Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
D-α-tocopherol polyethylene glycol succinate-based derivative nanoparticles as a novel carrier for paclitaxel delivery
Authors Wu Y, Chu Q, Tan S ![]() , Zhuang X, Bao Y, Wu T, Zhang Z
, Zhuang X, Bao Y, Wu T, Zhang Z
Received 13 February 2015
Accepted for publication 18 May 2015
Published 20 August 2015 Volume 2015:10(1) Pages 5219—5235
DOI https://doi.org/10.2147/IJN.S82847
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Professor Lei Yang
Yupei Wu,1,* Qian Chu,2,* Songwei Tan,1 Xiangting Zhuang,1 Yuling Bao,1 Tingting Wu,1 Zhiping Zhang1,3,4
1Tongji School of Pharmacy, 2Department of Oncology, Tongji Hospital, Tongji Medical School, 3Hubei Engineering Research Center for NDDS, 4National Engineering Research Center for Nanomedicine, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Abstract: Paclitaxel (PTX) is one of the most effective antineoplastic drugs. Its current clinical administration Taxol® is formulated in Cremophor EL, which causes serious side effects. Nanoparticles (NP) with lower systemic toxicity and enhanced therapeutic efficiency may be an alternative formulation of the Cremophor EL-based vehicle for PTX delivery. In this study, novel amphipathic 4-arm-PEG-TPGS derivatives, the conjugation of D-α-tocopherol polyethylene glycol succinate (TPGS) and 4-arm-polyethylene glycol (4-arm-PEG) with different molecular weights, have been successfully synthesized and used as carriers for the delivery of PTX. These 4-arm-PEG-TPGS derivatives were able to self-assemble to form uniform NP with PTX encapsulation. Among them, 4-arm-PEG5K-TPGS NP exhibited the smallest particle size, highest drug-loading efficiency, negligible hemolysis rate, and high physiologic stability. Therefore, it was chosen for further in vitro and in vivo investigations. Facilitated by the effective uptake of the NP, the PTX-loaded 4-arm-PEG5K-TPGS NP showed greater cytotoxicity compared with free PTX against human ovarian cancer (A2780), non-small cell lung cancer (A549), and breast adenocarcinoma cancer (MCF-7) cells, as well as a higher apoptotic rate and a more significant cell cycle arrest effect at the G2/M phase in A2780 cells. More importantly, PTX-loaded 4-arm-PEG5K-TPGS NP resulted in a significantly improved tumor growth inhibitory effect in comparison to Taxol® in S180 sarcoma-bearing mice models. This study suggested that 4-arm-PEG5K-TPGS NP may have the potential as an anticancer drug delivery system.
Keywords: 4-arm-PEG, TPGS, paclitaxel, nanoparticles, antitumor
Introduction
Nanotechnology has been widely applied to anticancer drug delivery with the advantages of high drug loading and encapsulation efficiency, enhanced cellular uptake, as well as improved therapeutic effects and reduced side effects of the formulated drugs.1–3 Many nanosized formulations, including nanoparticles (NP), liposomes, microspheres, polymer conjugates, dendritic polymers, and water-soluble prodrugs,4–9 have been investigated and shown remarkable therapeutic efficiency. Nanomedicine can penetrate through capillaries and be taken up by cells, leading to efficient drug accumulation at target sites. Moreover, sustained and controlled release of drugs at target sites can last over a period of days or even weeks, thereby offering the following enormous advantages, such as reduction of dosage, improvement on the pharmacokinetic/dynamic properties, protection of drugs against degradation, reduced side effects, etc.10 The developments of nanomedicine have the potential to solve many of modern medicine’s intractable problems, as evidenced from the fact that over 200 nanomedicine products are approved or in different stages of clinical trials.11
Paclitaxel (PTX), one of the internationally acknowledged anticancer drugs, has excellent therapeutic activities against a wide spectrum of cancers, including breast, brain, pancreatic, ovarian, and non-small cell lung cancers.12 However, PTX shows limitations in clinical application due to its poor aqueous solubility.13 Its current clinical administration, Taxol®, is formulated in Cremophor EL and dehydrated alcohol (1:1, v/v), which is diluted 5–20-fold in normal saline or glucose injection before administration. Unfortunately, Cremophor EL is not well tolerated and is associated with various severe side effects, such as hypersensitivity reactions, gastrointestinal toxicity, cardiotoxicity, and neurotoxicity.14 Hence, it is essential to develop a new carrier to solve the formulation problem of PTX.
D-α-tocopheryl polyethylene glycol succinate (vitamin E TPGS or simply TPGS), which has been approved by the Food and Drug Administration as a pharmaceutical ingredient, is a water-soluble derivative of natural vitamin E. As a PEGylated vitamin E, TPGS has an amphiphilic structure of lipophilic alkyl tail and hydrophilic polar head with a relatively low critical micelle concentration of 0.02% w/w.15 Its bulky structure and large surface area make it a safe pharmaceutic adjuvant such as absorption enhancer, emulsifier, solubilizer, and stabilizer.16 In addition, TPGS has also been utilized as a P-glycoprotein inhibitor to overcome multidrug resistance and to greatly improve the oral bioavailability of anticancer drugs.17–20 In the past decade, TPGS-based derivatives, which can significantly enhance the solubility and stability of the formulated drug and realize sustained, controlled, and targeted drug delivery, have been widely investigated.21 Nevertheless, the application of independent TPGS micelles for drug delivery is limited by the disadvantage that they were not stable enough in physiological environments.22 Furthermore, the polyethylene glycol (PEG) chain of TPGS is not long enough to ensure the micelles to prevent opsonin bindings and realize the extended blood circulation time.23 PEGylation is a well-used technology in the pharmaceutical industry due to the aqueous solubility, biocompatibility, and non-immunogenicity of PEG.24 Several new PEGylated TPGS-based micelles with improved physiological stability have been reported including TPGS2K, PLV2K, and PEG5K-VE2.23,25,26 Recently, 4-armed copolymers have been receiving great attention because of their unique properties.27,28 It has been reported that 4-armed copolymers presented a lower surface tension, greater stability, and higher drug entrapment efficiency.4,29 Hence, we designed and synthesized TPGS-based derivatives – 4-arm-PEG-TPGS – as nanoplatforms for hydrophobic drug PTX delivery.
In this study, novel derivatives based on 4-arm-PEG of different molecular weights and TPGS were synthesized and investigated. PTX-loaded 4-arm-PEG-TPGS NP (PTX-NP) were prepared and characterized by particle size, morphology, and drug loading efficiency. The release behavior and stability in vitro of the NP were also investigated. The cell cytotoxicity was carefully evaluated in human ovarian cancer A2780, non-small cell lung cancer A549, human breast cancer cells MCF-7, and mouse sarcoma tumor cell line S180. The cellular uptake, induction of apoptosis, and retardation of cell cycle of NP were studied against A2780 cells. The tumor inhibition effect was further evaluated in S180 sarcoma-bearing mice models.
Materials and methods
Materials
PTX of purity 99% was obtained from Jinhe Limited, People’s Republic of China. 4-arm-PEG (molecular weight of 5, 10, 20 kDa) were purchased from Sinopeg Biotech Co., People’s Republic of China. TPGS, succinic anhydride (SA), dicyclohexylcarbodiimide, propidium iodide (PI), RNase A, and trypsin-ethylenediaminetetraacetic acid (EDTA) were supplied by Sigma-Aldrich (St Louis, MO, USA). 4-dimethylamino pyridine (DMAP) was purchased from Aladdin, People’s Republic of China. RPMI-1640 medium was from Gibco BRL (Gaithersburg, MD, USA). Taxol® was obtained from Bristol-Myers Squibb Caribbean Company. Penicillin-streptomycin, fetal bovine serum (FBS), and trypsin without EDTA were obtained from Hyclone (Waltham, MA, USA). MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide) and Hoechst 33342 were purchased from Biosharp, South Korea. The annexin V-fluorescein isothiocyanate (FITC)/PI double staining assay kit was supplied by KeyGEN, People’s Republic of China. All of the solvents used were of analytical grade and were procured from Sinopharm, People’s Republic of China. Human ovarian cancer cell line A2780, human breast adenocarcinoma cell line MCF-7, non-small cell lung cancer cell line A549, and mouse sarcoma tumor cell line S180 were provided by the Shanghai Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences, People’s Republic of China. Kunming mice (female, 5–7 weeks old, 18–20 g) were obtained from Laboratory Animal Resources of Huazhong University of Science and Technology (Certificate No SCXK 2010-0009). The animals were maintained at 25°C±1°C and 60%±10% humidity under a 12-hours light–dark cycle during the experiments. All animals were maintained under the specific pathogen-free (SPF) condition in the Animal Center of Huazhong University of Science and Technology, People’s Republic of China. All animals were treated according to the regulations of Chinese law and the study was approved by the local Ethical Committee Quantita.
Synthesis and characterization of derivatives
4-arm-PEG5K-TPGS, 4-arm-PEG10K-TPGS, and 4-arm-PEG20K-TPGS were synthesized by a two-step conjugation method, shown in Figure 1. TPGS was first functionalized with a carboxylic acid group by esterification with SA, according to our previous work.30 The second step was the formation of ester bond between the primary groups hydroxy of 4-arm-PEG and carboxylic acid functions of activated TPGS-SA. Briefly, TPGS-SA (1.64 g, 1.0 mmol), 4-dimethylamino pyridine (0.12 g, 1.2 mmol), and dicyclohexylcarbodiimide (0.20 g, 1.2 mmol) were co-dissolved in 5 mL anhydrous dichloromethane (DCM) and reacted at room temperature for 24 hours. The turbid liquid was filtered to remove N, N-dicyclohexylurea and mixed with a 5 mL solution containing 0.1 mmol 4-arm-PEG. After 24 hours, the products were precipitated in diethyl ether and washed three times and dried under vacuum.
 | Figure 1 Synthetic route of 4-arm-PEG-TPGS. |
The structure of resultant TPGS-based derivatives 4-arm-PEG-TPGS were characterized by 1H-NMR spectra (Bruker AVANCE III 400 MHz NMR spectrometer, CDCl3) and Fourier transform infrared spectroscopy (FTIR) (Bruker VERTEX 70 FTIR spectrophotometer). Gel permeation chromatography (GPC) (Waters-2410 system) was also carried out to measure the molecular weights of 4-arm-PEG-TPGS. Tetrahydrofuran was used as the mobile phase at a flow rate of 1.0 mL/min. The molecular weights were calculated by using a calibration curve constructed using polystyrene as the standard. The solubility of the materials in water was estimated simply by visual determination.
Preparation and characterization of NP
The PTX-loaded NP were prepared by a solid dispersion method. Typically, PTX (1, 1.5, or 2 mg) and 4-arm PEG-TPGS (10 mg) were dissolved in 2 mL of DCM by sonication. The organic solvent was evaporated on a rotary evaporator under reduced pressure at 37°C to obtain a homogenous coevaporation PTX/copolymer film. Subsequently, the film was hydrated with 5 mL phosphate buffered saline (PBS), incubated at 37°C for 30 minutes. The resultant mixture was centrifuged at 3,000 rpm for 10 minutes to remove the nanoparticles and free PTX. The blank NP were prepared in a similar manner without PTX added.
The average size, size distribution, and ζ potential of the obtained NP were determined by dynamic light scattering (DLS) (ZetaPlus, Brookhaven Instruments, USA). Data were displayed as the mean value of at least three measurements ± standard deviation. The morphology of the NP was observed by transmission electron microscope (JEM-1230, Japan). The NP were diluted with distilled water and placed on a copper grid covered with nitrocellulose, and dried at room temperature before measurement.
A steady-state pyrene fluorescence method was used to determine the critical aggregate concentration (CAC) of the TPGS-based derivatives. Steady-state fluorescence spectra were obtained on a Hitachi F-4600 luminescence spectrometer. Fifty microliter of 4.8×10−5 M solution of pyrene in acetone was added in the centrifuge tube. Acetone was then evaporated and replaced with 4 mL solution of 4-arm PEG-TPGS with concentrations ranging from 0.01 to 1,000 μg/mL to get a final pyrene concentration of 6×10−7 M. The solution was incubated overnight. Excitation spectra of the sample solutions were obtained at an emission wavelength of 372 nm with excitation spectra (300–350 nm). The change of the fluorescence intensity ratio (I339/I335) was analyzed as a function of the CAC value.
The stability of the NP was investigated by measuring the sizes of samples at different time points. To examine the effect of serum on particle stability, the PTX-loaded NP samples were prepared with PBS at a concentration of 10 mg/mL, and then diluted with FBS or PBS by the ratio of 1:9. The changes in NP size were monitored by DLS.
Encapsulation efficiency and drug loading content
The amount of PTX encapsulated in the NP was measured by high-performance liquid chromatography (HPLC) (Hitachi 2000, Japan) equipped with an L-2130 pump, an L-2400 UV detector, and an Inertsil® ODS-3 C18 reversed phase column (150 mm ×4.6 mm, 5 μm) (Agilent, Santa Clara, CA, USA). Briefly, 2 mg of the freeze-dried NP powder was dissolved in 1 mL DCM in order to disrupt the NP structure and then the solution was dried under nitrogen. Three milliliter mobile phase (acetonitrile/water, 50:50, v/v) was added to dissolve the drugs. The solution was then filtered by 0.45 μm filter for HPLC analysis. The column effluent was detected at 227 nm with a UV detector. The mobile phase was pumped at a flow rate of 1.0 mL/min. The drug encapsulation efficacy (EE) was obtained by the following equations:
|
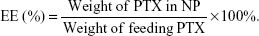
Hemolytic effect of 4-arm-PEG-TPGS
Fresh blood from Sprague Dawley rat was collected in heparinized tubes and washed three times with ice-cold 0.9% sodium chloride (NaCl) by centrifugation at 3,000 rpm for 5 minutes at 4°C. The obtained red blood cells (RBCs) were diluted to 2% (w/v) by ice-cold 0.9% NaCl containing various concentrations (0.001, 0.01, 0.1, 1.0, and 5.0 mg/mL) of 4-arm-PEG-TPGS and polyethylenimine (PEI) (25 kDa), respectively, and then incubated at 37°C in an incubator shaker for 4 hours. The samples were then centrifuged at 3,000 rpm for 10 minutes at 4°C, and 100 μL of supernatant from each sample was transferred into a 96-well plate. The absorbance of the supernatant was determined at 540 nm using a microplate reader (Multiskan MK3; Thermo Scientific, USA). RBCs treated with distilled water and 0.9% NaCl were considered as the positive (100% hemolysis) and negative (0% hemolysis) controls, respectively. The hemolytic effect of Cremophor EL-based vehicle (Cremophor EL and dehydrated alcohol, 1:1, v/v) was also assessed. The degree of hemolysis was determined by the following equation:
|
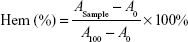
where A100 and A0 were the absorbances of the solution at 100% and 0% hemolysis, respectively.
In vitro release study
The drug release behavior of NP was investigated by using a dialysis method. Four milliliters of PTX-loaded 4-arm-PEG5K-TPGS NP (PTX-NP5K) was placed in a dialysis bag (Snakeskin, Pierce, USA) with a molecular weight cut-off of 2,000 Da. The dialysis bag was suspended in 50 mL of PBS (pH 7.4) or FBS and placed in a shaking water bath at 37°C with a shaking speed of 120 rpm. At every predetermined time, 10 mL of the solution was removed followed by an addition of 10 mL fresh PBS. PTX of the collected incubation medium was extracted by DCM. The drug concentration was determined by HPLC as described earlier.
Cell culture
All cell lines were cultured in RPMI-1640 containing 10% FBS and 1% penicillin-streptomycin in humidified environment at 37°C with 5% carbon dioxide (CO2). After the cells grew to 80%–90% confluence, they were trypsinized with 0.25% trypsin-EDTA.
In vitro cellular uptake
Cellular uptake was analyzed by confocal laser scanning microscopy (CLSM) (Leica TCSNT1, Germany) and coumarin-6, a widely used replacement fluorescent marker of hydrophobic drug, was used as the probe. Coumarin-6 loaded NP of 4-arm-PEG5K-TPGS (coumarin-6-NP5K) were prepared. A2780 cells were seeded onto a 24-well plate at a density of 1.0×104 cells/well. After 24 hours attachment, they were incubated with coumarin-6-M at a concentration of 25 μg/mL for 2 hours at 37°C. The wells were then rinsed carefully three times with cold PBS and fixed with 4% paraformaldehyde for 15 minutes. After being washed twice again with cold PBS, the cells were stained with Hoechst 33342 for 8 minutes and then mounted on a glass slide for observation by CLSM.
The cellular uptake was further studied by a flow cytometer (Becton Dickinson, San Jose, CA, USA). A2780 cells were seeded into six-well black plates at 5×105 cells/well; after the cells reached 80% confluence, the medium was changed to the suspension of coumarin-6-M at an NP concentration of 25 μg/mL and incubated for 0.5, 1, 2, and 4 hours, respectively. Cells treated with only medium were used as control. After incubation, the wells were rinsed three times with cold PBS, and then cells were collected by centrifugation and resuspended in 0.5 mL PBS. The amount of uptake was analyzed by flow cytometry.
In vitro cytotoxicity
The cytotoxicity of PTX formulated in 4-arm-PEG5K-TPGS NP was assessed with four types of cancer cell lines (A2780, MCF-7, A549, and S180) and compared to Taxol® formulation and free PTX (DMSO-dissolved, final DMSO concentration ≤0.1%). Briefly, A2780, MCF-7, and A549 cells in their logarithmic growth were seeded in 96-well plates at a density of 5,000 cells/well. Following overnight attachment, the culture medium in each well was carefully replaced with 100 μL of medium containing Taxol®, PTX-NP5K, or free PTX at PTX concentrations ranging from 0.025 to 100 μg/mL (n=8). Cells treated with only RPMI-1640 medium were tested as controls. S180 cell was promptly seeded with a density approximately 5,000 cells/well before assay. After treatment for 24, 48, and 72 hours, respectively, the relative cell viability was assessed by an MTT assay as described in our previous work.31 IC50 (concentration resulting in 50% inhibition of cell growth) value was determined by SPSS software (version 19.0). The experiment was repeated thrice.
Apoptosis analysis
The qualitative apoptosis of the A2780 cell line treated with different PTX formulation was determined by the Hoechst 33342 staining method. Specifically, A2780 cells were seeded onto a 24-well plate (104 cells/well). Following overnight attachment, the cells were then treated with medium containing Taxol®, PTX-NP5K, or free PTX at the same PTX concentration of 2.5 μg/mL. The control group was incubated with drug-free culture medium. After incubation for 24 hours, the wells were rinsed three times with cold PBS and then fixed with 200 μL of 4% paraformaldehyde for 15 minutes. The cells were further washed three times with 500 μL PBS, followed by staining with 200 μL Hoechst 33342 (10 μg/mL) for 8 minutes in the dark. After being triple-washed with PBS, the cells were finally observed by a fluorescence microscope (IX71; Olympus, Tokyo, Japan).
Annexin V-FITC/PI double staining is a sensitive method for detecting quantitative apoptosis. A2780 cells were seeded onto six-well plates at a density of 5×105 cells/well, followed by attachment for 24 hours. The cells were then incubated with Taxol®, PTX-NP5K, or free PTX at the PTX concentration of 2.5 μg/mL in culture medium; untreated cells were used as the control. After 24 hours incubation, the cells were trypsinized, collected, and resuspended in 300 μL of binding buffer. Thereafter, 3 μL of annexin V-FITC and 3 μL of PI were added and mixed for 30 minutes in the dark. The stained cells were analyzed using a flow cytometer. The quantitative apoptosis of S180 cells were detected in a similar way except that the cell was promptly seeded before assay.
Cell cycle distribution analysis
Cell cycle distribution analysis was further investigated. A2780 cells were seeded onto six-well plates (1.0×105 cells/well). After attachment overnight, the cells were exposed to Taxol®, PTX-NP5K, or free PTX (drug concentration of 2.5 μg/mL). Cells treated with only medium were used as controls. After 24 hours of incubation, the cells were washed twice with cold PBS and fixed overnight with 70% precooled alcohol at 4°C. The cells were washed twice with cold PBS to eliminate alcohol and then incubated with RNase A (100 μg/mL) for 15 minutes at 37°C, followed by staining with PI solution (50 μg/mL) for 30 minutes in the dark. The distribution of DNA content was analyzed by the flow cytometry method and the percentage of cells in each phase of the cell cycle was calculated using ModFit software (Verity Software House, Topsham, ME, USA).
In vivo therapeutic study
Tumor inhibition activity against a solid tumor model was evaluated using female Kunming mice. Kunming mice were subcutaneously injected at the right forelimb axilla with 0.2 mL S180 cell suspension containing 1×107 cells. After 48 hours of transplantation, all the tumor-bearing mice were divided randomly into four groups (n=5). Treatment started when the tumor volume of the mice reached 100–150 mm3 on average, and this was designated as day 1. Each group was treated by tail vein injection on days 1, 3, 5, and 7 with saline, PTX-NP5K (at a dosage of 10 mg/kg), PTX-NP5K (30 mg/kg), and Taxol® (10 mg/kg), respectively. Tumor sizes were measured every day to evaluate the antitumor efficiency. When the tumor length in the saline group was greater than 20 mm, all the mice were sacrificed and the tumors were extirpated and weighed. The tumor was finally cut into small histological sections and stained with hematoxylin and eosin for histological analysis by light microscopy with a CAD system.
Results and discussion
Synthesis and characterization of 4-arm-PEG-TPGS
Three types of 4-arm-PEG-TPGS derivatives were synthesized with various molecular weights of 4-arm-PEG (5, 10, 20 kDa). The products were investigated by 1H-NMR, FTIR, and GPC analysis to confirm the successful conjugation. As shown in Figure 2A, the newly appearing signals at 2.65–2.72 ppm were assigned to the -CH2CH2- part of the succinyl group of TPGS-SA, verifying the esterification of TPGS as compared to the TPGS spectrum.30 Taking 4-arm-PEG5K-TPGS as an example of the 4-arm-PEG-TPGS copolymers, the intensity of 3.65 ppm ascribed to -OCH2CH2- in the PEG chain was significantly increased compared to that of TPGS, proving the conjugation of TPGS with 4-arm-PEG. The TPGS contents in 4-arm-PEG-TPGS derivative were calculated on the basis of the peak area of 0.86 ppm and 3.65 ppm, which were 78%, 68%, and 57% (denoted as 4-arm-PEG5K-TPGS, 4-arm-PEG10K-TPGS, and 4-arm-PEG20K-TPGS), respectively. The structure of 4-arm-PEG-TPGS was further studied by FTIR (Figure 2B). There were no obvious differences among them. The characteristic peaks of TPGS exhibited in the derivative, such as the vibration peak of the C=O bond (νC=O) of the ester bond at 1,735 cm−1 and the C–O stretching vibration (νC–O) of PEG at 1,109 cm−1. However, the enhancement of the peak at 1,109 cm−1 can indicate the formation of 4-arm-PEG-TPGS.
 | Figure 2 Characterization of 4-arm-PEG-TPGS. |
GPC was also performed. As shown in Figure 3, the 4-arm-PEG-TPGS exhibited increased molecular weight with narrow molecular weight distribution and a significant peak shift compared with 4-arm-PEG. This proved the successful grafting of TPGS onto 4-arm-PEG. It should be noted that the 4-arm-PEG-TPGS derivatives showed drastic difference in solubility. The solubility of 4-arm-PEG5K-TPGS was >10 mg/mL, while for 4-arm-PEG10K-TPGS and 4-arm-PEG20K-TPGS, they were only approximately 1 mg/mL, which might limit their applications.
 | Figure 3 GPC results of 4-arm-PEG and TPGS-based derivative 4-arm-PEG-TPGS. |
Preparation and characterization of NP
The PTX-loaded NP were fabricated by a solid dispersion method in this study. The 4-arm-PEG-TPGS NP were prepared with spherical morphology and narrow size distribution (as shown in Figure 4A–F). The diameter observed from DLS was a little larger compared with the transmission electron microscope result. This may be attributed to the fact that the particle size measured by DLS was hydrodynamic diameter with a solvation layer on the surface of the particles.32 As seen from Table 1, the ζ potentials of 4-arm-PEG-TPGS NP were all negative, and higher than that of TPGS micelles. This may be caused by the different PEG densities on the NP surface.
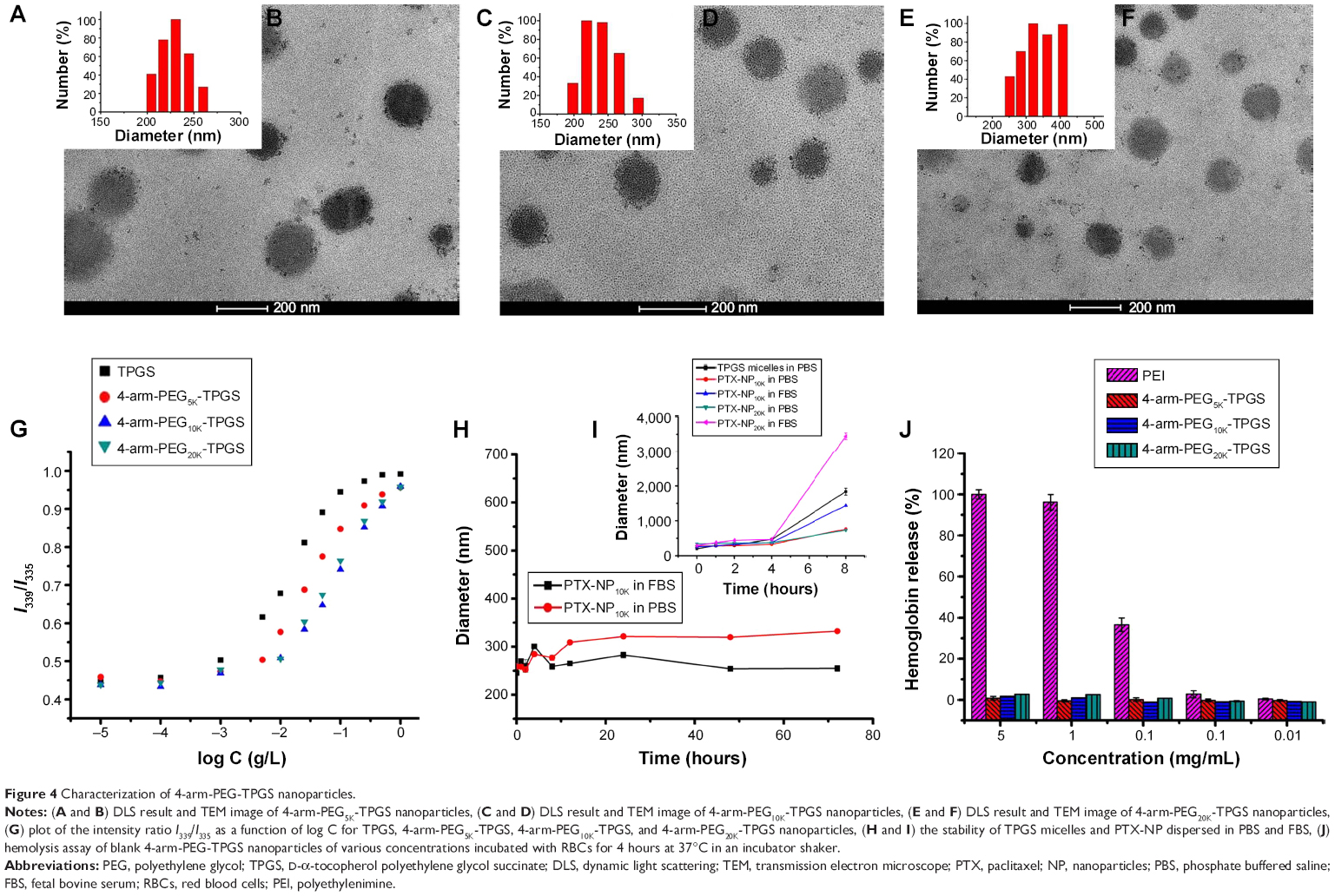 | Figure 4 Characterization of 4-arm-PEG-TPGS nanoparticles. |
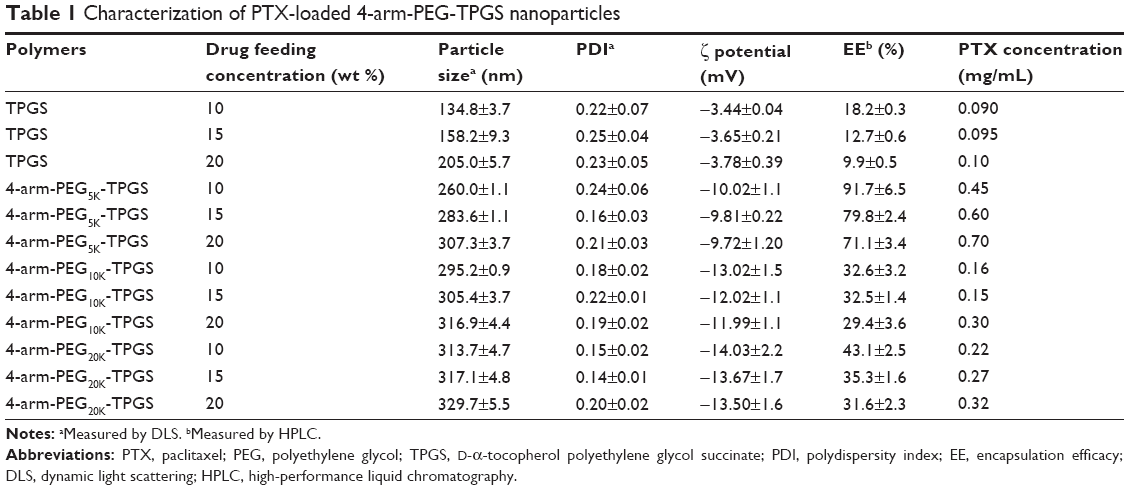 | Table 1 Characterization of PTX-loaded 4-arm-PEG-TPGS nanoparticles |
The CAC of 4-arm-PEG-TPGS was tested by the pyrene fluorescence probe method. The CAC values were obtained by plotting the I339/I335 ratio of each curve in the excitation spectra versus log concentration of the polymer. The CAC values were 3.3×10−3, 4.9×10−3, and 5.6×10−3 g/L for 4-arm-PEG5K-TPGS, 4-arm-PEG10K-TPGS, and 4-arm-PEG20K-TPGS, respectively (Figure 4G), which were approximately similar to that of surfactant TPGS.17 The CAC value was essential to evaluate the formation of NP. It was anticipated that the NP with low CAC value would be intact even on high dilution by a much larger volume of blood in vivo.23
The relationship between the drug encapsulation efficiency and the drug feeding ratio of TPGS and the three types of NP were further studied. As seen in Table 1, the EE of the NP were decreased along with the drug feeding ratio rising. TPGS micelles exhibited EE as low as 9.9% for 20% drug feeding ratio. The EE of 4-arm-PEG5K-TPGS could be up to 91.7%±6.5% for 10% drug feeding ratio and was much higher than 32.6%±3.2% for 4-arm-PEG10K-TPGS and 43.1%±2.5% for 4-arm-PEG20K-TPGS. This may be caused by the difference of the binding affinity between hydrophobic PTX and the hydrophobic core region from 4-arm-PEG-TPGS.33 It is worth noting that the PTX concentration in PTX-NP5K injection could be as high as 0.7 mg/mL. To summarize, 4-arm-PEG5K-TPGS was expected to be a better drug carrier with smaller particle size and higher drug loading capacity.
DLS was also used to assess the colloidal stability of the NP. As shown in Figure 4H, the mean diameter of PTX-NP5K did not remarkably change both in PBS and FBS. Moreover, no drug precipitation was observed during this period, indicating the colloidal stability of the NP under physiological conditions. The probable reason may be that the aggregation among NP and the binding between particles and plasma protein were prevented by the hydrophilic PEG shell.34 However, TPGS micelles, PTX-loaded 4-arm-PEG10K NP (PTX-NP10K), and PTX-loaded 4-arm-PEG20K NP (PTX-NP20K) exhibited contrary properties that may be related to the improper length of the hydrophilic PEG chain (Figure 4I). PTX-NP5K was thus chosen for further in vitro and in vivo cytotoxicity investigation.
PEI, a cationic polymer with potent cell surface activity, was used as positive contrast for examination of the hemolytic activity.26 As seen in Figure 4J, only a negligible level of hemolysis (<1%) was observed for RBCs treated with all of the three blank NP even exposed to high dose of 5 mg/mL. On the contrary, PEI treated RBCs exerted obvious hemolysis in the same dose. We further studied the hemolytic activity of Cremophor EL-based vehicle (Cremophor EL and dehydrated alcohol, 1:1, v/v), as shown in Figure S1, it exhibited a distinct level (>10%) of hemolysis at 25 μL/mL (diluted by saline). The negligible hemolytic activity of NP suggested that 4-arm-PEG-TPGS had no destructive effect on erythrocyte and would be safer than the Cremophor EL-based vehicles.
In vitro drug release of PTX-NP5K
The release profile of PTX from the PTX-NP5K under PBS (pH 7.4) and FBS was investigated (Figure 5). The NP exhibited the limited burst release (12.1%) in PBS after 24 hours. After 7 days, the PTX-NP5K had the accumulated amount of PTX released being approximately 31.5%±2.1% at pH 7.4. It indicated that the 4-arm-PEG5K-TPGS NP could not only solubilize the poorly soluble PTX but also exhibit sustained drug release behavior. The PTX-NP5K also showed high stability in FBS and only 18.8% of PTX was released during the 168 hours test. The PTX-NP5K may thus decrease drug leakage during circulation in the blood and reduce side effects of anticancer drug.
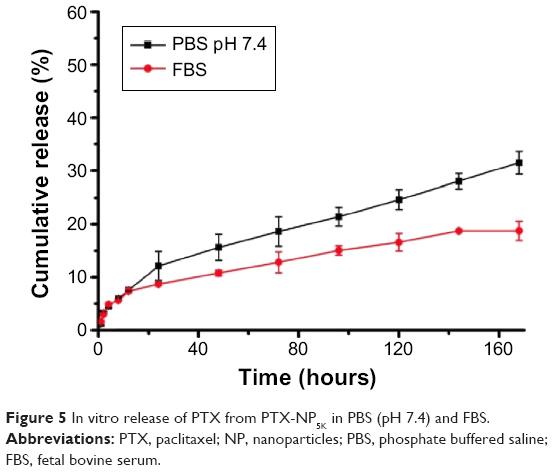 | Figure 5 In vitro release of PTX from PTX-NP5K in PBS (pH 7.4) and FBS. |
In vitro cellular uptake studies
It is known that many anticancer drugs including PTX take therapeutic effects only inside the tumor cells. Accordingly, the cellular uptake of coumarin-6-NP5K was qualitatively analyzed on A2780 cells by CLSM. After 2 hours incubation, NP (green) were intensively located around the nuclei (blue) (Figure 6A), which suggested the effective uptake of the NP by the cells.
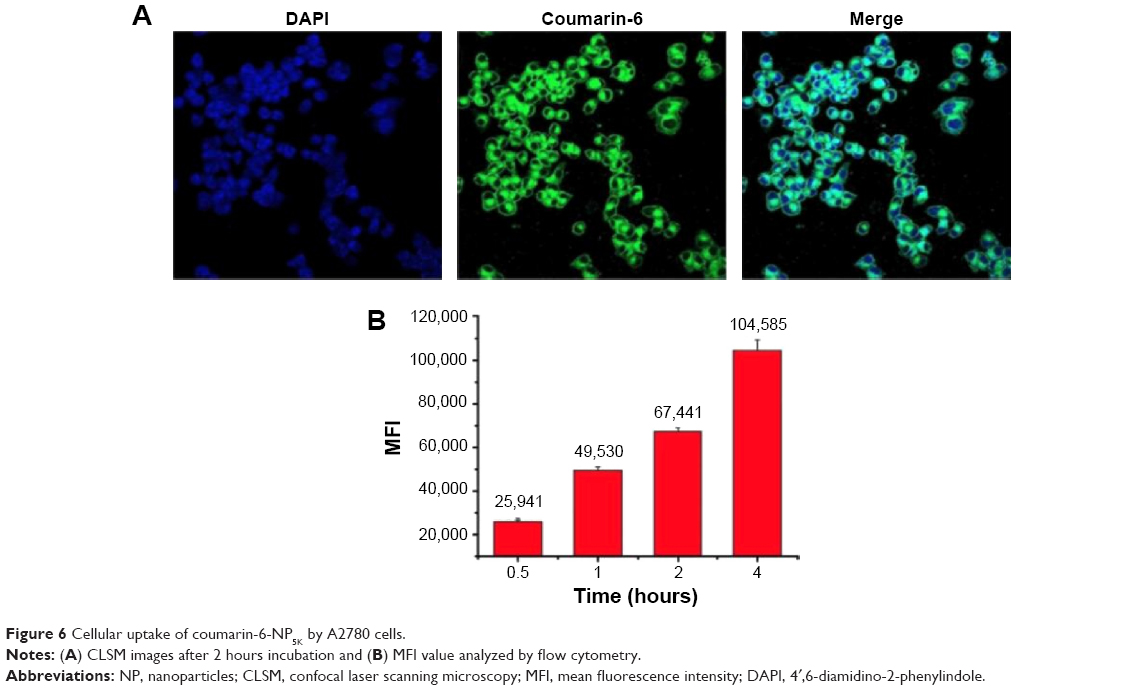 | Figure 6 Cellular uptake of coumarin-6-NP5K by A2780 cells. |
The cell uptake process of NP also exhibited the time-dependent on A2780 cells as shown in Figure 6B. After 4 hours of exposure, the MFI was 4.03-, 2.11-, 1.55-fold higher than that of 0.5, 1, and 2 hours, respectively. This effective cellular uptake of the 4-arm-PEG5K-TPGS NP may result in higher intracellular concentration of PTX than that of free PTX, leading to enhanced antineoplastic effect.
In vitro cell cytotoxicity
In order to evaluate cancer cell cytotoxicity of the PTX-NP5K, MTT assay was carried out against A2780, A549, MCF-7, and S180 cell lines as compared to Taxol® and free PTX. The cytotoxicities of PTX-NP5K and Taxol® were both concentration- and incubation time-dependent on all of the three cell lines (Figure 7). Meanwhile, the cytotoxicity of PTX-NP5K was lower than that of Taxol®, but much higher than that of free PTX. It implied that PTX-NP5K could somewhat enhance the cytotoxicity of PTX. Moreover, PTX-NP5K exhibited similar activity on S180 cells, especially in the concentrations of 25 and 10 μg/mL (Figure S2).
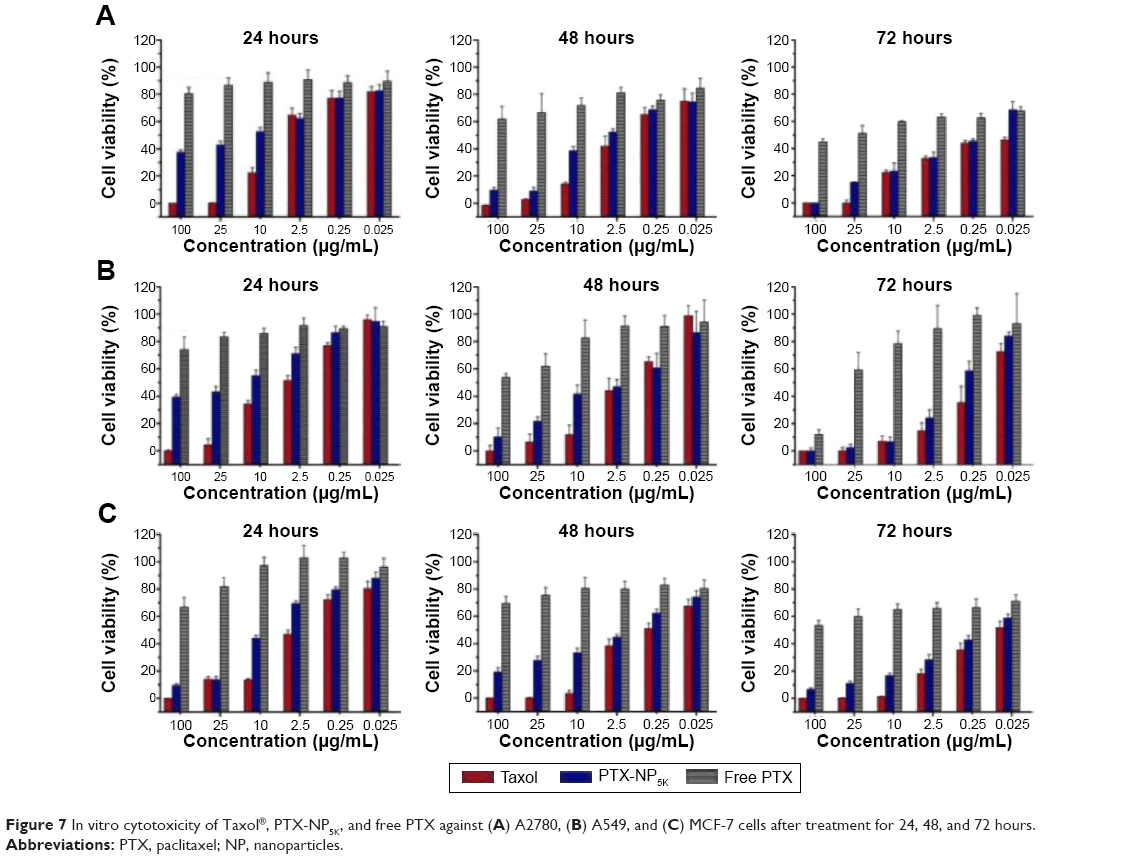 | Figure 7 In vitro cytotoxicity of Taxol®, PTX-NP5K, and free PTX against (A) A2780, (B) A549, and (C) MCF-7 cells after treatment for 24, 48, and 72 hours. |
The anticancer effects were further quantified by IC50 (illustrated in Table 2). The IC50 values of PTX-NP5K against A2780 cells were found to be 3.97±0.12, 1.12±0.01, and 0.19±0.07 μg/mL after 24, 48, and 72 hours of treatment, respectively, and were significantly lower than that of free PTX. A similar tendency was exhibited on the other two cell lines. Compared with Taxol®, the lower cytotoxicity of PTX-NP5K may result from the incomplete release of PTX from NP. However, it should be noted that the in vitro cytotoxicity of Taxol® could be partially caused by the vehicle Cremophor EL.34–36
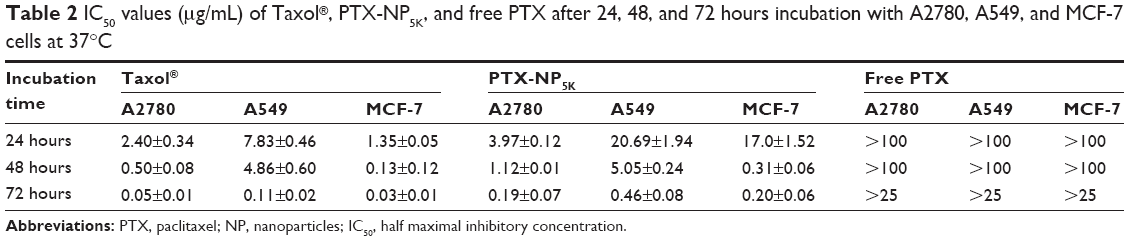 | Table 2 IC50 values (μg/mL) of Taxol®, PTX-NP5K, and free PTX after 24, 48, and 72 hours incubation with A2780, A549, and MCF-7 cells at 37°C |
Cell apoptosis assays
It has been widely reported that PTX kills cancer cells through the induction of apoptosis.37 The apoptosis-inducing ability of PTX-NP5K was qualitatively evaluated via Hoechst 33342 staining nuclei of A2780 cells. As observed under fluorescence microscopy, the cell nuclei showed a good integrity in the control group. However, some typical apoptotic features appeared in the PTX-NP5K and Taxol® groups, such as cell shrinkage, chromatin condensation, fragmentation of the nucleus, and apoptosis bodies (Figure 8A). Moreover, PTX-NP5K and Taxol® induced more cell apoptosis than free PTX, in accordance with the results of MTT assay.
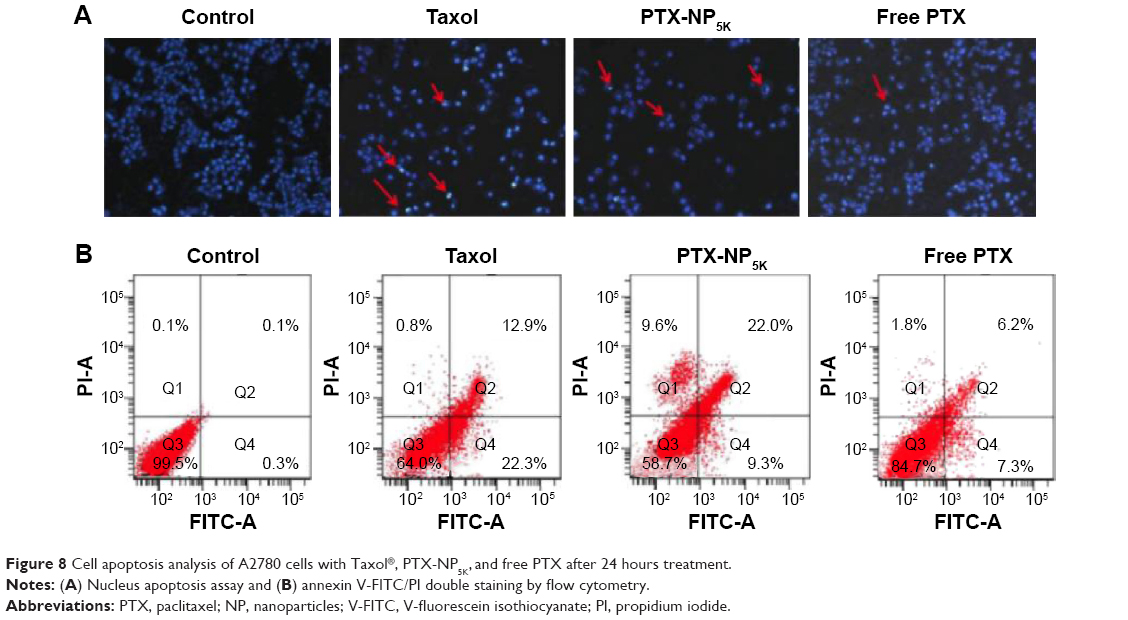 | Figure 8 Cell apoptosis analysis of A2780 cells with Taxol®, PTX-NP5K, and free PTX after 24 hours treatment. |
Annexin V-FITC/PI staining assay was carried out to quantitatively verify the cell apoptosis rate induced by different treatments. As shown in Figure 8B, after 24 hours treatment, the percentages of early apoptotic cells (Q4, annexin positive and PI negative) for Taxol®, PTX-NP5K, and free PTX were 22.3%, 9.6%, and 7.3%, while those of late apoptotic cells (Q2, annexin, and PI double positive) were 12.9%, 22.0%, and 6.2%, respectively. The quantitative apoptosis of S180 cells showed a similar tendency (Figure S3). Both the quantitative and qualitative results demonstrated that PTX-NP5K enhanced PTX-induced apoptosis compared with free PTX.
Cell cycle arrest assays
The antitumor efficacy of PTX is associated with mitosis inhibition and cell arrest in the G2/M phase. Increased G2/M phase arrest indicates the inhibition on cell division and restraint on cell growth.38 The cell cycle of A2780 cells treated with various formulation of PTX was examined to evaluate the therapeutic effects of PTX. As seen from Figure 9, the G2/M phase treated with PTX-NP5K for 24 hours was significantly increased to 70.8% compared with that of free PTX (43.7%). The cell cycle arrest effect in the G2/M phase from PTX-NP5K appeared to be consistent with the cell apoptosis analysis, demonstrating strong antitumor efficacy.
 | Figure 9 Cell cycle distribution in A2780 cells treated with various formulations. |
Antitumor activity
The in vivo antitumor efficiency of PTX-NP5K was evaluated in tumor-bearing mice. The mice were treated every other day with saline, PTX-NP5K (10 mg/kg and 30 mg/kg), and Taxol® (10 mg/kg), respectively. Both PTX-NP5K and Taxol® demonstrated tumor growth inhibition (Figure 10A–C). Tumors of saline, Taxol®, PTX-NP5K (10 mg/kg), and PTX-NP5K (30 mg/kg) groups were 0.59±0.26 g, 0.38±0.19 g, 0.29±0.11 g, and 0.21±0.04 g, respectively. Clearly, PTX-NP5K exhibited better therapeutic efficiency than Taxol® at the dose of 10 mg/kg. The tumor inhibition rates of Taxol® and PTX-NP5K were 36.4% and 50.8%, respectively. It is also worth noting that although PTX-NP5K showed a higher therapy property than Taxol® at the dose of 10 mg/kg, their tumor-inhibition result was not statistically significant. Another noteworthy fact is that the mice treated with Taxol® at a dosage above 20 mg/kg showed apathy and died 1 hour after injection. However, for PTX-NP5K, the dosage can be higher than 30 mg/kg with the inhibition rate of 71.2%, which is 1.57-fold higher than that treated with Taxol® (10 mg/kg). These results indicate that the NP offer advantages of decreased side effects and improved drug tolerance. It may suggest that the PTX-NP5K is a promising platform for safe and efficient cancer chemotherapy. The body weight of the mice was also monitored every day. As shown in Figure 10D, no significant variations in body weight were noticed in saline and the treatment groups with PTX dose of 10 mg/kg. The hematoxylin and eosin staining was further investigated (Figure 10E). In saline group, the tumor cells were polykaryocytes with large irregular karyons, rich cytoplasm, and more nuclear division. Nuclei apoptosis and spotty necrosis was observed in the tumor section after PTX treatment. These in vivo antitumor effects proved that 4-arm-PEG5K-TPGS was a good vehicle of PTX and could improve the chemotherapeutic efficacy of PTX. This might be accounted for the reason that 4-arm-PEG5K-TPGS NP increased the local accumulation concentration of PTX in the tumor tissue.
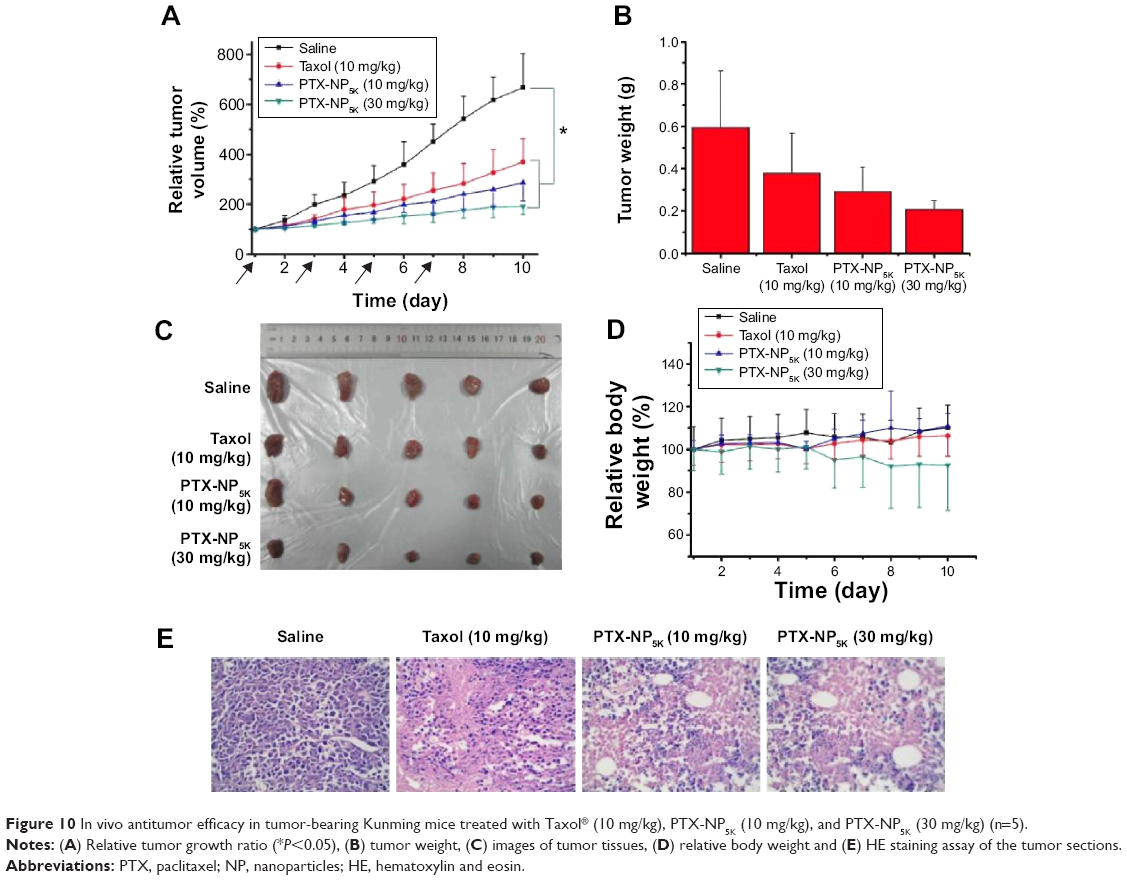 | Figure 10 In vivo antitumor efficacy in tumor-bearing Kunming mice treated with Taxol® (10 mg/kg), PTX-NP5K (10 mg/kg), and PTX-NP5K (30 mg/kg) (n=5). |
Conclusion
4-arm-PEG-TPGS copolymers with different PEG molecular weights were successfully synthesized and they could readily self-assemble into spherical nanosized NP. Among the three, 4-arm-PEG5K-TPGS drew our attention for its CAC value, solubility, and drug loading efficiency. The PTX-loaded 4-arm-PEG5K-TPGS NP showed good stability and a well-sustained drug release behavior in vitro. The NP could be effectively uptaken by the A2780 cell line with a time-dependent manner. Besides, PTX-NP5K could induce cell death via the apoptosis pathway and G2/M phase cell cycle arrest, in harmony with the results of the in vitro cytotoxicity assay. More importantly, the NP exhibited enhanced therapeutic efficacy. These findings indicate that 4-arm-PEG5K-TPGS may be an appropriate carrier for anticancer drug delivery in tumor.
Acknowledgments
This work was supported by the National Basic Research Program of China (2012CB932501), the National Natural Science Foundation of China (81373360), the Doctoral Fund of Ministry of Education of China (20120142120093), the Fundamental Research Funds for the Central Universities (2014TS091 and 2014QN134), Chutian Scholar Award, and 2013 Youth Scholar Award of HUST. We thank Prof Li-Qun Wang and Mr Fang Yuan in the Department of Polymer Science and Engineering, Zhejiang University, for their assistance in GPC measurement. The authors thank the Analytical and Testing Center of HUST for facilitating TEM and FTIR measurements.
Disclosure
The authors report no conflicts of interest in this work.
References
Calixto G, Fonseca-Santos B, Chorilli M, Bernegossi J. Nanotechnology-based drug delivery systems for treatment of oral cancer: a review. Int J Nanomedicine. 2014;9:3719–3735. | ||
Javad S, Zohre Z. Advanced drug delivery systems: nanotechnology of health design: a review. J Saudi Chem Soc. 2014;18(2):85–99. | ||
Steichen SD, Caldorera-Moore M, Peppas NA. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci. 2013;48(3):416–427. | ||
Wang K, Guo L, Xiong W, Sun L, Zheng Y. Nanoparticles of star-like copolymer mannitol-functionalized poly(lactide)-vitamin E TPGS for delivery of paclitaxel to prostate cancer cells. J Biomater Appl. 2014; 29(3):329–340. | ||
Chen M-X, Li B-K, Yin D-K, Liang J, Li S-S, Peng D-Y. Layer-by-layer assembly of chitosan stabilized multilayered liposomes for paclitaxel delivery. Carbohydr Polym. 2014;111:298–304. | ||
Park I-K, Ki YJ, Tran TH, Huh KM, Lee YK. Water-soluble heparin-PTX conjugates for cancer targeting. Polymer. 2010;51:3387–3393. | ||
Kojima C, Watanabe K, Nagayasu T, Nishio Y, Makiura R, Nakahira A. Preparation of hydroxyapatite-decorated poly(lactide-co-glycolide) microspheres for paclitaxel delivery. J Nanopart Res. 2013; 15(12):1. | ||
Ooya T. Effects of ethylene glycol-based graft, star-shaped, and dendritic polymers on solubilization and controlled release of paclitaxel. J Control Release. 2003;93(2):121–127. | ||
Mi Y, Zhao J, Feng S-S. Vitamin E TPGS prodrug micelles for hydrophilic drug delivery with neuroprotective effects. Int J Pharm. 2012;438(1–2):98–106. | ||
Liu Y, Miyoshi H, Nakamura M. Nanomedicine for drug delivery and imaging: a promising avenue for cancer therapy and diagnosis using targeted functional nanoparticles. Int J Cancer. 2007;120(12): 2527–2537. | ||
Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine. 2013; 9(1):1–14. | ||
Zhang Z, Lee SH, Gan CW, Feng S-S. In vitro and in vivo investigation on PLA-TPGS nanoparticles for controlled and sustained small molecule chemotherapy. Pharm Res. 2008;25(8):1925–1935. | ||
Huh KM, Lee SC, Cho YW, Lee J, Jeong JH, Park K. Hydrotropic polymer micelle system for delivery of paclitaxel. J Control Release. 2005;101(1–3):59–68. | ||
Liggins RT, Burt HM. Polyether-polyester diblock copolymers for the preparation of paclitaxel loaded polymeric micelle formulations. Adv Drug Deliv Rev. 2002;54:191–202. | ||
Zhang Z, Tan S, Feng S-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Zhang Z, Lin M, Feng S-S. Vitamin E d-a-tocopheryl polyethylene glycol 1000 succinate-based nanomedicine. Nanomedicine. 2012;7(11):1645–1647. | ||
Bao Y, Guo Y, Zhuang X, et al. D-α-tocopherol polyethylene glycol succinate-based redox-sensitive paclitaxel prodrug for overcoming multidrug resistance in cancer cells. Mol Pharm. 2014;11(9):3196–3209. | ||
Tang J, Fu Q, Wang Y, Racette K, Wang D, Liu F. Vitamin E reverses multidrug resistance in vitro and in vivo. Cancer Lett. 2013;336(1): 149–157. | ||
Zhao S, Tan S, Guo Y, et al. pH-sensitive docetaxel-loaded D-α-tocopheryl polyethylene glycol succinate-poly(β-amino ester) copolymer nanoparticles for overcoming multidrug resistance. Biomacromolecules. 2013;14(8):2636–2646. | ||
Zhao L, Feng S-S. Enhanced oral bioavailability of paclitaxel formulated in vitamin E-TPGS emulsified nanoparticles of biodegradable polymers: in vitro and in vivo studies. J Pharm Sci. 2010;99(8): 3552–3560. | ||
Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49(2):175–186. | ||
Zhang J, Li Y, Fang X, Zhou D, Wang Y, Chen M. TPGS-g-PLGA/Pluronic F68 mixed micelles for tanshinone IIA delivery in cancer therapy. Int J Pharm. 2014;476(1–2):185–198. | ||
Mi Y, Liu Y, Feng S-S. Formulation of docetaxel by folic acid-conjugated D-α-tocopheryl polyethylene glycol succinate 2000 (vitamin E TPGS2k) micelles for targeted and synergistic chemotherapy. Biomaterials. 2011;32(16):4058–4066. | ||
Jain A, Jain SK. PEGylation: an approach for drug delivery. A review. Crit Rev Ther Drug Carrier Syst. 2008;25(5):403–407. | ||
Wang J, Sun J, Chen Q, et al. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;33(28):6877–6888. | ||
Lu J, Huang Y, Zhao W, et al. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Mol Pharm. 2013;10(8):2880–2890. | ||
Ding H, Yong KT, Roy I, et al. Bioconjugated PLGA-4-arm-PEG branched polymeric nanoparticles as novel tumor targeting carriers. Nanotechnology. 2011;22(16):165101. | ||
Lv L, Shen Y, Li M, et al. Novel 4-arm poly(ethylene glycol)-block-poly(anhydride-esters) amphiphilic copolymer micelles loading curcumin: preparation, characterization, and in vitro evaluation. Biomed Res Int. 2013;2013:1–11. | ||
Zhang X, Cheng J, Wang Q, Zhong Z, Zhuo R. Miktoarm copolymers bearing one poly(ethylene glycol) chain and several poly(ε-caprolactone) chains on a hyperbranched polyglycerol core. Macromolecules. 2010;43(16):6671–6677. | ||
Guo Y, Chu M, Tan S, et al. Chitosan-g-TPGS nanoparticles for anticancer drug delivery and overcoming multidrug resistance. Mol Pharm. 2014;11(1):59–70. | ||
Song Q, Tan S, Zhuang X, et al. Nitric oxide releasing D-α-tocopheryl polyethylene glycol succinate for enhancing antitumor activity of doxorubicin. Mol Pharm. 2014;11(11):4118–4129. | ||
Du N, Song LP, Li XS, et al. Novel pH-sensitive nanoformulated docetaxel as a potential therapeutic strategy for the treatment of cholangiocarcinoma. J Nanobiotechnology. 2015;13(1):17. | ||
Zhang W, Sun J, Fang W, et al. Nanomicelles based on X-shaped four-armed PEGylated distearylglycerol as long circulating system for doxorubicin delivery. Eur J Pharm Sci. 2015;66:96–106. | ||
Xin H, Chen L, Gu J, et al. Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(ε-caprolactone) nanoparticles: in vitro and in vivo evaluation. Int J Pharm. 2010;402(1–2):238–247. | ||
Wei Z, Hao J, Yuan S, et al. Paclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization. Int J Pharm. 2009;376:176–185. | ||
Liebmann J, Cook JA, Teague CLD, Fisher J, Mitchell JB. The influence of Cremophor EL on the cell cycle effects of paclitaxel (Taxol) in human tumor cell lines. Cancer Chemother Pharmacol. 1994;33:331–339. | ||
Wang T-H, Wang H-S, Soong Y-K. Paclitaxel-induced cell death-where the cell cycle and apoptosis come together. Am Cancer Soc. 2000;88:2619–2628. | ||
Shen J, Yin Q, Chen L, Zhang Z, Li Y. Co-delivery of paclitaxel and survivin shRNA by pluronic P85-PEI/TPGS complex nanoparticles to overcome drug resistance in lung cancer. Biomaterials. 2012;33(33):8613–8624. |
Supplementary materials
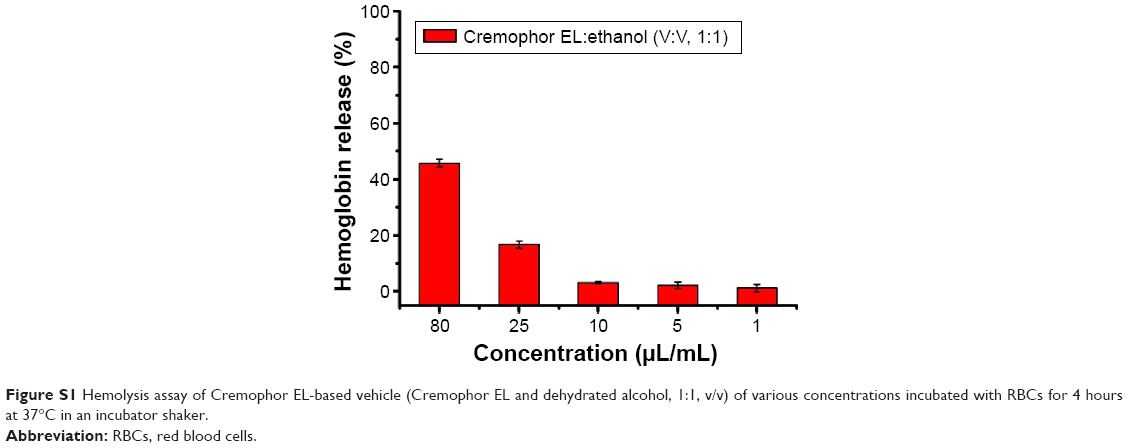 | Figure S1 Hemolysis assay of Cremophor EL-based vehicle (Cremophor EL and dehydrated alcohol, 1:1, v/v) of various concentrations incubated with RBCs for 4 hours at 37°C in an incubator shaker. |
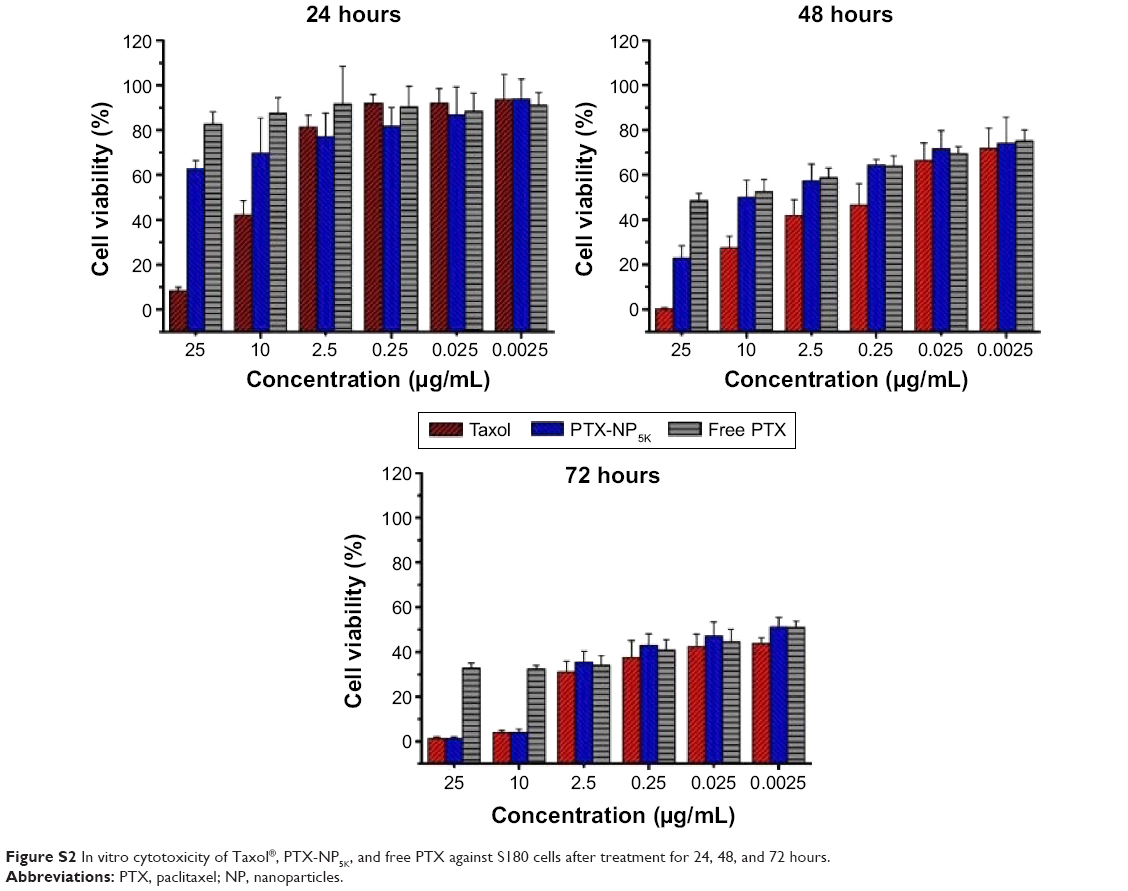 | Figure S2 In vitro cytotoxicity of Taxol®, PTX-NP5K, and free PTX against S180 cells after treatment for 24, 48, and 72 hours. |
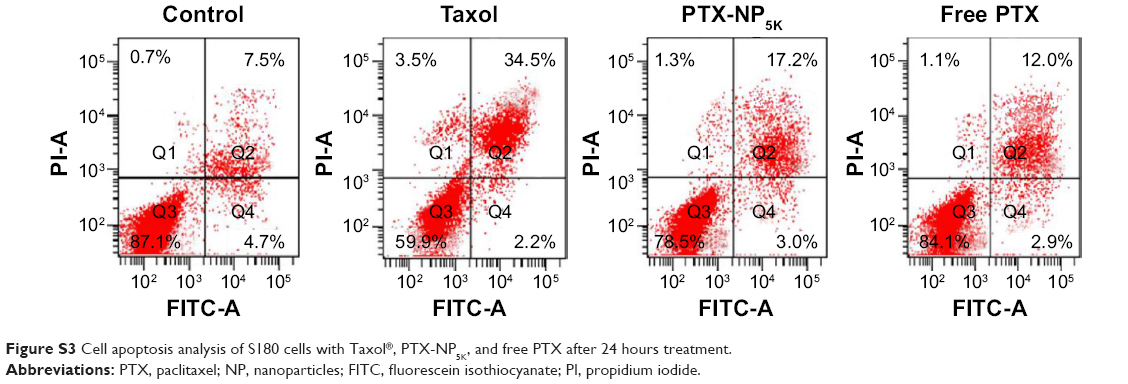 | Figure S3 Cell apoptosis analysis of S180 cells with Taxol®, PTX-NP5K, and free PTX after 24 hours treatment. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.