")
Back to Journals » OncoTargets and Therapy » Volume 7
Cyclooxygenase-2 inhibitor is a robust enhancer of anticancer agents against hepatocellular carcinoma multicellular spheroids
Authors Cui J, Guo Y, Zhang H, Jiang L, Ma J, Wang W, Wang M, Yang C, Nan K, Song L
Received 17 October 2013
Accepted for publication 16 December 2013
Published 24 February 2014 Volume 2014:7 Pages 353—363
DOI https://doi.org/10.2147/OTT.S56115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jie Cui,1,2 Ya-Huan Guo,3 Hong-Yi Zhang,4 Li-Li Jiang,1 Jie-Qun Ma,1 Wen-Juan Wang,1 Min-Cong Wang,1 Cheng-Cheng Yang,1 Ke-Jun Nan,1 Li-Ping Song5
1Department of Oncology, First Affiliated Hospital, College of Medicine of Xi'an Jiaotong University, Xi'an, 2Department of Oncology, Yan'an University Affiliated Hospital, Yan'an, 3Department of Oncology, Shaanxi Province Cancer Hospital, Xi'an, 4Department of Urology, Yan'an University Affiliated Hospital, Yan'an, 5Department of Radiotherapy, First Affiliated Hospital, College of Medicine of Xi'an Jiaotong University, Xi'an, People's Republic of China
Purpose: Celecoxib, an inhibitor of cyclooxygenase-2 (COX2), was investigated for enhancement of chemotherapeutic efficacy in cancer clinical trials. This study aimed to determine whether celecoxib combined with 5-fluorouracil or sorafenib or gefitinib is beneficial in HepG2 multicellular spheroids (MCSs), as well as elucidate the underlying mechanisms.
Methods: The human hepatocellular carcinoma cell line HepG2 MCSs were used as in vitro models to investigate the effects of celecoxib combined with 5-fluorouracil or sorafenib or gefitinib treatment on cell growth, apoptosis, and signaling pathway.
Results: MCSs showed resistance to drugs compared with monolayer cells. Celecoxib combined with 5-fluorouracil or sorafenib exhibited a synergistic action. Exposure to celecoxib (21.8 µmol/L) plus 5-fluorouracil (8.1 × 10-3 g/L) or sorafenib (4.4 µmol/L) increased apoptosis but exerted no effect on COX2, phosphorylated epidermal growth-factor receptor (p-EGFR) and phosphorylated (p)-AKT expression. Gefitinib (5 µmol/L), which exhibits no growth-inhibition activity as a single agent, increased the inhibitory effect of celecoxib. Gefitinib (5 µmol/L) plus celecoxib (21.8 µmol/L) increased apoptosis. COX2, p-EGFR, and p-AKT were inhibited.
Conclusion: Celecoxib combined with 5-fluorouracil or sorafenib or gefitinib may be superior to single-agent therapy in HepG2 MCSs. Our results provided molecular evidence to support celecoxib combination-treatment strategies for patients with human hepatocellular carcinoma. MCSs provided a good model to evaluate the interaction of anticancer drugs.
Keywords: hepatocellular carcinoma, celecoxib, multicellular spheroids, 5-fluorouracil, sorafenib, gefitinib
Introduction
Hepatocellular carcinoma (HCC) is the most common primary cancer of the liver. HCC, which shows increasing incidence, ranks as the fifth-most common malignancy worldwide.1 This disease is a relatively chemoresistant tumor highly refractory to cytotoxic chemotherapy. Therefore, novel agents or strategies to improve HCC treatment need to be evaluated.
Overexpression of the inducible isoform of the cyclooxygenase (COX)-2 enzyme has been observed in various malignant tumors.2,3 Induction of COX-2 promotes cell growth, inhibits apoptosis, and enhances cell motility and adhesion. Multiple studies have indicated that COX-2 inhibitors can inhibit tumor growth both in vitro and in vivo.4–6 These inhibitors are currently being tested in clinical trials as single-agent therapies or in combination with other agents for the management of several types of cancers.7–9
COX-2 is not frequently overexpressed, but can be detected in HCC.10 COX-2 expression is associated with a significantly reduced median survival time.11,12 COX-2 expression leads to a prosurvival effect; therefore, COX-2 inhibitors have been investigated for their potential to enhance chemotherapeutic efficacy. In the current study, we investigated the possible synergistic effect of the COX-2 inhibitor combined with 5-fluorouracil (5-FU; a cytotoxic agent for HCC treatment) and sorafenib, an oral multikinase inhibitor that is frequently used for HCC treatment.13
The epidermal growth-factor receptor (EGFR) is a receptor tyrosine kinase that is abnormally amplified or activated in various tumors, including liver cancer.14 Gefitinib and erlotinib, inhibitors of the tyrosine-kinase activity of EGFR (EGFR-TKI), have been extensively studied in patients with non-small-cell lung cancer. Both inhibitors compete with adenosine triphosphate to bind to the tyrosine-kinase pocket of the receptor.15–17 However, HCC exhibits primary resistance to TKI treatment. The EGFR and COX-2 pathways have been shown to interact at several levels, and the evaluation of simultaneous inhibition of both pathways has drawn interest.18–20 Thus, we formulated a hypothesis that the COX-2 inhibitor combined with gefitinib benefits HCC treatment.
In recent years, multicellular spheroids (MCSs) have been widely used for drug-sensitivity and molecular mechanism studies to investigate the difference in biological characteristics and phenotypic expression not provided in monolayer cells. Many studies have revealed that resistance in MSCs was more closely associated with the natural resistance observed in patient tumors than the monolayer cells and supported in vitro models for the study of cytotoxic drugs.21–24 The present study used human HCC HepG2 MCSs to investigate the differential effects of celecoxib, a selective COX-2 inhibitor,25 combined with 5-FU or sorafenib or gefitinib on cell growth, apoptosis, and signaling pathways.
Materials and methods
Drugs
Gefitinib provided by AstraZeneca (London, UK) was dissolved in dimethyl sulfoxide (DMSO) at 20 mM as a stock solution. Celecoxib purchased from Pfizer (New York, NY, USA) was dissolved in DMSO at 0.5 M. Sorafenib supplied by Pinnacle Pharmaceuticals (Cape Town, South Africa) was dissolved in DMSO at 10 mM. 5-FU (25 mg/mL) was purchased from Xudong Haipu Pharmaceutical Co., Ltd (Shanghai, People’s Republic of China). These drugs were diluted in a culture medium before use.
Cell line
The human HCC cell line used in the present study was HepG2 conserved at the Center of Molecular Biology at Xi’an Jiaotong University.
Monolayer cells and multicellular spheroid cultures
HepG2 cells (American Type Culture Collection, Manassas, VA, USA) were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin in 5% CO2/95% air at 37°C. The passage number was five. A HepG2 single-cell suspension in complete medium was seeded at 2×105 cells/mL in each culture flask. MCSs were obtained with a liquid-overlay technique.26 A single-cell suspension in a complete medium was seeded in each culture flask coated with 2% agarose. The culture condition of the MCSs was exactly the same as that of the monolayer cells, except for the presence of an agarose layer. After incubation for 3 or 4 days, MCSs were obtained from each culture flask.
Scanning electron microscopy
The MCSs were washed with phosphate-buffered saline and fixed in 2.5% glutaraldehyde for 2 hours. The cells were then postfixed on the plate with 1% OsO4 and dehydrated by graded ethanol. The cells were covered with gold palladium and examined by scanning electron microscopy (JSM-840; JEOL, Tokyo, Japan).
Growth-inhibition assay in vitro
Antiproliferative effects were determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay using a previously described method.27 The antiproliferative activity of the single-agent treatment was assessed in monolayer cells and MCSs. A total of 5,000 cells in either monolayer cells or MCSs in 200 μL of the maintenance medium were seeded into a 96-well plate. The half-maximal inhibitory concentration (IC50) was determined as the concentration resulting in 50% cell-growth inhibition by 48-hour exposure to drug compared with untreated control cells. We concurrently used 0.125, 0.25, 0.5, 1, and 2 times the IC50 dose of celecoxib and 5-FU or sorafenib for 48 hours to evaluate the antiproliferative effects of the combined treatment on MCSs and monolayer cells. The results of celecoxib combined with 5-FU or sorafenib were analyzed in accordance with the method used by Chou.28 The combination index (CI), a well-established index reflecting the interaction of two drugs,28 was calculated at different growth-inhibition levels with CalcuSyn software (Biosoft, Great Shelford, UK). CI values of <1, 1, and >1 indicate synergistic, additive, and antagonistic effects, respectively. Considering that gefitinib exhibits no growth-inhibitory effect as a single-agent treatment, we combined gefitinib (5 μmol/L) with celecoxib at 0.125, 0.25, 0.5, 1, and 2 times the IC50 dose concurrently for 48 hours to evaluate the antiproliferative effects of the combined treatment. All sample measurements were replicated five times.
Cell-apoptosis analysis
Cell apoptosis was analyzed by flow cytometry. A total of 105 cells of MCSs were seeded in six-well culture plates and cultured for 24 hours before incubation with the anticancer drug administered alone or combined with celecoxib, and all plates were incubated at 37°C. After 48 hours, MCSs were digested with trypsin, harvested, suspended, stained with propidium iodide (PI), and assayed for annexin V. Cells were briefly resuspended in a 200 μL solution containing fluorescein isothiocyanate-conjugated annexin V antibody (Beyotime Institute of Biotechnology, Shanghai, People’s Republic of China) and PI (50 μg/mL) for 15 minutes and analyzed by flow cytometry. The percentage of annexin V-positive/PI-negative apoptotic cell population was calculated using CellQuest (BD Biosciences, San Jose, CA, USA).
Western blot analysis
Cells were lysed with cell-lysis buffer. The timing of protein-sample extraction was 48 hours after drug exposure. Cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum. Equivalent amounts of protein were separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim milk and incubated overnight at 4°C with primary antibodies. Antibodies to COX-2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and phosphorylated Y1068 (p-Y1068) EGFR and phosphorylated (p)-AKT (serine 473) were obtained from Cell Signaling Technology (Beverly, MA, USA). The EGFR and AKT were purchased from Bioworld Technology, Inc. (St Louis Park, MN, USA), and β-actin was supplied by Sinopept (Beijing, People’s Republic of China). The blots were visualized with a horseradish peroxidase-conjugated secondary antibody (Sinopept) and an enhanced chemiluminescence-detection system (EMD Millipore). Western blots were repeated three times for each protein. ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used for performing densitometry analysis on Western blot. The HCC827 and H1975 cell lines were used as positive and negative controls of p-EGFR, respectively.
Statistical analysis
Data were reported as means ± standard error of at least three experiments. Student’s t-test and two-way analysis of variance were used to calculate the statistical differences, Tukey’s test was used for multiple comparison, and P≤0.05 was considered statistically significant.
Results
HepG2 MCS morphology
MCSs were observed under a scanning electron microscope (Figure 1). The MCSs were irregular, with diameters ranging from 100 μm to 200μm after 3 or 4 days. The cells were oval spheroids or polyhedrons with tight cell junctions. HepG2 MCSs exhibited resistance to 5-FU, sorafenib, and celecoxib compared with monolayer cells.
 | Figure 1 (A and B) Scanning electron microscopy images of HepG2 multicellular spheroids. The multicellular spheroids are irregular, with a diameter ranging from 100 μm to 200 μm. |
We evaluated the inhibitory effects in HepG2 MCSs and monolayer cells treated with 5-FU, sorafenib, gefitinib, and celecoxib for 48 hours. Figure 2 shows the concentration-dependent inhibitory effects of 5-FU, sorafenib, and celecoxib in MCSs and monolayer cells. Compared with monolayer cells, MCSs exhibited resistance. Table 1 summarizes the IC50 of these drugs in different culture models. The IC50 of 5-FU, sorafenib, and celecoxib in MCSs was higher than monolayer cells (P<0.05). The cell-culture method and drug concentrations significantly affected cell-growth inhibition (P<0.05) (Tables 2–4). Significant differences in growth inhibition were indicated among different concentrations of 5-FU, sorafenib, and celecoxib (P<0.05). However, no statistical difference in growth inhibition was observed between 12.5 and 25×10−3 g/L of 5-FU or 1.4 and 2.85 μmol/L of sorafenib (P>0.05). No statistical difference in growth inhibition was found between 6.25 and 12.5 μmol/L of celecoxib (P>0.05). Gefitinib as a single-agent therapy showed no growth-inhibitory activity at concentrations tested both in MCSs and monolayer cells.
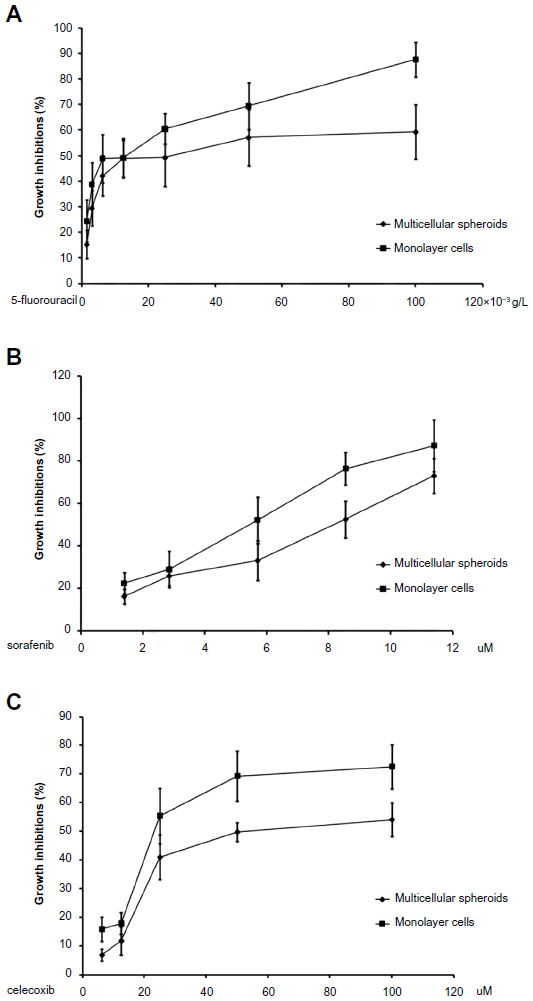 | Figure 2 (A–C) The antiproliferative effects of 5-fluorouracil, sorafenib, and celecoxib in HepG2 multicellular spheroids (MCSs) and monolayer cells were concentration-dependent. Compared with monolayer cells, MCSs became relatively resistant to 5-fluorouracil, sorafenib, and celecoxib. (A) 5-fluorouracil; (B) sorafenib; (C) celecoxib. A total of 5,000 cells in either monolayer cells or MCSs were seeded into a 96-well plate. Monolayer cells or MCSs were exposed to these anticancer drugs for 48 hours. All sample measurements were replicated five times. |
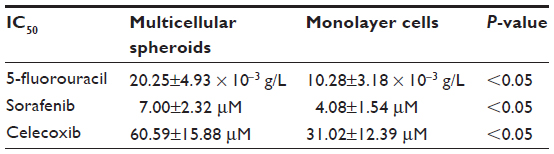 | Table 1 Half-maximal inhibitory concentration (IC50) values for the antiproliferative effects of 5-fluorouracil, sorafenib, and celecoxib on the growth of multicellular spheroids and monolayer cells in vitro |
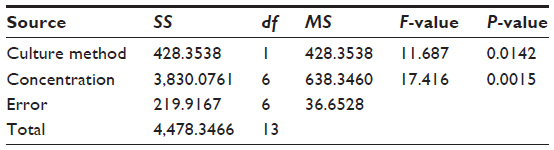 | Table 2 Analysis of variance for 5-fluorouracil |
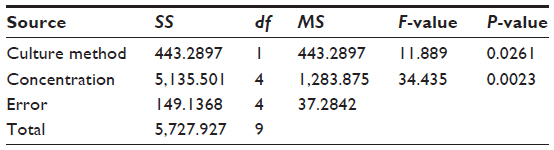 | Table 3 Analysis of variance for sorafenib |
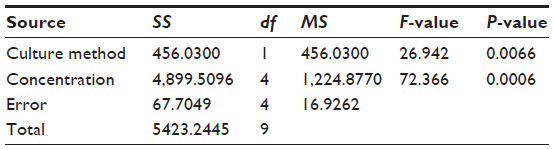 | Table 4 Analysis of variance for celecoxib |
Synergistic effects of celecoxib combined with 5-FU or sorafenib on the growth of HepG2 MCSs in vitro
To detect the inhibitory effects of celecoxib combined with 5-FU or sorafenib, MCSs were concurrently exposed to these anticancer drugs and celecoxib for 48 hours at a fixed ratio. The combined effect was evaluated based on the CI. The sensitivity of HepG2 MCSs to 5-FU or sorafenib increased when combined with celecoxib, and the interaction was identified as synergistic (CI <1). Each experiment was repeated in triplicate (Figure 3).
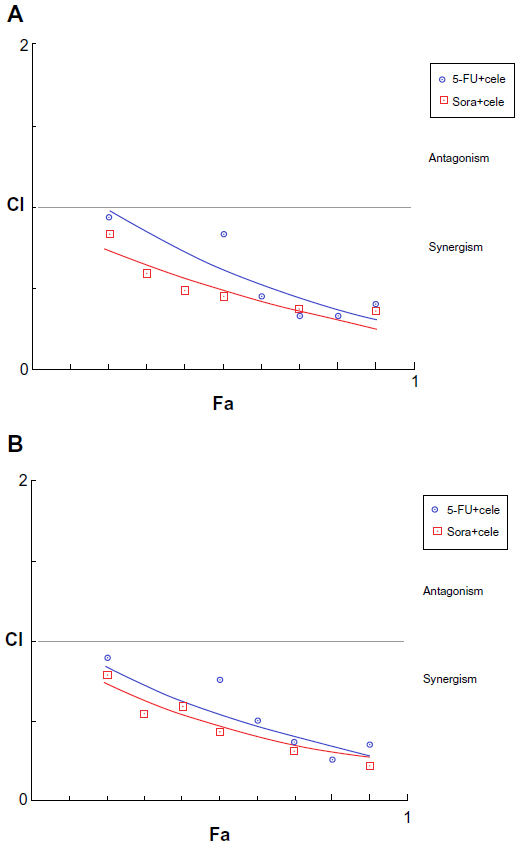 | Figure 3 (A and B) Effects of celecoxib (cele) combined with 5-fluorouracil (5-FU) or sorafenib (Sora) on the growth of HepG2 multicellular spheroids and monolayer cells in vitro. (A) HepG2 multicellular spheroids; (B) HepG2 monolayer cells. Multicellular spheroids and monolayer cells were concurrently exposed to these anticancer drugs and celecoxib for 48 hours at a fixed ratio, and then cell viability was measured. The interaction between celecoxib and 5-fluorouracil or sorafenib was evaluated based on the combination index (CI), which is plotted against the fraction of growth inhibition. These combinations exhibited synergistic effects. |
Gefitinib increased the inhibitory effects of celecoxib on the growth of HepG2 MCSs in vitro
To detect the inhibitory effect of celecoxib combined with gefitinib, MCSs were exposed to gefitinib (5 μM) and different concentrations of celecoxib concurrently for 48 hours. Figure 4 shows that gefitinib, which exhibits no growth-inhibition activity as a single-agent therapy, increased the inhibitory effect of celecoxib.
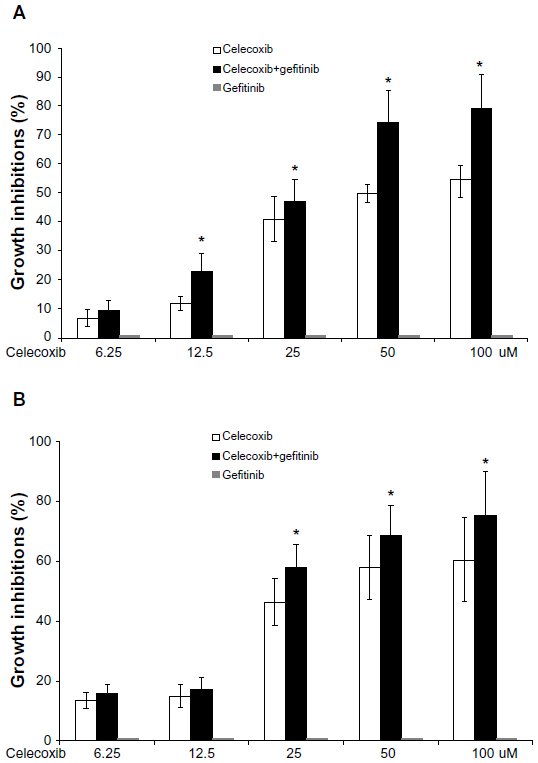 | Figure 4 (A and B) The antiproliferative effect of celecoxib combined with gefitinib was evaluated in HepG2 multicellular spheroids and monolayer cells. (A) HepG2 multicellular spheroids; (B) HepG2 monolayer cells. Gefitinib (5 μmol/L) increased the inhibitory effect of celecoxib. |
Celecoxib combined with 5-FU or sorafenib or gefitinib increased apoptosis in HepG2 MCSs
To understand the mechanisms of celecoxib combined with 5-FU or sorafenib or gefitinib, apoptosis induction was analyzed by flow cytometry at 48 hours after MCSs were treated with either 5-FU, sorafenib, gefitinib and celecoxib given alone, or celecoxib combined with 5-FU or sorafenib or gefitinib. 5-FU, sorafenib, and celecoxib as single-agent treatments clearly induced apoptosis in HepG2 MCSs. No marked apoptosis was observed in cells treated with gefitinib. Celecoxib plus 5-FU or sorafenib or gefitinib increased apoptosis, compared with celecoxib as a single agent (P<0.05) (Figure 5).
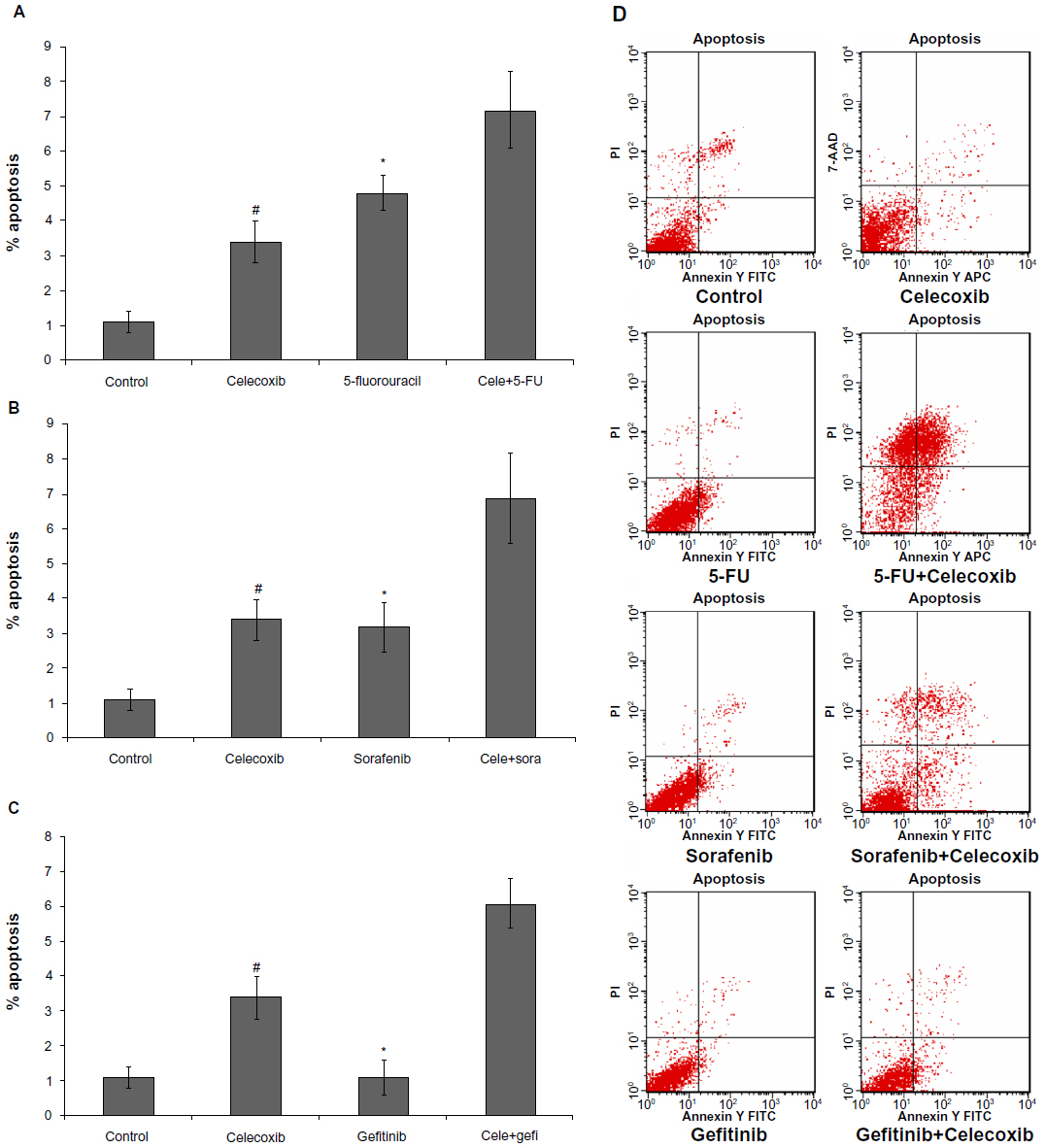 | Figure 5 (A–D) 5-fluorouracil (5-FU; 8.1×10−3 g/L), sorafenib (sora; 4.4 μmol/L), and celecoxib (Cele; 21.8 μmol/L), administered individually, clearly induced apoptosis of HepG2 multicellular spheroids. No marked apoptosis was observed in the multicellular spheroids treated with gefitinib (gefi; 5 μmol/L). Celecoxib combined with 5-fluorouracil, sorafenib, or gefitinib rather than administered as a single-agent treatment, increased apoptosis. (A) Celecoxib plus 5-fluorouracil; (B) celecoxib plus sorafenib; (C) celecoxib plus gefitinib; (D) representative data of flow cytometry. |
COX-2, EGFR, and the downstream signaling pathway were inhibited by combined celecoxib and gefitinib in HepG2 MCSs
We performed Western blot analysis to investigate COX-2, p-EGFR, and p-AKT expression in HepG2 MCSs treated with different agents. The protein-expression levels of COX-2, p-EGFR, and p-AKT were investigated under similar conditions. Celecoxib and gefitinib as single agents failed to inhibit COX2, p-EGFR, or p-AKT expression, whereas combined celecoxib and gefitinib markedly reduced their expression in MCSs (P<0.05) (Figure 6C). We used the HCC827 cell line, which was sensitive to gefitinib as a positive control, and H1975 cell line, which was resistant to gefitinib as a negative control.29 After 48-hour exposure with gefitinib (5 μM), p-EGFR protein expression was markedly decreased in the HCC827 cell line (P<0.05); however, p-EGFR protein expression was not affected in the H1975 cell line (Figure 7).
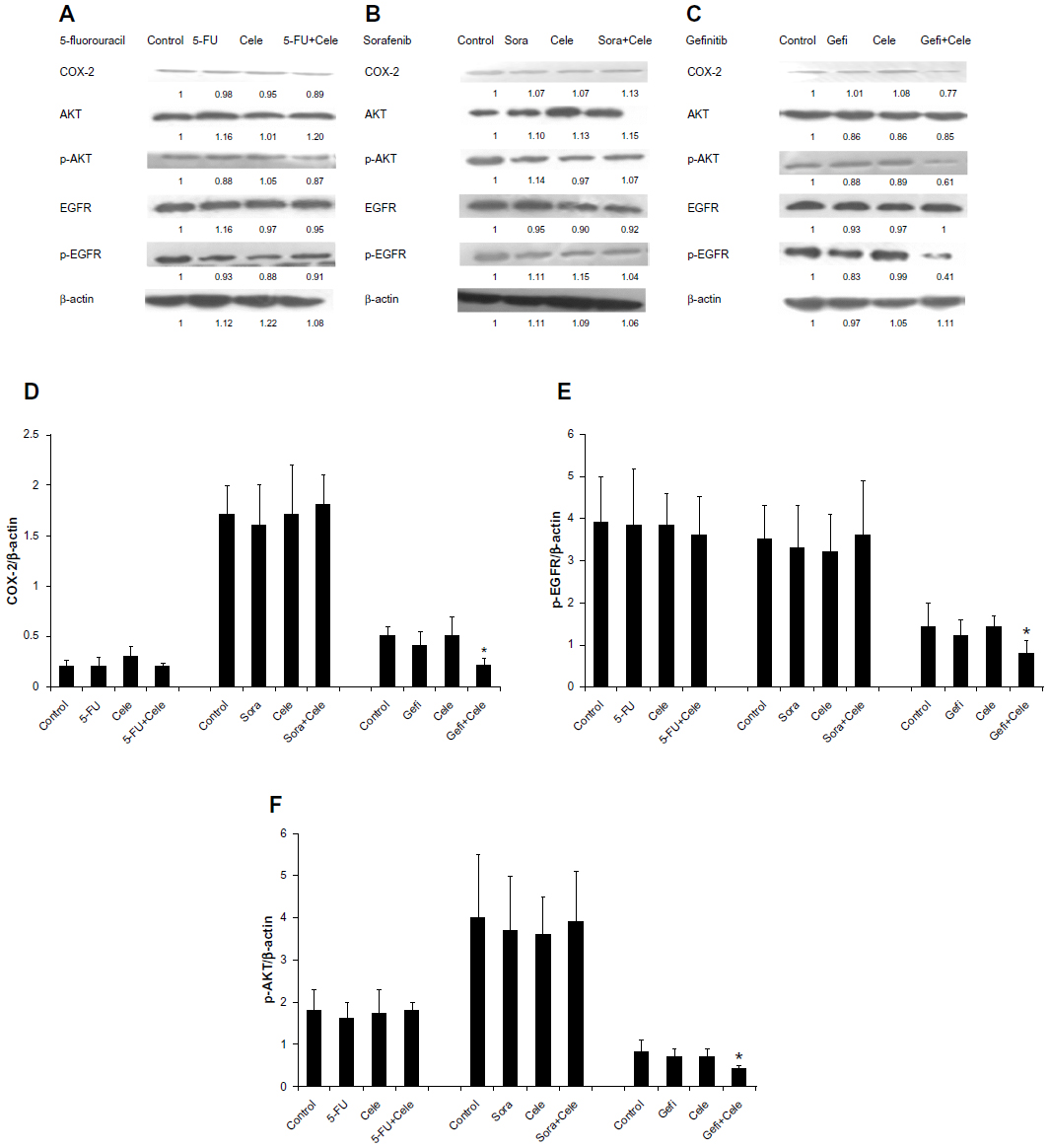 | Figure 6 (A–F) Protein expression of cyclooxygenase (COX)-2, phosphorylated epidermal growth-factor receptor (p-EGFR), and phosphorylated (p)-AKT in HepG2 multicellular spheroids determined by immunoblot analysis. After concurrent exposure of 48 hours with 5-fluorouracil (5-FU; 8.1×10−3 g/L), sorafenib (Sora; 4.4 μmol/L), and gefitinib (Gefi; 5 μM) with or without celecoxib (Cele; 21.8 μmol/L), the samples were collected. Samples incubated with only celecoxib were obtained 48 hours after celecoxib administration. (A–C) Immunoblots represent observations from one single experiment repeated three times. The integrated optical densities of (D) COX-2, (E) p-EGFR, and (F) p-AKT proteins were analyzed after normalization with β-actin (43 kDa) in each lane. (D–F) Means ± standard error of the mean of three separate experiments. |
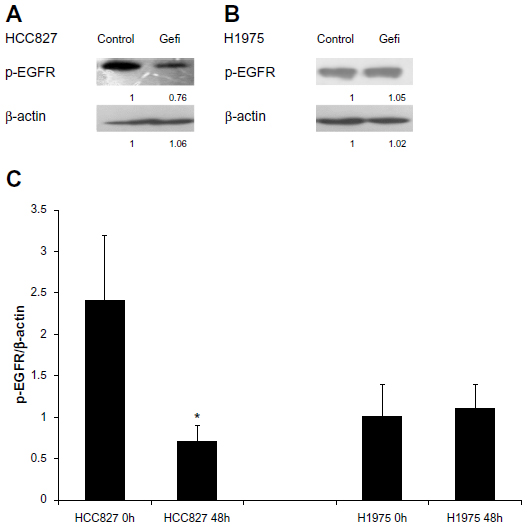 | Figure 7 (A–C) Protein expression of phosphorylated epidermal growth-factor receptor (p-EGFR) in hepatocellular carcinoma (HCC)827 and H1975 cells determined by immunoblot analysis. The samples were collected at 0 and 48 hours after exposure to gefitinib (Gefi; 5 μM). (A and B) Immunoblots represent the observations from one single experiment repeated three times. The integrated optical densities of (C) p-EGFR proteins were analyzed after normalization with β-actin (43 kDa) in each lane; means ± standard error of the mean of three separate experiments. |
COX-2, EGFR, and the downstream signaling pathway remained unaffected by celecoxib plus 5-FU or sorafenib in HepG2 MCSs
COX-2, p-EGFR, and p-AKT expression in HepG2 MCSs remained unaffected by either 5-FU, sorafenib alone, or celecoxib combined with 5-FU or sorafenib (Figure 6A and B).
Discussion
Wigle and Sutherland30 established MCSs as an in vitro model for the systematic study of tumor response to therapy. Tumor cells often form compact MCSs when maintained in a three dimensional (3-D) culture system. Compared with conventional monolayer cultures, cells in 3-D aggregates more closely resembled the in vivo characteristics, such as cell shape, cell microenvironment, and drug resistance, which is called multicellular resistance.31,32 In the present study, human HCC HepG2 cells were cultured with a liquid-overlay technique previously mentioned to form MCSs. The results indicated that the cells were oval spheroids or polyhedrons with diameter ranging from 100 μm to 200 μm after 3 or 4 days.
Compared with the monolayer cells, the MCS cells were less sensitive to 5-FU, sorafenib, and celecoxib. The results obtained in this study were consistent with those in the studies by Ponce de León and Barrera-Rodríguez,33 and Sutherland et al.34 Cells isolated from MCSs generally exhibit higher resistance to cytotoxic drugs than the same cells grown as monolayers.33,34 The higher chemoresistance observed in MCSs may be associated with increased deoxyribonucleic acid repair in spheroids, limited uptake and diffusion of drugs, and a specific microenvironment, which can directly or indirectly affect the activity of cytotoxic compounds by reducing the proliferation rate of the tumor cells.35,36
We investigated the inhibitory effects of celecoxib combined with 5-FU or sorafenib or gefitinib in HepG2 MCSs. We found that the sensitivity of HepG2 MCSs to 5-FU or sorafenib was increased upon combination with celecoxib, and the interaction was synergistic. Celecoxib combined with 5-FU or sorafenib enhanced the apoptotic effects without affecting COX-2, p-EGFR, or p-AKT protein expression. The most likely route of celecoxib to potentiate the efficacy of chemotherapy was considered via COX-2 inhibition. However, recent experiments have suggested that antitumor potency does not depend on whether celecoxib could inhibit COX-2 expression.37 This finding suggests that the inhibitory effect of celecoxib combined with 5-FU or sorafenib occurs neither by COX-2 inhibition nor p-EGFR and p-AKT inhibition in HepG2 MCSs. The mitogen-activated protein-kinase pathway is important for cell proliferation, apoptosis, and differentiation. Therefore, measurement of p-mitogen-activated protein kinases 1/2, p-p38, or p-c-Jun N-terminal kinase 1/2 protein-expression levels may be helpful for explaining the increased apoptotic effects of 5-FU or sorafenib upon celecoxib addition.
Gefitinib as a single-agent treatment failed to inhibit growth activity at the tested concentrations in MCSs and monolayer cells. However, gefitinib increased the inhibitory effect of celecoxib in MCSs. Increased apoptosis was observed when gefitinib was combined with celecoxib. COX-2, p-EGFR, and p-AKT protein expression were not affected by any single-agent treatment. However, celecoxib plus gefitinib markedly reduced COX-2, p-EGFR, and p-AKT protein expression. Our results suggested that combined inhibition of both the EGFR and the COX-2 pathways was beneficial in HepG2 MCSs. The combination of the EGFR and COX-2 inhibitors was previously evaluated in preclinical models. In pancreatic cancer cell lines, celecoxib can potentiate the growth-inhibitory effects of erlotinib.38 Another study on non-small-cell lung cancer cells with EGFR mutations demonstrated that the effectiveness of the addition of celecoxib to an EGFR-TKI is significantly greater than single drugs,39 which is consistent with our results. A previous study indicated that activation of the EGFR pathway promotes transcription of the COX-2 gene.40,41 Similarly, the COX-2 signaling pathway activates EGFR phosphorylation42 and transcription.43 Both the EGFR and the COX-2 pathways are involved in anticancer drug resistance. AKT is a master regulator involved in protein synthesis, antiapoptosis, cell survival, proliferation, and glucose metabolism.44 Also, a recent study showed that COX-2 was involved in apoptosis.45 Therefore, targeting both EGFR and COX-2 can potentially increase apoptosis and modulate both pathways and their downstream signaling, resulting in synergistic effects.
Some preclinical data contradict the claim that celecoxib can enhance the cytotoxic effects of anticancer drugs.46 This contradiction encourages in vivo testing of celecoxib combined with anticancer drugs for HCC, because some beneficial effects of celecoxib, such as inhibition of angiogenesis and metastasis, can only be demonstrated using in vivo models.
The highest concentration of 15 mg of 5-FU/kg administrated by intravenous drip was 7,625 ng/mL in systemic vein blood, which was similar to the concentration of 5-FU we used in our study (8.1×10−3 g/L).47 Plasma trough concentrations at 400 mg twice daily (3.75 mg/L) of sorafenib exceeded the IC30 for inhibition of tumor cell proliferation in vitro in our study (4.4 μmol/L).48 A gefitinib concentration of 5 μmol/L is similar to the achievable concentration in tumor tissue of treated humans.49 However, in our experiments, the applied concentrations of celecoxib induced 30% cell-growth inhibition in MCSs, indicating a 17-fold increase in the maximal concentration of celecoxib in the clinic.50 This scenario suggests that the translation of data to the clinic should be carefully handled.
In conclusion, we demonstrated that celecoxib combined with 5-FU or sorafenib exhibited a synergistic antiproliferative effect in HepG2 MCSs, and not via COX-2, p-EGFR, or p-AKT inhibition. Gefitinib increased the inhibitory effect of celecoxib, which was associated with the inhibition of COX-2, p-EGFR, and p-AKT by combined gefitinib and celecoxib. We also suggested that MCSs were good models to evaluate the interaction of anticancer drugs. This study is the first to report on growth-inhibitory effects on liver cancer MCSs. We intend to conduct animal studies to duplicate our in vitro findings, which warrant clinical evaluation.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Health statistics and health information systems: WHO mortality database. Available from: http://www.who.int/healthinfo/mortality_data/en. Accessed December 21, 2013. | |
Soslow RA, Dannenberg AJ, Rush D, et al. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000; 89(12):2637–2645. | |
Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55(1):115–122. | |
Jolly K, Cheng KK, Langman MJ. NSAIDs and gastrointestinal cancer prevention. Drugs. 2002;62(6):945–956. | |
Bastos-Pereira AL, Lugarini D, Oliveira-Christoff A, et al. Celecoxib prevents tumor growth in an animal model by a COX-2 independent mechanism. Cancer Chemother Pharmacol. 2010;65(2):267–276. | |
Groen HJ, Sietsma H, Vincent A, et al. Randomized, placebo-controlled phase III study of docetaxel plus carboplatin with celecoxib and cyclooxygenase-2 expression as a biomarker for patients with advanced non-small-cell lung cancer: the NVALT-4 study. J Clin Oncol. 2011;29(32):4320–4326. | |
Lustberg MB, Povoski SP, Zhao W, et al. Phase II trial of neoadjuvant exemestane in combination with celecoxib in postmenopausal women who have breast cancer. Clin Breast Cancer. 2011;11(4):221–227. | |
Halamka M, Cvek J, Kubes J, et al. Plasma levels of vascular endothelial growth factor during and after radiotherapy in combination with celecoxib in patients with advanced head and neck cancer. Oral Oncol. 2011;47(8):763–767. | |
Edelman MJ, Hodgson L, Wang X, et al. Serum vascular endothelial growth factor and COX-2/5-LOX inhibition in advanced non-small cell lung cancer: Cancer and Leukemia Group B 150304. J Thorac Oncol. 2011;6(11):1902–1906. | |
Um TH, Kim H, Oh BK, et al. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J Hepatol. 2011;54(5):939–947. | |
Czachorowski MJ, Amaral AF, Montes-Moreno S, et al. Cyclooxygenase-2 expression in bladder cancer and patient prognosis: results from a large clinical cohort and meta-analysis. PLoS One. 2012;7(9):e45025. | |
Kim HS, Moon HG, Han W, et al. COX-2 overexpression is a prognostic marker for stage III breast cancer. Breast Cancer Res Treat. 2012; 132(1):51–59. | |
Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62(6):394–399. | |
Sengupta B, Siddiqi SA. Hepatocellular carcinoma: important biomarkers and their significance in molecular diagnostics and therapy. Curr Med Chem. 2012;19(22):3722–3729. | |
Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small cell lung cancer (The IDEAL1 trial) [corrected]. J Clin Oncol. 2003;21(12):2237–2246. | |
Pérez-Soler R, Chachoua A, Hammond LA, et al. Determinants of tumor response and survival with erlotinib in patients with non-small-cell lung cancer. J Clin Oncol. 2004;22(16):3238–3247. | |
Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet. 2005;366(9496):1527–1537. | |
Giampaolo Tortora, Rosa Caputo, Vincenzo Damiano, et al. Combination of a selective cyclooxygenase-2 inhibitor with epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 and protein kinase A antisense causes cooperative antitumor and antiangiogenic effect. Clin Cancer Res. 2003;9(4):1566–1572. | |
Buchanan FG, Holla V, Katkuri S, Matta P, DuBois RN. Targeting cyclooxygenase-2 and the epidermal growth factor receptor for the prevention and treatment of intestinal cancer. Cancer Res. 2007;67(19):9380–9388. | |
Krishnaswamy N, Lacroix-Pepin N, Chapdelaine P, et al. Epidermal growth factor receptor is an obligatory intermediate for oxytocin-induced cyclooxygenase 2 expression and prostaglandin F2 alpha production in bovine endometrial epithelial cells. Endocrinology. 2010;151(3):1367–1374. | |
Hoffman RM. The three dimensional question: can clinically relevant tumor drug resistance be measured in vitro? Cancer Metastasis Rev. 1994;13(2):169–173. | |
Bokhari M, Carnachan RJ, Cameron NR, Przyborski SA. Novel cell culture device enabling three-dimensional cell growth and improved cell function. Biochem Biophys Res Commun. 2007;354(4):1095–1100. | |
Mueller-Klieser W. Three dimensional cell cultures: from molecular mechanisms to clinical applications. Am J Physiol. 1997;273(4 Pt 1):C1109–C1123. | |
Kim KU, Wilson SM, Abayasiriwardana KS, et al. A novel in vitro model of human mesothelioma for studying tumor biology and apoptotic resistance. Am J Respir Cell Mol Biol. 2005;33(6):541–548. | |
Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Control. 2002;9(Suppl 2):28–35. | |
Green SK, Francia G, Isidoro C, Kerbel RS. Antiadhesive antibodies targeting E-cadherin sensitize multicellular tumor spheroids to chemotherapy in vitro. Mol Cancer Ther. 2004;3(2):149–159. | |
Zhang X, Wang W, Yu W, et al. Development of an in vitro multicellular tumor spheroid model using microencapsulation and its application in anticancer drug screening and testing. Biotechnol Prog. 2005;21(4):1289–1296. | |
Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. | |
Okabe T, Okamoto I, Tsukioka S. Addition of S-1 to the epidermal growth factor receptor inhibitor gefitinib overcomes gefitinib resistance in non small cell lung cancer cell lines with MET amplification. Clin Cancer Res. 2009;15(3):907–913. | |
Wigle JC, Sutherland RM. Increased thermoresistance developed during growth of small multicellular spheroids. J Cell Physiol. 1985;122(2):281–289. | |
Desoize B, Jardilier J. Multicellular resistance: a paradigm for clinical resistance? Crit Rev Oncol Hematol. 2000;36(2–3):193–207. | |
Ma HL, Jiang Q, Han S, et al. Multicellular tumor spheroids as an in vivo-like tumor model for three-dimensional imaging of chemotherapeutic and nano material cellular penetration. Mol Imaging. 2012;11(6):487–498. | |
Ponce de León V, Barrera-Rodríguez R. Changes in P-glycoprotein activity are mediated by the growth of a tumour cell line as multicellular spheroids. Cancer Cell Int. 2005;5(1):20. | |
Sutherland RM, Eddy HA, Bareham B, Reich K, Vanantwerp D. Resistance to adriamycin in multicellular spheroids. Int J Radiat Oncol Biol Phys. 1979;5(8):1225–1230. | |
Orlandi P, Barbara C, Bocci G, et al. Idarubicin and idarubicinol effects on breast cancer multicellular spheroids. J Chemother. 2005;17(6):663–667. | |
dit Faute MA, Laurent L, Ploton D, Poupon MF, Jardillier JC, Bobichon H. Distinctive alterations of invasiveness, drug resistance and cell-cell organization in 3D-cultures of MCF-7, a human breast cancer cell line, and its multidrug resistant variant. Clin Exp Metastasis. 2002;19(2):161–168. | |
Zhang S, Da L, Yang X, et al. Celecoxib potentially inhibits metastasis of lung cancer promoted by surgery in mice, via suppression of the PGE2-modulated β-catenin pathway. Toxicol Lett. 2013;225(2):201–207. | |
Ali S, El-Rayes BF, Sarkar FH, Philip PA. Simultaneous targeting of the epidermal growth factor receptor and cyclooxygenase-2 pathways for pancreatic cancer therapy. Mol Cancer Ther. 2005;4(12):1943–1951. | |
Gadgeel SM, Ali S, Philip PA, Ahmed F, Wozniak A, Sarkar FH. Response to dual blockade of epidermal growth factor receptor (EGFR) and cycloxygenase-2 in nonsmall cell lung cancer may be dependent on the EGFR mutational status of the tumor. Cancer. 2007;110(12):2775–2784. | |
Elder DJ, Halton DE, Playle LC, Paraskeva C. The MEK/ERK pathway mediates COX-2-selective NSAID-induced apoptosis and induced COX-2 protein expression in colorectal carcinoma cells. Int J Cancer. 2002;99(3):323–327. | |
Sheng H, Shao J, DuBois RN. Akt/PKB activity is required for Ha-Ras-mediated transformation of intestinal epithelial cells. J Biol Chem. 2001;276(17):14498–14504. | |
Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8(3):289–293. | |
Kinoshita T, Takahashi Y, Sakashita T, Inoue H, Tanabe T, Yoshimoto T. Growth stimulation and induction of epidermal growth factor receptor by overexpression of cyclooxygenases 1 and 2 in human colon carcinoma cells. Biochim Biophys Acta. 1999;1438(1):120–130. | |
Green BD, Jabbour AM, Sandow JJ, et al. Akt1 is the principal Akt isoform regulating apoptosis in limiting cytokine concentrations. Cell Death Differ. 2013;20(10):1341–1349. | |
Piplani, Honit;Vaish, Vivek;Rana, Chandan;Sanyal, Sankar N. Up-regulation of p53 and mitochondrial signaling pathway in apoptosis by a combination of COX-2 inhibitor, celecoxib and dolastatin 15, a marine mollusk linear peptide in experimental colon carcinogenesis. Mol Carcinog. 2013;52(10):845–858. | |
Elrod, Heath A;Yue, Ping;Khuri, Fadlo R;Sun, Shi-Yong. Celecoxib antagonizes perifosine’s anticancer activity involving a cyclooxygenase-2-dependent mechanism. Mol Cancer Ther. 2009;8(9):2575–2585. | |
Almersjö OE, Gustavsson BG, Regårdh CG, Wåhlén P. Pharmacokinetic studies of 5-fluorouracil after oral and intravenous administration in man. Acta Pharmacol Toxicol (Copenh). 1980;46(5):329–336. | |
Minami H, Kawada K, Ebi H, et al. Phase I and pharmacokinetic study of sorafenib, an oral multikinase inhibitor, in Japanese patients with advanced refractory solid tumors. Cancer Sci. 2008;99(7):1492–1498. | |
Nakagawa K, Tamura T, Negoro S, et al. Phase I pharmacokinetic trial of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (‘Iressa’, ZD1839) in Japanese patients with solid malignant tumors. Ann Oncol. 2003;14:922–930. | |
Stempak D, Gammon J, Klein J, Koren G, Baruchel S. Single-dose and steady-state pharmacokinetics of celecoxib in children. Clin Pharmacol Ther. 2002;72(5):490–497. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.