Back to Journals » ImmunoTargets and Therapy » Volume 8
Cytokine Release Syndrome: Current Perspectives
Authors Murthy H ![]() , Iqbal M, Chavez JC, Kharfan-Dabaja MA
, Iqbal M, Chavez JC, Kharfan-Dabaja MA ![]()
Received 14 August 2019
Accepted for publication 7 October 2019
Published 29 October 2019 Volume 2019:8 Pages 43—52
DOI https://doi.org/10.2147/ITT.S202015
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Hemant Murthy,1 Madiha Iqbal,1 Julio C Chavez,2 Mohamed A Kharfan-Dabaja1
1Division of Hematology-Oncology and Blood and Marrow Transplantation Program, Mayo Clinic, Jacksonville, FL, USA; 2Department of Malignant Hematology, Moffitt Cancer Center, Tampa, FL, USA
Correspondence: Mohamed A Kharfan-Dabaja
Division of Hematology-Oncology and Blood and Marrow Transplantation Program, Mayo Clinic, 4500 San Pablo Road, Mangurian Bdg 3rd Floor, Jacksonville, FL 32224, USA
Tel +1 904 953-2000
Email [email protected]
Abstract: Chimeric antigen receptor T cell (CART) therapy represents a novel and a paradigm-shifting approach to treating cancer. Recent clinical successes have widened the applicability of CD19 CART cells for the treatment of relapsed/refractory B-cell NHL, namely tisagenleclucel and axicabtagene ciloleucel. Tisagenleclucel is also approved for relapsed and/or refractory B-ALL up to age 25. CART therapy is associated with unique and potentially life-threatening toxicities, notably cytokine release syndrome (CRS). A better understanding of the pathogenesis of CRS is crucial to ensure proper management. In this review, CRS definitions, profiles, risk factors and grading systems are discussed. Finally, current and novel investigational approaches and therapies for CRS are summarized.
Keywords: cytokine release syndrome, chimeric antigen receptor T-cell therapy
Introduction
Chimeric antigen receptor (CAR)-T (CART) cell therapy represents a novel and a paradigm shifting approach to treating cancer. Using genetically modified cytotoxic immune T cells to target tumor-specific antigens, this immunotherapy platform has resulted in durable remissions in relapsed and/or refractory B-cell non-Hodgkin lymphoma (NHL) and B-cell acute lymphoblastic leukemia (ALL)1–3 and is showing promising early results in multiple myeloma.4 Currently, there are two FDA-approved products for the treatment of relapsed/refractory B-cell NHL, namely tisagenleclucel and axicabtagene ciloleucel. Tisagenleclucel is also approved for relapsed and/or refractory pediatric B-ALL up to the age of 25 years.
Structurally, a CART consists of three essential components: an ectodomain, consisting of an extracellular, antibody-derived antigen recognition domain, typically a single-chain fragment variable (scFv) originating from a monoclonal antibody specific for the selected tumor antigen; an endodomain which contains an intracellular T cell receptor (TCR) derived, activating domains from CD3ζ or CD3γ.5 Additionally, second and subsequent generation CARTs also contain costimulatory domains such as CD28 and 4-1BB. These costimulatory domains are necessary for T cell activation, resulting in significant expansion, proliferation and persistence of the CART cells;5 Lastly, a transmembrane domain which connects the ectodomain to the endodomain.
Recent clinical successes have helped to thrust CART cells towards wider applicability, including clinical trials for other hematologic malignancies and even solid tumors. Moreover, there is an expectation to expand use of CART beyond specialized academic centers into the wider community practice at large. Use of CART cells has brought a unique set of toxicities such as cytokine release syndrome (CRS) and neurotoxicity. Here, we provide an extensive overview of CRS, including risk factors, emerging grading models, and current and emerging strategies for prevention and treatment of CRS.
Defining CRS
CRS represents a potentially serious complication of CART therapy. It is a cytokine-mediated systemic inflammatory response which occurs in concert with in vivo CART activation and expansion.6 The exact mechanism of CRS remains to be better understood. Cytokines are released when interaction between tumor and immune effector cell occurs; and it can originate not only from the CART cell but also from host immune cells such as macrophages, which respond in part to CART activation.7
Clinically, the CRS can present with fevers, myalgias, hypotension and hypoxia. They can be mild and self-limiting, or progress in severity to high-grade fevers, hemodynamic compromise requiring vasopressor support, capillary leak, and severe hypoxia requiring ventilator support. Moreover, clinical manifestations of CRS can also manifest as arrhythmias, renal failure, pleural effusions, transaminitis, coagulopathy, and hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS),8 though these typically are uncommon in the absence of hypotension, hypoxia or both.9,10 CRS can present either by itself or concurrently with immune effector cell-associated neurotoxicity syndrome (ICANS). CRS can vary in time of onset between 1 and 58 days post-CART infusion with median time of onset following CART infusion of 2–3 days.11–13 The duration of CRS can vary according to the CART construct, manufacturing or therapeutic interventions but typically resolution of CRS is seen within 2–3 weeks of CART infusion.
Cytokines Profile Of CRS
As the name implies, a number of cytokines released during CRS are found to be elevated. The main cytokines implicated in the pathogenesis of CRS include interleukin-6 (IL-6), interleukin-10 (IL-10), interferon (IFN)-γ, monocyte chemoattractant protein 1 (MCP-1) and granulocyte-macrophage colony-stimulating factor (GM-CSF); other cytokines, including tumor necrosis factor (TNF), IL-1, IL-2, IL-2–receptor-α, and IL-8 have also been reported during CRS.14,15
IL-6, likely arising from activation of endothelial cells, can cause capillary leakage, hypotension, activation of complement pathway and coagulation cascades, and myocardial dysfunction.16,17 IFN-γ can cause flu-like symptoms and can also trigger macrophage activation, leading to secretion of host cytokines such as IL-6, TNF-α, and IL-1015,18 which could further exacerbate CRS. Other biomarkers of endothelial cell activation, such as Angiopoietin-2 and von Willebrand factor, have also been described to predict CRS severity, before CART infusion and during CRS.19 Non-specific markers of inflammation including ferritin and C-reactive protein (CRP) are also elevated in patients with CRS.6,20,21 In more severe CRS, particularly those who develop HLH/MAS, additional cytokines such as IL-18, IL8, IP10, MCP1, MIG, and MIP1β have been reported to be elevated and appear to portend a poorer outcome.6
Risk Factors For CRS
With increasing utilization of CART and with a better understanding of CRS, there is an unmet need to identify clinical and biochemical factors to better predict CRS, particularly severe CRS cases. It is anticipated that any risk factor which portends in vivo CART expansion and activation would be predictive CRS severity.
Clinical factors include disease burden and marrow involvement, lymphodepletion with fludarabine/cyclophosphamide conditioning, and higher CART infusion doses.4,19,22,23 Other patient-specific factors such as pre-existent state of inflammation (baseline serum ferritin) and baseline endothelial activation (thrombocytopenia) appear to be predictive of higher grade CRS2,4,19,24,25 (Table 1).
 |
Table 1 Risk Factors For Development Of Severe CRS |
Efforts are underway to develop and standardize cytokine activation profiles which correlate with CRS severity, to help abrogate it at an earlier stage.6,24 The group at Memorial Sloan Kettering Cancer Center (MSKCC) showed a 75-fold increase in a panel of seven cytokines (IL-6, IL-5, IL-10, GM-CSF, IFN-γ, fractalkine, FLT-3L) from baseline in those with severe CRS.24 The group at University of Pennsylvania (UPenn) identified an increase in 24 cytokines (including IFNγ, IL6, IL8, sIL2Rα, sgp130, sIL6R, MCP1, MIP1α, MIP1β, and GM-CSF) in the first month after CART infusion to be highly associated with severe CRS.6 The two groups, however, found differing results predicting severe CRS with CRP peak elevations. The MSKCC group found significant elevations of CRP predictive of severe CRS, while the UPenn group did not find it correlating with severe CRS development although CRP elevation was noted in CRS development,6,24,26,27 One noteworthy limitation of a cytokine-defined intervention for CRS is the laboratory capabilities and turn-around time for results required for immediate action, particularly in potentially severe CRS. At this time, it is recommended to treat CRS based on clinical symptoms.10
Grading Models Of CRS
With widespread availability and use of T-cell directed therapies under clinical trials and as a standard of care, there has been several attempts to establish a consistent and accurate grading system for clinical management and also for trial reporting purposes. CRS is not a new concept. It has been described in the early 1990s. To our knowledge, the first case of CRS was described as a result of systemic inflammation caused by an anti CD3 monoclonal antibody used for organ transplantation.28 Several monoclonal antibodies (moAbs) have been associated with development of CRS.29 The prevailing assumption was that CRS could occur within minutes or few hours of moAb infusion, which is clinically different from cell therapies. Thus, the initial grading system proposed using the Common Terminology Criteria for Adverse Events version 4.03 (CTCAE v.4.03) was perhaps not optimal for grading of CART-related CRS.
In the earlier phases of CART cell development, there were several grading systems that were used including NCI Consensus Grading,30 the UPenn grading,31 and the MSKCC criteria25 as described previously.
An initial effort focused on enhancing the CTCAE v4.03 CRS30 to determine categories of mild, moderate, severe and life-threatening CRS. Under this system, grade 1 CRS consisted of presence of fever with or without constitutional symptoms but without organ dysfunction; grade 2 CRS entailed hypoxia (requiring up to 40% FiO2 supplementation), hypotension (responsive to intravenous fluids (IVF) or low-dose vasopressors) and up to grade 2 organ toxicity; for patients to develop grade 3 CRS, they should show higher oxygen requirements (FiO2>40%), use of higher doses of vasopressors (or multiple vasopressors), grade 3 organ toxicity (grade 4 transaminitis); and grade 4 CRS entailed life-threatening symptoms, requirement of ventilator support or grade 4 organ toxicity (except for transaminitis).
The MSKCC criteria25 had some differences from Lee’s criteria. For instance, while the definition of grade 2 or 3 CRS required the need for vasopressors as criteria (similar to Lee’s criteria), it was based on the duration of vasopressor use (<24 versus >24 hrs). Patients requiring higher doses of vasopressors for more than 3 hrs were considered as having grade 4 CRS.
In contrast, in the UPenn CRS grading,31 the definition of CRS grade 2 was less clear requiring “some signs of organ dysfunction” and intravenous fluids were permitted for management except when hypotension was present. If fluids or any dose of vasopressors (for any duration) were required for management of hypotension, then patients were classified as having grade 3 CRS.
These diverse grading systems led to differences in the reported incidence and even severity of CRS in clinical trials. To underscore this issue, for example, a patient with fevers and hypotension responsive to fluids and hypoxia requiring less than 40% FiO2 is classified as having grade 2 CRS (non-severe) per Lee criteria.10 However, by UPenn criteria, this CRS would be considered grade 3.
This difference was clearly emphasized after an analysis of the JULIET trial of tisagenlecleucel for DLBCL, where experts in the field “re-graded” patients with CRS using the Lee’s criteria and compared to UPenn criteria.30,31 In 31% of cases, the Lee’s grading yielded a lower score than the UPenn grading, in 61% it resulted in the same grading and in 8% of cases the Lee grading was higher than that of UPenn.32
Another grading model known as the CARTOX system is based on a three-step-based approach that consists of grading, assessment and treatment. The grading of CRS was mainly based on the Lee’s criteria using four parameters, namely temperature, presence of hypotension, oxygen requirements and organ toxicity. It specifically defined fever as a temperature >38°C and hypotension as a systolic pressure of less than 90 mmHg. The CARTOX grading also proposed that CRS represents a dynamic process and, accordingly, it required evaluation at least twice a day but more often if there was a justifying change in the clinical condition of the CART recipient.33
Most recently, The American Society for Transplantation and Cellular Therapy (ASTCT) developed consensus guidelines for grading CART toxicities including CRS; the authors also summarized previously described grading models.10 In this new grading system fever, hypotension and hypoxia remained the cardinal features of CRS. While fever was required for the diagnosis of CRS, it did not have to persist during the periods of CRS toxicity. One of the main goals of the ASTCT CRS grading system was to both harmonize and simplify the current grading of CRS in order to facilitate the reporting of cell therapy-related toxicities.
One of the most relevant changes in the ASTCT grading model was the removal of organ toxicity from the CRS grading as these changes occur concomitantly with hypotension and hypoxia and would not likely influence the decision to prescribe anti-IL6 based therapy. A patient with fevers only was considered a grade 1 CRS. Grade 2 CRS required the presence of fevers along with hypotension (without the use of vasopressors) and/or hypoxia requiring low flow oxygen. Grade 3 entailed the presence of hypotension requiring one vasopressor with/without vasopressin and/or hypoxia requiring high flow oxygen. Grade 4 CRS represented a life-threatening condition requiring multiple vasopressors (excluding vasopressin) and hypoxia requiring positive pressure ventilation systems (BIPAP, CPAP or mechanical ventilation); we refer the readers to the ASTCT consensus guidelines which also provides a summary and a side-by-side comparison of all CRS grading models.10
Table 2 summarizes the major studies and the grading criteria used.
 |
Table 2 Selected CAR-T Trials Reporting CRS Incidence, Grading And Treatment |
CRS Treatment And Prevention
CRS generally occurs within days after CART cell infusion. While identification of factors predictive of severe CRS continues to evolve, one mainstay of CRS treatment is to deploy anti-cytokine therapy early in the CRS course to prevent progression into severe life-threatening higher grade CRS.
As many symptoms of CRS can mimic other medical conditions such as sepsis, infection, or adrenal insufficiency, it is of utmost importance that a thorough workup is performed to rule them out. One major challenge remains to identify agents effective for CRS treatment that do not interfere with the cytokine-mediated anti-tumor effects of CART cells.
Tocilizumab
Tocilizumab is humanized IL-6 receptor antagonist moAb which functions by inhibiting both classic and trans-IL-6 signaling on immune effector cells.34 It works on both membrane-bound IL-6 receptor and soluble IL-6 receptor by competitively competing with IL-6 for binding to both receptors, leading to decrease in IL-6 signaling and reducing immune activation and inflammation.35
Given the central role IL-6 plays in CRS and since its earliest reported use in successfully dampening severe CRS in a pediatric CART recipient,36 the IL-6 antagonist tocilizumab represents an important therapy for CRS. It was approved by the FDA for the treatment of severe or life-threatening CART-cell-induced CRS in adults and pediatric patients ≥2 years old.37 Tocilizumab was later shown to reduce fevers and CRS symptoms without affecting CART levels in serum or bone marrow.24
Corticosteroids
Systemic corticosteroids are effective in dampening CRS due to its established anti-inflammatory properties. Due to early concern about steroids inhibiting CART activity and expansion,24 its use early in CRS onset was relatively restricted in an effort to preserve CART anti-tumor activity. Typically, corticosteroids are reserved for cases of CRS refractory to tocilizumab, or being administered concomitantly with tocilizumab for high-grade CRS or CRS that occurs with associated neurotoxicity.10
Preliminary data suggest using IL-6 receptor blockade and/or steroids do not result in higher rates of relapse.38,39 Moreover, prolonged steroid use (beyond 7 or more days) does not appear to adversely affect CART outcomes.40 These findings have helped consider intervening with tocilizumab plus corticosteroids earlier in the treatment course,41 and even as a prophylaxis against CRS.42 Yet, large prospective trials are certainly needed before widely adopting this approach.
Siltuximab
Siltuximab is a murine chimeric moAb which is an IL-6 antagonist. While currently not FDA approved for the treatment of CRS, several centers have reported prescribing it for CART-related CRS. According to a survey conducted by a special interest group, half of the respondents described using siltuximab to treat CRS, but all agreed that siltuximab should be considered for cases of tocilizumab-refractory CRS.43 One potential benefit of siltuximab, from the mechanistic standpoint, is the resulting increase in IL-6 levels following tocilizumab administration due to IL-6R binding, as opposed to siltuximab which binds directory to IL-6.44 This rise in IL-6 is thought to increase risk of neurotoxicity.14 We believe that more data are needed to better define the role of siltuximab in the first-line CRS treatment setting.
Anakinra
Anakinra blocks the biologic activity of IL-1 alpha and beta by competitively inhibiting IL-1 binding to the interleukin-1 type I receptor (IL-1RI), which is expressed in a wide variety of tissues and organs. Currently, it is FDA approved for the treatment for rheumatoid arthritis.45 Macrophage produced IL-1 has been linked with CRS and neurotoxicity from CART, hence suggesting a role for IL-1 blockade with anakinra.14,46 Anakinra has been described to have a therapeutic effect in hemophagocytic lymphohistiocytosis (HLH)47,48 although it has not been reported or studied in CAR T induced HLH/MAS.
Dasatinib
Dasatinib is a tyrosine kinase inhibitor approved for the treatment of chronic myelogenous leukemia and Philadelphia chromosome-positive ALL. It also has other effects including suppressing T-cell activation and inhibiting T cell signaling kinases including Src, Fyn and Lc.49
These effects have been studied in two separate preclinical models with CD19 CART, both demonstrating dasatinib reversibly suppressing cytolytic activity, cytokine production, and CD4+ and CD8+ antigen-induced proliferation of CART cells containing either CD28 or 4-1BB costimulatory modules.50,51 In addition, Weber et al demonstrated that not only can this dasatinib function-off state be sustained for several days without affecting T cell viability, but that it is dose dependent, allowing titration of dasatinib for partial or complete CART functional suppression. These pre-clinical findings are exciting and warrant prospective clinical trials to further investigate the role of dasatinib on CRS. Table 3 summarizes current clinic trials focusing on the treatment of CRS.
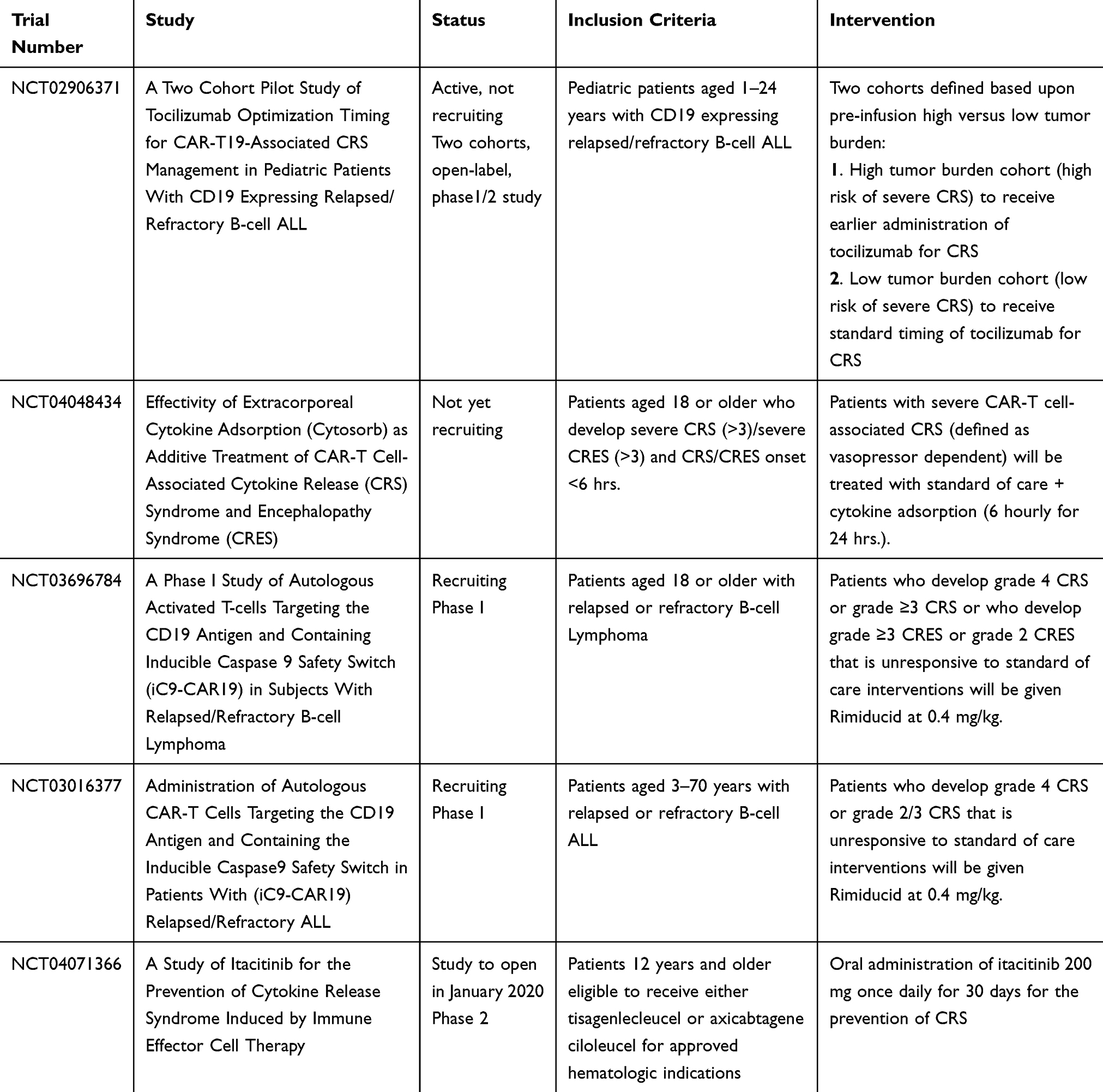 |
Table 3 Current Clinical Trials For Prevention Or Treatment Of CRS |
A3 Adenosine Receptor Agonists
A3 adenosine receptors are expressed on various immune cells and activation of A3AR has been correlated with anti-inflammation52 Binding of A3AR with A3AR agonists inhibit inflammatory cytokine production and release by inhibiting the production of inflammatory cytokines through downregulation of NF-κB, hence reducing production of pro-inflammatory cytokines including TNF-α, IL-1, and IL-6.53,54 Highly selective A3AR agonists such as namodenoson and piclidenoson could represent potential therapeutic options for CRS treatment, but require additional clinical investigation.53
JAK/STAT Inhibitors
IL-6 signaling occurs through two different mechanisms: via the higher-affinity membrane-bound receptor (classic IL-6 signaling) or via a soluble IL-6 receptor (sIL-6R; trans-IL-6 signaling). Both ultimately result in the activation of the JAK/STAT pathway. Ruxolitinib is a JAK/STAT pathway inhibitor that has resulted in a significant reduction of inflammatory cytokines in preclinical and clinical studies. It was investigated for prevention of CRS from a CD123-directed CART in an AML xenograft model. This pre-clinical model demonstrated not only the efficacy of ruxolitinib in prevention of CRS, but it did not appear to affect anti-tumor activity of the CD123-directed CART.55 Itacitinib, another JAK/STAT pathway inhibitor selective for JAK1, is being studied in a phase 2 study for the prevention of CRS. Further research is warranted regarding the role of JAK/STAT inhibition in the management and prevention of CRS (Table 2).
Lenlizumab
GM-CSF cytokine elevation was identified by both the MSKCC and UPenn groups as a cytokine with a large increase in severe CRS,6,24 while GM-CSF elevation was also observed in the development of severe grade 3 or 4 neurotoxicity.1 Lenzilumab is a human monoclonal antibody that neutralizes human GM-CSF. Preclinical studies showed prevention of CRS and reduction in neuroinflammation without affecting CD19-targeted CAR-T function and enhanced anti-tumor activity in vivo when compared to CD19-CART without lenzilumab.56 A clinical trial of axicabtagene ciloleucel with lenziumab is anticipated.
Suicide Gene
One approach to manage refractory toxicities such as CRS is to encode a conditional safety switch which can be used to eliminate the CART cells thus abrogating its immune effects. Suicide genes are one way to encode this safety switch into a CART. One of the best known safety switch mechanisms is the caspase 9 (iCasp9)/AP1903 suicide system. This system was first utilized in allogeneic stem cell transplantation, where exposure to rimiducid would eliminate iCasp9-expressing t cells, eliminating t-cell-mediated effects of graft-versus-host disease.57–59 Preclinical studies demonstrate the efficacy of this system when encoded in CART.60,61 Other suicide gene platforms are in development with safety switches such as EGFR62 and CD20.63
Discussion
CART cell therapy represents a success for the treatment of relapsed and/or refractory B-cell NHL and B-cell ALL. Early clinical experiences with CART cell products followed by large multi-center clinical trials, leading to their approval for wide commercial use, have highlighted the unique resulting toxicities. Although there are only two CD19 CART cell commercial products available, additional CART cell products targeting other novel antigens are anticipated to enter into clinical practice in the near future.64–66 Additionally, other T cell redirected therapies such as bi-specific T-cell-engaging antibodies (BiTEs) and TCR-gene therapies are also part of the emerging treatment landscape secondary to the common underlying principle of immune effector cell activation causing tumor cell death.67–69
A thorough understanding of the toxicities associated with these highly effective therapies is of paramount importance in advancing the field of cellular therapeutics. CRS is one the main toxicities associated with CART cell therapy.33 The underlying pathophysiology of CRS involves a supra-physiologic response of the immune system secondary to the activation of T cells, which further results in release of a multitude of cytokines and chemokines.70 This is reflected in the current definition of CRS proposed by ASTCT and applies to not only CART cell therapy but to any therapy using immune effector cell activation as its primary mechanism of action.10 As described above, signs and symptoms of CRS can be extremely variable ranging from mild symptoms to those requiring ICU care with cardiorespiratory support.30 The lack of specificity of CRS presentation requires exclusion of other clinical situations that may mimic CRS. Infection and sepsis, for example, may present with fevers, hypotension, and other signs and symptoms similar to CRS. It is recommended that workup for CRS entails a thorough investigation to exclude infectious etiology, including cultures, imaging and/or initiation of broad spectrum antibiotics whenever clinically indicated. Also, CRS can share many overlapping features with HLH/MAS such as elevated ferritin and C-reactive protein.1,6,33 This is not surprising given that both share the same underlying physiology of immune activation. However, features suggestive of HLH/MAS generally resolve with the resolution of CRS and it is therefore considered as part of the spectrum of CRS and not a separate entity.10
Early identification and accurate management is crucial in the treatment of toxicities associated with immune effector cell activating therapy. In this regard grading systems have been of vital importance and have evolved significantly from the original use of CTCAE v3 in early clinical trials to the current ASTCT consensus grading.10 The important features of the ASTCT consensus include elimination of laboratory parameters as a part of grading system given the lack of specificity of most biomarkers; and the difficulty in obtaining results in real-time.
The importance of supportive care in the management of patients with CRS cannot be undervalued. Close monitoring by experienced nursing staff who are well informed of the current grading systems is crucial. To date, the most commonly used therapy for systemic treatment of CRS remains tocilizumab. Multiple studies confirmed the correlation of peak IL-6 levels with the severity of CRS and this led to the approval of tocilizumab for treatment of CRS concurrent with the approval of tisagenlecleucel.23,24 The timing of administration of tocilizumab remains an area of debate. Initial clinical experience reserved tocilizumab to patients manifesting severe CRS.36 With increased clinical experience and reports showing no significant compromise on the efficacy of CART, we are now seeing a shift in clinical practice where tocilizumab is being prescribed earlier in the course of CRS.1,71 This is a practice approach which is also being supported by recent ASTCT guidelines.10 This is similar to the experience with use of corticosteroids where the impact on CART efficacy is of less concern nowadays.24,65,72
CART therapies have produced dramatic results and they will continue to change the therapeutic landscape of oncology practice. Further refinement of existing strategies and development of new therapies to prevent and treat unique and potentially life-threatening toxicities such as CRS will be important to ensure this treatment can be safely administered to patients everywhere.
Disclosure
Professor Mohamed A Kharfan-Dabaja reports consultancy for Daiichi Sankyo and Pharmacyclics, outside the submitted work. Julio C Chavez reports consultancy for Kite/Gilead and Novartis and consultancy for Genentech, Bayer, and Karyopharm and speaker Bureau for Genentech. The authors report no other conflicts of interest in this work.
References
1. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447
2. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. doi:10.1056/NEJMoa1804980
3. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866
4. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–1737. doi:10.1056/NEJMoa1817226
5. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–126. doi:10.1111/imr.12131
6. Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6(6):664–679. doi:10.1158/2159-8290.CD-16-0040
7. Gauthier J, Turtle CJ. Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr Res Transl Med. 2018;66(2):50–52. doi:10.1016/j.retram.2018.03.003
8. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20(2):119–122. doi:10.1097/PPO.0000000000000035
9. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–3330. doi:10.1182/blood-2016-04-703751
10. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–638. doi:10.1016/j.bbmt.2018.12.758
11. Novartis Pharmaceuticals corporation. Kymriah (tisagenlecleucel) [package insert]. U.S. Food and Drug Administration website. Available from: https://www.fda.gov/media/107296/download. Revised March 28, 2019. Accessed October 2019.
12. Kite Pharma, Incorporated. Yescarta (axicabtagene ciloleucel) [package insert]. U.S. Food and Drug Administration website. Available from: https://www.fda.gov/media/108377/download. Revised February 20, 2018. Accessed October 2019.
13. Kite Pharma, Incorporated. Yescarta (axicabtagene ciloleucel) [package insert- list of adverse reactions]. U.S. Food and Drug Administration website. Available from: https://www.fda.gov/media/108377/download. Revised February 20, 2018. Accessed October 2019.
14. Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi:10.1038/s41591-018-0036-4
15. Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res. 2018;6:4. doi:10.1186/s40364-018-0116-0
16. Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016;8(8):959–970. doi:10.2217/imt-2016-0020
17. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448–457. doi:10.1038/ni.3153
18. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi:10.1146/annurev.immunol.021908.132532
19. Hay KA, Hanafi L-A, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130(21):2295–2306. doi:10.1182/blood-2017-06-793141
20. Frey N, Porter D. Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biol Blood Marrow Transplant. 2019;25(4):e123–e127. doi:10.1016/j.bbmt.2018.12.756
21. Fitzgerald JC, Weiss SL, Maude SL, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017;45(2):e124–e131. doi:10.1097/CCM.0000000000002053
22. Turtle CJ, Hanafi L-A, Berger C, et al. Immunotherapy of non-hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8(355):355ra116. doi:10.1126/scitranslmed.aaf0746
23. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222
24. Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. doi:10.1126/scitranslmed.3008226
25. Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi:10.1056/NEJMoa1709919
26. Frey N. Cytokine release syndrome: who is at risk and how to treat. Best Pract Res Clin Haematol. 2017;30(4):336–340. doi:10.1016/j.beha.2017.09.002
27. Rouce RH, Heslop HE. Forecasting cytokine storms with new predictive biomarkers. Cancer Discov. 2016;6(6):579–580. doi:10.1158/2159-8290.CD-16-0493
28. Chatenoud L, Ferran C, Reuter A, et al. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected]. N Engl J Med. 1989;320(21):1420–1421. doi:10.1056/NEJM198905253202117
29. Bugelski PJ, Achuthanandam R, Capocasale RJ, Treacy G, Bouman-Thio E. Monoclonal antibody-induced cytokine-release syndrome. Expert Rev Clin Immunol. 2009;5(5):499–521. doi:10.1586/eci.09.31
30. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729
31. Porter DL, Hwang W-T, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi:10.1126/scitranslmed.aad3106
32. Schuster SJ, Maziarz RT, Ericson SG, Rusch ES. Consensus grading of Cytokine Release Syndrome (CRS) in adult patients with relapsed or refractory diffuse large B-cell lymphoma (r/r DLBCL) treated with tisagenlecleucel on the JULIET study. Blood. 2018;132:4190.
33. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. doi:10.1038/nrclinonc.2017.148
34. Kotch C, Barrett D, Teachey DT. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev Clin Immunol. 2019;15(8):1–10.
35. Nishimoto N, Kishimoto T. Humanized antihuman IL-6 receptor antibody, tocilizumab. Handb Exp Pharmacol. 2008;181:151–160.
36. Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi:10.1056/NEJMoa1215134
37. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome | FDA [WWW Document]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome. Accessed 14 July 2019.
38. Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T-cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433–444. doi:10.1200/EDBK_238691
39. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. doi:10.1056/NEJMra1706169
40. Karschnia P, Jordan JT, Forst DA, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood. 2019;133(20):2212–2221. doi:10.1182/blood-2018-12-893396
41. Topp MS, van Meerten T, Wermke M, et al. Preliminary results of earlier steroid use with axicabtagene ciloleucel (axi-cel) in patients with relapsed/refractory large B-cell lymphoma (R/R LBCL).
42. Locke FL, Neelapu SS, Bartlett NL, et al. Preliminary results of prophylactic tocilizumab after axicabtageneciloleucel (axi-cel; KTE-C19) treatment for patients with refractory, aggressive Non-Hodgkin Lymphoma (NHL). Blood. 2017.
43. Mahmoudjafari Z, Hawks KG, Hsieh AA, et al. American Society for Blood and Marrow Transplantation Pharmacy Special Interest Group survey on chimeric antigen receptor T cell therapy administrative, logistic, and toxicity management practices in the United States. Biol Blood Marrow Transplant. 2019;25(1):26–33. doi:10.1016/j.bbmt.2018.09.024
44. Chen F, Teachey DT, Pequignot E, et al. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J Immunol Methods. 2016;434:1–8. doi:10.1016/j.jim.2016.03.005
45. Amgen Inc. Kineret (anakinra) [package insert] U.S. Food and Drug Administration website. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/103950-0_Kineret.cfm. Revised April 2005. Accessed October 2019.
46. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–738. doi:10.1038/s41591-018-0041-7
47. Rajasekaran S, Kruse K, Kovey K, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children*. Pediatr Crit Care Med. 2014;15(5):401–408. doi:10.1097/PCC.0000000000000078
48. Wohlfarth P, Agis H, Gualdoni GA, et al. Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically ill adult patients with hemophagocytic lymphohistiocytosis. J Intensive Care Med. 2017;885066617711386.
49. Montero JC, Seoane S, Ocana A, Pandiella A. Inhibition of Src family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res. 2011;17(17):5546–5552. doi:10.1158/1078-0432.CCR-10-2616
50. Mestermann K, Giavridis T, Weber J, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. 2019;11(499):eaau5907. doi:10.1126/scitranslmed.aav5519
51. Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, Mackall CL. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019;3(5):711–717. doi:10.1182/bloodadvances.2018028720
52. Bar-Yehuda S, Silverman MH, Kerns WD, Ochaion A, Cohen S, Fishman P. The anti-inflammatory effect of A3 adenosine receptor agonists: a novel targeted therapy for rheumatoid arthritis. Expert Opin Investig Drugs. 2007;16(10):1601–1613. doi:10.1517/13543784.16.10.1601
53. Cohen S, Fishman P. Targeting the A3 adenosine receptor to treat cytokine release syndrome in cancer immunotherapy. Drug Des Devel Ther. 2019;13:491–497. doi:10.2147/DDDT.S195294
54. Varani K, Padovan M, Vincenzi F, et al. A2A and A3 adenosine receptor expression in rheumatoid arthritis: upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res Ther. 2011;13(6):R197. doi:10.1186/ar3527
55. Kenderian SS, Ruella M, Shestova O, et al. Ruxolitinib prevents cytokine release syndrome after car T-cell therapy without impairing the anti-tumor effect in a xenograft model. Biol Blood Marrow Transplant. 2017;23(3):S19–S20. doi:10.1016/j.bbmt.2016.12.003
56. Sterner RM, Sakemura R, Cox MJ, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697–709. doi:10.1182/blood-2018-10-881722
57. Di Stasi A, Tey S-K, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–1683. doi:10.1056/NEJMoa1106152
58. Zhou X, Dotti G, Krance RA, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. 2015;125(26):4103–4113. doi:10.1182/blood-2015-02-628354
59. Zhou X, Di Stasi A, Tey S-K, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123(25):3895–3905. doi:10.1182/blood-2014-01-551671
60. Budde LE, Berger C, Lin Y, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 2013;8(12):e82742. doi:10.1371/journal.pone.0082742
61. Diaconu I, Ballard B, Zhang M, et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther. 2017;25(3):580–592. doi:10.1016/j.ymthe.2017.01.011
62. Paszkiewicz PJ, Fräßle SP, Srivastava S, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. 2016;126(11):4262–4272. doi:10.1172/JCI84813
63. Valton J, Guyot V, Boldajipour B, et al. A versatile safeguard for chimeric antigen receptor T-cell immunotherapies. Sci Rep. 2018;8(1):8972. doi:10.1038/s41598-018-27264-w
64. Watanabe K, Terakura S, Martens AC, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. 2015;194(3):911–920. doi:10.4049/jimmunol.1402346
65. Ali SA, Shi V, Maric I, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128(13):1688–1700. doi:10.1182/blood-2016-04-711903
66. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi:10.1038/nm.4441
67. Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi:10.1126/science.1129003
68. Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi:10.1200/JCO.2010.32.2537
69. Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. doi:10.1016/S1470-2045(14)71170-2
70. Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–549. doi:10.1200/JCO.2014.56.2025
71. Teachey DT, Bishop MR, Maloney DG, Grupp SA. Toxicity management after chimeric antigen receptor T cell therapy: one size does not fit “ALL”. Nat Rev Clin Oncol. 2018;15(4):218. doi:10.1038/nrclinonc.2018.19
72. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi:10.1016/S0140-6736(14)61403-3
73. Abramson JS, Gordon LI, Palomba ML, Lunning MA. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J Clin Oncol.2018;36(15_suppl):7505-7505.
74. Nastoupil LJ, Jain MD, Spiegel JY, Ghobadi A, Lin Y. Axicabtagene ciloleucel (axi-cel) CD19 chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory large B-cell lymphoma: real world experience. Blood. 2018;132:91.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.