")
Back to Journals » OncoTargets and Therapy » Volume 12
Cyclin-dependent kinase inhibitor 2B gene is associated with the sensitivity of hepatoma cells to Sorafenib
Authors Weng X, Zeng L, Yan F, He M, Wu X, Zheng D
Received 30 November 2018
Accepted for publication 6 April 2019
Published 28 June 2019 Volume 2019:12 Pages 5025—5036
DOI https://doi.org/10.2147/OTT.S196607
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjay Singh
Xie Weng,* Lixian Zeng,* Feifei Yan, Mengxue He, Xiuqiong Wu, Dayong Zheng
Department of Cancer Center,TCM-Integrated Hospital of Southern Medical University, Guangzhou, Guangdong Province 510310, People’s Republic of China
*These authors contributed equally to this work
Purpose: The sensitivity of advanced hepatocellular carcinoma (HCC) to Sorafenib is low. The purpose of this study was to investigate the effects of cyclin-dependent kinase inhibitor 2B (CDKN2B) gene on the prognosis of HCC and the sensitivity of HCC cells to Sorafenib.
Patients and methods: Streptavidin-perosidase (SP) staining was performed to determine the expression of CDKN2B in HCC tissues and adjacent tissues. The cell counting kit-8 (CCK-8) assay was carried out to determine cell viability. CDKN2B mRNA and protein were tested by real-time quantitative polymerase chain reaction (RT-qPCR) and Western blot, respectively. CDKN2B gene was silenced or over-expressed in the cells by plasmid transfection technique. Flow cytometry was carried out to detect cell cycle and apoptosis.
Results: SP staining results showed that CDKN2B was positive in adjacent tissues and in HCC tissues from partial response (PR) patients, CDKN2B was slightly positive in stable disease (SD) patients, but negative in progression disease (PD) patients. The survival rate of patients with low expression of CDKN2B was low. Up-regulation of CDKN2B expression could promote the pro-apoptotic effect of Sorafenib and cell arrest in G1 phase. When the CDKN2B gene expression was down-regulated, the cell apoptosis rate and the proportion of cells treated with Sorafenib in G1 phase decreased. Silencing CDKN2B reversed CDKN2B overexpression caused by Sorafenib.
Conclusion: CDKN2B genes were lowly expressed in tumor tissues from HCC patients who were treated with Sorafenib and had a poor prognosis. Up-regulation of CDKN2B promoted sensitivity of HCC to Sorafenib, and similarly down-regulation of CDKN2B reduced the sensitivity.
Keywords: hepatocellular carcinoma, Sorafenib, cyclin-dependent kinase inhibitor 2B, prognosis
Introduction
The incidence of hepatocellular carcinoma (HCC) ranked fifth and its mortality is the third highest among all cancers.1 However, there is still no effective treatment for HCC. For HCC patients at an early stage, liver resection and liver transplantation are preferred treatment methods.2,3 Although the liver has a strong compensatory function, the early symptoms of HCC are not easily found and lack specificity, and most HCC patients are diagnosed at the late stage of the disease.4,5 Patients with advanced HCC are not suitable for surgery, and chemotherapy is the only strategy.6
Traditional chemotherapeutics such as Doxorubicin and Fluorouracil are not effective for HCC treatment and have obvious side-effects.6 With the rise of targeted drug therapy, some small molecule kinase inhibitor drugs have made considerable progress in the treatment of cancer.7 Sorafenib is currently the only clinical solution for treating HCC patients who are not suitable for surgery.8 Sorafenib is a Raf kinase inhibitor; on one hand, Sorafenib directly inhibits tumor growth by inhibiting the activity of RAF-1 and the B-RAF’s serine/threonine kinase, on the other hand, Sorafenib can inhibit tumor growth by inhibiting angiogenesis in tumors through tyrosine kinase activities of FLT-3, VGFR-2, VEGF-3, and PDGF receptors.9–12 However, over the past few years, it has been found that HCC had a significant resistance to Sorafenib, and only about 30% of patients with advanced HCC can benefit from it.13 In addition, a systematic analysis of multiple random clinical control test results found that, even among these beneficiaries, survival time prolonged by only 3 months.14,15 Therefore, it is highly important to find effective molecular targeted drugs against tumor drug resistance. Also, the researches on the mechanism of HCC resistance and the improvement of the sensitivity of HCC to Sorafenib have attracted much attention in recent years.
Cyclin-dependent kinase inhibitor 2A (CDKN2A) and cyclin-dependent kinase inhibitor 2B (CDKN2B) are located on chromosome 9. The deletion of the CDKN2B-CDKN2A gene cluster is the most common genetic event in cancer.16,17 The CDKN2B gene encodes p15Ink4b protein. The encoded protein forms a complex with CDK4 or CDK6 and prevents CDK kinase activated by cyclin D, indicating it acts as a cell growth regulator, thus inhibiting the progress of cell cycle G1. Studies on the relation between CDKN2A and tumors and the relation between CDKN2B and tumors have been gradually conducted in recent years. Studies have shown that the absence of CDKN2B promoted melanoma and renal cell carcinoma.18,19 It has been found that Cyclin E1 and RTK/RAS signaling could drive CDK inhibitor resistance by activating E2F and ETS in human ovarian carcinoma cells.20 In recent years, studies have found that, in SMMC-7721 cells, Verapamil (VER) can increase the tolerance of cells to Doxorubicin chemotherapy, and CDKN2B gene overexpression may be reversed by HCC chemotherapy resistance.21 These studies suggested that CDK inhibitors had an effect on drug resistance in cancer treatment. However, to the best of our knowledge, there is currently no study conducted on the relation between CDKN2B and the sensitivity of HCC to Sorafenib.
Therefore, this study mainly focused on the expressions of CDKN2B in HCC tissues from patients with different HCC prognosis. We also explored whether CDKN2B could regulate the sensitivity of HCC to Sorafenib in order to provide a new approach to improving the efficacy of Sorafenib on HCC.
Materials and methods
HCC patients and samples
From January 2013 to January 2016, 249 cases of HCC patients treated with Sorafenib were collected, and the Response Evaluation Criteria in Solid Tumors (RECIST) were applied to evaluate the efficacy of each patient. The follow-up deadline was March 1, 2018. The inclusion criteria were that HCC was a primary tumor and was not associated with other malignancies in a patient, while the exclusion criteria were that a patient received less than three cycles of chemotherapy, with use of other chemotherapy drugs, targeted drugs or radiotherapy, or the patients who drop out during treatment. There were 55 patients (CR 0 cases, PR 8 cases, SD 37 cases, and PD 10 cases) who met the criteria. HCC tissues and adjacent tissues of each patient were collected. According to the expression level of CDKN2B in HCC tissues, 55 patients were divided into the CDKN2B high expression group and CDKN2B low expression group. The study was approved by the Ethics Committee of TCM-Integrated Hospital of Southern Medical University, and all patients signed informed consent.
Cells culture and transfection
The human hepatocyte cell line THLE-2, and HCC cell lines Hep3B, SNU-182, SNU-387, SK-Hep1, and PLC/PRF/5 cells were purchased from ATCC (USA). The cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum in an incubator at 37°C in 5% CO2. Cell culture-related reagents were purchased from Gibco (USA). To explore whether CDKN2B could regulate the sensitivity of HCC to Sorafenib, drug intervention was performed on Hep3B and SNU-182 cells. Sorafenib was purchased from Bayer Corporation (West Haven, CT, USA), and dimethylsulfoxide (DMSO) was purchased from KeyGen Biotech Co., Ltd. (Nanjing, China). Sorafenib was dissolved in DMSO and diluted with RPMI 1640 to the desired concentration for in vitro studies. In the control group, DMSO was added to cultures as a vehicle.
The wild-type CDKN2B coding sequence was subcloned into pcDNA3.1 (Sangon Biotech, China) to construct a pcDNA-CDKN2B expression vector. CDKN2B transfections were performed using Lipofectamine 2000 (Invitrogen, USA). siCDKN2B was purchased from GenePharma (China), and the siRNA transfections were performed by Lipofectamine 2000. The transfection efficiencies were detected using real-time quantitative polymerase chain reaction (RT-qPCR) and Western blot.
Streptavidin-perosidase (SP) staining
SP staining was performed to detect the CDKN2B proteins expression levels in HCC tissues and adjacent tissues. Specimens were cut into 4-μm-thick sections and deparaffinized in xylene; 0.01 mol/L citrate buffer solution was used for antigen retrieval, and 50 μL peroxidase blocking solution was added to block endogenous peroxidase activity. The primary antibody was added and incubated at 4°C for 12 hours, according to the kit instructions. The secondary antibody was added and incubated at room temperature for 10 minutes. Then 100 μL DAB was added for visualization for 5 minutes, counterstained, and the staining was observed under a microscope.
Cell counting kit-8 (CCK-8) assay
The CCK-8 assay was used to detect the cell viability, and the kit was purchased from Tongren (Japan). Cells were pre-incubated at 37°C in 5% CO2 in an incubator, and then CCK-8 reagent were added to the cells and cultured at 37°C in 5% CO2 atmosphere for 4 hours. The optical density (OD) of each well at 450 nm was measured using a microplate reader (ELX 800, Bio-Teck, USA).
Flow cytometry
Flow cytometry was used to detect cell apoptosis, and the kits were purchased from BD Pharmingen (USA). Cells of 1×106 were washed with PBS at 4°C and resuspended to a concentration of 4×105 cells/mL. Annexin-V-PE and 7-aminoactinomycin D (7-AAD) were added, respectively. The samples were incubated at room temperature in the dark for 10 minutes. A flow cytometer (Becton Dickerson, SanJose, CA, USA) was applied to analyze the cell apoptosis. Flow cytometry was also applied to detect the cell cycle.
Real-time quantitative polymerase chain reaction (RT-qPCR) analysis
RT-qPCR was used to detect the CDKN2B mRNA expression. The cells were triturated and lysed. The RNAs were then extracted by CHCl3 (Aladdin, China) and dissolved in DEPC water (Sigma aliquots). Reverse transcription kit (TaKaRa, Japan) was used to synthesize cDNA. The reverse transcription reaction conditions were set at 37°C for 15 minutes, and the reverse transcription inactivation conditions were set at 85°C for 15 seconds. RT-qPCR was performed with the RT-qPCR kit (TaKaRa) by activating the DNA polymerase at 95°C for 5 minutes, followed by 40 cycles of two-step PCR (95°C for 10 seconds and 60°C for 30 seconds) and a final extension at 75°C for 10 minutes, and held at 4°C. RNase-free water was used as the templates of negative control experiences. All primers were obtained from Genewiz (Suzhou, Jiangsu, China) and are listed in Table 1. The formula 2-ΔΔCT was implemented to determine the mRNA expression levels.
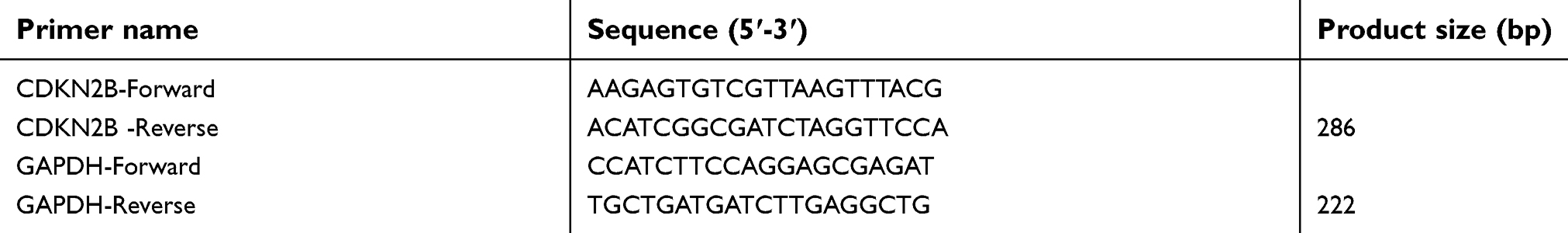 |
Table 1 The sequences of primers |
Western blot
Western blot was applied to detect CDKN2B protein expression levels. The cells were lysed and blocked with RIPA (Abmole, USA), and the supernatant was collected by centrifugation at 12,000 rpm at 4°C for 15 minutes. A bicinchoninic acid (BCA) method was used to determine the protein concentration. A 10% SDS-PAGE gel was prepared to electrophoresis. The PVDF membrane (Bio-Rad, USA) was transferred by a Trans-Blot Transfer Slot (Bio-Rad) and blocked with 5% fat-free milk for 2 hours at room temperature. According to the kit instructions, the primary antibody (anti-CDKN2B, Abcam, ab53034, dilution: 1:800) was added and shaken at room temperature for 2 hours and then incubated at 4°C for 12 hours. The secondary antibody (mouse anti-human IgG, Abcam, ab99823, 1:10,000) was added and incubated at room temperature for 1.5 hours. Chemiluminescence detection was carried out using ECL reagent (Huiying, Shanghai, China). GAPDH was used as the internal reference.
Statistical analysis
All the experimental data are shown as mean±standard deviation (SD). Statistical analysis was performed by applying SPSS 20 (SPSS, Inc., Chicago, IL, USA). The one-way analysis of variance (ANOVA) following Turkey’s multiple comparison was carried out to analyze the differences among the experimental groups. Kaplan-Meier survival analysis was used to compare differences in survival rates between the two groups. The statistical significant was expressed as P<0.05.
Results
CDKN2B expression levels in HCC tissues and adjacent tissues in patients with different therapeutic efficacy
The SP staining results showed that CDKN2B was positive in adjacent tissues and in HCC tissue of partial response (PR) patients. CDKN2B was slightly positive in stable disease (SD) patients, but negative in progression disease (PD) patients (Figure 1), suggesting that the level of CDKN2B expression in HCC tissue was down-regulated in patients with poor efficacy of Sorafenib.
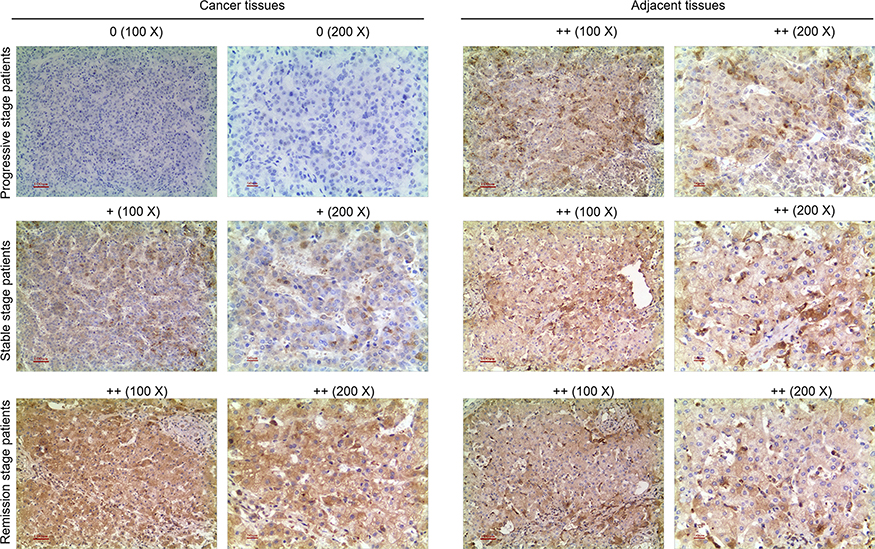 |
Figure 1 Streptavidin-perosidase (SP) staining results. SP staining was used to detect CDKN2B expression in tumor tissues and adjacent tissues of hepatocellular carcinoma (HCC) patients who had different prognoses and were treated with Sorafenib. |
CDKN2B mRNA expression levels and survival curves in 55 cases
RT-qPCR was applied to detect CDKN2B mRNA expression levels in 55 cases of HCC tissues and adjacent tissues. The results showed that the level of CDKN2B mRNA in 36 cases of HCC tissues was lower than that in adjacent tissues. The CDKN2B mRNA levels of HCC tissues in 29 PD patients were all lower than those in adjacent tissues (Figure 2A). According to the CDKN2B mRNA expression level in HCC tissues, 55 patients were divided into two groups. The survival rate of the group with high expression of CDKN2B was higher than that in the group with low expression of CDKN2B, and the difference was not statistically significant (P>0.05) (Figure 2B). This suggested that low expression of CDKN2B may indicate a poor prognosis, showing that a low expression of CDKN2B might affect the sensitivity of HCC to Sorafenib.
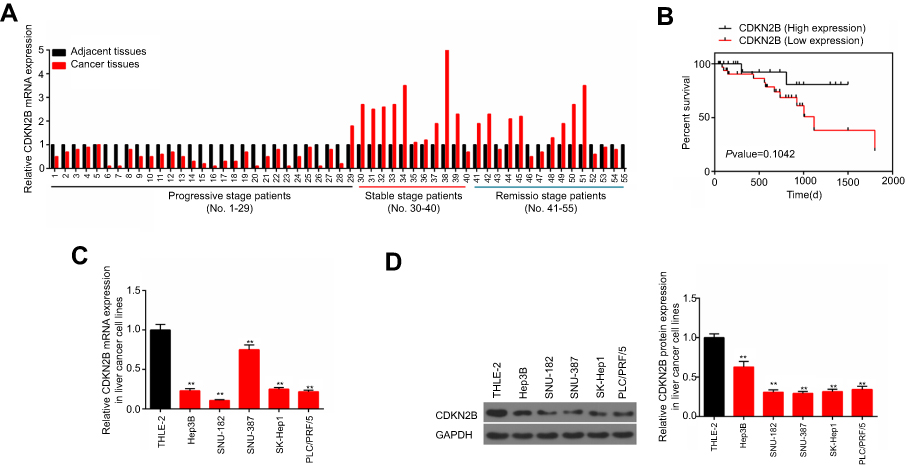 |
Figure 2 CDKN2B expressions in different hepatocellular carcinoma (HCC) patients and hepatic cell lines. (A) The mRNA levels of CDKN2B in tumor tissues and adjacent tissues of 55 HCC patients treated with Sorafenib were detected by RT-qPCR. (B) The survival curve of the CDKN2B high expression group and CDKN2B low expression group. (C, D) CDKN2B mRNA and protein expression levels in THLE-2, Hep3B, SNU-182, SNU-387, SK-Hep1, and PLC/PRF/5 cells were tested using RT-qPCR and Western blot, respectively. |
CDKN2B expression in different cell lines
Western blot and RT-qPCR experiments showed that CDKN2B was lowly expressed in HCC cell lines (Figures 2C and D). Hep3B and SNU-182 cell lines were selected for subsequent experiments.
Cell viability at different sorafenib concentrations
Hep3B and SNU-182 cells were cultured in the medium containing different concentrations of Sorafenib (0 μM, 1 μM, 2.5 μM, 5 μM, 10 μM, and 20 μM) for 48 hours. After that, cell viability of these cells was detected by CCK-8 assay. The results showed that cell viability decreased as the concentration of Sorafenib increased (Figures 3A and B).
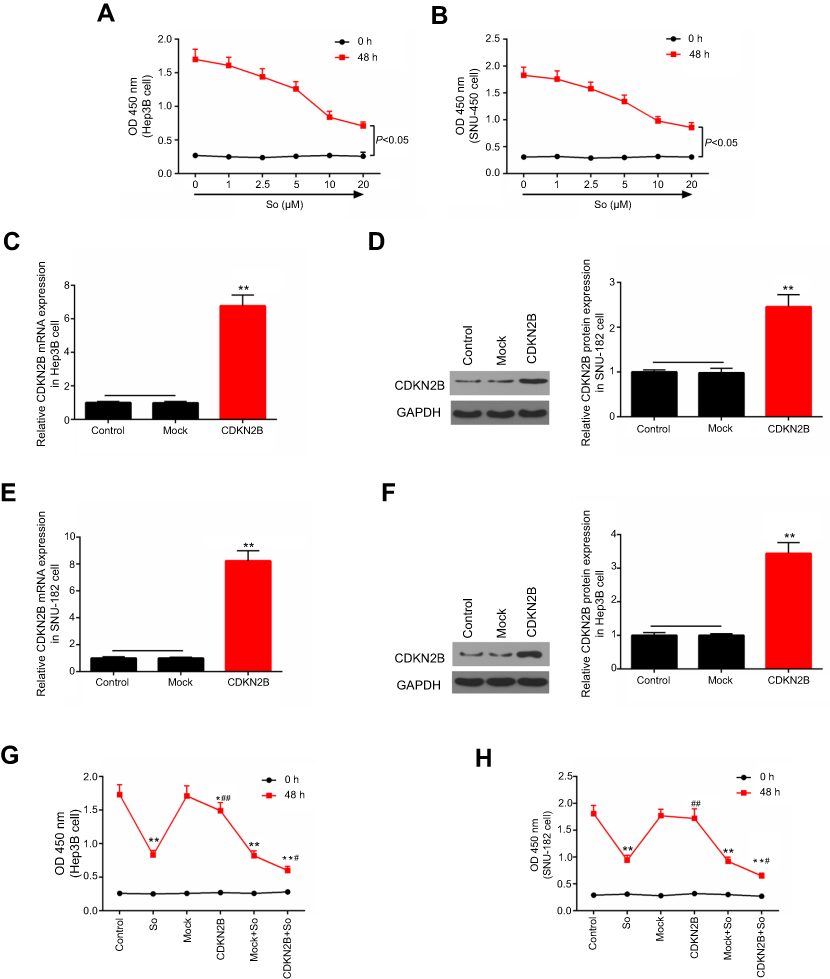 |
Figure 3 Effects of Sorafenib on cell viability of Hep3B and SNU-182 cells. (A, B) The cell viabilities of Hep3B and SNU-182 cells cultured with 0 μM, 1 μM, 2.5 μM, 5 μM, 10 μM, and 20 μM Sorafenib for 48 hours were detected by CCK-8 assay. (C–F) RT-qPCR and Western blot were performed to detect the expression of CDKN2B mRNA and protein in Hep3B and SNU-182 cells after CDKN2B overexpression plasmid transfection. (G, H) The cell viabilities of Hep3B and SNU-182 cells cultured with Sorafenib (So) or transfected with CDKN2B overexpression plasmid for 48 hours were detected by CCK-8 assay. *p<0.05, **p<0.01 versus control group; #p<0.05, ##p<0.01 versus So group; ^^p<0.01 versus Mock+So group. |
CDKN2B expression levels after transfection of overexpression plasmids
Western blot and RT-qPCR experiments showed that the expression levels of CDKN2B mRNA and protein in Hep3B and SNU-182 cells were significantly increased after transfection with overexpression plasmids (Figures 3C–F). This suggested that the overexpressing cell line was successfully constructed.
Effects of overexpression of CDKN2B on sensitivity of cells to sorafenib
The experimental groups were constructed as follows: control group, Sorafenib group (So group), NC group (Mock group), CDKN2B group, Mock+So group, and CDKN2B+So group, on the conditions of 10 μM concentration and 24 hours exposure time of Sorafenib. The results showed that the cell viability in the So group, CDKN2B group, Mock+So group, and CDKN2B+So group was lower than that in the control group and Mock group. The cell viability in the CDKN2B+So group was significantly lower than that in the Mock+So group (Figures 3G and H). This suggested that CDKN2B overexpression enhanced the sensitivities of Hep3B and SNU-182 cells to Sorafenib.
Effects of CDKN2B overexpression on apoptosis and cell cycle of Hep3B and SNU-182 cells to Sorafenib
Flow cytometry was used to detect apoptosis and cell cycle in the six groups to further explore the effects of CDKN2B expression on the sensitivity of cells to Sorafenib. The results showed that the apoptosis rate in the So group, CDKN2B group, Mock+So group, and CDKN2B+So group was higher than that in the control group and Mock group. The apoptosis rate in the CDKN2B+So group was significantly higher than that in the Mock+So group (Figures 4A–D). The proportion of G1 in the CDKN2B+So group was higher than that in the Mock+So group, and the proportion in S phase was significantly reduced (Figures 4E–H).
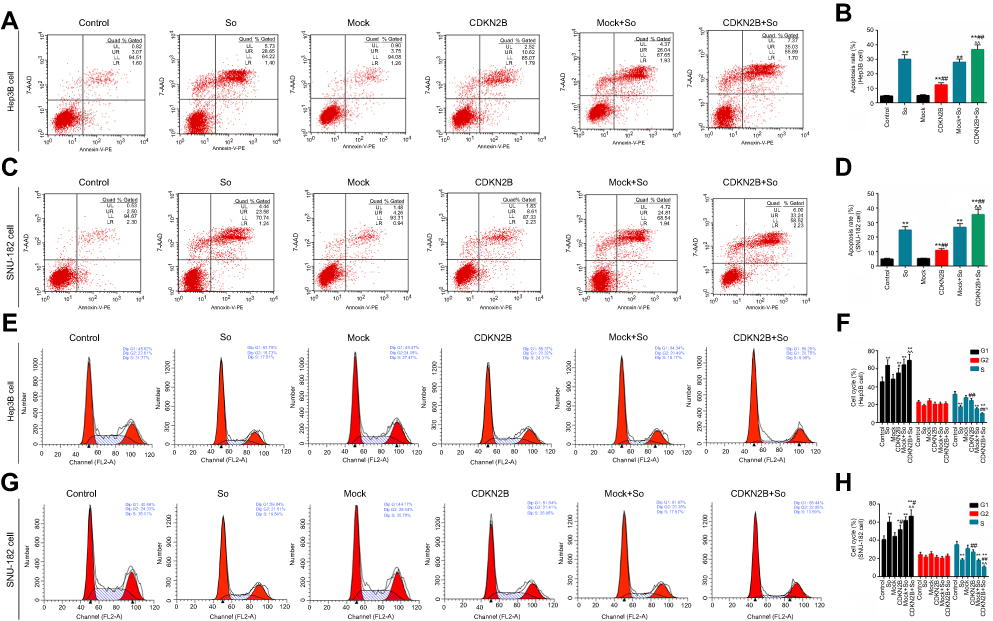 |
Figure 4 Effects of transfection of CDKN2B overexpression plasmid on sensitivity of cells to Sorafenib. (A–D) Flow cytometry was applied to detect apoptosis in Hep3B and SNU-182 cells in the control group, Sorafenib group, Mock group, CDKN2B group, Mock+So group, and CDKN2B+So group. (E–H) Flow cytometry was used to test the cell cycle in Hep3B and SNU-182 cells in the six groups. * p<0.05, p<0.01 versus control group; #p<0.05, ##p<0.01 versus So group; ^p<0.05, ^^p<0.01 versus Mock+So group. |
CDKN2B expression in the six groups
The expression level of CDKN2B gene was determined in the six groups in order to study the relation between the high expression level of CDKN2B gene and the sensitivity of HCC to Sorafenib. The results showed that Sorafenib up-regulated the expressions of CDKN2B in Hep3B and SNU-182 cells. In Hep3B cell line, the levels of CDKN2B mRNA and protein in CDKN2B group and CDKN2B+So group were significantly higher than those in the So group (Figures 5A and B). For SNU-182 cell line, CDKN2B mRNA and protein levels in the CDKN2B group and CDKN2B+So group were up-regulated, and, moreover, a significant higher tendency of those was observed in the CDKN2B+So group compared to the CDKN2B group and Mock group (Figures 5C and D). The expression level of the CDKN2B gene was consistent with the apoptotic conditions of the two cell lines. When the expression level of the CDKN2B gene was up-regulated, the apoptosis rate and the proportion of cells in the G1 phase were increased, suggesting that overexpression of CDKN2B could increase the sensitivity of HCC to Sorafenib.
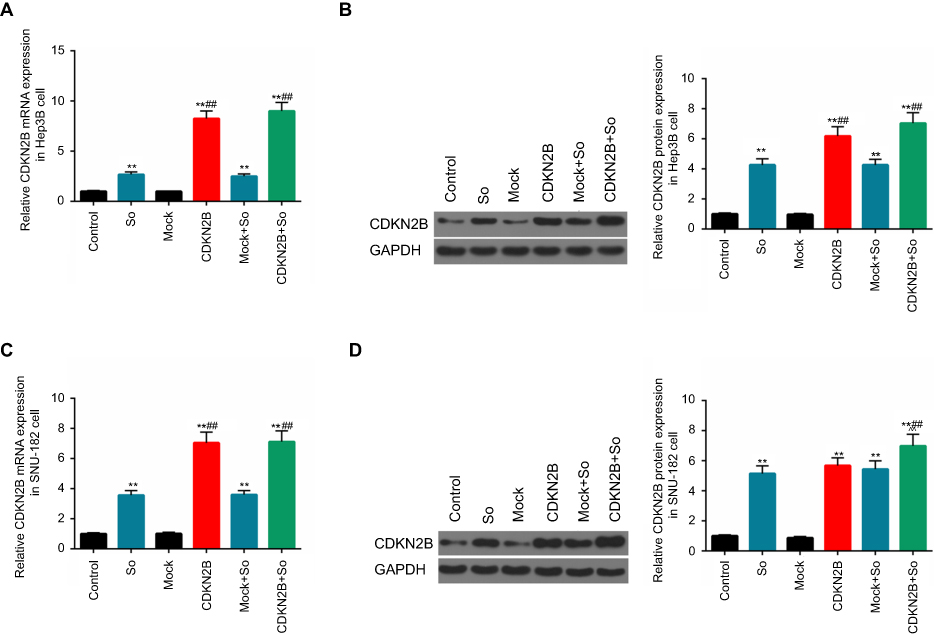 |
Figure 5 CDKN2B mRNA and protein expression levels in the six groups. (A–D) CDKN2B mRNA and protein in Hep3B and SNU-182 cells in the control group, Sorafenib group, Mock group, CDKN2B group, Mock+So group, and CDKN2B+So group were detected using RT-qPCR and Western blot, respectively.**p<0.01 versus control group; ## p<0.01 versus So group; ^^p<0.01 versus CDKN2B group. |
Effects of low-expression of CDKN2B on sensitivity of cells to Sorafenib
The experimental groups were constructed as follows: siNC group, siCDKN2B group, siNC+So group, and siCDKN2B+So group, on the condition of 10 μM concentration and 24 hours exposure time of Sorafenib. The results showed that the transfection of siCDKN2B plasmid had no significant effects on the cell viability of Hep3B and SNU-182 cells. The cell viability of the siCDKN2B+So group was significantly higher than that in the siNC+So group (Figures 6A and B), demonstrating that decreased expression of CDKN2B inhibited the effects of Sorafenib on cell viability.
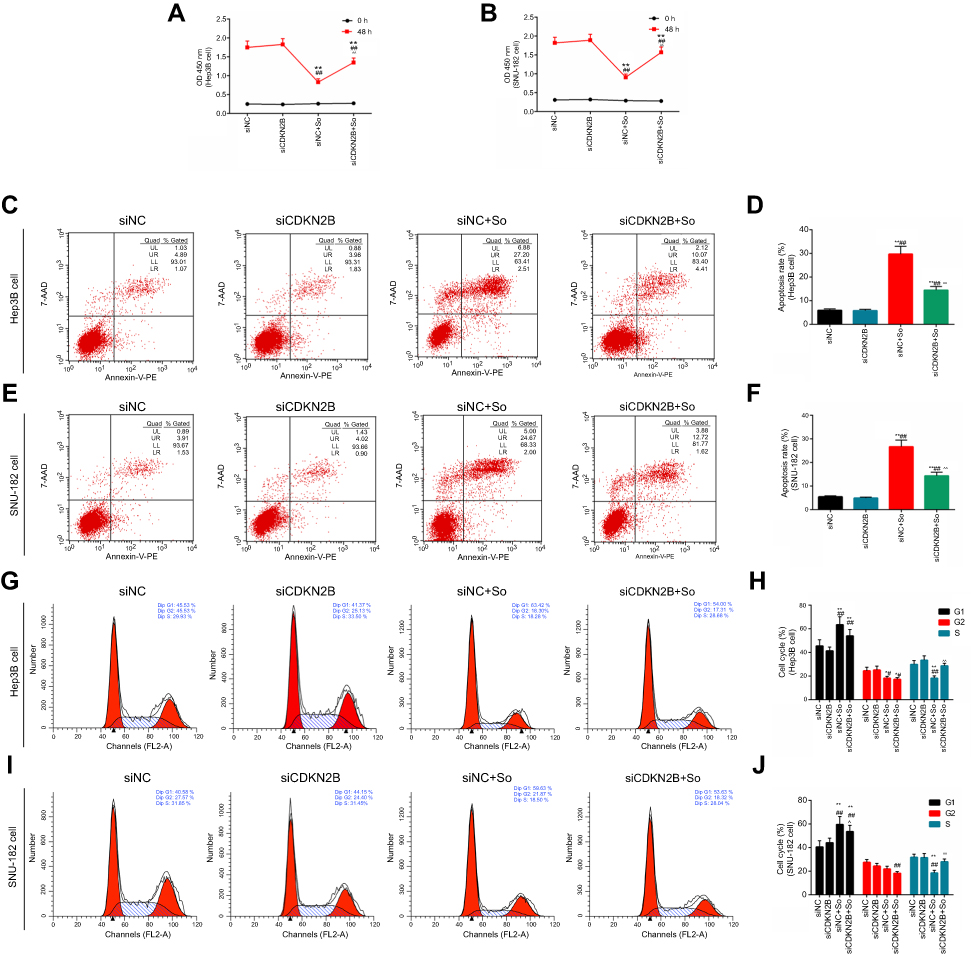 |
Figure 6 Effects of transfection of siCDKN2B plasmid on sensitivity of cells to Sorafenib. (A–B) The cell viabilities of Hep3B and SNU-182 cells with Sorafenib or transfected with siCDKN2B for 48 hours were detected by CCK-8 assay. (C-F) Flow cytometry was applied to detect apoptosis in Hep3B and SNU-182 cells in the control group, siCDKN2B group, siNC+So group, and siCDKN2B+So group. (G–J) Flow cytometry was used to test cell cycle in Hep3B and SNU-182 cells in the four groups.*p<0.05, **p<0.01 versus siNC group; #p<0.05, ##p<0.01 versus siCDKN2B group; ^^p<0.01 versus siNC+So group. |
To further explore the effects of low-expression of CDKN2B on sensitivity of Hep3B and SNU-182 cells to Sorafenib, flow cytometry was used to detect apoptosis and cell cycle in the four groups. The results showed that the apoptosis rate in the siCDKN2B+So group was lower than that in the siNC+So group (Figures 6C–F), and that the proportion in siCDKN2B+So group cells in the G1 phase was also lower than that in the siNC+So group (Figures 6G–J). This suggested that decreased expression of CDKN2B inhibited the effects of Sorafenib on cell apoptosis.
Effects of low-expression of CDKN2B on the sensitivity to sorafenib
To investigate the relation between the low expression level of CDKN2B gene and the sensitivity of HCC to Sorafenib, the expression level of the CDKN2B gene was determined in the four groups. This shows that the levels of CDKN2B mRNA and protein in the siCDKN2B+So group were lower than those in the siNC+So group (Figures 7A–D). This suggested that Sorafenib could induce elevated levels of CDKN2B mRNA and protein, whereas silencing CDKN2B reversed CDKN2B overexpression caused by Sorafenib.
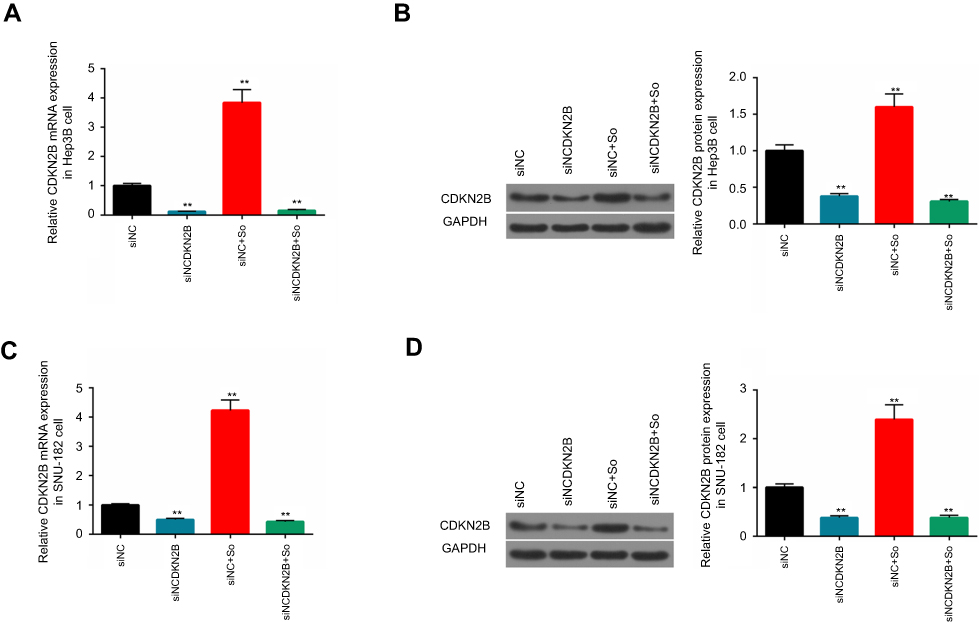 |
Figure 7 CDKN2B mRNA and protein expression levels in the four groups. (A–D) CDKN2B mRNA and protein in Hep3B and SNU-182 cells in the control group, siCDKN2B group, siNC+So group, and siCDKN2B+So group were detected by RT-qPCR and Western blot, respectively. **p<0.01 versus siNC group. |
Discussion
Studies have shown that the incidence of HCC is increasing. Although it may be related to hepatitis virus infection and alcohol intake, the cause of HCC is still unknown.22 For advanced HCC, Sorafenib is the first-line standard treatment program, but the remission rate is low, patients benefit from it only for a short period of time, and it is highly susceptible to drug resistance.23,24 Long-term exposure to anti-neoplastic drugs can lead to decreased sensitivity of HCC cells to drugs.24,25 The mechanism of drug resistance in HCC has not yet been widely studied. Previous studies have suggested that several mechanisms of Sorafenib acquired drug resistance including cross-reactions of the PI3K/Akt and JAK-STAT pathways, hypoxia-inducible pathways, and epithelial-mesenchymal transitions.26–29 There are few studies conducted on the mechanism of the above pathways or transformation changes. Therefore, it is of great significance to find new approaches to increasing the sensitivity of HCC to Sorafenib.
CDKN2B is a tumor suppressor gene, encoded p15INK4B protein, which can inhibit the tumor via Rb pathway.30 Studies have found that CDKN2B gene-deleted mice could cause pancreatic cancer in recent years.31 At the same time, a new study found that the CDKN2B gene may be associated with HCC resistance.21 It has also been found that the copy number loss of CDKN2B gene was associated with poor overall survival in pulmonary squamous cell carcinoma.32 To explore the relation between CDKN2B gene and the efficacy of Sorafenib on HCC, we compared the expression of CDKN2B in HCC tumor tissues of different efficacy patients treated with Sorafenib. SP staining results showed that CDKN2B was negative in PD patients. RT-qPCR experiments further found that, in 55 patients with HCC, 36 patients had low expressions of CDKN2B in hepatocellular carcinoma, and all PD patients had low expressions. This suggests that decreased expression of CDKN2B was associated with poor prognosis. Further comparisons also showed that the survival rate of patients with low CDKN2B expression was lower than that with high expression of CDKN2B. There was no statistical difference in survival between the two groups, which might be explained by our small sample size. However, the survival rates of the two groups were still different. This suggested that the expression level of CDKN2B affected the efficacy of Sorafenib in the treatment of HCC. In future studies, we will extend the sample size to further clarify the association between CDKN2B expression and patient prognosis.
To further investigate the effect of CDKN2B expression on sensitivity of HCC to Sorafenib, we examined the effects of Sorafenib on apoptosis and cell cycle of Hep3B and SNU-182 cells under different levels of CDKN2B expression. The results showed that Sorafenib up-regulated CDKN2B levels, and that CDKN2B overexpression promoted apoptosis and arrested cells in G1 phase and elevated expression of CDKN2B promoted the proapoptotic capacity of Sorafenib and increased the proportion of cells in the G1 phase. This suggested that elevated expression of CDKN2B might increase the sensitivity of HCC to Sorafenib. Meanwhile, the results also showed that, after the down-regulation of CDKN2B, the apoptosis rate and the proportion of cells in the G1 phase under Sorafenib treatment were significantly reduced. This suggested that low expression of CDKN2B could reduce the sensitivity of HCC cells to Sorafenib. These results suggest that the decreased sensitivity of HCC cells to Sorafenib may be related to the low expression of CDKN2B.
CDKN2B belongs to the INK4 protein family. The CDKN2B-encoded p15INK4B protein binds to and inhibits cyclin-dependent kinases 4 (CDK4) and cyclin-dependent kinases 6 (CDK6), thereby promoting apoptosis and arresting cells in the G1 phase.33,34 Previous studies showed that, when CDKN2B expression was upregulated, unbound Cyclin D was degraded by the ubiquitin-dependent proteasomal degradation pathway, causing cells to be arrested in the G1 phase.35,36 In addition, CDKN2B allele deletion and hypermethylation of promoters are present in a variety of hematological malignancies, including leukemia, lymphoma, and myelodysplastic syndromes.37 It was found that overexpression of CDKN2B gene promoted apoptosis in cells treated with VER and Doxorubicin, and that the CDKN2B gene was involved in VER-mediated tumor cell apoptosis.21 This suggested that up-regulation of the CDKN2B gene could increase the sensitivity of HCC to Sorafenib and improve the prognosis of Sorafenib treatment of HCC. However, this study has some limitations, for example, the expression of CDKN2B in HCC tissues needs to be further investigated in the absence of Sorafenib treatment. In addition, increasing the detection of IC50 and p-gp expression in cells was also required. Our future studies will explore the effect of CDKN2B on Sorafenib resistance in HCC and the associated signal transduction pathways.
Conclusion
In conclusion, CDKN2B genes were lowly expressed in tumor tissues of HCC patients who were treated with Sorafenib and had a poor prognosis. Up-regulation of CDKN2B expression levels promoted the sensitivity of HCC to Sorafenib and decreased CDKN2B expression reduced sensitivity, suggesting that the efficacy of Sorafenib on HCC could be improved by up-regulating CDKN2B.
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. No animals are involved in this research.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. doi:10.1002/hep.24199 [published Online First: Epub Date].
2. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63(5):844–855. doi:10.1136/gutjnl-2013-306627 [published Online First: Epub Date].
3. Hsueh KC, Lee TY, Kor CT, et al. The role of liver transplantation or resection for patients with early hepatocellular carcinoma. Tumour Biol. 2016;37(3):4193–4201. doi:10.1007/s13277-015-4243-z [published Online First: Epub Date].
4. Giannini EG, Cucchetti A, Erroi V, Garuti F, Odaldi F, Trevisani F. Surveillance for early diagnosis of hepatocellular carcinoma: how best to do it? World J Gastroenterol. 2013;19(47):8808–8821. doi:10.3748/wjg.v19.i47.8808 [published Online First: Epub Date].
5. Song P, Gao J, Inagaki Y, et al. Biomarkers: evaluation of screening for and early diagnosis of hepatocellular carcinoma in Japan and china. Liver Cancer. 2013;2(1):31–39. doi:10.1159/000346220 [published Online First: Epub Date].
6. Hollebecque A, Malka D, Ferte C, Ducreux M, Boige V. Systemic treatment of advanced hepatocellular carcinoma: from disillusions to new horizons. Eur J Cancer. 2015;51(3):327–339. doi:10.1016/j.ejca.2014.12.005 [published Online First: Epub Date].
7. Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016;21(1):5–10. doi:10.1016/j.drudis.2015.07.008 [published Online First: Epub Date].
8. Gauthier A, Ho M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: an update. Hepatol Res. 2013;43(2):147–154. doi:10.1111/j.1872-034X.2012.01113.x [published Online First: Epub Date].
9. Cervello M, Bachvarov D, Lampiasi N, et al. Molecular mechanisms of sorafenib action in liver cancer cells. Cell Cycle. 2012;11(15):2843–2855. doi:10.4161/cc.21193 [published Online First: Epub Date].
10. Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66(24):11851–11858. doi:10.1158/0008-5472.can-06-1377 [published Online First: Epub Date].
11. Ulivi P, Arienti C, Amadori D, et al. Role of RAF/MEK/ERK pathway, p-STAT-3 and Mcl-1 in sorafenib activity in human pancreatic cancer cell lines. J Cell Physiol. 2009;220(1):214–221. doi:10.1002/jcp.21753 [published Online First: Epub Date].
12. Wilhelm A, Aldridge V, Haldar D, et al. CD248/endosialin critically regulates hepatic stellate cell proliferation during chronic liver injury via a PDGF-regulated mechanism. Gut. 2016;65(7):1175–1185. doi:10.1136/gutjnl-2014-308325 [published Online First: Epub Date].
13. Nakano M, Tanaka M, Kuromatsu R, et al. Sorafenib for the treatment of advanced hepatocellular carcinoma with extrahepatic metastasis: a prospective multicenter cohort study. Cancer Med. 2015;4(12):1836–1843. doi:10.1002/cam4.548 [published Online First: Epub Date].
14. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. doi:10.1016/s0140-6736(11)61347-0 [published Online First: Epub Date].
15. Kyrochristos ID, Glantzounis GK, Ziogas DE, et al. From clinical standards to translating next-generation sequencing research into patient care improvement for hepatobiliary and pancreatic cancers. Int J Mol Sci. 2017;18(1). doi:10.3390/ijms18010180 [published Online First: Epub Date].
16. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.cd-12-0095 [published Online First: Epub Date].
17. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088 [published Online First: Epub Date].
18. McNeal AS, Liu K, Nakhate V, et al. CDKN2B loss promotes progression from benign melanocytic nevus to melanoma. Cancer Discov. 2015;5(10):1072–1085. doi:10.1158/2159-8290.cd-15-0196 [published Online First: Epub Date].
19. Jafri M, Wake NC, Ascher DB, et al. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell carcinoma. Cancer Discov. 2015;5(7):723–729. doi:10.1158/2159-8290.cd-14-1096 [published Online First: Epub Date].
20. Taylor-Harding B, Aspuria PJ, Agadjanian H, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget. 2015;6(2):696–714. doi:10.18632/oncotarget.2673 [published Online First: Epub Date].
21. Zhang T, Ma K, Huang J, et al. CDKN2B is critical for verapamil-mediated reversal of doxorubicin resistance in hepatocellular carcinoma. Oncotarget. 2017;8(66):110052–110063. doi:10.18632/oncotarget.22123 [published Online First: Epub Date].
22. Yuan JM, Govindarajan S, Arakawa K, Yu MC. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer. 2004;101(5):1009–1017. doi:10.1002/cncr.20427 published Online First: Epub Date].
23. Kim HY, Park JW, Nam BH, et al. Survival of patients with advanced hepatocellular carcinoma: sorafenib versus other treatments. J Gastroenterol Hepatol. 2011;26(11):1612–1618. doi:10.1111/j.1440-1746.2011.06751.x [published Online First: Epub Date].
24. Ogasawara S, Chiba T, Ooka Y, et al. Post-progression survival in patients with advanced hepatocellular carcinoma resistant to sorafenib. Invest New Drugs. 2016;34(2):255–260. doi:10.1007/s10637-016-0323-1 [published Online First: Epub Date].
25. Chow AK, Ng L, Lam CS, et al. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS One. 2013;8(11):e78675. doi:10.1371/journal.pone.0078675 [published Online First: Epub Date].
26. Chen J, Jin R, Zhao J, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015;367(1):1–11. doi:10.1016/j.canlet.2015.06.019 [published Online First: Epub Date].
27. Chen W, Wu J, Shi H, et al. Hepatic stellate cell coculture enables sorafenib resistance in Huh7 cells through HGF/c-Met/Akt and Jak2/Stat3 pathways. Biomed Res Int. 2014;2014:764981. doi:10.1155/2014/764981 [published Online First: Epub Date].
28. Zhang PF, Li KS, Shen YH, et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016;7:e2201. doi:10.1038/cddis.2015.324 [published Online First: Epub Date].
29. Liu F, Dong X, Lv H, et al. Targeting hypoxia-inducible factor-2alpha enhances sorafenib antitumor activity via beta-catenin/C-Myc-dependent pathways in hepatocellular carcinoma. Oncol Lett. 2015;10(2):778–784. doi:10.3892/ol.2015.3315 [published Online First: Epub Date].
30. Roussel MF. The INK4 family of cell cycle inhibitors in cancer. Oncogene. 1999;18(38):5311–5317. doi:10.1038/sj.onc.1202998 [published Online First: Epub Date].
31. Tu Q, Hao J, Zhou X, et al. CDKN2B deletion is essential for pancreatic cancer development instead of unmeaningful co-deletion due to juxtaposition to CDKN2A. Oncogene. 2018;37(1):128–138. doi:10.1038/onc.2017.316 [published Online First: Epub Date].
32. Zhao Y, Li Y, Lu H, Chen J, Zhang Z, Zhu ZZ. Association of copy number loss of CDKN2B and PTCH1 with poor overall survival in patients with pulmonary squamous cell carcinoma. Clin Lung Cancer. 2011;12(5):328–334. doi:10.1016/j.cllc.2011.02.007 [published Online First: Epub Date].
33. Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15(5):2672–2681.
34. Xia DY, Liu L, Hao MW, Liu Q, Chen RA, Liang YM. A combination of STI571 and BCR-ABL1 siRNA with overexpressed p15INK4B induced enhanced proliferation inhibition and apoptosis in chronic myeloid leukemia. Braz J Med Biol Res. 2014;47(12):1096–1101.
35. Elisei R, Shiohara M, Koeffler HP, Fagin JA. Genetic and epigenetic alterations of the cyclin-dependent kinase inhibitors p15INK4b and p16INK4a in human thyroid carcinoma cell lines and primary thyroid carcinomas. Cancer. 1998;83(10):2185–2193.
36. Campa D, Pastore M, Gentiluomo M, et al. Functional single nucleotide polymorphisms within the cyclin-dependent kinase inhibitor 2A/2B region affect pancreatic cancer risk. Oncotarget. 2016;7(35):57011–57020. doi:10.18632/oncotarget.10935 [published Online First: Epub Date].
37. Girgis AH, Iakovlev VV, Beheshti B, et al. Multilevel whole-genome analysis reveals candidate biomarkers in clear cell renal cell carcinoma. Cancer Res. 2012;72(20):5273–5284. doi:10.1158/0008-5472.can-12-0656 [published Online First: Epub Date].
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.