")
Back to Journals » International Journal of Nanomedicine » Volume 11
Cytotoxicity of CdTe quantum dots in human umbilical vein endothelial cells: the involvement of cellular uptake and induction of pro-apoptotic endoplasmic reticulum stress
Authors Yan M , Zhang Y, Qin H, Liu K, Guo M, Ge Y, Xu M, Sun Y, Zheng X
Received 2 August 2015
Accepted for publication 9 November 2015
Published 2 February 2016 Volume 2016:11 Pages 529—542
DOI https://doi.org/10.2147/IJN.S93591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Ming Yan,1,* Yun Zhang,2,* Haiyan Qin,3 Kezhou Liu,1 Miao Guo,1 Yakun Ge,1 Mingen Xu,1 Yonghong Sun,4 Xiaoxiang Zheng4
1Department of Biomedical Engineering, College of Life Information Science and Instrument Engineering, Hangzhou Dianzi University, Hangzhou, 2Basic Medical Sciences, College of Medicine, Shaoxing University, Shaoxing, 3Department of Chemistry, Zhejiang University, 4Zhejiang Provincial Key Laboratory of Cardio-Cerebral Vascular Detection Technology and Medicinal Effectiveness Appraisal, Department of Biomedical Engineering, Zhejiang University, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Cadmium telluride quantum dots (CdTe QDs) have been proposed to induce oxidative stress, which plays a crucial role in CdTe QDs-mediated mitochondrial-dependent apoptosis in human umbilical vein endothelial cells (HUVECs). However, the direct interactions of CdTe QDs with HUVECs and their potential impairment of other organelles like endoplasmic reticulum (ER) in HUVECs are poorly understood. In this study, we reported that the negatively charged CdTe QDs (–21.63±0.91 mV), with good dispersity and fluorescence stability, were rapidly internalized via endocytosis by HUVECs, as the notable internalization could be inhibited up to 95.52% by energy depletion (NaN3/deoxyglucose or low temperature). The endocytosis inhibitors (methyl-β-cyclodextrin, genistein, sucrose, chlorpromazine, and colchicine) dramatically decreased the uptake of CdTe QDs by HUVECs, suggesting that both caveolae/raft- and clathrin-mediated endocytosis were involved in the endothelial uptake of CdTe QDs. Using immunocytochemistry, a striking overlap of the internalized CdTe QDs and ER marker was observed, which indicates that QDs may be transported to ER. The CdTe QDs also caused remarkable ER stress responses in HUVECs, confirmed by significant dilatation of ER cisternae, upregulation of ER stress markers GRP78/GRP94, and activation of protein kinase RNA-like ER kinase-eIF2α-activating transcription factor 4 pathway (including phosphorylation of both protein kinase RNA-like ER kinase and eIF2α and elevated level of activating transcription factor 4). CdTe QDs further promoted an increased C/EBP homologous protein expression, phosphorylation of c-JUN NH2-terminal kinase, and cleavage of ER-resident caspase-4, while the specific inhibitor (SP600125, Z-LEVD-fmk, or salubrinal) significantly attenuated QDs-triggered apoptosis, indicating that all three ER stress-mediated apoptosis pathways were activated and the direct participation of ER in the CdTe QDs-caused apoptotic cell death in HUVECs. Our findings provide important new insights into QDs toxicity and reveal potential cardiovascular risks for the future applications of QDs.
Keywords: quantum dots, human umbilical vein endothelial cells, endocytosis, ER stress, apoptosis
Introduction
Semiconductor quantum dots (QDs), which are as small as 2–10 nm in diameter, are new types of fluorescent nanomaterials. They have important luminescent advantages over existing fluorescent dyes: they are up to 1,000 times brighter, more photostable, and have broader excitation ranges with narrower emissions. Besides these exceptional properties, their spectral emission tunability by size variations allows spectral multiplexing. Therefore, these nanoparticles (NPs) can be utilized as ideal agents for intracellular tracking, biomedical imaging, drug delivery, and diagnosis.1,2 For example, several research groups reported the successful labeling of blood vasculature in dermal, adipose, lymphatic, or tumor tissues by intravenous injections of QDs or peptide-conjugated QDs.3–6 These injected QDs can highlight morphological abnormalities in vasculature, trace biodistribution of drugs, and monitor the blood circulation dynamics. Moreover, because of the multicolor nature of QDs, researchers can investigate separate vascular systems in a multiplexed manner that could provide insight into the intricate blood circulation networks within organs and tissues. Despite QDs-based vascular imaging has shown great promise to improve our understanding of vascular physiological and pathological processes, to safely use QDs, we are urgent to understand their potential toxicity and the interactions with the toxin-susceptible vascular system. Especially, the vascular endothelial cells (ECs) that line the luminal surface of blood vessels may be the primary attack sites during QDs-caused vascular injury because QDs delivered to vascular may directly interact with and impair these cells.
Vascular ECs, which present a dynamic interface between the circulatory system and nonvascular tissues, are well recognized to play an important role in the regulation of vascular homeostasis. For instance, to supply nutrients or to transduct cell signals, ECs always internalize various plasma molecules and deliver these cargoes to different intracellular destinations or transendothelially to the intima of arteries.7 Nonetheless, under some circumstances, the undesirable uptake of toxicants (eg, viruses, bacteria, and some NPs) by ECs can also be observed, and it may result in subsequent organellar or cellular dysfunction.8–11 Hence, internalization of heavy metal-based QDs would be expected to alter the activity of cellular organelles of ECs and even these cells per se. Furthermore, ECs also maintain vascular homeostasis through the synthesis and secretion of a variety of vasoactive agents which regulate vasomotor tone, angiogenesis, thrombosis, and atherogenesis.12–14 ER, which serves as a site of secretory protein synthesis and modification, is thus crucial for vascular functions. However, a range of cytotoxic or pathological conditions (heavy metal exposure, high glucose, and hypoxia) could induce accumulation of unfolded or misfolded proteins in the ER and disturb the normal ER functions, leading to ER stress. Under physiological conditions, ER stress could trigger an evolutionarily conserved adaptive program called unfolded protein response (UPR). This cellular stress response activates ER-resident proteins, such as protein kinase RNA-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1. These ER membrane-resident effectors will start their respective downstream signaling cascade to reprogram transcription and translation in a concerted manner and restore cell survival.15–17 If ER stress is severe and prolonged, the stressed cells would be irreversibly damaged and eliminated by apoptosis.18 Previously, we demonstrated that QDs could impair mitochondria,19 lysosome, and plasma membrane (unpublished data), and they induced pro-apoptotic oxidative injuries in human umbilical vein ECs (HUVECs), but how the cadmium telluride quantum dots (CdTe QDs) enter into HUVECs and their hazards on ER homeostasis in HUVECs remain unknown.
In this in vitro study, we investigated the potential endothelial cytoxicity of red, mercaptosuccinic acid (MSA)-capped CdTe QDs using primary cultured HUVECs. All procedures in this study were performed in accordance with protocols approved by the Research and Ethics Committee of Shaoxing University. We mainly focused on the cellular uptake mechanism of CdTe QDs and the possible adverse effects of these CdTe QDs on ER in HUVECs. Our results clearly demonstrate that CdTe QDs could be incorporated into HUVECs through both clathrin- and caveolae/rafts-dependent endocytosis. These as-prepared CdTe QDs could activate UPR signaling cascade and ultimately lead to pro-apoptotic ER stress in HUVECs. Our results provide new insights into QDs toxicology and helpful guidance on the future safe use of QDs. It may also be useful for the eventual manipulation of QDs to make them more suitable in nanomedicine.
Materials and methods
Chemicals
Cadmium chloride (CdCl2, 99.0%) and tellurium powder (Te, 99.5%) were from Alfa Aesar (Ward Hill, MA, USA). Sodium borohydride (96%), hydrochloric acid (HCl), and sodium hydroxide (NaOH) were from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People’s Republic of China). MSA was from Sigma-Aldrich Corp. (St Louis, MO, USA). All chemicals were used without further purification. Solutions were prepared with Milli-Q water (Millipore, Darmstadt, Germany) as the solvent.
Medium 200 (M200), low-serum growth supplement (LSGS), Vybrant® Apoptosis Assay Kit, and Hoechst 33342 were from Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA). Salubrinal (SAL), SP600125, Z-LEVD-fmk, Z-VAD-fmk, and antibody against KDEL (which detects glucose-regulated protein, GRP78 and GRP94) were from Enzo Life Sciences, Inc. (Farmingdale, NY, USA). Other antibodies were purchased from the following companies: anti-human caspase-4, anti-human β-actin, and anti-human transcription factor C/EBP homologous protein (CHOP) (Santa Cruz Biotechnology, Dallas, TX, USA), PERK, eIF2α, phos-eIF2α, c-JUN NH2-terminal kinase (JNK), phos-JNK (Cell Signaling Technology, Inc., Beverly, MA, USA), phos-PERK and ATF4 (Beijing Biosynthesis Biotechnology, Beijing, China). 2′,7′-Dichlorofluorescein diacetate and one-step terminal deoxyribonucleotide transferase-mediated dUTP nick end labeling (TUNEL) apoptosis assay kit were from Beyotime Institute of Biotechnology (Jiangsu, People’s Republic of China). The WESTAR SUPERNOVA ECL reagents were from Cyanagen Srl (Bologna, Italy). Pierce® BCA assay kit was from Thermo Fisher Scientific (Waltham, MA, USA). The secondary antibodies and other reagents were from Sigma-Aldrich Corp.
Preparation and characterization of CdTe QDs
The CdTe QDs were synthesized and purified as described in our previous reports.19,20 To characterize the structural and optical properties of these QDs, the high-resolution transmission electron microscopy (HRTEM) image and absorption/emission spectra were obtained using a JEM 2010 microscope (JEOL, Tokyo, Japan; acceleration voltage at 200 kV) and UV2550 scanning spectrophotometer (Shimadzu, Kyoto, Japan)/F-2500 spectrofluorometer (Hitachi, Tokyo, Japan) over the range from 300 nm to 800 nm, respectively. The particle size and zeta potential of QDs in phosphate-buffered saline (PBS; pH 7.4) were concurrently measured using particle characterizer Zetasizer Nano ZS90 (Malvern Instruments Ltd., Malvern, UK). To measure their photostability in physiological environment, the stock solution of our as-prepared QDs was diluted 1:10,000 with PBS and added into a black 96-well microplate. The emission peak intensities were recorded from top at 405 nm excitation and 599 nm emission every 20 minutes for 4 hours with a fluorescence microplate reader (Flexstation 3, Molecular Device, Sunnyvale, CA, USA). All the measurements were performed at room temperature.
Cell culture
Primary HUVECs were obtained from human umbilical cord veins according to previously described procedure.21 Briefly, the vein was cannulated, filled, and digested with 0.1% type I collagenase at 37°C for 15 minutes. The obtained pellets were centrifuged and resuspended in M200 with LSGS at a concentration of 105 cells/mL. The cells were maintained in a humidified atmosphere containing 5% CO2. For all experiments, HUVECs were used between passages 3 and 5.
Assessment of the internalization and the subcellular location of CdTe QDs in HUVECs
Based on the fluorescence of QDs, the internalization of CdTe QDs in HUVECs was directly analyzed by flow cytometry (FCM). To determine the effect of concentration of CdTe QDs on endothelial uptake, the confluent HUVEC monolayer was incubated with M200 containing 2% fetal bovine serum (FBS) for 1 hour at 37°C in the presence of CdTe QDs at the concentrations ranging from 1 μg/mL to 50 μg/mL. In a separate experiment, to study the effect of incubation time on CdTe QDs’ uptake, the complete growth medium was replaced with 10 μg/mL suspension of CdTe QDs in M200 containing 2% FBS, and the plate was incubated for 1–48 hours. HUVECs incubated with CdTe QDs-free medium (also containing 2% FBS) served as untreated controls. After treatment, cells were rinsed three times with ice-cold PBS and harvested by trypsinization, and the fluorescence of internalized CdTe QDs was collected in FL-2 channel (585±21 nm). For analysis of FCM data, we initially set a region on the forward/side scatter dot plots to exclude most noncellular debris (shown in Figure 1A). The positive populations (HUVECs holding CdTe QDs) were identified as cells that expressed fluorescence activity above the autofluorescence of the untreated controls.
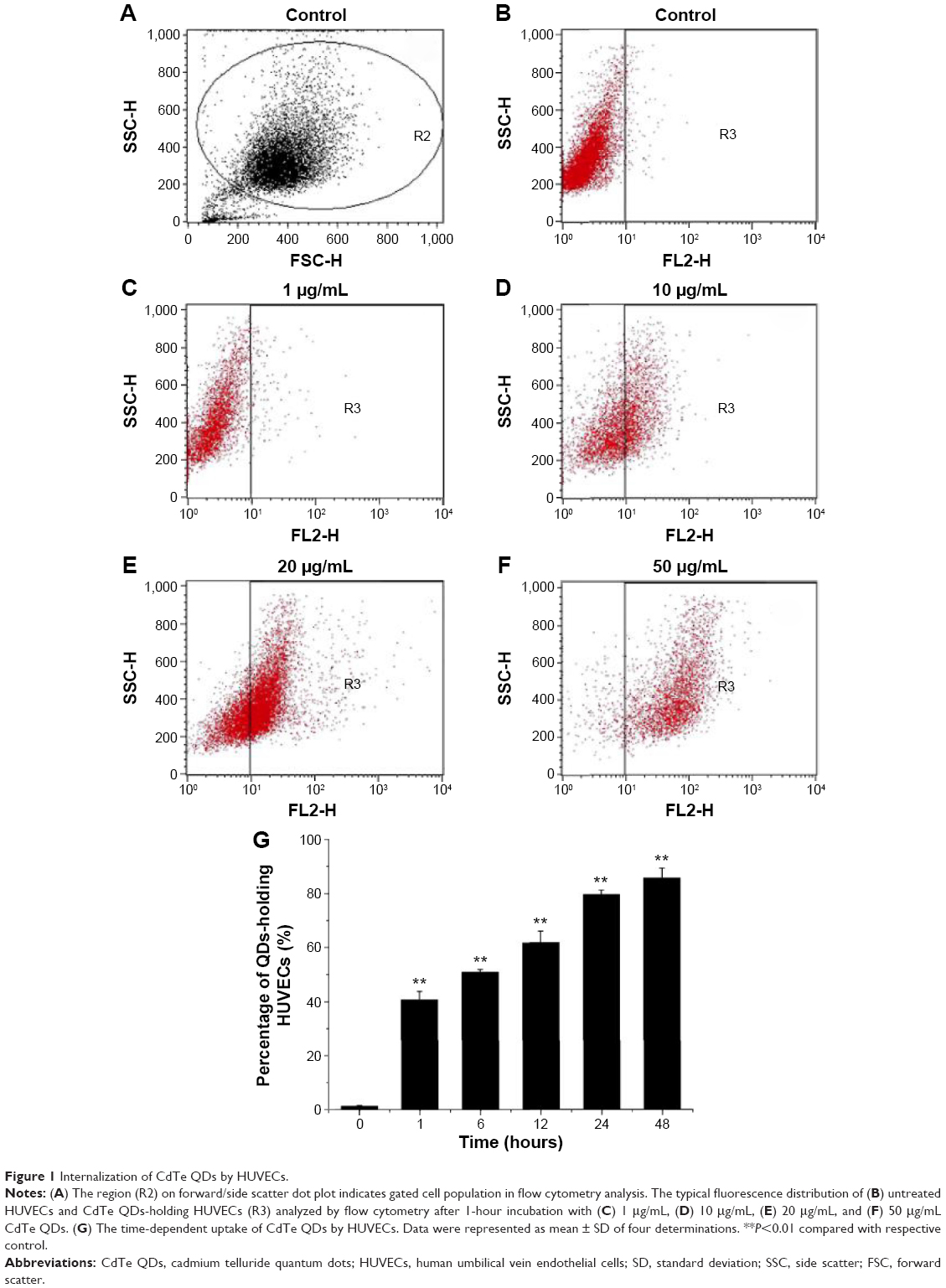 | Figure 1 Internalization of CdTe QDs by HUVECs. |
To study whether the uptake of CdTe QDs into HUVECs was energy-dependent, the cells were incubated with 0.1% (w/v) sodium azide and 50 mM 2-deoxyglucose (2-DG) for 1 hour prior to the addition of CdTe QDs (10 μg/mL) to HUVECs. The effect of temperature on CdTe QDs uptake was examined after pre-incubation of cells for 1 hour at 4°C and then incubation of cells with 10 μg/mL CdTe QDs suspension for an additional 1 hour at 4°C. To further clarify the specific endocytotic pathways by which HUVECs internalize CdTe QDs, HUVECs were treated with the inhibitors, methyl-β-cyclodextrin (5 mM), genistein (0.1 mM), sucrose (0.45 M), chlorpromazine hydrochloride (20 μM), cytochalasin B (1 μM), and colchicine (1 mM), in normal culture medium for 60 minutes at 37°C. CdTe QDs (10 μg/mL) were then added and incubated for 1 hour. Subsequently, the cells were washed three times with ice-cold PBS and analyzed by FCM.
To track the subcellular distribution of CdTe QDs, cells cultured on coverslips were firstly treated with 10 μg/mL CdTe QDs for 24 hours. The lysosomes and nuclei were then stained with 0.5 μM Lysotracker Green and Hoechst 33342 for 10–15 minutes at 37°C, respectively. ER was labeled with anti-KDEL (ER marker) antibody, and then incubated with fluorescein isothiocyanate (FITC)-conjugated secondary antibodies, following fixation with 3.7% polyoxymethylene. Confocal microscope (LSM 510, Carl-Zeiss, Oberkochen, Germany) and inverted fluorescent microscope (IX81, Olympus, Tokyo, Japan) were used to monitor the intracellular location of QDs.
Ultrastructural evaluation of endoplasmic recticulum
HUVECs exposed to 10 μg/mL CdTe QDs for 24 hours were washed with PBS and fixed for 1 hour at 4°C in 2.5% glutaraldehyde in phosphate buffer (pH 7.4). Thereafter, the cells were transferred into 1% osmium tetroxide in Milli-Q water for 1 hour at room temperature. Dehydration was through ascending concentrations of ethanol with six changes before embedding in Epon. Ultrathin sections of cells of interest were stained with uranyl acetate and lead citrate before viewing in a JEM-1200EX (JEOL) transmission electron microscope.
Immunoblot detection of ER signaling proteins
HUVECs were exposed to 10 μg/mL CdTe QDs for 24 hours, and then washed with PBS. Total cell lysates were prepared by solubilizing HUVECs in cold radioimmune precipitation assay buffer (1% Triton X-100, 50 mM Tris/HCl, pH 7.4, 300 mM NaCl, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM sodium orthovanadate, and 2 mg/mL iodoacetamide). Protein concentration was quantified by the Pierce® BCA assay. The whole-cell lysates (50 μg of protein) were subjected to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and subsequently transferred onto polyvinylidene difluoride membranes (Millipore). The membranes were blocked with nonfat dry milk (5% in Tris-buffered saline containing 0.1% Tween-20) and probed with primary antibodies including anti-CHOP (1:300), anti-caspase-4 (1:500), anti-phos-PERK (1:500), anti-ATF4 (1:500), anti-β-actin (1:5,000), anti-GRP78/94 (1:1,000), anti-PERK (1:1,000), anti-eIF2α (1:1,000), anti-phos-eIF2α (1:1,000), anti-JNK (1:1,000), and anti-phos-JNK (1:1,000), followed by incubation with horse radish peroxidase-conjugated secondary antibodies. Proteins were visualized with a WESTAR SUPERNOVA ECL kit.
FCM analysis of apoptosis of HUVECs upon exposure of CdTe QDs
To measure CdTe QDs-induced apoptosis, HUVECs were firstly treated with 10 μg/mL CdTe QDs for 24 hours and collected by trypsinization. The pellets were then resuspended and stained with 5 μg/mL FITC-conjugated Annexin-V for 15 minutes in dark. The fluorescence of Annexin-V was measured with FACSCalibur flow cytometer on the FL-1 detector (530 nm). Alternatively, using a commercial one-step TUNEL apoptosis assay kit, in situ detection of DNA fragments was also performed to detect the apoptotic cell death of treated HUVECs. Briefly, cells were trypsinized and fixed with 3.7% polyoxymethylene. Following treatment with 0.1% Triton X-100, the cells were incubated with TUNEL reaction mixture for 1 hour and then washed twice with PBS for FCM analysis. The FITC-labeled TUNEL-positive cells were measured on the FL-1 detector. To clarify the role of ER in CdTe QDs-induced endothelial apoptosis, the specific ER stress inhibitors – SAL (25 μM), Z-LEVD-fmk, and SP600125 – were added into HUVECs 1 hour before the incubation of CdTe QDs, respectively. Then, after 24-hour incubation of HUVECs with CdTe QDs, the apoptotic HUVECs were detected by Annexin-V assay on a FACSCalibur flow cytometer.
Statistical analysis
The data were presented as mean ± SD and evaluated for statistical significance by one-way analysis of variance, followed by least significance difference post hoc test (SPSS13.0; SPSS Inc., Chicago, IL, USA). Values of P<0.05 were considered statistically significant.
Results
Characteristics of CdTe QDs
As shown in Figure 2A and B, the HRTEM image illustrates that the average size of CdTe QDs is approximately 4 nm, conforming a highly crystalline nature and good dispersity. The size distribution of CdTe QDs is 3.492±0.87 nm (Figure 2C) measured by dynamic light scattering. The zeta potential data (Figure 2D) showed that CdTe QDs with carboxyl group were highly negatively charged (−21.63±0.91 mV). Moreover, these QDs had symmetric emission spectra: the full width at half maximum was approximately 60 nm, and the emission peak was at 599 nm (Figure 2E). The fluorescence intensity of these QDs at physiological pH was slowly declined in a time-dependent manner during 4-hour periods (Figure 2F): the average emission peak intensity only decreased by 8.87% during the test cycle. These results suggest that CdTe QDs present promising prospects in the applications of bioimaging.
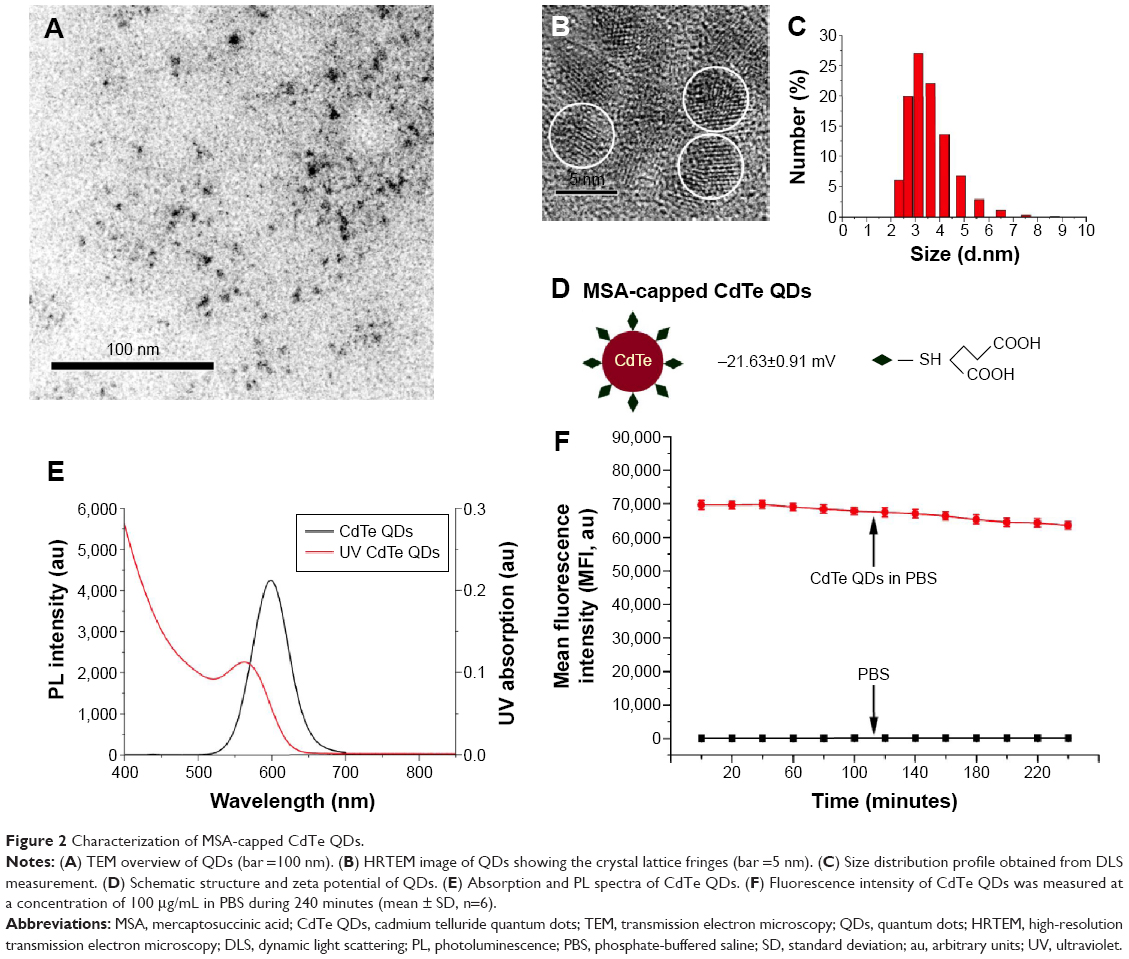 | Figure 2 Characterization of MSA-capped CdTe QDs. |
Internalization of CdTe QDs by HUVECs
Figure 1 shows that HUVECs internalized CdTe QDs to a great extent, and this internalization increased in both dose-dependent and time-dependent manner. After 1-hour exposure to 0 μg/mL, 1 μg/mL, 10 μg/mL, 20 μg/mL, and 50 μg/mL CdTe QDs, the percentage of QDs-holding HUVECs was 0.91%, 3.64%, 42.85%, 69.06%, and 92.89%, respectively. The uptake of CdTe QDs in HUVECs was gradually increased during the incubation time (Figure 1G). Upon 1-hour incubation with 10 μg/mL CdTe QDs, there were ~41% of QDs-holding HUVECs. This percentage reached to 50.81%, 61.63%, and 79.53% after 6-hour, 12-hour, and 24-hour treatment, respectively, but only increased to 6.11% after additional 24-hour incubation.
To assess the underlying mechanism involved in CdTe QDs internalization, the cellular uptake of CdTe QDs at a low temperature or in an energy-depleted environment was examined by FCM and is shown in Figure 3A. The low temperature or NaN3/2-DG induced marked decreases in the endothelial internalization of CdTe QDs (95.52% or 54.17% of untreated cells, respectively). Subsequently, using various biochemical inhibitors, the contribution of different endocytotic pathways in the uptake of CdTe QDs was characterized. As shown in Figure 3B, the inhibitor of macropinocytosis/phagocytosis cytochalasin B (1 μM) did not affect the entry of CdTe QDs into HUVECs, whereas the internalization was dramatically decreased by 44.83% or 23.81% in the presence of 5 mM cholesterol depletor methyl-β-cyclodextrin (to disrupt caveolae/rafts) or 0.1 mM genistein that could specifically inhibit caveolae-mediated uptake. The uptake of CdTe QDs in 0.45 M hypertonic sucrose (to disperse clathrin lattices) or 20 μM chlorpromazine (an inhibitor of the clathrin-coated pit-pathway)-pretreated HUVECs was also significantly reduced to 32.07% or 75.58% of the cells without pretreatment. Additionally, inhibition of microtubules by colchicine (1 mM) resulted in a 32.5% decrease in CdTe QDs uptake, indicating that microtubules may also be implicated in controlling intracellular uptake of CdTe QDs.
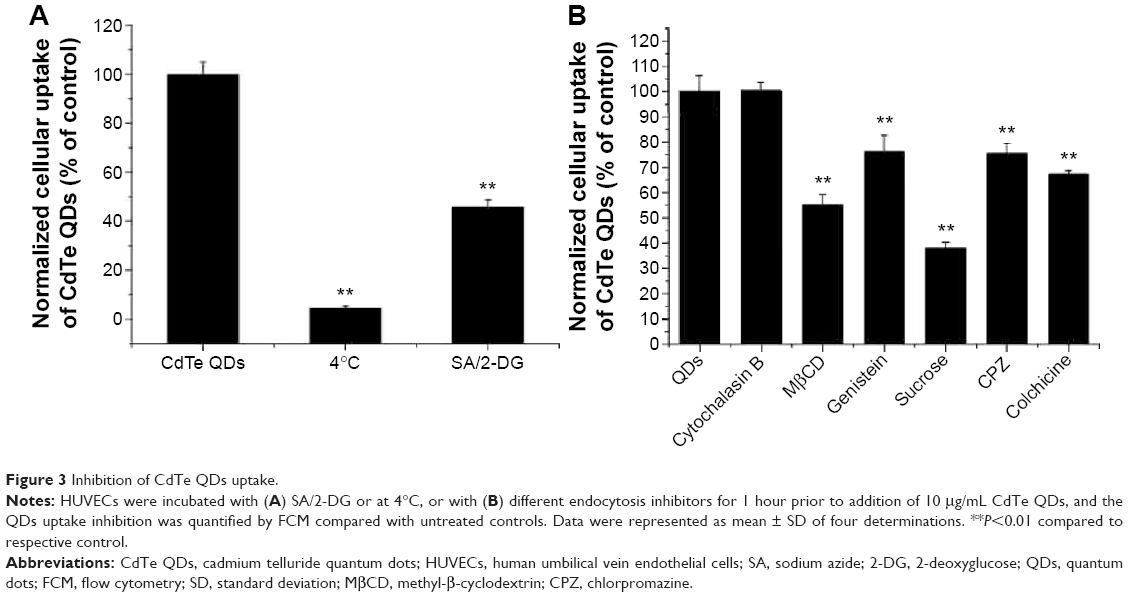 | Figure 3 Inhibition of CdTe QDs uptake. |
Intracellular distribution of CdTe QDs in HUVECs
The colocalization between CdTe QDs and organelles was explored by laser confocal microscopy after the treatment of HUVECs with 10 μg/mL CdTe QDs for 24 hours. The nuclei were labeled with Hoechst 33342 (blue fluorescence, Figure 4A). Figure 4B and C shows that the CdTe QDs (red fluorescence) were only distributed throughout the cytoplasm of HUVECs but did not enter into nuclei within 24 hours. The lysosomes labeled with by Lysotracker Green showed green fluorescence (Figure 4D), and the CdTe QDs in HUVECs presented red fluorescence (Figure 4E). The merged fluorescent image (Figure 4F) revealed that partial CdTe QDs were aggregated and entrapped within the peripheral cytoplasmic vesicles (yellow fluorescence), which suggests that only part of internalized CdTe QDs were localized in lysosomes. In addition, Figure 4G and H shows the immunofluorescence of ER and the fluorescent CdTe QDs in HUVECs, respectively. The merged image (Figure 4I) revealed that the fluorescent areas of CdTe QDs (red) and ER (green) had large overlap in the QDs-treated group. This result suggests that a portion of CdTe QDs were located in ER, where the QDs possibly affect the morphology and function of ER.
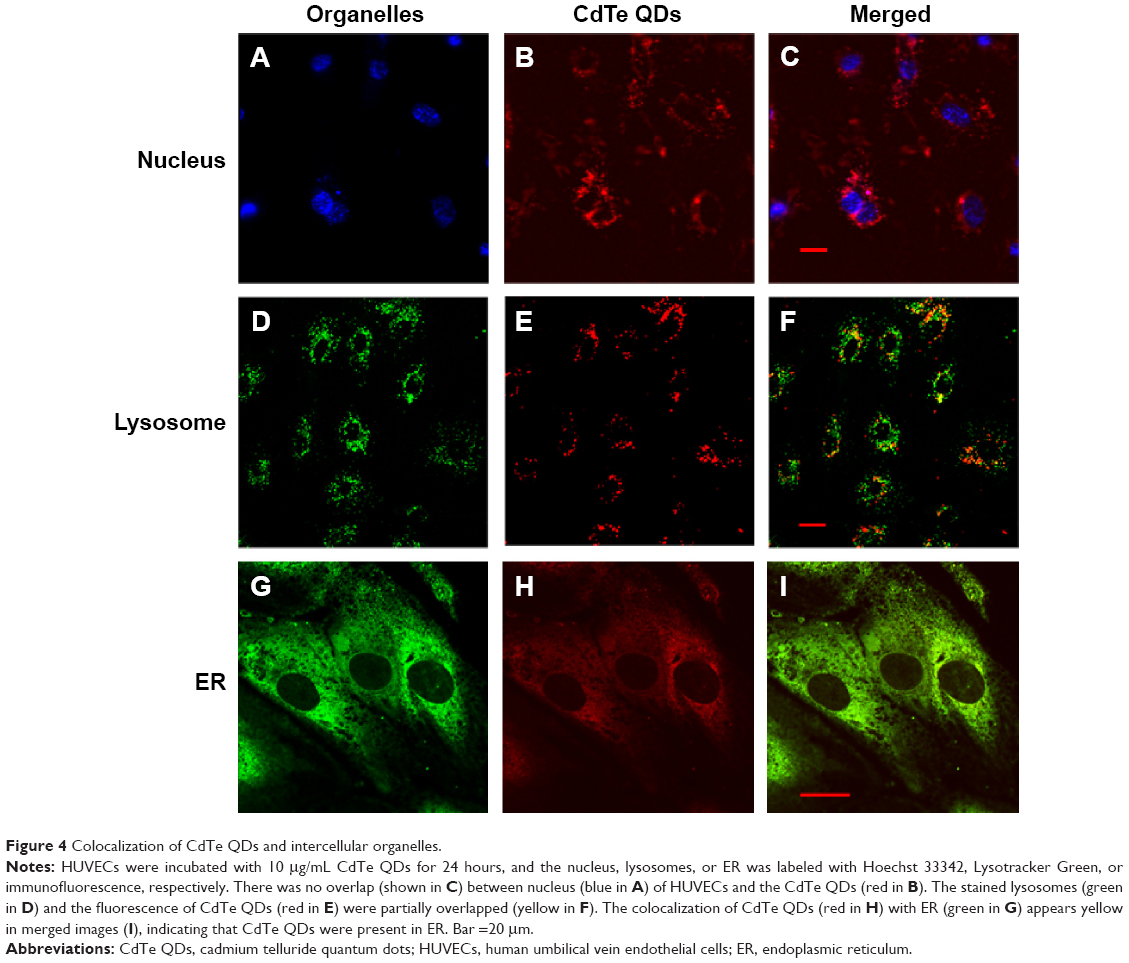 | Figure 4 Colocalization of CdTe QDs and intercellular organelles. |
CdTe QDs induces ER stress in HUVECs
We measured the impact of CdTe QDs on the activation of ER stress sensors and downstream effectors, including KDEL (GRP78 and GRP94) and PERK-eIF2α-ATF4 pathway proteins, by Western blotting. As shown in Figure 5A, the CdTe QDs treatment resulted in increased levels of GRP78 and GRP94 in a dose-dependent manner. The GRP78 expression was increased by 46.39% in 1 μg/mL group and by 68.83% in 10 μg/mL group compared with the untreated control. Similarly, 0.1 μg/mL, 1 μg/mL, and 10 μg/mL CdTe QDs also dose-dependently raised the level of GRP94 to 160.59%, 170.93%, and to 229.36% of the untreated control, respectively (Figure 5B). This outcome indicates that CdTe QDs could trigger significant ER stress in HUVECs. To dissect the molecular pathways involved in CdTe QDs-induced ER stress, we found that PERK was phosphorylated and eIF2α was heavily phosphorylated after QDs treatment for 24 hours (Figure 5C). The incubation of HUVECs with CdTe QDs also led to a significant increase in ATF4, the downstream target protein of phos-eIF2α (Figure 5C).
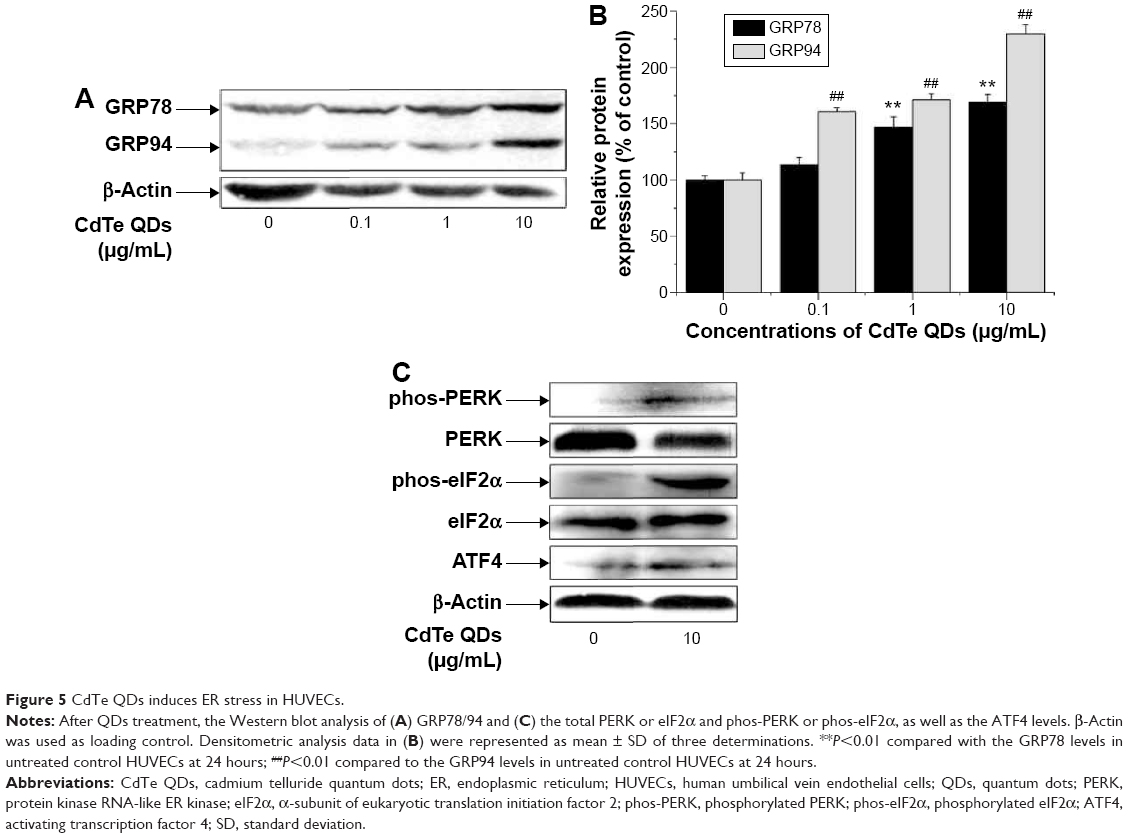 | Figure 5 CdTe QDs induces ER stress in HUVECs. |
To observe cellular changes in response to ER stress, ultrastructure was analyzed by TEM. In untreated control cells, the ER consisted of an extensive network of branching tubules distributed throughout the cytosol (Figure 6A, arrows). After 24-hour treatment with 10 μg/mL QDs, the most prominent change is that the bulk of the rough ER with narrow cisternae was dilated (Figure 6B, arrows).
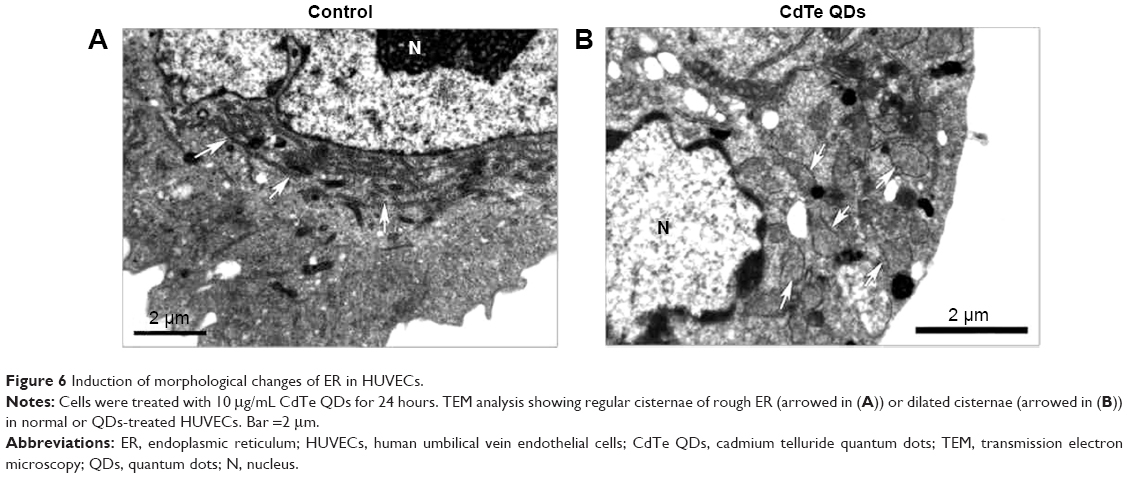 | Figure 6 Induction of morphological changes of ER in HUVECs. |
CdTe QDs cause ER stress-induced apoptosis
Since the persistent ER stress can also trigger apoptotic cell death, we hereby proceed to determine whether the CdTe QDs directly induce apoptotic changes in HUVECs. The TUNEL assay confirmed that 10 μg/mL CdTe QDs caused 14.07%±1.58% of treated HUVECs to undergo apoptosis (Figure 7A and C). Using an Annexin-V apoptosis detection kit, we also observed that the percentage of apoptotic HUVECs was dramatically increased by 462.75% when the cells were incubated with 10 μg/mL CdTe QDs (Figure 7B and D).
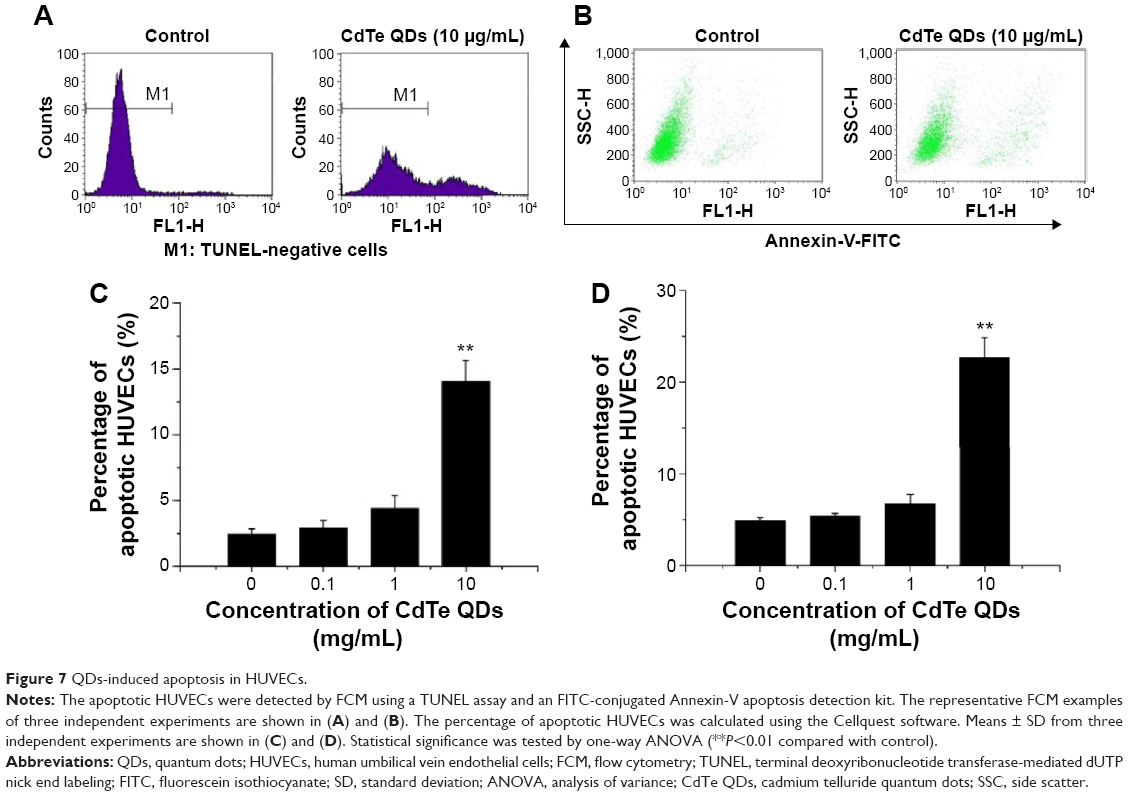 | Figure 7 QDs-induced apoptosis in HUVECs. |
In a step toward deciphering the underlying mechanisms of QDs-induced apoptosis of HUVECs, we firstly found that 10 μg/mL CdTe QDs caused a 1.9-fold increase in the CHOP (also called DDIT3/GADD153) expression in HUVECs after 24-hour incubation. Treatment with CdTe QDs also significantly promoted the phosphorylation of JNK46/54 without changing the total JNK levels. In addition, densitometric analysis indicated that the amount of pro-caspase-4 in HUVECs was substantially decreased by 34.34%, whereas the active caspase-4 was increased by 42.28% detectable in the CdTe QDs-treated HUVECs (Figure 8A and B).
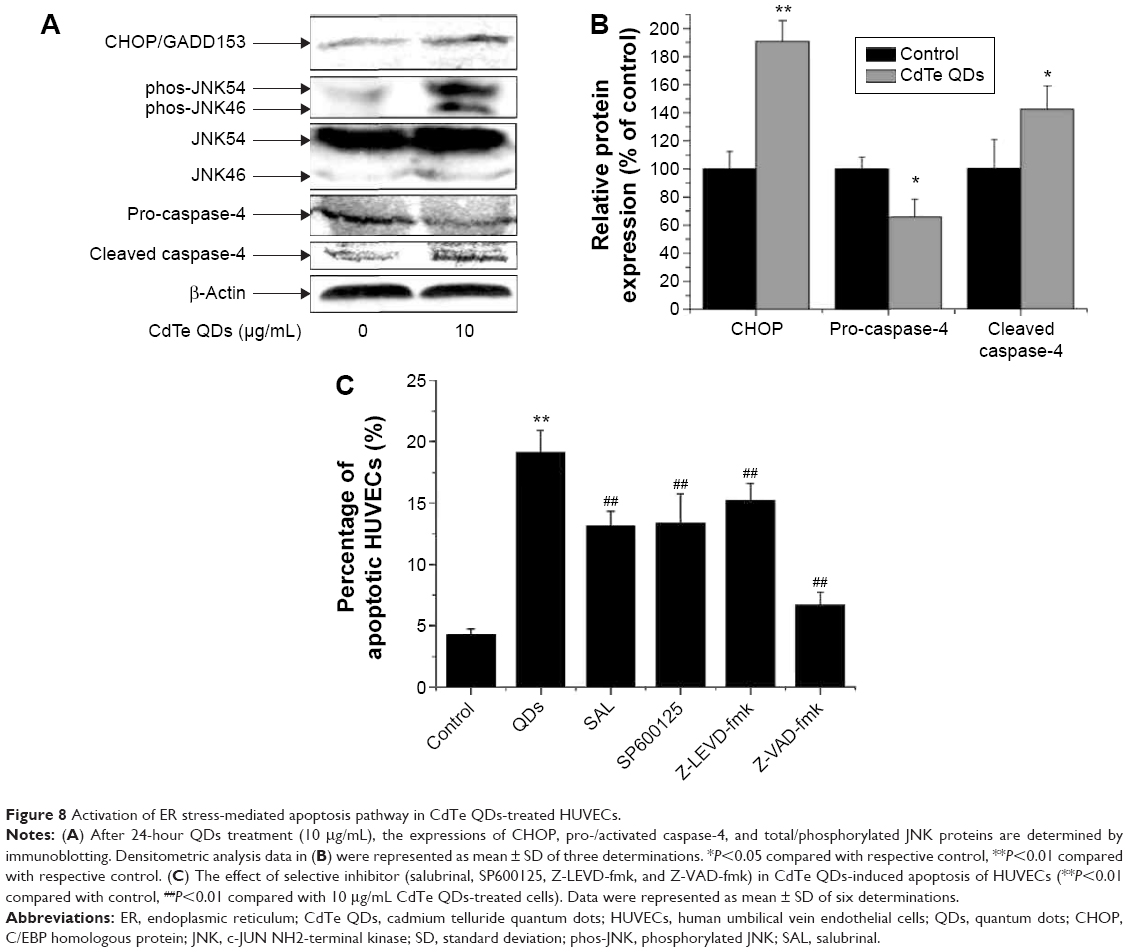 | Figure 8 Activation of ER stress-mediated apoptosis pathway in CdTe QDs-treated HUVECs. |
Chemical inhibitors of JNK (SP600125, 20 μM), eIF2α (SAL, 25 μM), or caspase-4 (Z-LEVD-fmk, 10 μM) were found protecting HUVECs from CdTe QDs-induced apoptosis to a certain extend as did a broad-spectrum caspase inhibitor (Z-VAD-fmk; Figure 8B). The ratio of apoptotic HUVECs reduced by 30.26%, 31.34%, 20.60%, and 53.12% in the presence of SP600125, Z-LEVD-fmk, SAL, and Z-VAD-fmk (50 μM), respectively (Figure 8C). These results strongly suggest that ER stress may be one of the driving forces for apoptosis in the CdTe QDs-treated HUVECs.
Discussion
Vascular ECs, which line the inner side of the vessels, play key roles in the regulation of vascular homeostasis. Therefore, aberrant EC function induced by nanomaterials may result in pathological processes such as atherogenesis and bleeding disorders. So far, cultured vascular ECs are widely used as in vitro model systems for nanotoxicology research, although ECs display structural and functional heterogeneity in different organs and vascular beds.22 As one of vascular ECs, HUVECs, which have typical endothelial phenotype, are relatively easy to isolate and culture.23 Hence, they are the most commonly used primary ECs and are well characterized for the specific study of endothelial toxicity. In this study, using HUVECs, we report that MSA-capped CdTe QDs could be uptaken by HUVECs through several endocytotic pathways. It also caused significant cellular injury in HUVECs by induction of pro-apoptotic ER stress. The CdTe QDs-triggered apoptotic cell death in HUVECs can be partly inhibited by specific inhibitors of ER stress pathways, suggesting that the impairment of ER probably contributes to cell death caused by these QDs.
The first important finding is that both caveolae/raft- and clathrin-mediated endocytosis were moderately involved in the uptake of CdTe QDs. The clathrin-mediated endocytosis was found to be responsible for the entry of anionic CdSe/ZnS QDs (size in PBS was ~27 nm) in human prostatic carcinoma (PC3-PSMA).24 But similar CdSe/ZnS QDs with carboxy group coating (<−30 mV and diameter >10 nm) were capable of entering human epidermal keratinocytes only by lipid rafts-dependent endocytosis.25 The cellular uptake and the cytotoxicity of various NPs could be regulated by surface charge as well as the particle size;26,27 the different mechanistic results of QDs uptake may thus arise from their different physical properties of QDs. On the other hand, caveolae, as little caves in the plasma membrane, are particularly abundant in vascular ECs.28 Therefore, they are believed to play a major role in endothelial endo-/transcytosis.29 For example, the uptake and transport of negative albumin (isoelectric point, pI 4.7–4.9) were defected in caveolae-deficient ECs.30 The MSA-capped CdTe QDs may be thereby uptaken through caveolar endocytosis pathway due to the negative surface charge (−21.63±0.91 mV, Figure 2), similar to albumin. However, whether the caveolar endocytosis of CdTe QDs by HUVECs is cell type specific will need further detailed evaluation.
The intracellular fate of the internalized QDs in cells is a complicated issue. Classically, cargoes that entered cells through clathrin-mediated endocytosis are delivered into lysosomes,31 where they are degraded by lysosomal hydrolytic enzymes. Lysosomes also participate in intracellular organelle and macromolecule turnover via autophagy. Thus, the internalized nanomaterials-induced autophagy may be an attempt to degrade what is perceived by the cell as foreign or aberrant.32,33 Likewise, we also found that considerable CdTe QDs were localized in lysosomes (Figure 4D–F) and triggered increase in the autophagic marker LC3 (data not shown) after 24-hour treatment which are consistent with previous reports.34–36 Moreover, our preliminary results showed that the intracellular cadmium dose in the 10 μg/mL CdTe QDs-treated HUVECs was much lower than the concentration (1 μmol/L) that did not cause endothelial injury like lactate dehydrogenase leakage.37,38 Taking together, we speculated that some amount of the endocytotic CdTe QDs were slowly degraded in lysosomes, but only the Cd2+ released from CdTe QDs is insufficient to induce severe cellular malfunction during our tested period. These results also prompt that encapsulating CdTe QDs core with appropriate coating may effectively protect them from lysosomal degradation and herein reduce their cytotoxicity.
It is noteworthy that there was a striking colocalization between CdTe QDs and ER networks (Figure 4G–I), which was rarely reported in the literature. Studies have revealed that cholera toxin (CTX), simian virus 40 (SV40), and autocrine motility factors that undergo caveolae-dependent endocytosis are transported to the ER or Golgi.39–41 Therefore, one reasonable explanation for the colocalization of CdTe QDs and ER in HUVECs would be that these CdTe QDs were sorting to ER through the caveolae/rafts-dependent endocytosis. After entering to the ER, both CTX and SV40 could take advantage of the protein folding and quality control machinery in the ER for unfolding/uncoating and membrane translocation so as to induce disease.42,43 However, whether the CdTe QDs sequestrated in ER were broken down or transported to other position in HUVECs remains unknown. It is thus necessary to determine the final fate of CdTe QDs in ER as well as the probable disturbance of ER or even cellular metabolism.
ER, which synthesizes and modifies secretory proteins, is crucial for the secretory functions of ECs. However, pathological stimuli (eg, oxidized low-density lipoprotein) could disrupt protein folding reactions and cause an ER-initiated UPR in vascular ECs.44 It is known that the silver/silica NPs and other toxicants could elevate GRP78 and GRP94, which are important hallmarks of ER stress, in mammalian cells.16,45 Similarly, we observed that low-dose CdTe QDs (0.1 μg/mL and 1 μg/mL) caused an increase in these two chaperones in HUVECs (Figure 5A), whereas the high-concentration CdTe QDs (10 μg/mL) triggered not only the upregulation of GRP78 and GRP94 (Figure 5A) but also the dilation of ER ultrastructure (Figure 6). This dose-dependent ER stress response showed a similar increase pattern as the CdTe QDs-induced endothelial apoptosis (Figure 7). In conclusion, our results validate that high dose of CdTe QDs (10 μg/mL, close to the TC50 obtained from our previous results19) might cause severe pro-apoptotic ER stress response. Combined with previous report,45,46 our data also suggest that NPs-induced ER stress may be a common cellular response to nanomaterials in various cell types. In addition, the lower dosage could be considered as a warning to the following realistic damage outcomes.
Importantly, if serious ER stress was launched by ischemia, radiation, or NPs, the GRP78 that is physiologically occupied with the ER-transmembrane effector PERK will activate this molecule.47 Then, the eIF2α could be directly phosphorylated by activated PERK to inhibit protein translation.48,49 However, ATF4 can bypass the eIF2α phosphorylation-caused translation inhibition and lead to expression of apoptosis genes like ATF3 and CHOP/GADD153.50 Likewise, in response to CdTe QDs-caused ER stress, PERK was first activated and eIF2α was phosphorylated, accompanying the increased levels of ATF4 (Figure 5). These results suggest that CdTe QDs activated PERK/eIF2α/ATF4-dependent ER stress signaling pathway in HUVECs. Moreover, CHOP, as one of the downstream targets of the PERK/eIF2α/ATF4 UPR pathway, is a crucial component of the ER stress-mediated apoptosis pathway. The JNK and the ER-associated caspase-12 (caspase-4 in human) were also involved in ER stress-mediated apoptotic event.50,51 Recently, it was reported separately that significant activation of caspase-12,52 JNK,53 or CHOP52,54 occurred in the many types of NPs-treated cells. In this study, we observed an increase in CHOP expression, the stimulated caspase-4 processing alongside with the phosphorylation of JNK in QDs-treated HUVECs (Figure 8A). The specific inhibitors of caspase-4 (Z-LEVD-fmk) and JNK (SP600125) partly attenuated endothelial apoptotic death (Figure 8C). The eIF2α phosphorylation inhibitor SAL that can reduce CHOP expression55 also effectively protects cells against ER stress-mediated apoptosis (Figure 8C). These findings demonstrate the direct participation of ER in the CdTe QDs-caused apoptotic cell death in HUVECs, since all three ER stress-mediated apoptosis pathways were involved. Our results of QDs-induced pro-apoptotic ER stress signaling pathways therefore complement the knowledge of the potential mechanism of QDs or even nanomaterials’ toxicity in previous investigations. Furthermore, because ER stress has a deleterious role in atherogenesis by induction of inflammatory cytokine expression, disturbance of ER calcium metabolism, and triggering apoptosis in ECs,56 the CdTe QDs which induced pro-apoptotic ER stress in HUVECs can be interpreted as a potential risk for the development of cardiovascular diseases. Although important findings have been presented by this study, because of the extreme complexity of in vivo systems, further in vivo studies are required to accurately assess toxicity of QDs, particularly with regard to the cardiovascular system.
Conclusion
In summary, our data showed that CdTe QDs could be internalized by HUVECs via both caveolae/raft- and clathrin-dependent endocytosis. We also provided the first evidence that endothelial ER may be a target of QDs toxicity because the CdTe QDs treatment caused significant ER stress, activated both the PERK-eIF2α-ATF4 UPR pathway and all of the three downstream ER stress-mediated apoptosis pathways, and ultimately triggered ER stress-induced apoptotic cell death in HUVECs (summary in Figure 9). Our study highlighted the understanding of the toxic effects from QDs, especially the potential safety risks for their future applications of CdTe QDs. The unexpected cytotoxicity of bare core CdTe QDs suggests researchers to improve the manipulation of CdTe QDs to make them nontoxic fluorescent probes for future scientific and clinical usage.
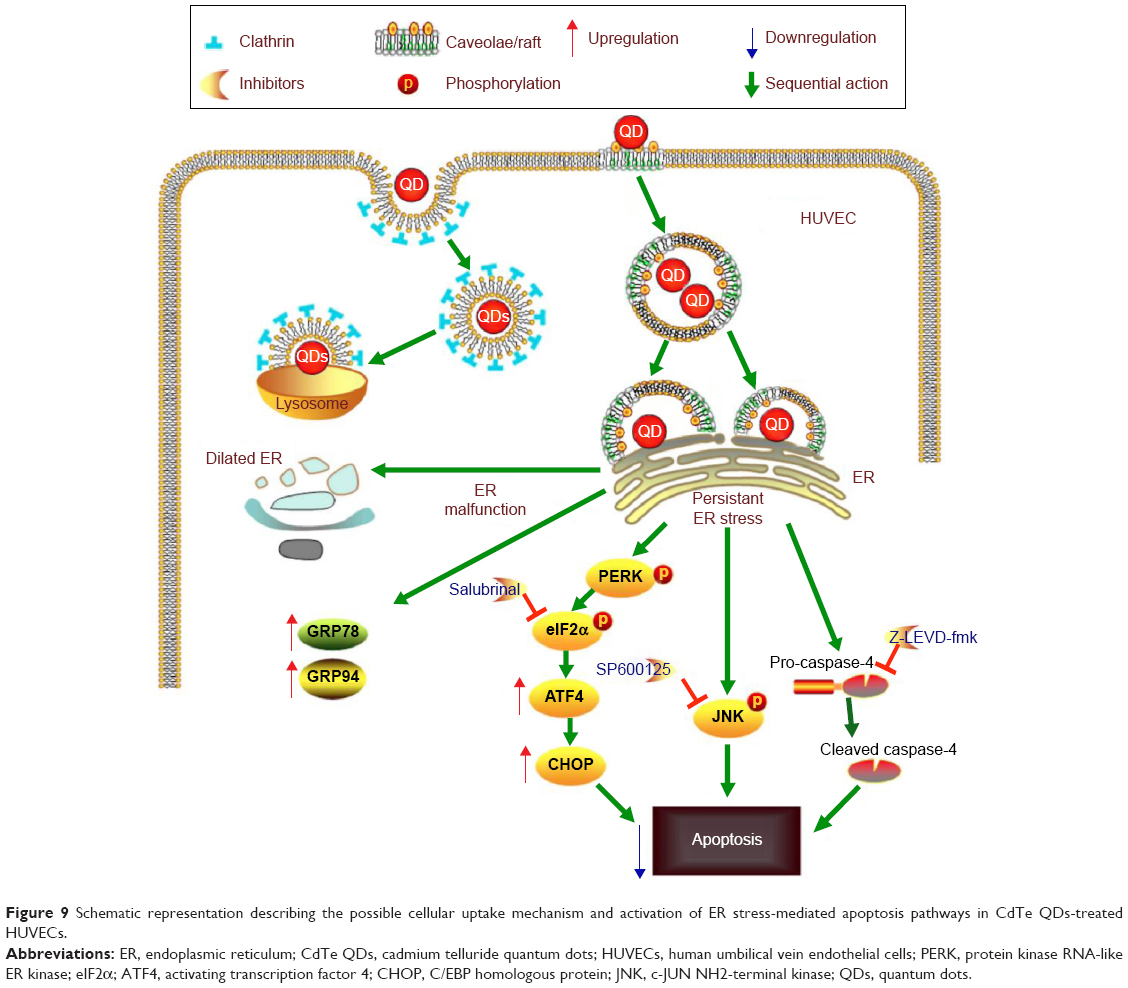 | Figure 9 Schematic representation describing the possible cellular uptake mechanism and activation of ER stress-mediated apoptosis pathways in CdTe QDs-treated HUVECs. |
Acknowledgments
The work was supported by Zhejiang Provincial Natural Science Foundation (No.’s LY15H180012, LY13H060003, LY14C100004, and LY13C100003) and National Natural Science Foundation of China (No. 30900301).
Disclosure
The authors report no conflicts of interest in this work.
References
Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Quantum dots versus organic dyes as fluorescent labels. Nat Methods. 2008;5(9):763–775. | ||
Michalet X, Pinaud FF, Bentolila LA, et al. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 2005;307(5709):538–544. | ||
Larson DR, Zipfel WR, Williams RM, et al. Water-soluble quantum dots for multiphoton fluorescence imaging in vivo. Science. 2003;300(5624):1434–1436. | ||
Cai W, Shin DW, Chen K, et al. Peptide-labeled near-infrared quantum dots for imaging tumor vasculature in living subjects. Nano Lett. 2006;6(4):669–676. | ||
Bhang SH, Won N, Lee TJ, et al. Hyaluronic acid-quantum dot conjugates for in vivo lymphatic vessel imaging. ACS Nano. 2009;3(6):1389–1398. | ||
Hu R, Yong KT, Roy I, et al. Functionalized near-infrared quantum dots for in vivo tumor vasculature imaging. Nanotechnology. 2010;21(14):145105. | ||
Azizi PM, Zyla RE, Guan S, et al. Clathrin-dependent entry and vesicle-mediated exocytosis define insulin transcytosis across microvascular endothelial cells. Mol Biol Cell. 2015;26(4):740–750. | ||
Bergmann S, Schoenen H, Hammerschmidt S. The interaction between bacterial enolase and plasminogen promotes adherence of Streptococcus pneumoniae to epithelial and endothelial cells. Int J Med Microbiol. 2013;303(8):452–462. | ||
DuRose JB, Li J, Chien S, Spector DH. Infection of vascular endothelial cells with human cytomegalovirus under fluid shear stress reveals preferential entry and spread of virus in flow conditions simulating atheroprone regions of the artery. J Virol. 2012;86(24):13745–13755. | ||
Alkilany AM, Shatanawi A, Kurtz T, Caldwell RB, Caldwell RW. Toxicity and cellular uptake of gold nanorods in vascular endothelium and smooth muscles of isolated rat blood vessel: importance of surface modification. Small. 2012;8(8):1270–1278. | ||
Duan J, Yu Y, Li Y, Zhou X, Huang P, Sun Z. Toxic effect of silica nanoparticles on endothelial cells through DNA damage response via Chk1-dependent G2/M checkpoint. PLoS One. 2013;8(4):e62087. | ||
Dmitrieva NI, Burg MB. Secretion of von Willebrand factor by endothelial cells links sodium to hypercoagulability and thrombosis. Proc Natl Acad Sci U S A. 2014;111(17):6485–6490. | ||
Ju R, Zhuang ZW, Zhang J, et al. Angiopoietin-2 secretion by endothelial cell exosomes: regulation by the phosphatidylinositol 3-kinase (PI3K)/Akt/endothelial nitric oxide synthase (eNOS) and syndecan-4/syntenin pathways. J Biol Chem. 2014;289(1):510–519. | ||
Zhou J, Li YS, Nguyen P, et al. Regulation of vascular smooth muscle cell turnover by endothelial cell-secreted microRNA-126: role of shear stress. Circ Res. 2013;113(1):40–51. | ||
Chakrabarti S, Davidge ST. High glucose-induced oxidative stress alters estrogen effects on ER alpha and ER beta in human endothelial cells: reversal by AMPK activator. J Steroid Biochem Mol Biol. 2009;117(4–5):99–106. | ||
Shinkai Y, Yamamoto C, Kaji T. Lead induces the expression of endoplasmic reticulum chaperones GRP78 and GRP94 in vascular endothelial cells via the JNK-AP-1 pathway. Toxicol Sci. 2010;114(2):378–386. | ||
Paris S, Denis H, Delaive E, et al. Up-regulation of 94-kDa glucose-regulated protein by hypoxia-inducible factor-1 in human endothelial cells in response to hypoxia. FEBS Lett. 2005;579(1):105–114. | ||
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. | ||
Yan M, Zhang Y, Xu K, Fu T, Qin H, Zheng X. An in vitro study of vascular endothelial toxicity of CdTe quantum dots. Toxicology. 2011;282(3):94–103. | ||
Fu T, Qin HY, Hu HJ, Hong Z, He S. Aqueous synthesis and fluorescence-imaging application of CdTe/ZnSe core/shell quantum dots with high stability and low cytotoxicity. J Nanosci Nanotechnol. 2010;10(3):1741–1746. | ||
Li H-B, Ge Y-K, Zhang L, Zheng X-X. Astragaloside IV improved barrier dysfunction induced by acute high glucose in human umbilical vein endothelial cells. Life Sci. 2006;79(12):1186–1193. | ||
Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med. 2012;2(1):a006429. | ||
Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52(11):2745–2756. | ||
Barua S, Rege K. Cancer-cell-phenotype-dependent differential intracellular trafficking of unconjugated quantum dots. Small. 2009;5(3):370–376. | ||
Zhang LW, Monteiro-Riviere NA. Mechanisms of quantum dot nanoparticle cellular uptake. Toxicol Sci. 2009;110(1):138–155. | ||
Albanese A, Tang PS, Chan WCW. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–16. | ||
Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. | ||
Sowa G. Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front Physiol. 2012;2012(2):120–120. | ||
Frank PG, Pavlides S, Lisanti MP. Caveolae and transcytosis in endothelial cells: role in atherosclerosis. Cell Tissue Res. 2009;335(1):41–47. | ||
Schubert W, Frank PG, Razani B, Park DS, Chow CW, Lisanti MP. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem. 2001;276(52):48619–48622. | ||
Liu P, Sun Y, Wang Q, Li H, Duan Y. Intracellular trafficking and cellular uptake mechanism of mPEG-PLGA-PLL and mPEG-PLGA-PLL-Gal nanoparticles for targeted delivery to hepatomas. Biomaterials. 2014;35(2):760–770. | ||
Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9:20. | ||
Khan MI, Mohammad A, Patil G, Naqvi SA, Chauhan LK, Ahmad I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials. 2012;33(5):1477–1488. | ||
Stern ST, Zolnik BS, McLeland CB, Clogston J, Zheng J, McNeil SE. Induction of autophagy in porcine kidney cells by quantum dots: a common cellular response to nanomaterials? Toxicol Sci. 2008;106(1):140–152. | ||
Cho SJ, Maysinger D, Jain M, Roder B, Hackbarth S, Winnik FM. Long-term exposure to CdTe quantum dots causes functional impairments in live cells. Langmuir. 2007;23(4):1974–1980. | ||
Zhang Y, Mi L, Xiong R, et al. Subcellular localization of Thiol-Capped CdTe quantum dots in living cells. Nanoscale Res Lett. 2009;4(7):606–612. | ||
Dong Z, Wang L, Xu J, et al. Promotion of autophagy and inhibition of apoptosis by low concentrations of cadmium in vascular endothelial cells. Toxicol In Vitro. 2009;23(1):105–110. | ||
Kaji T, Suzuki M, Yamamoto C, Mishima A, Sakamoto M, Kozuka H. Severe damage of cultured vascular endothelial cell monolayer after simultaneous exposure to cadmium and lead. Arch Environ Contam Toxicol. 1995;28(2):168–172. | ||
Le PU, Nabi IR. Distinct caveolae-mediated endocytic pathways target the Golgi apparatus and the endoplasmic reticulum. J Cell Sci. 2003;116(6):1059–1071. | ||
Nichols BJ. A distinct class of endosome mediates clathrin-independent endocytosis to the Golgi complex. Nat Cell Biol. 2002;4(5):374–378. | ||
Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol. 2001;3(5):473–483. | ||
Schelhaas M, Malmström J, Pelkmans L, et al. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell. 2007;131(3):516–529. | ||
Chinnapen DJ, Chinnapen H, Saslowsky D, Lencer WI. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol Lett. 2007;266(2):129–137. | ||
Sanson M, Augé N, Vindis C, et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells prevention by oxygen-regulated protein 150 expression. Circ Res. 2009;104(3):328–U102. | ||
Christen V, Fent K. Silica nanoparticles and silver-doped silica nanoparticles induce endoplasmatic reticulum stress response and alter cytochrome P4501A activity. Chemosphere. 2012;87(4):423–434. | ||
Zhang R, Piao MJ, Kim KC, et al. Endoplasmic reticulum stress signaling is involved in silver nanoparticles-induced apoptosis. Int J Biochem Cell Biol. 2012;44(1):224–232. | ||
Szegezdi E, Duffy A, O’Mahoney ME, et al. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;349(4):1406–1411. | ||
Badiola N, Penas C, Miñano-Molina A, et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149. | ||
Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2alpha in caspase-3/7-deficient cells. Oncogene. 2010;29(22):3241–3251. | ||
Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. | ||
Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7(9):880–885. | ||
Chen R, Huo L, Shi X, et al. Endoplasmic reticulum stress induced by zinc oxide nanoparticles is an earlier biomarker for nanotoxicological evaluation. ACS Nano. 2014;8(3):2562–2574. | ||
Lao F, Chen L, Li W, et al. Fullerene nanoparticles selectively enter oxidation-damaged cerebral microvessel endothelial cells and inhibit JNK-related apoptosis. ACS Nano. 2009;3(11):3358–3368. | ||
Pauksch L, Hartmann S, Rohnke M, et al. Biocompatibility of silver nanoparticles and silver ions in primary human mesenchymal stem cells and osteoblasts. Acta Biomater. 2014;10(1):439–449. | ||
Komoike Y, Inamura H, Matsuoka M. Effects of salubrinal on cadmium-induced apoptosis in HK-2 human renal proximal tubular cells. Arch Toxicol. 2012;86(1):37–44. | ||
Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31(12):2792–2797. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.