archived Journals » Metalloproteinases In Medicine » Volume 4
dCas9-mediated transcriptional activation of tissue inhibitor of metalloproteinases
Authors Duellman T, Doll A, Chen X, Wakamiya R, Yang J
Received 19 July 2017
Accepted for publication 30 August 2017
Published 19 September 2017 Volume 2017:4 Pages 63—73
DOI https://doi.org/10.2147/MNM.S146752
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yoshifumi Itoh
Tyler Duellman, Andrea Doll, Xi Chen, Rie Wakamiya, Jay Yang
Department of Anesthesiology, University of Wisconsin SMPH, Madison, WI, USA
Abstract: Selective gene activation with the dCas9 (deactivated clustered regularly interspaced short palindromic repeats [CRISPR] associated protein 9)/CRISPR targeting of a transcriptional activator effector is now well established. However, the optimal targeting of guide RNA (gRNA) for a given gene is largely a matter of trial and error. We explored the optimal targeting site for tissue inhibitor of metalloproteinases (TIMPs) by first screening multiple gRNA target sites using a luciferase-based promoter-reporter system and next confirmed the effective TIMP induction in the mouse motor neuron-like neuron-enriched spinal cord 34 (NSC34) cells. Screening of many gRNAs targeting the 1–1.9 kB promoter regions of TIMP1–3 identified several hot-spots for optimal gene induction, however, no general pattern defining the optimal target site with respect to the proximity of known transcription factor binding sites or distance from the start ATG was apparent. TIMP2 with a larger basal transcriptional activity showed a greater fold-induction with gRNA compared with TIMP1 or 3 supporting the importance of an open-chromatin for best gRNA-mediated transcriptional induction. The rank order of induction potency for different gRNA identified in the promoter-reporter screening held true for the NSC34 cells. Co-activation with multiple gRNAs greatly increased the gene induction.
Keywords: TIMP, promoter, CRISPR, transcriptional activation, gRNA
Introduction
Matrix metalloproteinases (MMPs) constitute a family of zinc-dependent proteases with pleotropic roles in normal biology and diseases. Because of the promiscuous role in various diseases, the development of small molecule selective inhibitors of MMPs has been an active area of investigation for some time.1,2 The MMP family is divided into subfamilies according to the substrate specificity and structural similarities. In addition to the traditional role of regulating the extracellular matrix, it is now clear that MMPs play a broader role in modulating inflammation and a role in overall neuronal survival and toxicity within the nervous system.3,4 MMPs are also recognized as playing a key role in essential activities such as neural plasticity and associative learning5 and a broad-spectrum inhibition of MMP is undesirable.6 In most cells excluding the neutrophils, the expression of MMPs is largely regulated at the transcriptional level, although post-transcriptional and epigenetic regulations have been reported (reviewed by Vandooren et al).7 Once translated and activated through proteolytic cleavage of the regulatory N-terminus, the active MMP protein is further regulated by circulating α-microglobulin and the tissue-resident tissue inhibitor of metalloproteinase (TIMP) proteins.8
Selective transcriptional activation of a desired gene can be accomplished with the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system where the gRNA targeted effector is replaced from the native nuclease Cas to enzymatically deactivated CRISPR associated proteins (dCas) fused to a heterologous transcriptional activator.9–12 The CRISPR/dCas system is particularly attractive since multiple gRNA can simultaneously target the effector to multiple sites along the genome to maximize transcriptional activation of a single gene or co-activate several different genes appropriate for the biological goal. While the concept of targeting the transactivating effector to a regulatory sequence is now well established, the optimal location of gRNA targeting for maximal transcriptional activation of any given gene is mostly a matter of trial and error since no universal rule for optimal targeting has been reported.
We explored the possibility of selective transcriptional activation of TIMP in the neuron-enriched spinal cord 34 (NSC34) motoneurons-like cells13 using the CRISPR/Cas system with the goal of developing a tool that may be useful in selectively inhibiting MMP activity in the spinal motoneurons implicated in the pathogenesis of amyotrophic lateral sclerosis (ALS).14 Specifically, we systematically explored the optimal site of gRNA targeting within the reported regulatory region of mouse TIMPs first using a promoter-reporter luciferase assay in transfected human embryonic kidney (HEK)293 cells, then comparing the efficacy of several dCas effectors reported in the literature, and next confirming the selective transcriptional activation of TIMPs within the normal genomic context of an intact cell.
Material and methods
Plasmid constructs: Mouse TIMP promoter-reporters (luciferase): A 1900 bp TIMP-1 promoter sequence spanning from 1026 bp upstream of the start ATG through the 9 bp of exon 2 was synthesized (GenScript, Piscataway, NJ, USA) and subcloned into the pGL4.10 (Promega, Madison, WI, USA) promoter-less firefly luciferase reporter vector. A similar promoter sequence driving the expression of a lacZ reporter was shown to reproduce the spatial and temporal expression patterns of TIMP1 gene in a mouse embryo.15 TATA-containing 1158 bp TIMP-2 and 1154 bp TIMP-3 promoter sequences located immediately upstream of the start codon reported to exhibit promoter activity16,17 were synthesized and subcloned upstream of firefly luciferase reporter vector. The promoter sequence for mouse TIMP-2 was deduced from the human sequence. The 3 TIMP promoter sequences are listed in Table S1. The 5xGAL4 sequence polymerase chain reaction (PCR) amplified from the pG5luc vector (Promega) flanked by the Mlu I and Bam HI restriction enzyme sites was subcloned upstream of the minCMV.mCherry sequence in the pAAV-minCMV-mCherry vector (Addgene #27,970) to create the control GAL4-mCherry reporter.
Determination of guide RNA target sites and creation of gRNA: gRNA targeting loci were manually chosen in an unbiased manner by scanning the promoter sequence for SpCas9 PAM sequences. Several target loci were chosen to obtain an equal representation of bottom and top targeting gRNA throughout the length of the promoter sequence (Table S2).
Various Cas-effectors: VP64 and VP160 sequences containing 4 or 10 copies of the nucleotides encoding VP16 minimal transactivator sequence DALDDFDLDML18 (Table S3) were synthesized (GenScript) and subcloned in the N-terminus of dCas9 in the pCI-neo vector (Promega). The dCas9. VP64-p65-Rta (VPR) construct19 which served as the template for the various PCR amplifications was purchased from Addgene (#63,798; Cambridge, MA, USA). All VP-based constructs were subcloned into the pCI/neo expression vector (Promega) for uniformity. The Cas-effector with synthetic activation motif (SAM) targeting the mouse TIMP1 gene was purchased from Santa Cruz Biotechnology (sc-423402-ACT, Santa Cruz, CA, USA).
Cell culture and transfection: HEK293 (#CRL-1572; ATCC, Manassas, VA, USA) and mouse motor neuron-like NSC34 cells (a kind gift from Dr. Arnold Ruoho, University of Wisconsin) were grown in Dulbecco’s Modification of Eagle’s Medium (DMEM) (Mediatech Inc, Manassas, VA, USA) with 4.5 g/L glucose, L-glutamine, sodium pyruvate, and supplemented with 10% heat inactivated fetal bovine serum (FBS) (Mediatech Inc), 100 U/mL penicillin (Mediatech Inc), and 0.1 mg/mL streptomycin (Mediatech Inc). Transfection was performed using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. The use of the cell lines was approved by the University of Wisconsin Office of Biological Safety.
Quantitative real-time polymerase chain reaction (qRT-PCR): Total RNA was isolated with RNeasy (Qiagen, Germantown, MD, USA) per the manufacturer’s instructions. cDNA was reverse transcribed from 750 ng of total RNA using iScript Reverse Transcriptase (BioRad, Hercules, CA, USA). Quantitative PCR was performed in a 20 µL reaction volume containing 10 µL of Eva Green qPCR Master mix (MIDSCI, Valley Park, MO, USA; #BEQPCR-LR), 0.2 µM of a mixture of forward and reverse primers, 1 µL of cDNA template, and H2O using the MX3000P instrument (Stratagene, San Diego, CA, USA) with the following amplification conditions: 95°C for 10 minutes, 40 cycles of 30 seconds at 95°C and 60 seconds at 60°C. PCR specificity was confirmed by verifying that the melting curve demonstrated a clear single peak and agarose gel analysis of the PCR amplification product. Relative expression was quantified using the ∆∆Ct method, in which glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal standard. Validated qPCR primers were obtained from Qiagen: mouse GAPDH (PPM02946E), mouse TIMP1 (PPM03693F), mouse TIMP2 (PPM03614C), and mouse TIMP3 (PPM03453F). Experiments were performed from 4 biological replicates with 2 assay duplicates for all conditions.
Western blot: Cell pellets were lysed in radioimmunoprecipitation assay buffer (10 mM Tris-Cl pH 7.5, 50 mM NaCl, 1 mM sodium orthovanadate, 30 mM sodium pyrophosphate, 50 mM NaF, 1% NP40, 0.1% sodium dodecyl sulfate, 1 mM phenylmethylsulfonyl fluoride, 1% Triton X-100, and 0.5% sodium deoxycholate) and collected for analysis. After bicinchoninic acid quantitation, 20 mg lysate/lane was run on a 10% polyacrylamide gel and transferred onto a 0.45 mm nitrocellulose membrane. Membranes were probed with anti-hemagglutinin (HA) (Covance, Princeton, NJ, USA) at 1:2000 and anti-mouse horseradish peroxidase (Jackson Labs, Bar Harbor, ME, USA) at 1:5000 followed by imaging using the Quality One software (BioRad) with a Chemi-Doc XRS + Molecular Imager (BioRad). Densitometric quantitation of Western blots was performed using Image Lab 4.1 software (BioRad).
Promoter-reporter assay: The mouse TIMP1, 2, and 3 promoter-luciferase reporters were transfected along with a Renilla luciferase expressing plasmid (pGL4.75, Promega) into HEK293 cells in OPTI-MEM (Life Technologies). Six hours post-transfection, cells were seeded in triplicate in 1% FBS/DMEM in a 96-well plate. Preliminary time course experiments indicated adequate reporter expression at 24 hours after transfection (Figure S2). Therefore, all subsequent assays were done at 24 hours post-transfection. Cells were analyzed for the promoter activity (firefly luciferase) and normalized for potential transfection variability by Renilla luciferase using a Dual-Glo Luciferase Reporter kit (Promega) following the kit’s instructions using the Synergy 2 microplate reader (BioTek Inc, Winooski, VT, USA).
MMP activity assays: Inhibition of MMP activity by the cell culture supernatant was assessed by a fluorogenic substrate assay and reverse zymography. MMP fluorogenic substrate (Enzo Life Sciences, Farmingdale, NY, USA; BMP-P128–0001) diluted to a final concentration of 20 µM in 100 µL of 2X assay buffer (100 mM HEPES, 20 mM CaCl2, 0.1% Brij-35, pH 7.0) was mixed 1:1 with the cell culture supernatant and kinetic measurements (360/40 nm excitation; 460/40 emission) obtained every minute for 30 minutes on a Synergy 2 microplate reader (BioTek Inc.). The cell culture supernatant of NSC34 failed to exhibit endogenous MMP catalytic activity and the reaction was supplemented with 100 nM recombinant MMP9 catalytic core (Enzo Life Sciences; BML-SE360-0010) to allow assessment of inhibitor activity in the culture supernatant. The slope of the fluorescence vs time plot was taken as the measure of relative catalytic activity. For reverse zymography, a 12% polyacrylamide gel supplemented with 0.1% gelatin and 160 ng/mL recombinant MMP2 loaded with 15 µL cell culture supernatant and 3 µL non-reducing loading buffer was separated by electrophoresis. The enzyme in the gel was renatured for 1 hour in Triton X-100 (2.5% v/v), developed overnight at 37°C in a buffer (50 mM Tris, 200 mM NaCl, 50 mM CaCl2, 5 µM ZnCl2, 0.02% Brij-35, pH 7.5), stained (0.5% coomassie blue in 30% methanol, 10% acetic acid) for 2 hours at room temperature, and destained (30% methanol, 10% acetic acid) until the bands were visible. Gels were placed on a white light box and images captured on a Chemi-Doc XRS + Molecular Imager (BioRad).
Fluorescence microscopy: dCas9.2A.eGFP (enhanced green fluorescent protein) constructs were transfected into HEK293 cells. Twenty-four hours post-transfection, cells were imaged with a Sensicam camera (PCO-Tech Inc, Romulus, MI, USA) mounted on an Olympus IX50 microscope (Olympus Corporations, Tokyo, Japan) using the IPLab3.6 software (Spectra Services, Ontario, NY, USA). Pseudo-color for eGFP (green) was accomplished using Adobe Photoshop CS5.1 (Mountain View, CA, USA).
Immunocytochemistry: 24 hours after HEK293 cells were transfected with dCas9.HA.2A.eGFP, they were fixed in 4% paraformaldehyde (in 0.1 M phosphate buffer, pH 7.4) blocked in 10% donor goat serum/PBST (PBS with 0.2% Triton X-100) and immuno-stained with anti-HA (1:200; Covance) in PBST with 2% normal goat serum for 2 hours followed by an Alexa 594-conjugated secondary antibody (1:250; Life Technologies) for 1 hour. Nuclear staining was performed using 500 ng/mL Hoechst stain and washed with PBST. Images were captured and analyzed with fluorescence microscopic images.
Results
Previous reports suggested the placement of the transactivation domain on the C-terminus of the dCas9 protein.9–11 However, prior to initiating the study, we wanted to re-explore the optimal location for placing the transactivator domain within the dCas protein to attain maximum gene activation. Starting from the PCR-amplified dCas9-eGFP vector insert subcloned into a eukaryotic expression vector, we placed the VP64 module on the N-terminal, C-terminal, or both ends of the dCas9, with dCas9 alone with no VP64 serving as a negative control construct (Figure S1A). All constructs expressed well but dCas9 with 2 VP64 only expressed at half the level of the other 3 and dCas9.nVP64 with placement of the transactivation domain on the N-terminus of dCas9 exhibited the best fold-induction (Figure S1B–D). Placement of the VP64 on the N-terminus did not interfere with the nuclear localization of the dCas9. Therefore, we chose the N-terminal placement of the transactivating domain for the remainder of the study. Figure 1A is a drawing of the dCas9.nVP64 construct and the control dCas9 vector without the transactivating domain. Both constructs have an HA-epitope tag, nuclear localization sequence for driving the dCas9 into the nucleus, and an eGFP reporter for easy visualization of transfected cells. Both constructs express well with the expected nuclear localization of dCas confirmed by anti-HA immunohistochemistry, while the post-translationally cleaved eGFP reporter shows a diffuse cytoplasmic localization (Figure 1B–D). Transactivation of the mCherry reporter by the dCas9.nVP64-eGFP construct targeting the GAL4 region of GAL4-mCherry control reporter system was also confirmed (Figure 1 E–F), although the induction by the dCas9.nVP64-eGFP appeared weaker than the control induction by the positive-control MyoD-VP16 2-hybrid inducer.
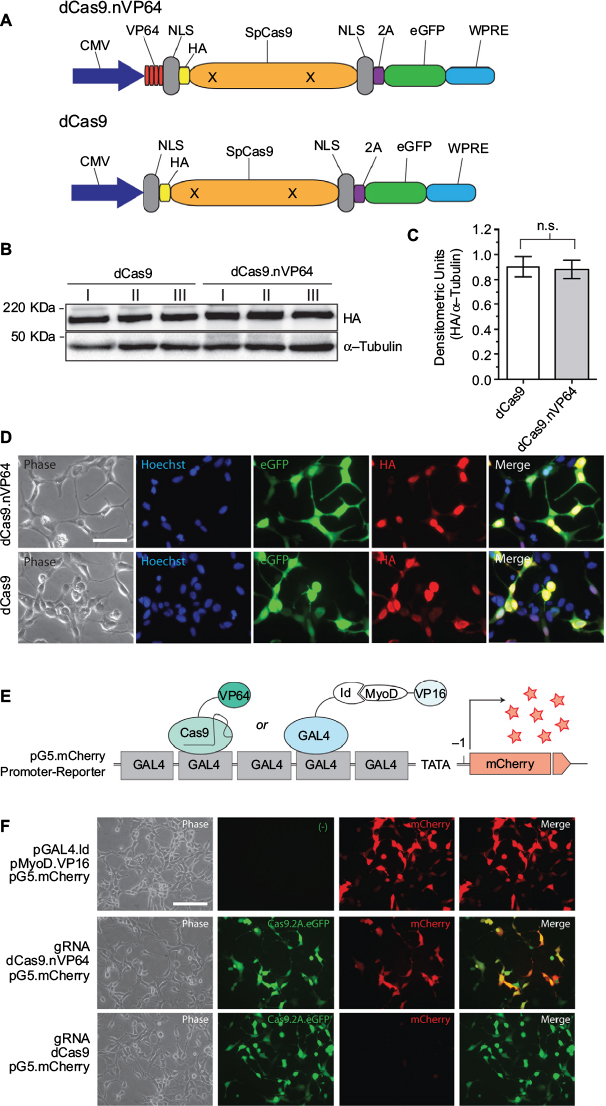 | Figure 1 Expression and transactivation of dCas9.nVP64. Notes: (A) Cartoon illustration of the HA-tagged deactivated-form of dCas9.nVP64 was engineered at the N-terminus in the transactivating construct (dCas9.nVP64) while the non-transactivating construct was unlabeled (dCas9). Note that both dCas9 are HA-epitope tagged and the eGFP follows the 2A sequence resulting in cleavage of this reporter from the dCas9. (B) Western blot of overexpressed dCas9 from 3 replicate experiments. Anti-HA was used to detect dCas9 expression and anti-α-tubulin was used as a loading control. (C) Quantification of HA band intensity normalized by the α-tubulin loading control band intensity, mean±standard error of the mean, unpaired t-test, n=3. (D) Immunocytochemistry of cells expressing dCas9.nVP64 (top) or dCas9 (bottom). Phase images are on the left. The Hoechst nuclear stain (middle left, blue), the eGFP signal (middle, green), and the HA signal (middle right, red), are shown as a merged image on the right. Scale bar=50 mm. (E) Illustration of the reporter system used to detect transactivation. A 5xGAL4 promoter sequence is upstream of the mCherry reporter in the pG5.mCherry vector. As a control, GAL4-Id protein is expressed along with MyoD-VP16. GAL4 binds to the GAL4 promoter sequence and recruits VP16 upstream of the transcription start site (arrow) via the Id: MyoD interaction (CheckMate Two Hybrid, E2440; Promega, Madison, WI, USA). gRNA was engineered to target to the GAL4 promoter loci recruiting the dCas9.nVP64 upstream of the TSS driving expression of mCherry. (F) Fluorescence microscopy of cells expressing the pG5.mCherry promoter-reporter vector along with pGAL4.Id and pMyoD.VP16 (top), GAL4.gRNA and dCas9.nVP64 (middle), or GAL4.gRNA and dCas9 (bottom). Phase images are on the left. eGFP images indicate equal expression of both dCas9 and dCas9.nVP64 (middle left). mCherry signal, indicating the level of transactivation (middle right) while the merge analysis shows the dCas9.nVP64 transactivation is specific only to cells expressing the dCas9.nVP64 construct (right). Scale bar=100 mm. Abbreviations: CMV, cytomegolovirus; eGFP, enhanced green fluorescent protein; HA, hemagglutinin; NLS, nuclear localization sequence; n.s., not significant; WPRE, Woodchuck hepatitis virus post-transcriptional regulatory element. |
Since no rule exists for identifying the most effective gRNA target for dCas-mediated gene activation, we followed the 20N-NGG rule (https://www.addgene.org/crispr/guide/) for identifying gRNA targets appropriate for the Streptococcus pyogenes Cas9 effector within the promoter sequence of TIMPs. Both top and bottom strands were considered and several gRNAs spanning the entire promoter sequences were created and tested for promoter activation using the luciferase-reporter system. Figure 2A–C summarizes the results demonstrating variable luciferase induction by different gRNAs for all 3 TIMPs. The strength of induction varied from 20-fold to minimal for TIMP1, 12-fold to minimal for TIMP2, and only 4-fold to minimal for TIMP3 gRNA targets. In general, the strength of induction was the greatest for TIMP1 > TIMP2 > TIMP3 but with all inductions well above the negative control baseline where no gRNA was co-transfected with the dCas.nVP64 effector.
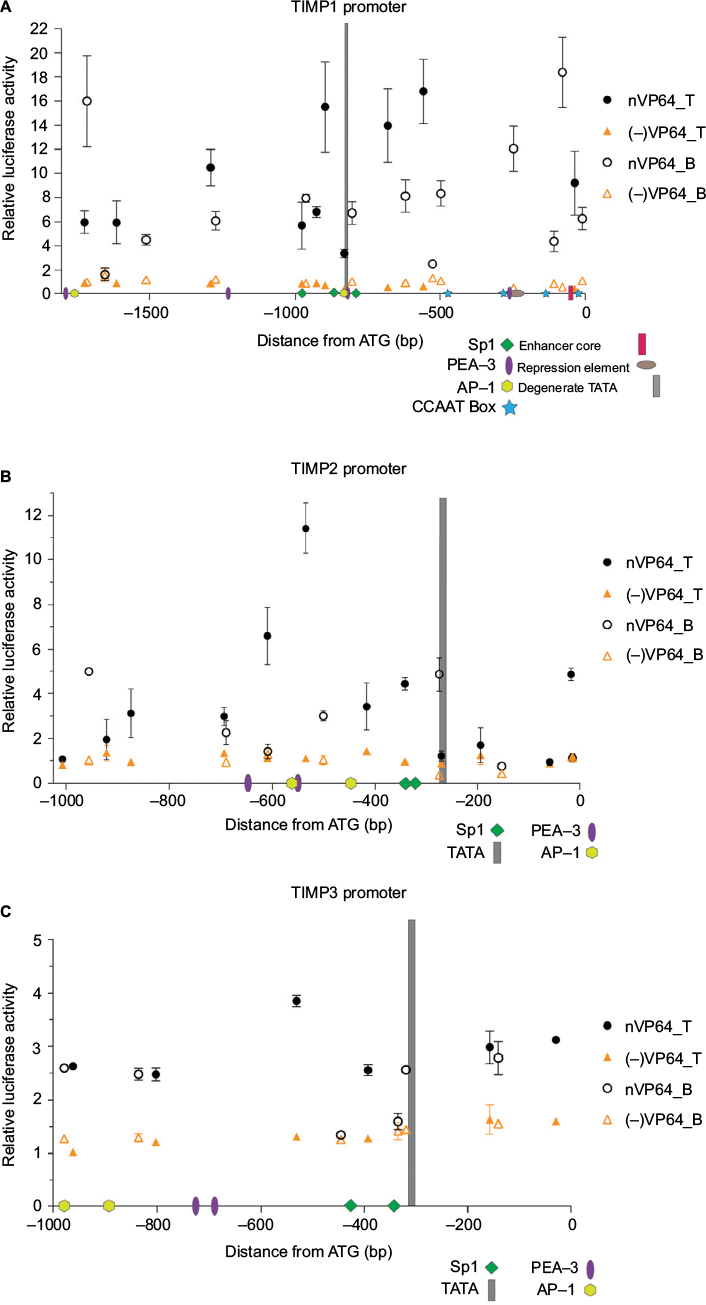 | Figure 2 Loci-specific transactivation of the mouse TIMP promoters using dCas9.nVP64. Notes: Fold-induction over the no gRNA transfection controls using dCas9.nVP64 (black circle) or dCas9 (orange triangle) and the relative gRNAs targeting loci within the (A) TIMP1, (B) TIMP2, or (C) TIMP3 core promoters. Closed symbols represent targeting of the top strand (also indicated by T) while open symbols represent targeting of the bottom strand (indicated by B) of the promoter DNA. X-axis shows the relative distance from the start codon and the numerous transcription factor-binding sites located within the mouse TIMP promoters. Data from Clark et al.24 Mean±standard error of the mean , n=3. Abbreviation: TIMP, tissue inhibitor of metalloproteinases. |
Most transcriptional activators are derived from the VP16 gene (VP) of the herpes virus where the carboxy-terminal 78 residues convey the transcription enhancing activity.20 Multiple replicates of the core activation domain were concatenated to create the VP64 and up toVP160 transcriptional activators with greater transactivation potency. However, the more traditional VP-based transactivators proved inefficient for dCas-mediated applications showing only a modest increase in the mRNA levels and synthetic transcriptional activators such as SAM and VPR, which recruit transcription factors that show greater gene activation compared with the VP-based constructs were created.19,21 We compared the 3 VP-based (VP64, VP160, VPR) (Figure 3A) and a commercially available SAM-based transactivator dCas-effector for TIMP1 and 2 activations using our luciferase promoter-reporter system. The rank order of TIMP induction by different gRNAs remained unchanged independent of the effectors, but the VPR transactivator demonstrated the greatest induction of promoter activity for both TIMP1 and 2 (Figure 3B–E).
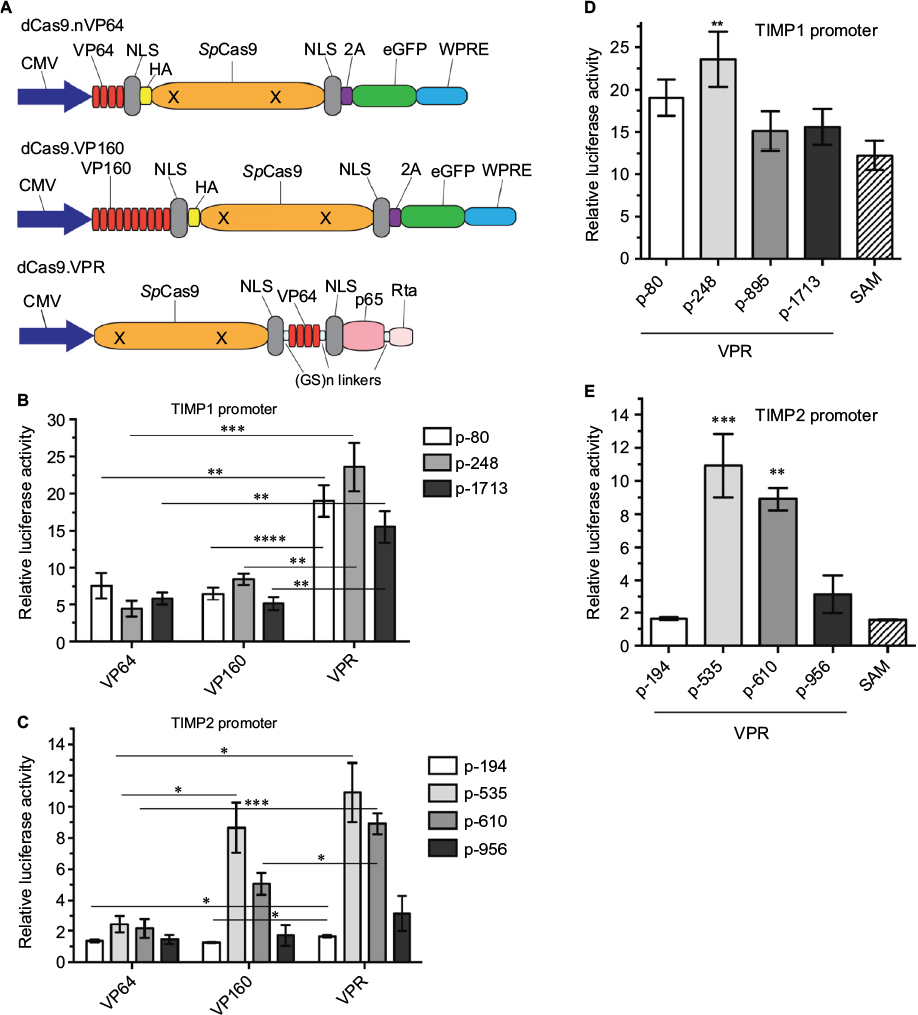 | Figure 3 Comparison of the transactivation efficiency of TIMP1 and TIMP2 promoters using different transcriptional activating effectors. Notes: (A) Illustration of the different dCas9 constructs used. (B, C) Fold luciferase induction over no gRNA transfection controls comparing VP64-, VP160-, or VPR.dCas9 effector constructs targeting 3 loci within the mouse TIMP1 (B) or 4 loci within the mouse TIMP2 (C) promoter. Mean±standard error of the mean (SEM), *P<0.025, **P<0.005, ***P<0.0005, ****P<0.0001, unpaired t-test, n=4. (D, E) Fold luciferase induction over no gRNA transfection comparing VPR targeting 4 loci in the mouse TIMP1 (D) and mouse TIMP2 (E) promoter to SAM targeting between 0 and 250 bps upstream of the transcription start site. Mean±SEM, **P<0.01, ***P<0.005, one-way analysis of variance with multiple comparisons, n=4. Abbreviations: CMV, cytomegolovirus; eGFP, enhanced green fluorescent protein; HA, hemagglutinin; NLS, nuclear localization sequence; SAM, synthetic activation motif; TIMP, tissue inhibitor of metalloproteinases; VPR, VP64-p65-Rta; WPRE, Woodchuck hepatitis virus post-transcriptional regulatory element. |
We confirmed that the TIMP promoter-reporters we utilized consisting of a short immediate upstream sequence from the start codon responded to a biological stimulus to the phorbol ester phorbol-12-myristate-13-acetate (PMA). As reported previously, the short promoter region contained the phorbol ester-responsive element for TIMP122 and TIMP3 but not for TIMP2 (Figure 4A). The inability of PMA to stimulate TIMP2 has been reported in a cellular context as well,23 indicating that the promoter-reporter constructs captured the essence of TIMP regulation in a biological context. Nevertheless, we asked whether the gRNAs identified by an in vitro promoter-reporter assay showed effective TIMP induction in the context of intact cells. Because of our interest in motoneuron diseases, we chose to study gRNA dCas-VPR induction of TIMPs in the motoneuron-like NSC34 cells. Since transcriptional activation of genes by gRNA-dCas system could depend on the basal state of the chromatin,21 we examined the basal expression of TIMPs by qRT-PCR. mRNA for TIMP2 > TIMP3 > TIMP1 in NSC34 cells showed an ~10-fold difference in the levels of expression for the respective TIMPs (Figure 4B), indicating a large difference in the basal state of the chromatin for these genes. In contrast to the results from the promoter-reporter assays, dCas-VPR transcriptional activation of TIMPs by co-transfection of NSC34 cells with the effector and gRNAs previously identified showed little induction of TIMP1 even with multiple gRNAs co-stimulation (Figure 4C). The gRNAs targeting TIMP2 showed a significant increase in the mRNA-level consistent with the promoter-reporter assay (Figure 4D). The efficacy of TIMP2 induction by different gRNAs followed the rank order previously observed in the promoter-reporter assay (i.e., p956~p610>p535>p194), and co-activation of multiple target sites with co-administration of multiple gRNAs resulted in an additive effect, increasing the TIMP2 mRNA over 10-fold over the already high baseline. The specificity of gRNA remained robust in that no increase in TIMP1 or 3 mRNA was observed with transfection of TIMP2-specific gRNAs.
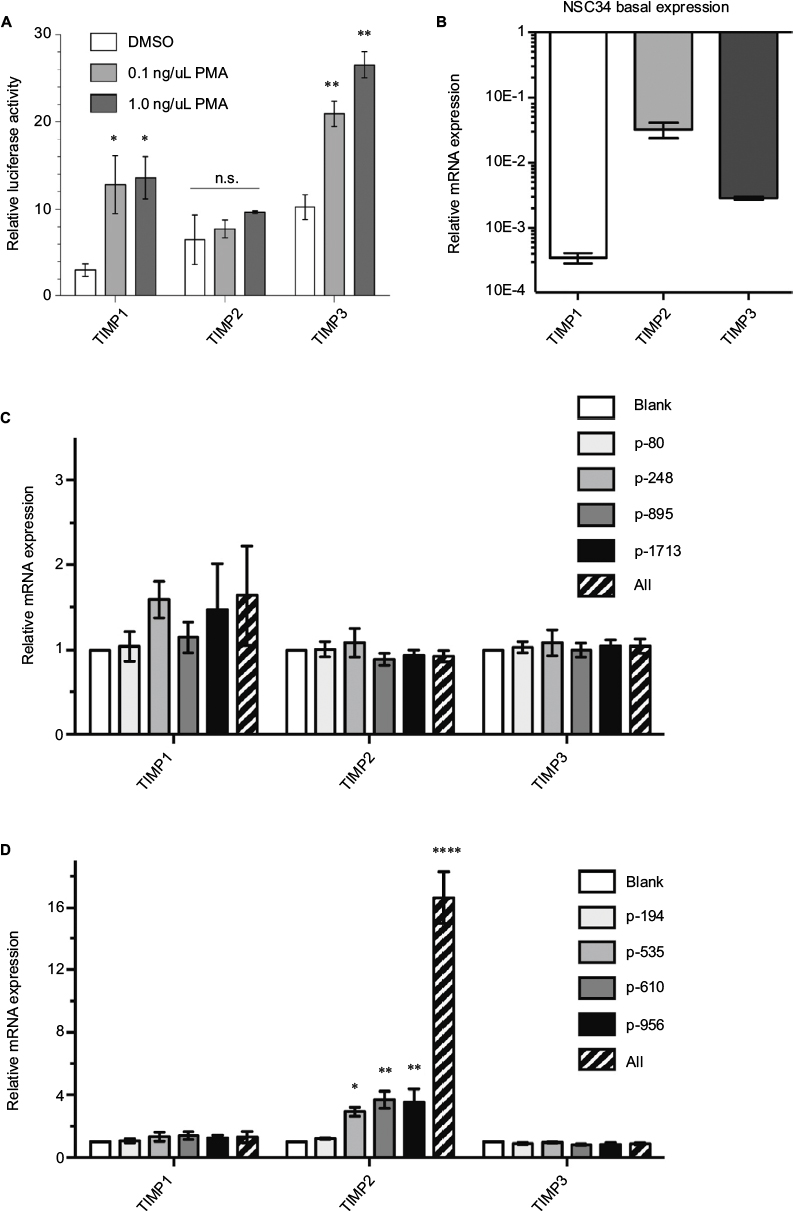 | Figure 4 Endogenous TIMP gene activation in mouse NSC34 cells. Notes: (A) PMA stimulation enhances promoter-reporter activity for TIMP1 and 3. TIMP promoter-reporter constructs were transfected into HEK293 cells. The promoter activity (luciferase) was assessed by the Dual-Glo assay 12 hours after DMSO or PMA stimulation at the indicated concentrations. Mean±standard error of the mean (SEM), *P<0.05, **P<0.01 or n.s., n=3. (B) Endogenous TIMP mRNA basal expression in unstimulated NSC34 cells relative to GAPDH. Mean±SEM, n=6–11. All bars were significantly different (P<0,001) from each other by a pair-wise two-tailed t-test. Specific endogenous Cas9-VPR mediated transactivation using gRNAs targeting the mouse TIMP1 (C) and mouse TIMP2 (D) promoters. Dashed bar indicates the expression of all 4 gRNAs simultaneously. Mean±SEM, *P<0.05, **P<0.01, one-way analysis of variance with multiple comparisons, n=4. The “All” condition was analyzed separately; mean±SEM, ****P<0.0001, unpaired t-test, n=4. Abbreviations: DMSO, dimethylsulfoxide; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; n.s., not significant; PMA, phorbol-12-myristate-13-acetate; TIMP, tissue inhibitor of metalloproteinases; VPR, VP64-p65-Rta. |
That the gRNA-mediated transcriptional activation of TIMP2 in NSC34 cells resulted in the production of functional TIMP2 protein was confirmed by reverse zymography (Figure 5A). With this technique, the inhibition of gelatinolytic activity of MMP embedded within the polyacrylamide gel by an inhibitor appears as a visible band remaining after digestion of the background gelatin. Comparison of the molecular mass of the MMP activity inhibiting molecule found in the cell culture supernatant with the control recombinant TIMP2 confirmed identical apparent molecular mass. An assay for MMP9 activity with the fluorogenic substrate assay also confirmed inhibition of the enzymatic activity only in the cell culture supernatant from gRNA-mediated TIMP2-induced condition (Figure 5B, C). Therefore, gRNA-mediated transcriptional activation of TIMP2 not only resulted in mRNA increase but also in the production of functional TIMP2 protein by NSC34 cells and secretion into the cell culture medium.
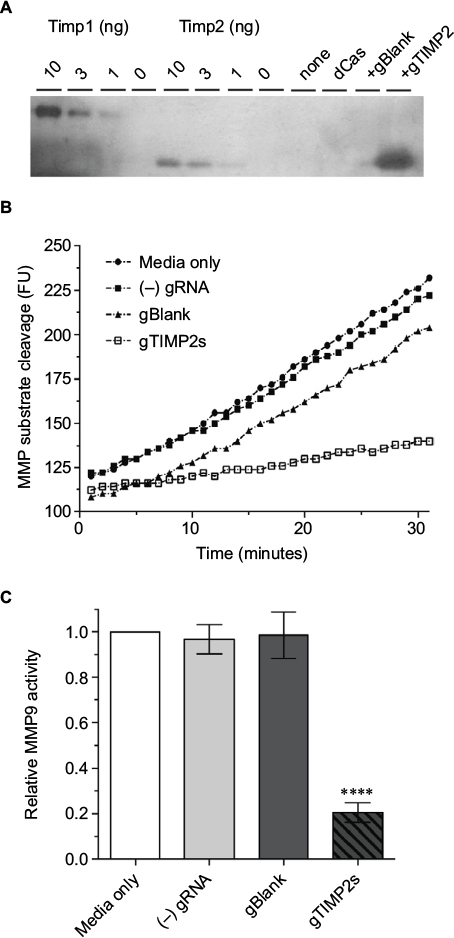 | Figure 5 Production of functional TIMP2 protein by gRNA-induced NSC34 cells. Notes: (A) Reverse zymography of cell culture supernatant. The lanes were loaded with control recombinant TIMP1 or 2 (10, 3,1, or 0 ng), or cell culture supernatant from NSC34 cells transfected with nothing, dCas-VPR alone, dCas-VPR with a negative control blank gRNA, or dCas-VPR with all 4 (p-194, p-535p, p-610, and p-956) TIMP2 gRNAs. The transfection condition was identical to that for mRNA assay except that the culture supernatant was harvested 48 hours after transfection. The blot is representative of 3 biological replicates. (B) Time course of increase in fluorescence due to cleavage of the fluorogenic MMP substrate. The slope of the fluorescence vs time plot was taken as a measure of the relative catalytic activity. The plot is representative of 3 biological replicate experiments. (C) A bar plot of relative MMP9 catalytic activity (mean±standard error of the mean, n=3). Note significant inhibition (****P<0.0001) of the catalytic activity by cell culture supernatant from wells transfected with all 4 TIMP2 gRNAs. Abbreviations: MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinases; VPR, VP64-p65-Rta. |
While NSC34 cells are relatively receptive to transfection, the transfection efficiency using Lipofectamine 2000 was ~50% at best. Since the qRT-PCR assay for TIMP mRNA is necessarily an average report of all cells, both transfected and non-transfected, we wanted to assess the true mRNA induction by the dCas-VPR/gRNA. We chose the TIMP2-targeting p535 gRNA and assayed for mRNA induction in pooled cells from the entire well and in selected transfected cells identified by the eGFP reporter co-expressed from the dCas-VPR plasmid. In a preliminary experiment, individual eGFP-positive cells were identified under an epifluorescent microscope and collected by aspiration into a glass capillary tube. Five cells were pooled, mRNA amplified by a single-cell transcriptome method, and qRT-PCR performed. The TIMP2 mRNA induction from eGFP-positive cells was approximately twice of that observed in an assay from the whole well (Figure S3A), consistent with the limited transfection efficiency of the NSC34 cells (Figure S3B). However, even with this level of gRNA p535 induction of TIMP2 in successfully transfected cells, the level of mRNA increase is approximately half of the 12-fold induction observed in the promoter-reporter luciferase assay for this gRNA, indicating potential suppression of gene expression by genomic elements outside of the immediate ~1 kB promoter upstream of the start ATG used in the reporter assay. Access of the dCas-VPR complex to the promoter region of the TIMP2 gene could be restricted in the cellular context, although the large amount of basal TIMP2 mRNA level would suggest an active and open chromatin. Nevertheless, based on the induction level observed when multiple gRNAs are employed, we estimate that the dCas-VPR/gRNA induction of TIMP2 could be as high as 30-fold induction over the already high basal level in the NSC34 cells.
Discussion
Multiple transcription factor binding motifs have been identified in the TIMP promoters with many confirmed as functional by the traditional promoter-reporter assays in mediating constitutive expression and stimulus-mediated gene induction (reviewed by Clark et al).24 However, which of these transcription factor binding motifs are amenable to dCas-mediated transcriptional activation and whether there are interactions between the endogenous transcription factors and the synthetic transactivator is unclear. A systematic targeting of gRNA along the TIMP promoters demonstrated that some sites were hot-spots leading to significant induction of promoter activity while others were not. Furthermore, gRNA targeting the top- or the bottom-strand of the same promoter region made a large difference in the level of induction. Overall, no apparent rule emerged that allowed identification of the best gRNA target sites for induction of the TIMP promoters with regard to proximity to known transcription factor binding motifs, affirming the necessity of adopting a trial and error approach in identifying the optimal gRNA target site. Nevertheless, a systematic survey of the gRNA targets along the TIMP promoters identified several candidates capable of TIMP induction by the promoter-reporter assay and recapitulated in a cellular context.
Examination of gRNA induction of TIMP gene in intact NSC34 cells confirmed the successful induction of TIMP2 gene mostly correlating with the rank-order potency of induction observed in the promoter-reporter screening (i.e., p194<p956~p610), although p533 showed greater induction by promoter-reporter, whereas this gRNA was only as effective as p956 and p610 in intact cells. Simultaneous targeting of the promoter region by multiple gRNA greatly increased TIMP2 induction as reported previously for other gene targets.9,11 In contrast, the gRNAs demonstrating induction of TIMP1 in the promoter-reporter assay failed to activate the promoter within the context of the intact NSC34 cells. A systematic study of 10 genes suggested an inverse correlation between the basal activity of the gene and the inducibility by the dCas-transactivator.21 The basal mRNA level of TIMP2 >> TIMP1 indicated open chromatin and enhanced transcription factor access to the TIMP2 promoter under the basal condition and our ability to induce TIMP2 was greater than for TIMP1. Although gRNA targeting of dCas to an inactive gene within closed chromatin has been reported to open the chromatin, allow local transcription factor interaction with the promoter, and enhanced gene activity,25 this appears not to be the case for TIMP1 where gRNA targeting of dCas-VPR failed to significantly activate this gene. Whether the differences in the genomic organization with the TIMP1 gene nested within an intron of syntaxin quite distinct from the TIMP2 gene26 plays a role in the difference in dCas-mediated transcriptional activation is not known.
A side-by-side comparison of the different transactivator motifs reported in the literature (VP64, VP160, VPR, and SAM) in our system demonstrated the greatest induction by VPR. A quantitative difference in the fold-induction observed between promoter-reporter and in intact cells for the same gRNA is partly an artifact of the limited transfection efficiency, since RNA harvested from the entire well includes cells successfully transfected by the plasmids and promoter activity induced as well as untransfected cells. However, even the 6-fold TIMP2 induction confirmed in isolated transfected cells is only half of the promoter activity observed in the promoter-reporter assay. Such quantitative discrepancy between the promoter-reporter assay and cell assay with an intact genome can well be expected since the promoter-reporter only captures a very short DNA segment out of the entire gene regulatory domain, including enhancers most likely spanning kilo-bases or even mega-bases away from the transcription start site.27 Nevertheless, a simple promoter-reporter assay serves well as an initial screening tool for identifying viable gRNA targets for dCas9-mediated transactivation.
We targeted the motoneuron-like NSC34 cells as our cellular model because a systematic search for biomarkers differentially expressed in neurodegeneration-sensitive and -insensitive motoneuron populations identified MMP9 as a potential gene correlating with sensitivity to neuronal death in ALS.14 While the exact cellular mechanism on how MMP9 might modulate motoneuron death in ALS remains unresolved, specific and selective inhibition of MMP9 activity might result in enhanced survival. Cas9-mediated induction of TIMPs and inhibition of MMP9 activity in the susceptible neuronal population may provide a novel approach for the treatment of ALS without the potential complications of a broad-spectrum inhibition of the metalloproteinases. Likewise, dCas-mediated induction of TIMPs and a selective inhibition of MMPs using the gRNA identified in the present study might prove to be a useful tool for the treatment of other diseases such as cancer and inflammatory disorders where overactive MMPs have been implicated.2
Acknowledgments
This study was supported by NIH RO1 GM105665, GM107054, and the Bamforth Chair Endowment Funds from the University of Wisconsin, Department of Anesthesiology.
Disclosure
The authors report no conflicts of interest in this work.
References
Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6(6):480–498. | ||
Mohan V, Talmi-Frank D, Arkadash V, Papo Niv, Sagi I. Matrix metalloproteinase protein inhibitors: highlighting a new beginning for metalloproteinases in medicine. Metalloprot Medicine. 2016;3:31–47. | ||
Brkic M, Lalusu S, Libert C, Vandenbroucke RE. Friends or foes: matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediators Inflam. 2015;2015:620581. | ||
Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nature Rev Immunol. 2013;13:649–665. | ||
Vafadari B, Salamian A, Kaczmarek L. MMP-9 in translation: from molecule to brain physiology, pathology, and therapy. J Neurochem. 2016;Suppl 2:91–114. | ||
Wright JW, Harding JW. Contributions of matrix metalloproteinases to neural plasticity, habituation, associative learning and drug addiction. Neural Plast. 2009;2009:579382. | ||
Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol. 2013;48(3):222–272. | ||
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophy Acta. 2010;1803(1):55–71. | ||
Cheng AW, Wang H, Yang H, et al. Multiplexed activation of endogenous gene by CRISPR-on an RNA-guided transcriptional activator system. Cell Res. 2013;23(10):1163–1171. | ||
Mali P, Aach J, Stranges PB, et al. Cas9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31(9):833–838. | ||
Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10(10):977–979. | ||
Lin S, Ewen-Campen B, Ni X, Housden BE, Perrimon N. In vivo transcriptional activation using CRISPR/Cas9 in Drosophila. Genetics. 2015;201(2):433–442. | ||
Cashman NR, Durham HD, Blusztajn JK, et al. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194(3):209–221. | ||
Kaplan A, Spiller KJ, Towne C, et al. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. 2014;81(2):333–345. | ||
Flenniken AM, Williams BR. Developmental expression of the endogenous TIMP gene and a TIMP-lacZ fusion gene in transgenic mice. Genes Dev. 1990;4(7):1094–1106. | ||
De Clerek YA, Darville MI, Eeckhout Y, Rousseau GG. Characterization of the promoter of the gene encoding human tissue inhibitor of metalloproteinases-1 (TIMP2). Gene. 1994;139:185–191. | ||
Sun Y, Hegamyer G, Kim H, et al. Molecular cloning of mouse tissue inhibitor of metalloproteinases-3 and its promoter. J Biol Chem. 1995;270(33):19312–19319. | ||
Seipel K, Georgiev O, Schaffner W. A minimal transcription activation domain consisting of a specific array of aspartic acid and leucine residues. Biol Chem Hoppe-Seyler. 1994;375(7):463–470. | ||
Chavez A, Scheiman J, Vora S, et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12(4):326–328. | ||
Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335(6190):563–564. | ||
Konermann S, Brigham MD, Trevino AE, et al. Genome-scale transcriptional activation by an engineered CRISPR/Cas9 complex. Nature. 2015;517(7536):583–588. | ||
Cambell CE, Flenniken AM, Skup D, Williams BR. Identification of a serum- and phorbol ester-responsive element in the murine tissue inhibitor of metalloproteinase gene. J Biol Chem. 1991;266(11):7199–7206. | ||
Sampieri CL, Nuttall RK, Young DA, Goldspink D, Clark IM, Edwards DR. Activation of p38 and JNK MAPK pathways abrogates requirement for new protein synthesis for phorbol ester mediated induction of select MMP and TIMP genes. Matrix Biol. 2008;27(2):128–138. | ||
Clark IM, Swingler TE, Sampieri CL, Edwards DR. The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol. 2008;40(6–7):1362–1378. | ||
Barkal AA, Srinivasan S, Hashimoto T, Gifford DK, Sherwood RI. Cas9 functionally opens chromatin. PLoS One. 2016;11(3):e0152683. | ||
Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12(11):233. | ||
Kim TK, Shiekhattar R. Architectural and functional commonalities between enhancers and promoters. Cell. 2015;162:948–959. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.